

Abstract
Naturally occurring smallpox has been eradicated but remains a considerable threat as a biowarfare/bioterrorist weapon (F. Fleck, Bull. World Health Organ. 81:917–918, 2003). While effective, the smallpox vaccine is currently not recommended for routine use in the general public due to safety concerns (http://www.bt.cdc.gov/agent/smallpox/vaccination). Safe and effective countermeasures, particularly those effective after exposure to smallpox, are needed. Currently, SIGA Technologies is developing the small-molecule oral drug, tecovirimat (previously known as ST-246), as a postexposure therapeutic treatment of orthopoxvirus disease, including smallpox. Tecovirimat has been shown to be efficacious in preventing lethal orthopoxviral disease in numerous animal models (G. Yang, D. C. Pevear, M. H. Davies, M. S. Collett, T. Bailey, et al., J. Virol. 79:13139–13149, 2005; D. C. Quenelle, R. M. Buller, S. Parker, K. A. Keith, D. E. Hruby, et al., Antimicrob. Agents Chemother., 51:689–695, 2007; E. Sbrana, R. Jordan, D. E. Hruby, R. I. Mateo, S. Y. Xiao, et al., Am. J. Trop. Med. Hyg. 76:768–773, 2007). Furthermore, in clinical trials thus far, the drug appears to be safe, with a good pharmacokinetic profile. In this study, the efficacy of tecovirimat was evaluated in both a prelesional and postlesional setting in nonhuman primates challenged intravenously with 1 × 108 PFU of Variola virus (VARV; the causative agent of smallpox), a model for smallpox disease in humans. Following challenge, 50% of placebo-treated controls succumbed to infection, while all tecovirimat-treated animals survived regardless of whether treatment was started at 2 or 4 days postinfection. In addition, tecovirimat treatment resulted in dramatic reductions in dermal lesion counts, oropharyngeal virus shedding, and viral DNA circulating in the blood. Although clinical disease was evident in tecovirimat-treated animals, it was generally very mild and appeared to resolve earlier than in placebo-treated controls that survived infection. Tecovirimat appears to be an effective smallpox therapeutic in nonhuman primates, suggesting that it is reasonably likely to provide therapeutic benefit in smallpox-infected humans.
INTRODUCTION
Smallpox is a contagious disease caused by Variola virus (VARV), which belongs to the family Poxviridae, the subfamily Chordopoxvirinae, and the genus Orthopoxvirus. Humans are believed to be the only reservoirs of VARV. Until its global eradication in 1980, due to an aggressive global surveillance, vaccination, and containment campaign conducted by the World Health Organization (WHO) from 1966 to 1977, smallpox was endemic in 31 countries in the 20th century. As recently as 1967, WHO estimated that 10 to 15 million people contracted smallpox yearly, and more than 2 million died of smallpox each year (1). Although mortality rates varied between outbreaks and by disease type and are dependent on age, the overall mortality rate is estimated at 30% for unvaccinated individuals (2). Survivors may be left with long-term side effects, such as disfiguring skin scars from pockmarks, blindness from corneal infections, arthritis from infection of the metaphysis of growing bones, and infertility in males. It is believed that previous exposure provides lifelong immunity to the disease (3, 4). The highest rates of morbidity and mortality occur in young children, elderly individuals, pregnant women, and seemingly immunocompromised individuals (5).
Currently, known VARV stocks are held only in two high-security, maximum-containment WHO reference laboratories. Although it is extinct in nature, smallpox is still feared as a potentially catastrophic epidemic disease due to the potential for a deliberate release of VARV as an act of war or bioterrorism (6, 7). Based on a Material Threat Assessment by the U.S. Department of Homeland Security, smallpox poses a serious risk to the security of the United States, as it may be used as a biological weapon. In addition, other zoonotic orthopoxviruses, such as monkeypox and cowpox, occurring in nature, intermittently infect human populations and pose serious health threats.
The U.S. smallpox response plan (8–12), developed by the U.S. Centers for Disease Control and Prevention, would be implemented in the event of an outbreak. Currently, the plan primarily seeks to contain an outbreak using surveillance, containment, and vaccination since there is no FDA-approved effective postexposure treatment for smallpox. Confirmed and suspected cases of smallpox would be isolated, and contacts of the infected would be traced and vaccinated, followed by vaccination of contacts of contacts in what is referred to as a “ring vaccination” strategy. Historically, familial contacts were at greatest risk of disease acquisition. The vaccine was greater than 95% effective against smallpox (13) if administered prophylactically, and there is limited evidence that it provides postexposure protection from major morbidity and death if administered to asymptomatic individuals within 4 days of infection (13). In order to ensure the welfare and security of the general population that is largely unvaccinated and immunologically naive, an effective therapeutic, capable of treating disease after the onset of clinical symptoms, is urgently needed.
Tecovirimat (previously known as ST-246) was identified via a high-throughput screen of a chemically diverse library consisting of >350,000 unique compounds. Tecovirimat was found to be specific for orthopoxviruses, including vaccinia, cowpox, ectromelia, rabbitpox, and the clinically relevant monkeypox and VARV, which cause serious human disease, without evidence of any activity against other DNA and RNA viruses against which it was tested (14). Tecovirimat's mechanism of action is distinct from that of cidofovir, a nucleoside analog that inhibits viral DNA replication, as it is fully active against cidofovir-resistant cowpox virus. The target of tecovirimat is a highly conserved virally encoded protein (commonly referred to as p37) present in all orthopoxviruses, yet with no mammalian homolog. The role of p37 in the orthopoxvirus replication cycle is to mediate, in concert with other viral and cellular proteins, the formation of enveloped virions (EV), which are egress competent and facilitate viral release from the infected cell (15, 16) and dissemination within the host (7, 17). Mutant viruses defective for EV production are avirulent in vivo (16, 18–20). Considering the role of p37 in the formation of EV, and the significance of EV in orthopoxvirus virulence, it represents a viable target for drugs capable of treating smallpox or other pathogenic orthopoxvirus diseases.
It would be neither feasible nor ethical to conduct clinical trials to evaluate the efficacy of tecovirimat against smallpox in humans. Other orthopoxviruses, such as vaccinia or cowpox, which occasionally infect humans, are not appropriate as surrogates for smallpox considering that they are much less pathogenic than smallpox. Furthermore, the mechanism of smallpox virulence is not fully understood, making it very difficult to assert the relevance of these surrogates.
