

Abstract
To facilitate studies of hepatitis C virus (HCV) NS4A, we aimed at developing J6/JFH1-based recombinants with genotype 1- to 7-specific NS4A proteins. We developed efficient culture systems expressing NS4A proteins of genotypes (isolates) 1a (H77 and TN), 1b (J4), 2a (J6), 4a (ED43), 5a (SA13), 6a (HK6a), and 7a (QC69), with peak infectivity titers of ∼3.5 to 4.5 log10 focus-forming units per ml. Except for genotype 2a (J6), growth depended on adaptive mutations identified in long-term culture. Genotype 1a, 1b, and 4a recombinants were adapted by amino acid substitutions F772S (p7) and V1663A (NS4A), while 5a, 6a, and 7a recombinants required additional substitutions in the NS3 protease and/or NS4A. We demonstrated applicability of the developed recombinants for study of antivirals. Genotype 1 to 7 NS4A recombinants showed similar responses to the protease inhibitors telaprevir (VX-950), boceprevir (Sch503034), simeprevir (TMC435350), danoprevir (ITMN-191), and vaniprevir (MK-7009), to alpha interferon 2b, and to the putative NS4A inhibitor ACH-806. The efficacy of ACH-806 was lower than that of protease inhibitors and was not influenced by changes at amino acids 1042 and 1065 (in the NS3 protease), which have been suggested to mediate resistance to ACH-806 in replicons. Genotype 1a, 1b, and 2a recombinants showed viral spread under long-term treatment with ACH-806, without acquisition of resistance mutations in the NS3-NS4A region. Relatively high concentrations of ACH-806 inhibited viral assembly, but not replication, in a single-cycle production assay. The developed HCV culture systems will facilitate studies benefitting from expression of genotype-specific NS4A in a constant backbone in the context of the complete viral replication cycle, including functional studies and evaluations of the efficacy of antivirals.
INTRODUCTION
Chronic hepatitis C virus (HCV) infection is a major public health burden. HCV is an enveloped virus with a positive-strand RNA genome with 5′- and 3′-untranslated regions (UTRs) and an open reading frame (ORF) encoding a single polyprotein, which is cleaved into structural proteins (core, E1, and E2), p7, and the nonstructural (NS) proteins: NS2, NS3, NS4A, NS4B, NS5A, and NS5B (1). The NS3 protein consists of a serine protease (NS3P), responsible for downstream cleavages, and an RNA helicase (NS3H) domain. The function of NS3P is dependent on the NS4A protein. Due to great genetic heterogeneity, HCV was classified into seven major genotypes, which differ in about 30% of the sequence (2).
For the last decade, combination therapy with pegylated alpha interferon 2 (IFN-alpha2) and ribavirin was the standard of care, but it was characterized by various contraindications and severe side effects. Efficacy of combination therapy depended on host factors as well as HCV genotype, with sustained viral response rates of 40 to 50% for genotype 1 and ∼80% for genotypes 2 and 3 and intermediate response rates for patients infected with genotypes 4 to 6 (1, 3). Various genome regions were suspected to confer resistance to interferon (1). Therefore, development of directly acting antivirals is a major research focus; the multifunctional NS3P protein and its cofactor, NS4A, are important targets. NS3P mediates cleavage of NS4A, NS4B, NS5A, and NS5B from the HCV polyprotein and cleavage of adaptor proteins, leading to inhibition of innate antiviral responses and IFN induction (1, 4). NS4A interacts with NS3P and NS3H and modulates replication, presumably by interaction with other nonstructural proteins (5–7). Furthermore, NS3 and NS4A apparently play a role in viral assembly, interacting with p7, NS2, and NS5A (8, 9). In 2011, the first two directly acting antivirals targeting HCV NS3P, telaprevir and boceprevir, were licensed for treatment of chronic genotype 1 infection (10). Several other protease inhibitors are advanced in clinical trials, while inhibitors of HCV NS4A are in preclinical development (10–13). We and others have reported that HCV recombinants with genotype-specific NS3P/NS4A show differential responses to protease inhibitors (14–16).
Identification of new drug candidates with efficacy against all HCV genotypes and functional studies of HCV proteins in a genotype-specific manner are hampered by the limited availability of in vitro culture systems for the major HCV genotypes. At the outset of this study, among available culture systems recapitulating the complete viral replication cycle, genotype 1 full-length systems showed low infectivity (17, 18), while efficient systems relied on the genotype 2a isolate JFH1 (19, 20). In this study, by exploiting the replication capacity of J6/JFH1 (20), our aim was to develop culture systems expressing genotype-specific NS4A proteins. We used the J6/JFH1-based NS4A recombinants to study whether genotype-specific NS4A influenced sensitivity to lead compound protease inhibitors, IFN-alpha2b, and a putative NS4A inhibitor.
MATERIALS AND METHODS
Plasmids.
We replaced NS4A (nucleotides [nt] 5313 to 5474; amino acids [aa] 1658 to 1711; nt and aa positions are given as absolute H77 [GenBank accession number AF009606] reference numbers) of pJ6/JFH1 (20) with the respective sequence of pCV-H77C (AF011751) (21), pHC-TN (EF621489) (22), pCV-J4L6S (AF054247) (23), pJ6CF (AF177036) (24), pS52 (GU814264) (25), or pED43 (GU814266) (25). SA13 and HK6a consensus NS4A sequences were amplified by reverse transcription-PCR (RT-PCR) from respective plasma pools, followed by analysis of multiple clones (26). QC69 (accession number EF108306) sequences were synthesized (Genscript). Alignments of NS4A aa sequences used in this study are shown in Fig. S1 in the supplemental material. Original plasmids and plasmids with point mutations were constructed using fusion PCR and restriction enzyme-based cloning. The HCV sequences of final DNA preparations (Qiagen plasmid maxikit) were confirmed (Macrogen). Plasmids encoding genotype 1a (isolate TN) and genotype 2a (isolate J6) full-length recombinants, i.e., TNcc (27) and J6cc (28), were previously described.
Transfection and viral passage in Huh7.5 cell cultures.
Generation of RNA transcripts, RNA transfection of Huh7.5 cells by use of Lipofectamine, viral passage by infection of naive cells with culture supernatant, and culture conditions were described previously (29). Huh7.5 cells were provided by C. Rice (30). The percentage of HCV NS5A antigen-positive cells was determined by immunostaining with anti-NS5A 9E10 primary antibody, provided by C. Rice (20), and with an Alexa Fluor 594 goat anti-mouse IgG(H+L) secondary antibody (Invitrogen) (29). Culture supernatant infectivity titers were determined as numbers of focus-forming units (FFU)/ml: 6 × 103 cells per well, plated the previous day on poly-d-lysine-coated 96-well plates (Nunc), were infected with serially diluted supernatant (lowest dilution, 1:2). Forty-eight hours after infection, cells were fixed, and HCV NS5A was immunostained with anti-NS5A 9E10 primary antibody and an enhanced chemiluminescence (ECL) anti-mouse IgG horseradish peroxidase (HRP)-linked whole antibody as the secondary antibody (GE Healthcare Amersham) and visualized with a DAB substrate kit (Dako) (29). FFU were counted with an ImmunoSpot series 5 UV analyzer (CTL Europe GmbH) (25), unless otherwise indicated. Means of FFU counts for 6 negative-control wells (always <15 FFU/well) were subtracted from counts for experimental wells. Counts above the lower limit of detection (mean for 6 negative-control wells plus 3 standard deviations plus 3) and below 200 FFU/well were accepted, as these were in the linear range of test dilution series and comparable to manual counts. Supernatant HCV RNA titers were measured by 5′UTR-based quantitative RT-PCR (29).
ORF sequencing of cell culture-produced HCV.
Methods for RNA extraction from culture supernatants, RT-nested PCR, and direct sequence analysis were described previously (29). Primers used for NS4A recombinants were the same as those described for J6/JFH1 (29).
HCV treatment experiments and statistical analysis.
A total of 5 × 103 cells per well, plated the previous day on poly-d-lysine-coated 96-well plates (Nunc), were infected with HCV stocks as specified. Cells were treated at 24 and 48 h postinfection with dilution series of VX-950, Sch503034, and IFN-alpha2b. Subsequently, extensive pilot studies revealed that treatment at only 24 h postinfection led to results similar to those for treatment at 24 and 48 h postinfection for treatments with various antivirals, including VX-950, Sch503034, TMC435350, ITMN-191, MK-7009, and ACH-806. Thus, in experiments with TMC435350, ITMN-191, and MK-7009, antivirals were administered only at 24 h postinfection. In experiments with ACH-806, the compound was administered as specified. Directly acting antivirals were purchased from Acme Bioscience and dissolved in dimethyl sulfoxide (DMSO). IFN-alpha2b was purchased from Schering. Cells were fixed at 72 h postinfection and stained as described for infectivity titration; for staining of full-length recombinants, a mixture of monoclonal anti-core antibody C7-50 (Enzo Life Sciences) and anti-NS5A 9E10 was used as primary antibody (27). Single HCV-positive cells were counted with an ImmunoSpot series 5 UV analyzer (CTL Europe GmbH) with customized software. The mean of counts for 12 noninfected, nontreated control wells was subtracted from experimental counts. Each antiviral concentration was tested in triplicate; counts from treated wells were related to the mean of counts from 6 infected, nontreated wells. Sigmoidal concentration-response curves [y = top/(1 + 10[logEC50−x] × Hillslope)] were fitted after logarithmic transformation of x values in GraphPad Prism. Log 50% effective concentrations (log EC50s) and standard errors of the means (SEM) for log EC50s from replicate experiments were used to calculate the inverse variance weighted mean of log EC50s, with SEM and the 95% confidence interval (CI). Mean differences between log EC50 values for genotype 2a(JFH1) and other recombinants, with SEM and 95% CI, were calculated from the inverse variance weighted mean log EC50 values for the compared viruses. Inverse logarithmic transformation rendered the median EC50 with 95% CI and the median fold difference with 95% CI. P values were determined by the Z test, and P values of <0.0001 were considered significant. Cell viability was monitored by the CellTiter 96 AQueous One Solution cell proliferation assay (Promega) (31).
Long-term treatment experiment for induction of HCV escape.
A total of 3.6 × 105 cells per well, plated the previous day in 6-well plates, were infected at a multiplicity of infection (MOI) of 0.05 with specified HCV stocks. Cells were treated with 1,000 nM ACH-806 at 24 h postinfection. Subsequently, the treatment was added 3 times per week, when cells were split. Control cultures were maintained without ACH-806.
Single-cycle production assay.