In order to develop medical countermeasures for those agents that cannot be tested in humans, the FDA Animal Rule was enacted (see 21 Code of Federal Regulations [CFR] 314.600 for drugs or 21 CFR 601.90 for biological products; online at http://www.gpo.gov/fdsys/pkg/FR-2002-05-31/html/02-13583.htm). The Animal Rule provides a mechanism by which the FDA may approve drugs (or vaccines) based on efficacy data from animal studies coupled with safety and pharmacokinetic data from human trials. Animal efficacy data allow the selection of the human dose. If the drug is shown to be safe and plasma exposure levels in humans are comparable to those with efficacious dosing in animals, then one may reasonably conclude that the drug will be efficacious in humans.
For most new therapeutics against select agents such as VARV, protective efficacy must be demonstrated following a lethal challenge. In cynomolgus macaques, an intravenous (i.v.) challenge dose of 1 × 109 PFU of VARV (Harper strain) results in 100% lethality, although progression of disease does not mimic typical smallpox disease in humans (21). This “lethal model” for human smallpox more closely recapitulates the hemorrhagic form of the disease, which is rare in humans (<3%) and has almost a 100% mortality rate. In the lethal model, also referred to as the hemorrhagic model, infected animals most often die within 3 to 6 days postchallenge and develop ordinary-type smallpox lesions only in the rare event they survive past day 7 postexposure. Animals dying within 3 to 6 days may develop petechial rash and mucosal lesions that do not fully develop into typical smallpox lesions prior to death. Clinically and pathologically, numerous features of hemorrhagic disease are present, but that feature may not be entirely dependent on VARV infection, as secondary bacterial infection also appears to contribute to outcome (22). Additional studies to explore the impact of challenge dose on outcome resulted in the development of a disease model referred to as the “ordinary” or “lesional model” for human smallpox (21). Following infection with1 × 108 PFU VARV, animals experience limited viral replication but consistently develop numerous lesions distributed centrifugally, as is typical of smallpox in humans. Focal lesions first become apparent between days 3 and 5 postinfection and progress to affect most skin surfaces, peaking in severity and number between days 7 and 11. Lesions progress through stages of development and resolution that are typical for human smallpox.
Intravenous challenge with 1 × 108 PFU VARV is not uniformly or reproducibly lethal. In a study conducted by Jahrling et al., mortality following IV challenge with 1 × 108 PFU VARV (Harper strain) was reported to be 33% (21). Therefore, survival as a primary endpoint for antiviral efficacy studies in this model is not feasible due to capacity limitations in biosafety level 4 (BSL-4) animal facilities approved to work with VARV. Fisher exact power calculations (data not shown) estimate that treatment groups containing 20 or more animals would be necessary to demonstrate a significant impact on survival (Fisher exact P value of <0.05, ≥90% power) based on estimations of 30% mortality in untreated animals, while much smaller group sizes (6 animals) would be necessary to demonstrate significant impact on lesion number and viremia. Thus, in this study, the lesional model was used to evaluate primarily the impact of tecovirimat treatment on lesion formation and secondarily its impact on viremia, virus shedding, and survival.
The objective of this double-blind, randomized, placebo-controlled, repeat-dose study was to determine the protective efficacy of orally administered tecovirimat against intravenous challenge with VARV in cynomolgus macaques. Tecovirimat treatment was delayed until day 2 or day 4 postchallenge to determine efficacy in a prelesional and postlesional disease setting.
MATERIALS AND METHODS
Ethical treatment of animals.
The study protocol was approved by the CDC Institutional Animal Care and Use Committee. Treatment of animals adhered to U.S. Government “Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Education,” the Guide for the Care and Use of Laboratory Animals, the Animal Welfare Act, and other applicable public laws and regulations.
Animals.
Eighteen male cynomolgus monkeys (Macaca fascicularis, strain Mauritius) were obtained from the USAMRIID colony, shipped to the CDC, quarantined, and acclimated to the animal biosafety level 4 (ABSL-4) animal facility 7 days prior to the start of the study. The animals averaged 5 years of age (range, 2.9 to 6.5 years) and 5.9 kg (range, 4.8 to 7.1 kg) at the start of the study. The animals were housed individually in standard stainless steel enclosures. The animal room temperature was monitored for targeted conditions: temperature of 64 to 84°F, humidity of 30 to 70%, and a photoperiod of 12 h of light and 12 h of dark, for which no deviations were observed. Animals received an appropriate diet of certified feed and tap water ad libitum. Study personnel provided biscuits and fruit to each animal after each daily physical/observation during the study. Food consumption was documented throughout the study. PRANG (oral rehydration drink) was offered ad libitum to all animals at the onset of treatment and continued until animals recovered, died, or were euthanized at the end of the study. CDC Animal Resources Division personnel observed animals a minimum of once daily for husbandry conditions and humane treatment. Prior to inclusion in the study, animals were tested for antibodies to vaccinia and monkeypox, as well as simian immunodeficiency virus (SIV), simian T-cell leukemia virus (STLV), and simian retrovirus, and all were found to be negative. All animal work was conducted at the Centers for Disease Control and Prevention in Atlanta, GA.
Virus preparation and challenge.
All virus manipulations were performed in the BSL-4 facility at the CDC. VARV Harper strain was cultured as previously described (21). Briefly, VARV was initially passed in chorioallantoic membranes followed by three passages in BSC40 cells. Infected cells and culture supernatant were collected after displaying >95% virus-induced cytopathic effect. The cells were lysed by multiple freeze-thaw cycles, after which the cellular debris was pelleted by low-speed centrifugation and the resulting supernatant was aliquoted and stored in liquid nitrogen in the BSL-4 facility at the CDC. For infection of animals, virus was diluted to 1 × 108 PFU/ml in Eagle's minimal essential medium (EMEM) and 1% fetal bovine serum (FBS) and administered as a single injection via the femoral or saphenous vein using a 21- to 25-gauge needle or butterfly, followed by a flush of 1 ml of normal saline.
Tecovirimat/control preparation and administration.