A total of 4 × 105 CD81-deficient S29 cells, originally provided by S. Emerson (32), were plated per well of a 6-well plate. After 24 h, cells were transfected with HCV RNA as described previously (29), except that the growth medium was replaced by Opti-MEM (Invitrogen) during a 4-h incubation with RNA-Lipofectamine transfection complexes. After the 4-h transfection period, cells were either treated with ACH-806 or maintained untreated. To determine intracellular HCV core levels at 4 and 48 h posttransfection, cells of replicate cultures were trypsinized. At 4 h posttransfection, all cells, and at 48 h posttransfection, 25% of the cells were centrifuged at 1,000 × g for 5 min at 4°C and washed in cold phosphate-buffered saline (PBS). Cells were lysed in cold radioimmunoprecipitation assay (RIPA) buffer (Thermo Scientific) supplemented with protease inhibitor cocktail set III (Calbiochem). Lysates were clarified at 20,000 × g for 15 min at 4°C. HCV core levels in clarified lysates were determined using the Architect HCV Ag assay (Abbott Diagnostics) according to the manufacturer's recommendations. For determination of intracellular infectivity titers, the remaining 75% of cells harvested at 48 h posttransfection were centrifuged at 1,000 × g for 5 min at 4°C and resuspended in 100 μl of growth medium. Cells were subjected to four freeze-thaw cycles, using liquid nitrogen and a 37°C water bath. Supernatants were clarified by two centrifugations at 1,500 × g for 5 min at 4°C. For determination of extracellular core levels and infectivity titers, supernatants were collected 48 h after transfection. No cytotoxicity was observed in this system at the applied ACH-806 concentrations.
Nucleotide sequence accession numbers.
The sequences of NS4A recombinants 1a(H77), 1a(TN), 1b(J4), 2a(J6), 4a(ED43), 5a(SA13), 6a(HK6A), and 7a(QC69) were deposited in GenBank under accession numbers KF693776 to KF693783.
RESULTS
We constructed J6/JFH1-based recombinants with NS4A proteins of reference isolates of genotypes (isolates) 1a (H77 and TN), 1b (J4), 2a (J6), 3a (S52), 4a (ED43), 5a (SA13), 6a (HK6a), and 7a (QC69) (26, 31) (a deduced NS4A aa alignment is shown in Fig. S1 in the supplemental material). In the following, recombinants are named according to the genotype(isolate) of the inserted NS4A sequence, e.g., 1a(H77); for consistency, genotype 2a reference recombinant J6/JFH1 (20) is designated 2a(JFH1). After transfection of Huh7.5 cells with RNA transcripts of the various constructs, we evaluated viability by determining the percentage of HCV NS5A-expressing cells and the culture supernatant infectivity titers. Adaptive mutations were identified by direct sequencing of the complete ORF of viruses passaged in naive Huh7.5 hepatoma cells and by reverse genetic studies.
Viability of genotype 1 to 7 NS4A recombinants.
The 1a(H77), 1a(TN), 1b(J4), 2a(JFH1), 2a(J6), 4a(ED43), and 7a(QC69) recombinants showed various percentages of HCV NS5A-expressing cells during the first 6 days posttransfection (Table 1; Fig. 1). In contrast, during this period, no apparent replication was observed for 3a(S52), 5a(SA13), and 6a(HK6a) (Table 1). 2a(J6) spread comparably to 2a(JFH1), infecting the complete culture on day 3. Long-term culture (23 to 70 days) led to spread of 1a(H77), 1a(TN), 1b(J4), and 4a(ED43) in 2/2 transfection experiments, whereas viral spread of 5a(SA13) was observed in only 1/4 transfections (Fig. 1). In contrast, long-term culture did not lead to spread of 3a(S52) and 6a(HK6a), with no evidence of replication throughout the experiment, or of 7a(QC69), despite initial evidence of replication, which was lost during follow-up. Passaged 1a(H77), 1a(TN), 1b(J4), 2a(J6), and 4a(ED43) yielded peak infectivity titers of 4.2 to 4.6 log10 FFU/ml and HCV RNA titers of 6.4 to 7.5 log10 IU/ml, while peak titers of 5a(SA13) were 3.6 log10 FFU/ml and 7.5 log10 IU/ml (Table 2).
Table 1.
Cell culture viability of J6/JFH1-based recombinants with genotype-specific NS4A, estimated by peak percentages of HCV-positive cells on days 1 to 6 following transfection of Huh 7.5 cells
| NS4A genotype | Isolate | Cell culture viabilitya | Peak % HCV NS5A-positive cells (range) | No. of replicate expt |
|---|---|---|---|---|
| 1a | H77 | ++* | 5–20 | 2 |
| TN | ++* | 5–20 | 2 | |
| 1b | J4 | +* | 1 | 2 |
| 2a | JFH1 | +++* | 90 | 6 |
| J6 | +++* | 90 | 2 | |
| 3a | S52 | − | 0 | 2 |
| 4a | ED43 | −/+* | 0–1 | 2 |
| 5a | SA13 | −* | 0 | 4 |
| 6a | HK6a | − | 0 | 4 |
| 7a | QC69 | + | 1 | 2 |
Cell culture viability was determined by the % of HCV NS5A-positive Huh7.5 cells during the first 6 days posttransfection: +++, approximately 90% HCV NS5A-positive cells detected in all replicate cultures; ++, 5 to 20% HCV NS5A-positive cells detected in all replicate cultures; +, approximately 1% HCV NS5A-positive cells detected in all replicate cultures; −, no HCV NS5A-positive cells detected in all replicate cultures; −/+, in replicate cultures, either a low percentage of (<1%) or no HCV NS5A-positive cells were detected. *, subsequent viral spread occurred in long-term culture in at least 1 replicate experiment; detailed spread kinetics of these cultures are given in Fig. 1.
Fig 1.
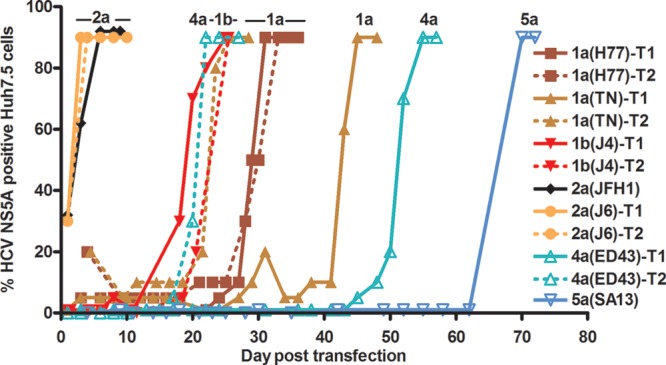
Viability of J6/JFH1-based recombinants with genotype-specific NS4A following transfection of Huh7.5 cells. The percentage of HCV NS5A antigen-positive cells was determined by immunostaining. T1 and T2, 1st and 2nd transfection experiments. Data shown are for transfections leading to viral spread (genotype 1a, 1b, 2a, 4a, and 5a recombinants). An overview of cell culture viability, evaluated by the range of peak percentages of HCV NS5A-positive cells in replicate cultures during the first 6 days posttransfection, is shown in Table 1. Not shown are data for transfections not leading to viral spread: 2 transfections for 3a(S52), 3 for 5a(SA13), 4 for 6a(HK6a), and 2 for 7a(QC69) (Table 1). For 3a(S52), 5a(SA13), and 6a(HK6a), no replicating cells were observed during the observation period. 3a(S52) cultures were closed on days 29 and 34, 5a(SA13) cultures on days 21 to 34, and 6a(HK6a) cultures on days 21 to 42. For 7a(QC69), no replicating cells were observed following day 6, and cultures were closed on days 28 and 39. ORF sequences of viruses derived from subsequent viral passage experiments are shown in Tables S1 to S8 in the supplemental material.
Table 2.
Characteristics of cell culture-derived J6/JFH1 viruses with NS4A proteins of genotype 1a, 1b, 2a, 4a, 5a, 6a, and 7a isolatesa
| Genotype | Isolate | Engineered adaptive mutations | Locations of mutations | Peak titer |
Passage (day) | Reference to supplemental material | |
|---|---|---|---|---|---|---|---|
| Infectivity (log10 FFU/ml) | RNA (log10 IU/ml) | ||||||
| Recombinants used for cell culture adaptation | |||||||
| 1a | H77 | None | 4.2 | 7.0 | 1st (4) | Table S2 | |
| 1a | TN | None | 4.2 | 6.8 | 1st (4) | Table S3 | |
| 1b | J4 | None | 4.3 | 6.4 | 1st (3) | Table S4 | |
| 2a | J6 | None | 4.6 | 7.5 | 1st (7) | Table S1 | |
| 4a | ED43 | None | 4.6 | 7.4 | 1st (10) | Table S5 | |
| 5a | SA13 | None | 3.6 | 7.5 | 1st (3) | Table S6 | |
| 6a | HK6a | F772S, V1663A | p7, NS4A | 4.0 | 7.1 | 1st (3) | Table S7 |
| 7a | QC69 | F772S, V1663A | p7, NS4A | 3.7 | 6.8 | 1st (10) | Table S8 |
| Most efficient cell culture-adapted recombinants with engineered mutations | |||||||
| 1a | H77 | F772S, V1663A | p7, NS4A | 3.8 | 7.1 | 2nd (7)* | Table S2 |
| TN | F772S, V1663A | p7, NS4A | 3.8 | 6.9 | 2nd (7)* | Table S3 | |
| 1b | J4 | F772S, V1663A | p7, NS4A | 4.3 | 7.2 | 2nd (7)* | Table S4 |
| 4a | ED43 | F772S, V1663A | p7, NS4A | 3.9 | 7.3 | 1st (6) | Table S5 |
| ED43 | I879T, V1663A | NS2, NS4A | 4.3 | 6.8 | 1st (9) | Table S5 | |
| 5a | SA13 | F772S, V1663A, V1683F | p7, NS4A, NS4A | 3.7 | 7.0 | 1st (9) | Table S6 |
| 6a | HK6a | F772S, E1105A, V1663A, V1677A | p7, NS3P, NS4A, NS4A | 3.6 | 7.6 | 1st (8) | Table S7 |
| 7a | QC69 | F772S, I1061V, V1663A, S1679C | p7, NS3P, NS4A, NS4A | 3.8 | 7.8 | 1st (13) | Table S8 |
RNA transcripts were transfected into Huh7.5 cells, and transfection culture supernatants from the peak of infection were used to infect naive cells. HCV infectivity and RNA titers were determined in supernatants from the 1st or 2nd viral passage as described in Materials and Methods. Infectivity titer, highest representative titer among supernatants obtained from several passage experiments; RNA titer, determined with the same supernatant for which infectivity titer is shown. Peak titers of all passage experiments in which the viral ORF was sequenced are shown in Tables S1 to S8. Recombinants used for cell culture adaptation acquired the mutations indicated in Tables S2 to S8; 2a(J6) did not acquire cell culture-adaptive mutations (see Table S1). Cell culture-adapted recombinants with engineered mutations did not acquire mutations in addition to the inserted mutations (see Tables S2 to S8). All aa positions are annotated with H77 (GenBank accession number AF009606) absolute reference numbers. *, 2nd-passage virus was derived from the 1st passage, indicated in the respective supplemental table. Among 2nd-passage viruses, NS3P, NS3H, and NS4A were shown to be genetically stable by direct sequence analysis.
Development of efficient recombinants with NS4A proteins of genotypes 1a, 1b, 2a, 4a, 5a, 6a, and 7a.