Tecovirimat consists of the active ingredient 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl-benzamide formulated as a 3.3-mg/ml liquid suspension in (vehicle) 1% (wt/vol) hydroxypropyl methylcellulose with 0.5% (wt/vol) Tween 80 in sterile water for injection. Placebo-treated animals were treated with vehicle plus 4.0% (wt/vol) Avicel in sterile water for injection in order to provide a similar appearance between tecovirimat and the placebo treatment to maintain blinding. For tecovirimat and placebo administration, animals were anesthetized with tiletamine-zolazepam (Telazol). Based on body weights acquired 1 day prior to exposure (day −1), animals were administered either 10 mg tecovirimat/kg of body weight or an equal volume/weight dose of placebo by oral gavage, followed by 5 ± 0.5 ml/kg of a 30% suspension of hydrated homogenized monkey biscuits, via a 7-10 French (2.3 to 3.3 mm diameter) red rubber feeding tube through the oral cavity. The hydrated monkey chow is provided to ensure that the tecovirimat is administered in a “fed” state to facilitate optimal absorption. Six animals were randomly assigned to each of the three treatment groups. The control group received placebo treatment daily from days 2 through 17. The group treated with tecovirimat from days 2 through 15 received two doses of placebo on days 16 and 17, while the group treated with tecovirimat from days 4 through 17 received two doses of placebo on days 2 and 3 prior to starting tecovirimat treatment. This was done to maintain study blindness.
Anesthesia.
All animals were anesthetized with 50 mg of tiletamine HCl and 50 mg of zolazepam per ml prior to handling. Anesthesia was used for VARV challenge, oral gavage, physicals, weights, temperature, phlebotomy, and lesion counting. Up to 3 mg/kg (0.03 ml of a 100-mg/ml solution) was injected intramuscularly (i.m.) in the caudal thigh muscle using a 21- to 27-gauge, 3/8- to 1-inch needle and 1-ml tuberculin syringe. The baseline weight established prior to any study procedures (day −3) was used to calculate drug dosage. Food was not provided in the morning until after completion of any scheduled anesthesia event. Following anesthesia, the principal investigator or the study personnel closely observed animals until they were fully recovered to an upright conscious position.
Clinical evaluations.
Study personnel performed physicals according to the study schedule, i.e., daily from day −1 to day 28. Monkeys were anesthetized with tiletamine-zolazepam prior to being weighed. Rectal temperature was taken in conjunction with body weight. Other observations were performed at least once daily and included, but were not limited to, monitoring of recumbency, dehydration, dyspnea, cough, eating, nasal discharge, ocular discharge, and edema. Each of these is recorded according to the severity of the observation ranking from 0 (absent) to 1 (mild), 2 (moderate), and 3 (severe). Scored observations are summed per nonhuman primate (NHP) on each day in order to generate a clinical score (see Fig. S1 in the supplemental material). Clinical scores were not a factor in determining the need for humane euthanasia for those animals experiencing severe disease (see euthanasia criteria below).
Lesion counts.
Characteristic lesions were counted once daily from day 0 through study termination at day 28, with the exception that counts were not performed on day 1 postchallenge (prior to any lesions forming) and on day 18 postchallenge. Dermal pox lesions can be broadly grouped into four types by lesion development stage: rash initiating (macules/papules/vesicles), ulcerating (pustules/umbilicated), resolving (scabbing), and healing (desquamating). For an assessment day, the total lesion count is the sum of all lesions regardless of stage across all regions of the body (head, chest, back, tail, arms, and legs). For some animals with lesions considered too numerous to count for a particular anatomical region, lesion counts for that region were estimated based on the percentage of the skin covered by lesions.
Euthanasia.
Animals were euthanized by exsanguination while under deep anesthesia either when moribund or at scheduled euthanasia at study termination. Tiletamine-zolazepam was administered at 9 mg/kg to achieve deep anesthesia, and then the NHP was exsanguinated by removal of greater than 30% of blood volume (>18 ml/kg of blood) via a peripheral blood vessel or cardiac puncture. Death was confirmed by loss of the corneal reflex.
Whenever possible, moribund animals were euthanized before succumbing on their own to reduce suffering and distress. For analysis purposes, euthanized animals were counted as “death due to severe smallpox disease” and were handled for analysis in the same fashion as those animals found dead. In order to provide a uniform and reproducible set of criteria for determining that an animal is moribund and euthanasia was warranted for humane reasons, the following criteria were used: (i) persistent prostration (for a period of 4 h or longer) and unresponsive to gentle prodding through the bottom of the cage or (ii) persistent prostration (for a period of 4 h or longer) but responsive to a gentle prodding through the bottom of the cage and having a rectal temperature of ≤34°C. These criteria did not preclude the PI, the attending veterinarian, or the veterinary technician from performing humane euthanasia for other unforeseen complications from smallpox disease. Animals that survived to day 28 were euthanized to allow for the evaluation of any possible treatment-induced sequelae. For analysis, these animals were considered surviving.
Blood collection for hematology, clinical chemistries, and evaluation of viral load.
Blood was collected on day −3, postinfection days 0, 2, and every subsequent 3rd day, and at the time of euthanasia for assessment of hematology, clinical chemistries, and viremia (real-time quantitative PCR [qPCR]).
(i) Hematology.
For hematological analysis, blood was collected from femoral or saphenous veins or other peripheral available blood vessels as appropriate, using a 21- to 25-gauge needle or butterfly. Drawn blood volumes did not exceed maximum volumes allowed by the animal care and use guidelines. The following parameters were measured: white blood cells (WBC), red blood cells (RBC), hemoglobin (Hgb), hematocrit (Hct), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), and platelet count (PLT).
(ii) Serum chemistry.
The following parameters were measured from the daily blood samples taken for serum chemistry: albumin (ALB), alkaline phosphatase (ALP), creatinine (CRE), alanine aminotransferase (ALT), aspartate aminotransferase (AST), glucose (GLU), amylase (AMY), total bilirubin (TBIL), blood urea nitrogen (BUN), total protein (TP), calcium (CA), gamma-glutamyl transpeptidase (GGT), and uric acid (UA).
(iii) Viral load.
Viral loads were measured from blood samples (collected in Li+ heparin tubes) collected every third day starting prior to infection, with one sample taken immediately following infection to evaluate if infection was successful. DNA was quantitated using a real-time qPCR method for detection of orthopoxvirus genomes in peripheral blood as previously described with adaptions by Huggins et al. (23). DNA was extracted from fresh or frozen blood samples using the QIAamp DNA blood minikit from Qiagen (Valencia, CA), and qPCR analysis was performed using the Roche Light cycler.
Throat swabs.