The 2a(J6) recombinant did not seem to require cell culture-adaptive mutations, because ORF sequencing of passaged viruses revealed genetic stability in one experiment and two quasispecies in another experiment (see Table S1 in the supplemental material). In contrast, passaged 1a(H77), 1a(TN), 1b(J4), 4a(ED43), and 5a(SA13) acquired various mutations; common mutations were located in p7 (coding for an F772S substitution) and NS4A (coding for a V1663A substitution) (see Tables S2 to S6).
(i) Genotype 1a, 1b, and 4a NS4A recombinants were adapted by mutations in p7 and NS4A (coding for F772S and V1663A substitutions).
F772S and V1663A substitutions led to efficient cell culture adaptation of 1a(H77), 1a(TN), 1b(J4), and 4a(ED43), with fast spread kinetics and peak infectivity titers of 3.6 to 4.6 log10 FFU/ml following transfection (Fig. 2A). First-passage viruses had peak infectivity titers of 3.8 to 4.3 log10 FFU/ml (Table 2) and were apparently genetically stable (see Tables S2 to S5 in the supplemental material). When we introduced the F772S substitution individually into 1a(H77), 1a(TN), 1b(J4), and 4a(ED43), spread occurred after an eclipse phase (see the legend to Fig. 2A), and passaged viruses had acquired the V1663A substitution (see Tables S2 to S5). For 4a(ED43)V1663A, passaged viruses acquired the F772S substitution (see Table S5). We also tested 4a(ED43)I879T,V1663A with a combination of mutations identified in a 2nd 4a(ED43) transfection experiment. Transfection resulted in fast spread and peak infectivity titers of ∼4.3 log10 FFU/ml; the virus was genetically stable following passage into naive cells (Fig. 2A; see Table S5). Thus, for 4a(ED43), I879T substitution in NS2, when combined with V1663A substitution in NS4A, could substitute for F772S substitution in p7.
Fig 2.

Identification of adaptive amino acid substitutions leading to efficient growth of NS4A recombinants. Supernatant infectivity titers are shown for cultures transfected with RNA transcripts of genotype 1a, 1b, and 4a (A), genotype 5a and 6a (B), and genotype 7a (C) recombinants (data are means for 3 replicates with SEM; the lower cutoff was 2.4 log10 FFU/ml, as indicated by the column break). ◆, culture stopped because of cell death due to viral infection (29); ●, not done; #, supernatants from peak infection were used for viral passage. ORF sequences and peak infectivity titers of passaged viruses are shown in Tables S2 to S8 in the supplemental material. Amino acid positions are annotated with H77 (accession number AF009606) absolute reference numbers. (A) Amino acid substitutions in p7 and NS4A (F772S and V1663A) were sufficient for adaptation of 1a, 1b, and 4a recombinants. The recombinants shown infected the complete culture at no later than day 6 posttransfection: 1a(H77)F772S,V1663A, day 4; 1a(TN)F772S,V1663A, day 6; 1b(J4)F772S,V1663A, day 4; 4a(ED43)V1663A, day 6; 4a(ED43) F772S,V1663A, day 4; and 4a(ED43)I879T,V1663A, day 3. Additional recombinants infected the complete culture after >6 days: 1a(H77)F772S, 20 days; 1a(TN)F772S, 20 days; 1b(J4)F772S, 13 days; 4a(ED43)F772S, 17 and 23 days (2 replicate experiments); and 4a(ED43)F772S,V1666A, 15 days. (B) 5a and 6a recombinants required adaptive amino acid substitutions in NS3P and/or NS4A in addition to the F772S and V1663A substitutions. The recombinants shown infected the complete culture at no later than 8 days posttransfection: 5a(SA13)F772S,V1663A,V1683F, day 4; 6a(HK6a)F772S,V1663A,V1677A, day 8; 6a(HK6a)F772S,E1105A,V1663A, day 6; and 6a(HK6a) F772S,E1105A,V1663A,V1677A, day 3. Two additional recombinants infected the complete culture after >8 days: 5a(SA13)F772S,V1663A, 18 days; and 6a(HK6a)F772S,V1663A, 39 and 22 days (2 replicate experiments). Finally, cultures of 5a(SA13)F772S and 6a(HK6a)F772S, showing no replicating cells throughout the experiment, were stopped on day 29. (C) 7a recombinants required adaptive aa substitutions in NS3P and NS4A in addition to the F772S and V1663A substitutions. Shown 7a(QC69) viruses infected the complete culture on day 10 posttransfection. In addition, the following recombinants were tested: 7a(QC69)F772S,V1663A, 90% infected cells on day 48; and 7a(QC69)F772S,I1061V,V1663A, terminated on day 16, with 50% infected cells.
(ii) Genotype 5a, 6a, and 7a NS4A recombinants required amino acid substitutions in NS3P and/or NS4A in addition to F772S/V1663A substitutions for cell culture adaptation.
Passaged 5a(SA13) acquired nt changes coding for F772S and V1663A substitutions and four additional nt changes, coding for M405T (E2), K750E (p7), V1683F (NS4A), and I2270M (NS5A) substitutions (see Table S6 in the supplemental material). 5a(SA13)F772S,V1663A,V1683F showed fast spread and reached peak infectivity titers of almost 4.0 log10 FFU/ml in transfection and 1st viral passage experiments (Fig. 2B; Table 2). Passaged virus was genetically stable (see Table S6). We also tested 5a(SA13) with only F772S and V1663A substitutions, and it replicated; however, spread to most culture cells did not occur until day 18 (see the legend to Fig. 2B).
Introduction of nt changes coding for F772S and V1663A substitutions conferred replication capacity to 6a(HK6a), resulting in infection of most culture cells on days 22 and 39 in 2 independent transfections (see the legend to Fig. 2B and Table 2). Mutations in NS3P (coding for E1105A substitution) and NS4A (coding for V1677A substitution), found in 1st-passage viruses (see Table S7 in the supplemental material), were introduced into 6a(HK6a)F772S,V1663A, individually and in combination. 6a(HK6a)F772S,E1105A,V1663A,V1677A showed the fastest spread and highest peak infectivity titers (4.0 log10 FFU/ml) (Fig. 2B); 1st-passage virus was genetically stable, with a peak infectivity titer of 3.6 log10 FFU/ml (Table 2; see Table S7).
Introduction of nt changes coding for F772S and V1663A substitutions conferred increased replication capacity to 7a(QC69), resulting in spread to most cells on day 48 (see the legend to Fig. 2C and Table 2). We tested additional mutations in NS3P (coding for I1061V substitution) and NS4A (coding for S1679C substitution) that were found in 1st-passage viruses (see Table S8 in the supplemental material). Only 7a(QC69)F772S,I1061V,V1663A,S1679C produced infectivity titers of >2.4 log10 FFU/ml, with a peak titer of 3.5 log10 FFU/ml (Fig. 2C); 1st-passage virus was genetically stable, with a peak infectivity titer of 3.8 log10 FFU/ml (Table 2; see Table S8).
(iii) Adaptation of genotype 3a(S52) NS4A recombinant was not achieved.
We could not confer replication capacity to 3a(S52) with F772S substitution, F772S and V1663A substitutions, or addition of the following aa substitutions to F772S and V1663A substitutions: V1683F (NS4A), found in the 5a(SA13) NS4A recombinant; I1061V (NS3P), found in the 1a(TN), 4a(ED43), 5a(SA13), and 7a(QC69) NS4A recombinants; or A1042T and V1065A (both in NS3P), changing residues putatively interacting with NS4A to 3a(S52)-specific residues. Finally, a 3a(S52) recombinant with G868S, L1663A, and E2267G aa changes, adapting a J6/JFH1-based recombinant with 3a(S52) NS3P/NS4A (14), did not replicate in 2 independent transfection experiments.
J6/JFH1 was attenuated when alanine at position 6 of NS4A was changed to a non-genotype 2-specific residue.
Given the importance of the V1663A substitution for adaptation of J6/JFH1 recombinants with genotype-specific NS4A, we investigated the effect of mutating aa 1663 of 2a(JFH1) from A, specific to genotype 2a, to V, found in genotype 1a, 1b, 4a, 5a, 6a, and 7a isolates, or to L, found in genotype 3a isolates (see Fig. S1 in the supplemental material). After transfection, 2a(JFH1)A1663V was attenuated, with ∼1-log10 lower infectivity titers than those of 2a(JFH1) on days 4 and 6 posttransfection. Attenuation of 2a(JFH1)A1663L was more pronounced, with 1.7- and 1.8-log10 lower titers than those of 2a(JFH1) on days 4 and 6, respectively (see Fig. S2A). However, 1st-passage viruses were genetically stable, with peak infectivity titers of ∼3.8 log10 FFU/ml. Attenuation was confirmed in a 2nd-passage kinetic experiment; differences were most pronounced on day 5, with an ∼1.5-log10 lower infectivity titer for 2a(JFH1)A1663V and 2a(JFH1)A1663L than that for 2a(JFH1) (see Fig. S2B). Sequencing of 2nd-passage viruses showed that 2a(JFH1)A1663V was genetically stable, while for 2a(JFH1)A1663L, L had reverted to A. Thus, optimal growth characteristics of 2a(JFH1) depended on the genotype 2-specific A at aa position 1663.
Recombinants with genotype-specific NS4A did not show major differences in susceptibility to lead compound protease inhibitors and IFN-alpha.
With a 96-well plate-based high-throughput assay, we determined concentration-response profiles with EC50s for the developed NS4A recombinants, using protease inhibitors and IFN-alpha2b. Overall, compared to 2a(JFH1), recombinants with genotype-specific NS4A did not show major differences in sensitivity to the lead compound protease inhibitors VX-950, Sch503034, TMC435350, ITMN-191, and MK-7009 (Table 3; Fig. 3) (10). However, for several recombinants and compounds, small but statistically significant differences were found. This was the case for 1b(J4) and 6a(HK6a) treated with the linear inhibitors VX-950 and Sch503034, as they were up to 3.2-fold more sensitive than 2a(JFH1) (Fig. 3A and B and Table 3). Furthermore, 1a(H77) was 2.1-fold less sensitive and 7a(QC69) was 1.8-fold less sensitive to VX-950 than 2a(JFH1). 5a(SA13) was 1.6-fold more sensitive to Sch503034 than 2a(JFH1). Under treatment with macrocyclic inhibitors, significant differences were found only for 7a(QC69) treated with TMC435350, which was 2.7-fold more sensitive than 2a(JFH1), and for 6a(HK6a) treated with ITMN-191, which was 4.6-fold more sensitive than 2a(JFH1); no significant differences were observed for MK-7009 (Fig. 3C to E and Table 3). Similarly, when treating recombinants with genotype-specific NS4A with IFN-alpha2b, no major differences in sensitivity were observed; 7a(QC69) showed an ∼2-fold-increased sensitivity compared to the other recombinants (Table 3 and Fig. 3F).
Table 3.