Throat swabs of the oropharynx were obtained on days 0, 2, and 4 and every subsequent 3rd day for animals in each of the three treatment groups. Virus was eluted from swabs, and throat swab titers (PFU/ml) were measured by standard plaque assay on VERO E6 cells.
Statistical methods.
SAS versions 9.1 and 9.3 were utilized to perform the statistical analyses of efficacy and related measures. The proportions of animals that survived through day 28 were compared for each pair of the three treatment groups using the Fisher exact test (2-sided). Kaplan-Meier (K-M) survival analysis was performed to analyze the time in days from infection day (day 0) to the day of either death or euthanasia due to moribund condition; the tecovirimat treatment groups combined were compared with the placebo group, and each pair of the three groups was compared using the log rank test. Lesion counts observed over the treatment period were analyzed using an analysis of covariance (ANCOVA) model, which included treatment (the effect of interest) as the effect and the animal's body weight at baseline as the covariate. Pairwise comparisons of the lesion counts were conducted for the three treatment groups using a model-based t test procedure. Missing daily lesion counts over the treatment period due to inability to collect data on a particular animal or animal death were imputed with the last observation carried forward (LOCF). The average of the log10 viral DNA levels over the treatment period and daily change from day 2 in log10 viral DNA levels at each posttreatment assessment time point through day 28, as well as the maximum value of log10 viral DNA levels observed from days 2 through 28 (inclusive), were analyzed using an ANCOVA model and the procedure of the pairwise comparisons described for lesion counts. The ANCOVA model included the three treatment groups and the log10 viral load of day 2 as the covariate. Median throat swab titer by day was calculated for each of the three treatment groups, and pairwise comparisons of the daily titers were conducted using the Wilcoxon rank sum test. Additionally, the Wilcoxon signed-rank test was used to examine the difference in titer from day 4 to day 7 within each treatment group. Statistical significance was considered at the level of P values of <0.05.
RESULTS
The objective of this double-blind, randomized, placebo-controlled, repeat-dose study was to determine the protective efficacy of orally administered tecovirimat (10 mg/kg/day) against intravenous (i.v.) challenge with VARV in NHP. Eighteen male cynomolgus macaques were infected by i.v. injection with 1 × 108 PFU VARV and assigned to three treatment groups (Table 1). Tecovirimat treatment was delayed until day 2 or day 4 postchallenge to determine efficacy in a prelesional and postlesional disease setting. The endpoints of the study were to monitor survival, lesion counts, viral load, clinical symptoms of disease, oropharyngeal virus shedding, and hematological/blood chemistry parameters throughout the 28-day monitoring period postchallenge.
Table 1.
Group assignment and treatment of monkeys
| Group | No. of PFU VARV (i.v. administration) at challenge day 0 | Treatment (dose in mg/kg) | Treatment course |
|---|---|---|---|
| Placebo | 1 × 108 | Placebo (0) | Days 2–17, placebo |
| Day 2 tecovirimat | 1 × 108 | Tecovirimat (10) | Days 2–15, tecovirimat; days 16 and 17, placebo |
| Day 4 tecovirimat | 1 × 108 | Tecovirimat (10) | Days 2 and 3, placebo; days 4–17, tecovirimat |
Survival.
Following challenge, three of six animals in the placebo-treated group succumbed to infection on days 13 (1 animal) and 14 (two animals), while all tecovirimat-treated animals survived to study termination at day 28, as shown in Fig. 1. There was no significant difference in survival probability between placebo-treated animals and either of the tecovirimat-treated groups when analyzed separately, although there was a significantly higher probability of survival for tecovirimat-treated animals when both tecovirimat treatment groups were combined (n = 12) than for placebo-treated animals (P = 0.0079). The survival rate was higher for all tecovirimat-treated animals combined than for placebo-treated animals (P = 0.0245).
Fig 1.
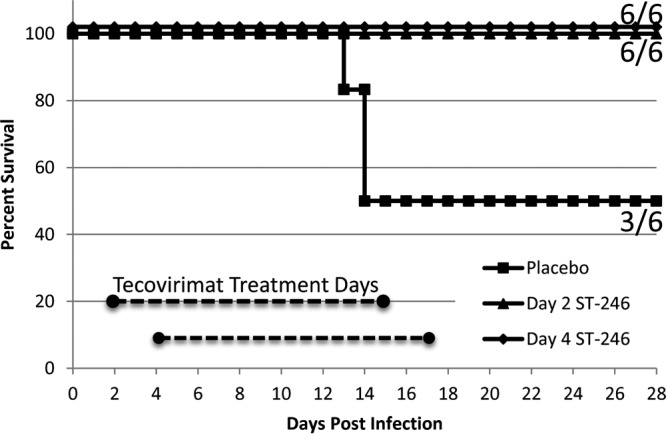
Survival following VARV challenge by treatment group. Cynomolgus macaques were challenged with 1 × 108 PFU VARV by the intravenous route on day 0 and were monitored for survival and signs of disease for 28 days postchallenge. All nonsurviving animals in the placebo group were preemptively humanely euthanized when moribund on days 13 and 14.
Lesion counts.
Lesions were apparent on all animals by day 3 postchallenge, first appearing focally, and then progressing to all body areas prior to death or resolution. The placebo-treated group formed an average of approximately 1,500 lesions over the total body surface area at peak by day 10 (Fig. 2), which declined thereafter as animals succumbed from disease or resolved infection. Tecovirimat treatment resulted in significant reductions in total lesion counts by day 4 (P < 0.02) for the animals beginning tecovirimat treatment on day 2 and by day 6 (P < 0.04) for animals beginning treatment on day 4. Significant differences were maintained between tecovirimat-treated groups and the placebo-treated group until day 22 postchallenge (P < 0.05), at which time survivors in the placebo-treated group began to resolve disease. Closed-test pairwise comparisons demonstrate significant reductions in maximum lesion counts compared to those in the placebo treatment group when tecovirimat treatment was started on day 2 or day 4 postchallenge (P = 0.0004 and 0.003, respectively). The difference in the maximum lesion counts was not significant between the two tecovirimat treatment groups (P = 0.23). Animals that were started on tecovirimat on day 2 postchallenge formed a maximum average of 225 lesions by day 6, which were at the macule/papule/vesicle stage. From this point, the majority of lesions resolved without fully progressing through the typical pustule/umbilicated stage prior to resolution (days 10 to 12). Animals that were started on tecovirimat on day 4 postchallenge formed an average of 440 lesions at peak by day 6 (macule/papule/vesicle), which progressed further to pustules/umbilicated lesions than those of animals that began treatment on day 2 but were much less severe than those of the placebo-treated animals. The majority of lesions were fully resolved by days 13 to 15. Differences between the two tecovirimat treatment groups never achieved statistical significance, although there is an indication that total lesion counts are reduced and that progression to pustules/umbilicated lesions is interrupted by earlier treatment. Animals in the placebo-treated group did not fully resolve their lesions by study termination (day 28), although the majority had progressed to late stages of scabbing and desquamation.