Efficacies of NS3 protease inhibitors and IFN-alpha 2b against recombinants with genotype-specific NS4A
| Drug and genotypea | Isolatea | EC50b |
Fold difference from 2a(JFH1)b |
P valuec | ||
|---|---|---|---|---|---|---|
| Median | 95% CI | Median | 95% CI | |||
| VX-950 (nM) | ||||||
| 2a | JFH1 | 514 | 474–557 | NA | NA | NA |
| 1a | H77 | 1,060 | 930–1,207 | 2.1 | 1.8–2.4 | <0.0001 |
| 1a | TN | 687 | 601–784 | 1.3 | 1.1–1.6 | 0.0003 |
| 1b | J4 | 377 | 347–410 | 0.7 | 0.7–0.8 | <0.0001 |
| 2a | J6 | 533 | 460–618 | 1.0 | 0.9–1.2 | 0.6740 |
| 4a | ED43 | 535 | 431–663 | 1.0 | 0.8–1.3 | 0.7337 |
| 5a | SA13 | 529 | 462–606 | 1.0 | 0.9–1.2 | 0.7121 |
| 6a | HK6a | 254 | 225–286 | 0.5 | 0.4–0.6 | <0.0001 |
| 7a | QC69 | 932 | 860–1,010 | 1.8 | 1.6–2.0 | <0.0001 |
| Sch503034 (nM) | ||||||
| 2a | JFH1 | 620 | 568–676 | NA | NA | NA |
| 1a | H77 | 696 | 560–865 | 1.1 | 0.9–1.4 | 0.3337 |
| 1a | TN | 552 | 478–637 | 0.9 | 0.8–1.1 | 0.1732 |
| 1b | J4 | 317 | 281–359 | 0.5 | 0.4–0.6 | <0.0001 |
| 2a | J6 | 567 | 502–642 | 0.9 | 0.8–1.1 | 0.2474 |
| 4a | ED43 | 598 | 533–671 | 1.0 | 0.8–1.1 | 0.6210 |
| 5a | SA13 | 377 | 331–430 | 0.6 | 0.5–0.7 | <0.0001 |
| 6a | HK6a | 196 | 167–231 | 0.3 | 0.3–0.4 | <0.0001 |
| 7a | QC69 | 507 | 444–578 | 0.8 | 0.7–1.0 | 0.0122 |
| TMC 435350 (nM) | ||||||
| 2a | JFH1 | 167 | 131–214 | NA | NA | NA |
| 1a | H77 | 179 | 155–207 | 1.1 | 0.8–1.4 | 0.6446 |
| 1a | TN | 134 | 111–160 | 0.8 | 0.6–1.1 | 0.1456 |
| 1b | J4 | 119 | 100–140 | 0.7 | 0.5–1.0 | 0.0220 |
| 2a | J6 | 123 | 98–155 | 0.7 | 0.5–1.0 | 0.0738 |
| 4a | ED43 | 218 | 165–289 | 1.3 | 0.9–1.9 | 0.1624 |
| 5a | SA13 | 207 | 171–252 | 1.2 | 0.9–1.7 | 0.1761 |
| 6a | HK6a | 341 | 225–518 | 2.0 | 1.3–3.3 | 0.0039 |
| 7a | QC69 | 63 | 56–70 | 0.4 | 0.3–0.5 | <0.0001 |
| ITMN-191 (nM) | ||||||
| 2a | JFH1 | 76 | 56–105 | NA | NA | NA |
| 1a | H77 | 87 | 63–120 | 1.1 | 0.7–1.8 | 0.5770 |
| 1a | TN | 71 | 54–93 | 0.9 | 0.6–1.4 | 0.7233 |
| 1b | J4 | 47 | 37–58 | 0.6 | 0.4–0.9 | 0.0123 |
| 2a | J6 | 83 | 64–107 | 1.1 | 0.7–1.6 | 0.6815 |
| 4a | ED43 | 27 | 15–46 | 0.3 | 0.2–0.7 | 0.0012 |
| 5a | SA13 | 44 | 25–78 | 0.6 | 0.3–1.1 | 0.0986 |
| 6a | HK6a | 17 | 10–27 | 0.2 | 0.1–0.4 | <0.0001 |
| 7a | QC69 | 33 | 25–45 | 0.4 | 0.3–0.7 | 0.0002 |
| MK-7009 (nM) | ||||||
| 2a | JFH1 | 133 | 91–195 | NA | NA | NA |
| 1a | H77 | 204 | 142–294 | 1.5 | 0.9–2.6 | 0.1133 |
| 1a | TN | 56 | 25–126 | 0.4 | 0.2–1.0 | 0.0577 |
| 1b | J4 | 70 | 52–94 | 0.5 | 0.3–0.8 | 0.0084 |
| 2a | J6 | 172 | 110–268 | 1.3 | 0.7–2.3 | 0.3963 |
| 4a | ED43 | 97 | 69–137 | 0.7 | 0.4–1.2 | 0.2319 |
| 5a | SA13 | 84 | 58–122 | 0.6 | 0.4–1.1 | 0.0912 |
| 6a | HK6a | 90 | 63–129 | 0.7 | 0.4–1.1 | 0.1431 |
| 7a | QC69 | 83 | 64–107 | 0.6 | 0.4–1.0 | 0.0443 |
| IFN-alpha2b (IU/ml) | ||||||
| 2a | JFH1 | 0.8 | 0.7–1.0 | NA | NA | NA |
| 1a | H77 | 1.0 | 0.8–1.3 | 1.2 | 0.9–1.7 | 0.2617 |
| 1a | TN | 0.8 | 0.7–1.1 | 1.0 | 0.8–1.4 | 0.8357 |
| 1b | J4 | 1.1 | 0.9–1.4 | 1.4 | 1.0–1.9 | 0.0596 |
| 2a | J6 | 0.9 | 0.6–1.2 | 1.1 | 0.7–1.6 | 0.7540 |
| 4a | ED43 | 0.9 | 0.7–1.2 | 1.1 | 0.8–1.5 | 0.6107 |
| 5a | SA13 | 0.9 | 0.7–1.4 | 1.1 | 0.8–1.8 | 0.5195 |
| 6a | HK6a | 0.8 | 0.6–0.9 | 0.9 | 0.7–1.3 | 0.6327 |
| 7a | QC69 | 0.4 | 0.3–0.5 | 0.5 | 0.4–0.6 | <0.0001 |
Genotype and isolate of NS4A in J6/JFH1-based recombinants with genotype-specific NS4A. Identities of virus stocks of NS4A recombinants were as follows. The 2a(JFH1) reference virus was a 2nd-passage virus stock without ORF mutations. The 1a(H77)F772S,V1663A, 1a(TN)F772S,V1663A, 1b(J4)F772S,V1663A, 2a(J6), 4a(ED43)F772S,V1663A, and 5a(SA13) F772S,V1663A,V1683F viruses were 2nd-passage viruses derived from 1st passages shown in Tables S1 to S6 in the supplemental material. For these stocks, the NS3-NS4A sequence was determined and found to be unchanged. 6a(HK6a)F772S,E1105A,V1663A was from the 1st passage (day 20) shown in Table S7. 7a(QC69) F772S,V1663A was from the 1st passage (day 10) shown in Table S8.