Fig 2.
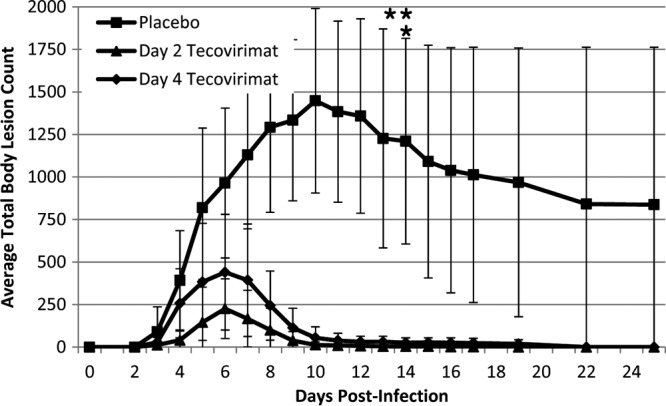
Total body lesion counts following VARV challenge by treatment group. Cynomolgus macaques were challenged with 1 × 108 PFU VARV by the intravenous route on day 0. Lesions were counted daily over the entire body surface area and totaled for 28 days postchallenge. Treatment group averages are shown. For the placebo-treated group, values from days 13 to 22 are imputed (last observation carried forward) for animals that succumbed to disease on days 13 and 14 (indicated by asterisks) prior to study termination. Error bars indicate standard deviations.
Viremia (qPCR of viral DNA in blood).
All animals were challenged successfully as determined by qPCR in blood samples taken within 2 min postchallenge (Fig. 3, day 0 values). All animals also experienced a viral “eclipse,” as shown by the approximately 1-log reduction in viremia from day 0 to day 2. Thereafter, viral load increased in the placebo group, reaching a maximum by day 7 (∼2 × 107 genome copies/ml of blood) and declining to below the limit of quantitation (104 copies/ml of blood) by day 16. The decline may suggest that the moribund animals (deaths on days 13 and 14, well after the peak of viremia) were either clearing virus from the blood at the time of death or possibly that there is a correlation between decreases in viral load and the decline of WBCs (i.e., less virus being circulated via an intracellular mechanism). For animals started on tecovirimat on day 2, the viremia levels did not increase appreciably after day 2 (∼2 × 106 GC/ml), remained steady on day 4, and declined to below the limit of quantitation by day 10. For animals started on tecovirimat on day 4, viremia increased from day 2 to day 4 and reached a maximum on day 7 (∼1 × 107 GC/ml). After day 7, viremia steadily declined to below the limit of quantitation by day 13. In evaluating daily percent change from baseline (i.e., day 2 log10 viral DNA load), tecovirimat treatment significantly impacted the change from baseline viral load after day 2 for both tecovirimat treatment groups in comparison to the placebo group by day 7 (P < 0.04), and the effect remained significant until day 13. Tecovirimat treatment initiated on day 2 resulted in significant reductions in maximum log10 viral load compared to that of the placebo group (P = 0.02), although the average log10 viral load was not significantly reduced. Treatment initiated on day 4 did not significantly impact maximum or average log10 viral load. This is notable considering that treatment initiation on day 4 resulted in significant reductions in lesion counts. This suggests that later treatment initiation may not immediately reduce circulating virus but that the virus is nevertheless prevented from disseminating to the dermis or replicating in the skin. Differences between the two tecovirimat treatment groups when comparing daily percent change from baseline, maximum log10 viral DNA load, or average log10 viral DNA load did not achieve statistical significance, although there is an indication that earlier treatment initiation resulted in earlier clearance.
Fig 3.
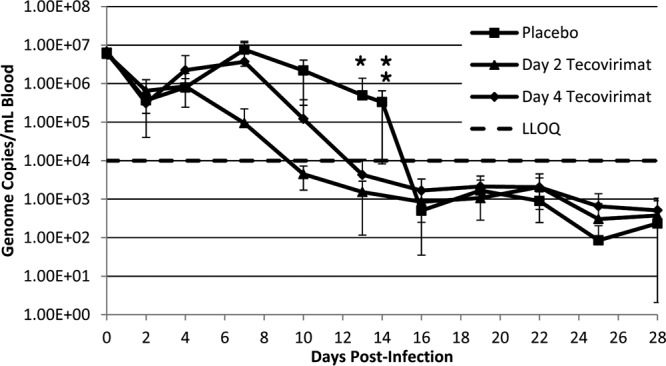
Viremia (qPCR of viral DNA in blood) following VARV challenge by treatment group. Cynomolgus macaques were challenged with 1 × 108 PFU VARV by the intravenous route on day 0. Every third day (and at unscheduled moribund euthanasia; see data point at day 14), DNA from blood samples was evaluated by qPCR for the presence and quantitation of viral genomes. Treatment group averages are shown. Error bars indicate standard deviations. In some instances, negative error is not displayed when the error extends to negative values (less than 0), due to plotting y axis data on a logarithmic scale. Unscheduled deaths (euthanasia due to moribund condition) in the placebo group are indicated by asterisks. Plotted data for the placebo group after day 14 are representative of the three surviving animals in the group. LLOQ, lower limit of quantitation (10,000 GC/ml of blood).
Virus shedding in the oropharynx.
Virus shedding in the oropharynx was first observed on day 4 in placebo-treated and tecovirimat-treated animals (Fig. 4). Compared to each tecovirimat treatment group, the median throat swab titer was higher for the placebo-treated animals on days 7 (both P = 0.002) and 13 (both P = 0.01). Compared to both tecovirimat treatment groups combined, the titer for placebo-treated animals was higher on days 7 (P < 0.0001), 10 (P = 0.008), and 13 (P < 0.0001). No daily titers were significantly different between the two tecovirimat treatment groups. Titers decreased from day 4 to day 7 for animals starting tecovirimat treatment on day 2 (P = 0.03) and for both tecovirimat treatment groups combined (P = 0.004), while no significant difference was found for placebo-treated animals (P = 1.0) or the day 4 tecovirimat treatment group (P = 0.09). Comparison of daily throat swab titers for days 4 to 13 suggested higher throat swab titers in placebo-treated animals that succumbed to illness than in placebo-treated animals that survived, although the results were not statistically significant.
Fig 4.
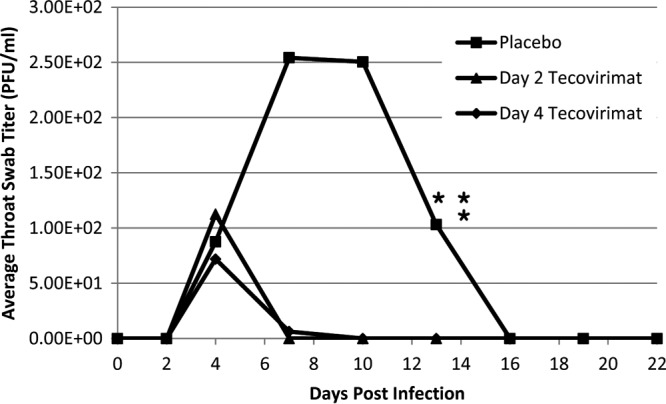
Throat swab plaque assay by treatment group. Cynomolgus macaques were challenged with 1 × 108 PFU VARV by the intravenous route on day 0. Oropharyngeal swabs were taken on days 0, 2, 4, 7, 10, 13, 16, 19, and 22 and evaluated by plaque assay for live virus. Treatment group median values are shown. Wide variability within groups was observed, and error bars are omitted to prevent obscuring the data. Unscheduled deaths (euthanasia due to moribund condition) in the placebo group are indicated by asterisks. No samples were positive after day 13, and day 22 is the last day for which samples were evaluated.
Temperature and weight.
All animals experienced an increase in temperature, peaking on average approximately 2°C above baseline, between days 3 and 5 postinfection, which, for survivors in all groups, returned to normal ranges between days 10 and 13 postinfection (data not shown). Three animals in the placebo-treated group experienced mild to severe (−1° to −7°) hypothermia prior to succumbing to infection on days 13 and 14. Placebo-treated animals experienced a steady decline in weight from day 2 postinfection to day 15, reaching a nadir averaging 10% below group starting weight (range, 3 to 13%). Following the loss of three animals in this group to disease, the surviving animals slowly began recovering weight, although only one animal recovered to its baseline weight (data not shown). Animals in both tecovirimat-treated groups experienced mild weight loss (∼2%), reaching a nadir between days 8 and 13 postinfection, and recovered to their baseline weights between days 16 and 17, without significant differences noted between the two tecovirimat groups.
Clinical observations and disease monitoring.
Daily cage-side observations were recorded to monitor animal health and disease progression. Observations included disease symptoms such as recumbency, unresponsiveness, dyspnea, cough, stool condition, nasal discharge, rash, edema, bleeding, lymphadenopathy, and dehydration. Additionally, appetite was evaluated based on biscuit and fruit consumption. A severity score was assigned to each observation. Clinical disease became evident by day 3 postchallenge for the placebo-treated group (see Fig. S1 in the supplemental material). Disease severity increased dramatically by day 4 for all placebo-treated animals and continued to increase until a maximum severity was observed on day 7. Although the group average showed a decline in disease severity, in fact, the data varied widely between survivors and nonsurvivors in the group. Surviving animals steadily resolved disease following peak severity at day 7, while the nonsurvivors showed increased disease severity, spiking on days 13 and 14, when they were humanely euthanized in moribund condition. Surviving animals in the placebo-treated group generally resolved disease by day 17. For the day 2 tecovirimat treatment group, clinical disease was not evident until day 4, which was the maximum for this group yet lower than the placebo or day 4 tecovirimat treatment groups. After day 4, clinical signs in the day 2 tecovirimat treatment group decreased steadily until fully resolved by day 12 or 13. For the day 4 tecovirimat treatment group, clinical signs were evident by day 3, which increased to their maximum on day 4. On day 5, clinical signs declined and then increased slightly on day 6, followed by steady decrease until full resolution by day 11.
Blood chemistry and hematological parameters.
Most blood chemistry and hematology parameters were not affected by variola disease or treatment (see Fig. S2 and S3 in the supplemental material). Exceptions include anemia for all animals in all groups, evident by day 4 postinfection and remaining low from days 7 through 14, with slow but incomplete recovery by the end of the observation period. Additional observations include transient increased white blood cell counts for most animals in all treatment groups, an initial decrease in platelet count followed by an increase for most animals (notably, the placebo-treated animals that succumbed did not fully recover after the initial decrease), decreased albumin levels for all animals, which was most dramatic for the animals that succumbed in the placebo treatment group, and increased alkaline phosphatase, increased blood urea nitrogen, and decreased total protein only in those animals that succumbed in the placebo treatment group.
DISCUSSION
Smallpox is an eradicated human disease but continues to be of concern because of its biothreat potential. The development of effective antiviral agents has been a U.S. government strategy to prevent morbidity and mortality should disease be reintroduced. Demonstrating efficacy has been a conundrum. It is currently not feasible nor would it be ethical to conduct clinical trials to evaluate the efficacy of tecovirimat against smallpox in humans. Hence, efficacy evaluations have been conducted in animal models, according to the Animal Rule (21 CFR 314.600 and 21 CFR 601.90, http://www.gpo.gov/fdsys/pkg/FR-2002-05-31/html/02-13583.htm). Tecovirimat has been used with other drugs or biologics in four compassionate emergency use protocols, two involving adverse events following smallpox vaccination (24, 25). In all cases, tecovirimat treatment was associated with clearance of the virus and resolution of disease with no adverse events associated with dosing. In addition, tecovirimat has been evaluated clinically for both safety and pharmacokinetics (26–28). In these trials, there was a very low incidence of treatment-emergent adverse events (TEAEs): rarely, headache and mild nausea were associated with tecovirimat treatment. There were no clinically significant results from laboratory assessments, vital sign measurements, physical examinations, or electrocardiograms. Human clinical dosing at 400 to 600 mg/day resulted in blood exposure levels comparable to levels associated with efficacy in monkeys, suggesting that dosing at this level would be effective in treating smallpox disease in humans.
SIGA Technologies has conducted, sponsored, or collaborated on more than 50 studies in animals to evaluate the efficacy and safety of tecovirimat: in mice infected with vaccinia, cowpox, or ectromelia viruses (14, 29–31), in prairie dogs and golden ground squirrels infected with monkeypox virus (32), in rabbits infected with rabbitpox virus (33), and in cynomolgus monkeys infected with monkeypox virus or VARV (23, 34). In each model, an efficacious dose was determined that prevented mortality and prevented or significantly reduced clinical symptoms of disease. Nonhuman primates, particularly cynomolgus macaques infected with monkeypox virus, have served as the most relevant model for human smallpox. The VARV challenge model utilized in the study presented here has the advantage over other models in that it represents challenge with the authentic etiological agent of smallpox. The drawback to the lesional model used in this study is that unlike in the high-dose (109 PFU) lethal model, uniform mortality rates have not been consistently achieved. That being said, the lethal model would be representative of only a minority of smallpox cases, whereas the lesional model (108 PFU) has clinical attributes of typical smallpox. Neither of the VARV challenge models is sufficiently well characterized so as to predict a response in humans, one of the requirements of the Animal Rule. The gaps in efficacy data therefore must come from other animal models, and the results from all animal models must be triangulated in order to assess the impact of tecovirimat treatment on all aspects of orthopoxvirus disease, which in their totality provide evidence that tecovirimat is reasonably likely to be effective in smallpox-afflicted humans.
The results presented here demonstrate the efficacy of tecovirimat in both a prelesional and postlesional setting in an NHP model of VARV disease. Both the 2-day delay and 4-day delay in tecovirimat treatment resulted in significant enhancement of survival and reductions in lesional disease, viremia, and virus shedding in the oropharynx, as well as reduction in the time necessary to resolve disease. While all tecovirimat-treated animals survived, and three of six placebo-treated animals succumbed to disease, the survival advantage was not statistically significant when comparing individual groups due to small group sizes (i.e., 6 animals per group). But when comparing all tecovirimat-treated animals (combining both tecovirimat treatment groups; n = 12) to placebo-treated animals, the survival advantage was shown to be statistically significant. Tecovirimat treatment as pre- or postlesional therapy effectively reduces the burden of disease and improves survival in the lesional NHP model for human smallpox. Observations of the reduction of virus shed in oropharyngeal secretions suggest that use of tecovirimat may also reduce disease transmission.
Taken together, these results are supportive of tecovirimat as an antiviral which can be used to treat smallpox disease in early stages and prevent disease if given in the incubation period.
Supplementary Material
ACKNOWLEDGMENTS
Douglas W. Grosenbach and Dennis E. Hruby are employed by SIGA Technologies. SIGA Technologies holds commercial interest in tecovirimat and provided tecovirimat for the study. Other authors have no conflict of interest.
The work described herein was supported by the Defense Threat Reduction Agency (DTRA). Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army or the Department of Defense.
Footnotes
Published ahead of print 7 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00977-13.
REFERENCES
- 1. Barquet N, Domingo P. 1997. Smallpox: the triumph over the most terrible of the ministers of death. Ann. Intern. Med. 127:635–642 [DOI] [PubMed] [Google Scholar]
- 2. Fenner F, Henderson DA, Arita I, Jezek Z, Ladnyi ID. 1988. Smallpox and its eradication. WHO, Geneva, Switzerland [Google Scholar]
- 3. Breman JG, Henderson DA. 2002. Diagnosis and management of smallpox. N. Engl. J. Med. 346:1300–1308 [DOI] [PubMed] [Google Scholar]
- 4. Phadke AM, Samant NR, Dewal SD. 1973. Smallpox as an etiologic factor in male infertility. Fertil. Steril. 24:802–804 [DOI] [PubMed] [Google Scholar]
- 5. Fulginiti VA, Papier A, Lane JM, Neff JM, Henderson DA. 2003. Smallpox vaccination: a review, part II. Adverse events. Clin. Infect. Dis. 37:251–271 [DOI] [PubMed] [Google Scholar]
- 6. Henderson DA. 1999. The looming threat of bioterrorism. Science 283:1279–1282 [DOI] [PubMed] [Google Scholar]
- 7. Smith GL, Vanderplasschen A, Law M. 2002. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 83:2915–2931 [DOI] [PubMed] [Google Scholar]
- 8. Kretzschmar M, van den Hof S, Wallinga J, van Wijngaarden J. 2004. Ring vaccination and smallpox control. Emerg. Infect. Dis. 10:832–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaplan EH. 2004. Preventing second-generation infections in a smallpox bioterror attack. Epidemiology 15:264–270 [DOI] [PubMed] [Google Scholar]
- 10. Legrand J, Viboud C, Boelle PY, Valleron AJ, Flahault A. 2004. Modelling responses to a smallpox epidemic taking into account uncertainty. Epidemiol. Infect. 132:19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaplan EH, Craft DL, Wein LM. 2002. Emergency response to a smallpox attack: the case for mass vaccination. Proc. Natl. Acad. Sci. U. S. A. 99:10935–10940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meltzer MI, Damon I, LeDuc JW, Millar JD. 2001. Modeling potential responses to smallpox as a bioterrorist weapon. Emerg. Infect. Dis. 7:959–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fenner F, Henderson D, Arita I, Jezek Z, Ladnyi I. 1988. The pathogenesis, pathology, and immunology of smallpox and vaccinia, p 122–167 In Smallpox and its eradication. World Health Organization, Geneva, Switzerland [Google Scholar]
- 14. Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, Barone L, Burns C, Rhodes G, Tohan S, Huggins JW, Baker RO, Buller RL, Touchette E, Waller K, Schriewer J, Neyts J, DeClercq E, Jones K, Hruby D, Jordan R. 2005. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus challenge. J. Virol. 79:13139–13149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen Y, Honeychurch KM, Yang G, Byrd CM, Harver C, Hruby DE, Jordan R. 2009. Vaccinia virus p37 interacts with host proteins associated with LE-derived transport vesicle biogenesis. Virol. J. 6:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blasco R, Moss B. 1991. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-dalton outer envelope protein. J. Virol. 65:5910–5920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Payne LG. 1980. Significance of extracellular enveloped virus in the in vitro and in vivo dissemination of vaccinia. J. Gen. Virol. 50:89–100 [DOI] [PubMed] [Google Scholar]
- 18. Gurt I, Abdalrhman I, Katz E. 2005. Pathogenicity and immunogenicity in mice of vaccinia viruses mutated in the viral envelope proteins A33R and B5R. Antiviral Res. 69:158–164 [DOI] [PubMed] [Google Scholar]
- 19. McIntosh AA, Smith GL. 1996. Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J. Virol. 70:272–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wolffe EJ, Isaacs SN, Moss B. 1993. Deletion of the vaccinia virus B5R gene encoding a 42-kilodalton membrane glycoprotein inhibits extracellular virus envelope formation and dissemination. J. Virol. 67:4732–4741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jahrling PB, Hensley LE, Martinez MJ, Leduc JW, Rubins KH, Relman DA, Huggins JW. 2004. Exploring the potential of variola virus infection of cynomolgus macaques as a model for human smallpox. Proc. Natl. Acad. Sci. U. S. A. 101:15196–15200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wahl-Jensen V, Cann JA, Rubins KH, Huggins JW, Fisher RW, Johnson AJ, de Kok-Mercado F, Larsen T, Raymond JL, Hensley LE, Jahrling PB. 2011. Progression of pathogenic events in cynomolgus macaques infected with variola virus. PLoS One 6:e24832. 10.1371/journal.pone.0024832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huggins J, Goff A, Hensley L, Mucker E, Shamblin J, Wlazlowski C, Johnson W, Chapman J, Larsen T, Twenhafel N, Karem K, Damon IK, Byrd CM, Bolken TC, Jordan R, Hruby D. 2009. Nonhuman primates are protected from smallpox virus or monkeypox virus challenges by the antiviral drug ST-246. Antimicrob. Agents Chemother. 53:2620–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lederman ER, Davidson W, Groff HL, Smith SK, Warkentien T, Li Y, Wilkins KA, Karem KL, Akondy RS, Ahmed R, Frace M, Shieh WJ, Zaki S, Hruby DE, Painter WP, Bergman KL, Cohen JI, Damon IK. 2012. Progressive vaccinia: case description and laboratory-guided therapy with vaccinia immune globulin, ST-246, and CMX001. J. Infect. Dis. 206:1372–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vora S, Damon I, Fulginiti V, Weber SG, Kahana M, Stein SL, Gerber SI, Garcia-Houchins S, Lederman E, Hruby D, Collins L, Scott D, Thompson K, Barson JV, Regnery R, Hughes C, Daum RS, Li Y, Zhao H, Smith S, Braden Z, Karem K, Olson V, Davidson W, Trindade G, Bolken T, Jordan R, Tien D, Marcinak J. 2008. Severe eczema vaccinatum in a household contact of a smallpox vaccinee. Clin. Infect. Dis. 46:1555–1561 [DOI] [PubMed] [Google Scholar]
- 26. Chinsangaram J, Honeychurch KM, Tyavanagimatt SR, Bolken TC, Jordan R, Jones KF, Marbury T, Lichtenstein I, Pickens M, Corrado M, Landis P, Clarke JM, Frimm AM, Hruby DE. 2012. Pharmacokinetic comparison of a single oral dose of polymorph form I versus form V capsules of the antiorthopoxvirus compound ST-246 in human volunteers. Antimicrob. Agents Chemother. 56:3582–3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chinsangaram J, Honeychurch KM, Tyavanagimatt SR, Leeds JM, Bolken TC, Jones KF, Jordan R, Marbury T, Ruckle J, Mee-Lee D, Ross E, Lichtenstein I, Pickens M, Corrado M, Clarke JM, Frimm AM, Hruby DE. 2012. Safety and pharmacokinetics of the anti-orthopoxvirus compound ST-246 following a single daily oral dose for 14 days in human volunteers. Antimicrob. Agents Chemother. 56:4900–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jordan R, Chinsangaram J, Bolken TC, Tyavanagimatt SR, Tien D, Jones KF, Frimm A, Corrado ML, Pickens M, Landis P, Clarke J, Marbury TC, Hruby DE. 2010. Safety and pharmacokinetics of the antiorthopoxvirus compound ST-246 following repeat oral dosing in healthy adult subjects. Antimicrob. Agents Chemother. 54:2560–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berhanu A, King DS, Mosier S, Jordan R, Jones KF, Hruby DE, Grosenbach DW. 2009. ST-246(R) inhibits in vivo poxvirus dissemination, virus shedding, and systemic disease manifestation. Antimicrob. Agents Chemother. 53:4999–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grosenbach DW, Berhanu A, King DS, Mosier S, Jones KF, Jordan RA, Bolken TC, Hruby DE. 2010. Efficacy of ST-246 versus lethal poxvirus challenge in immunodeficient mice. Proc. Natl. Acad. Sci. U. S. A. 107:838–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quenelle DC, Buller RM, Parker S, Keith KA, Hruby DE, Jordan R, Kern ER. 2007. Efficacy of delayed treatment with ST-246 given orally against systemic orthopoxvirus infections in mice. Antimicrob. Agents Chemother. 51:689–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sbrana E, Jordan R, Hruby DE, Mateo RI, Xiao SY, Siirin M, Newman PC, Da Rosa AP, Tesh RB. 2007. Efficacy of the antipoxvirus compound ST-246 for treatment of severe orthopoxvirus infection. Am. J. Trop. Med. Hyg. 76:768–773 [PubMed] [Google Scholar]
- 33. Nalca A, Hatkin JM, Garza NL, Nichols DK, Norris SW, Hruby DE, Jordan R. 2008. Evaluation of orally delivered ST-246 as postexposure prophylactic and antiviral therapeutic in an aerosolized rabbitpox rabbit model. Antiviral Res. 79:121–127 [DOI] [PubMed] [Google Scholar]
- 34. Jordan R, Goff A, Frimm A, Corrado ML, Hensley LE, Byrd CM, Mucker E, Shamblin J, Bolken TC, Wlazlowski C, Johnson W, Chapman J, Twenhafel N, Tyavanagimatt S, Amantana A, Chinsangaram J, Hruby DE, Huggins J. 2009. ST-246 antiviral efficacy in a nonhuman primate monkeypox model: determination of the minimal effective dose and human dose justification. Antimicrob. Agents Chemother. 53:1817–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.