Median EC50s and median fold differences with 95% CI were calculated from replicate experiments as described in Materials and Methods. Representative experiments are shown in Fig. 3. NA, not applicable.
P values of <0.0001 are shown in bold.
Fig 3.

HCV NS4A recombinants do not show major differences in sensitivity to protease inhibitors and IFN-alpha2b. Cell cultures in 96-well plates were infected with recombinants with NS4A proteins of the indicated genotypes (isolates); virus stocks are detailed in Table 3, footnote a. VX-950 (telaprevir) (A), Sch503034 (boceprevir) (B), TMC435350 (simeprevir) (C), ITMN-191 (danoprevir) (D), MK-7009 (vaniprevir) (E), and IFN-alpha2b (F) were administered at the indicated concentrations; each concentration was tested in triplicate. Cells were immunostained for HCV NS5A, and single positive cells were counted automatically. Percentages of HCV NS5A-positive cells were calculated by comparison to the mean count for six nontreated cultures. After logarithmic transformation of x values, a sigmoidal concentration-response curve was fitted as described in Materials and Methods. Error bars indicate SEM for 3 replicates. No cellular toxicity was observed for the given concentration range. Median EC50 values from these and replicate experiments are shown in Table 3.
Similar efficacies of a putative NS4A inhibitor against recombinants with genotype-specific NS4A.
The putative NS4A inhibitor ACH-806 (11) showed similar efficacies against the developed NS4A recombinants (Fig. 4A). However, antiviral effects of treatment with the highest noncytotoxic concentrations of ACH-806 were less pronounced than antiviral effects of treatment with relatively high concentrations of protease inhibitors (Fig. 3 and 4A). While a concentration of 10,000 nM ACH-806 caused only a slight decrease in cellular proliferation (90% cell viability compared to nontreated controls), we observed cytotoxicity at 30,000 nM (52% cell viability) (see Fig. S3 in the supplemental material).
Fig 4.
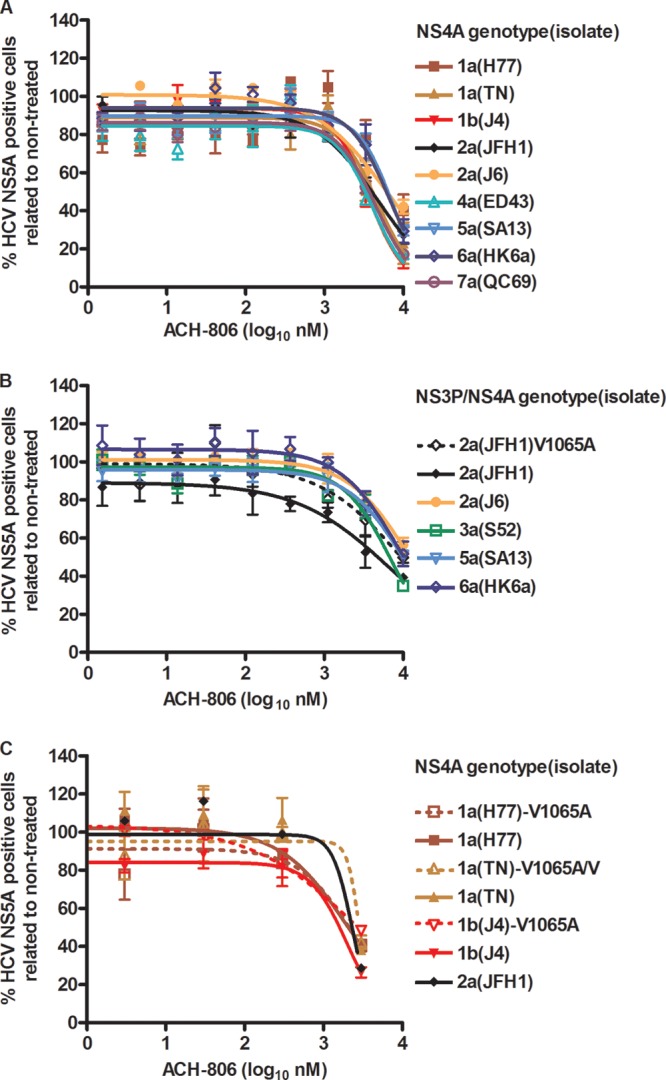
HCV NS4A and NS3P/NS4A recombinants show similar sensitivities to a putative NS4A inhibitor. Cell cultures in 96-well plates were infected with recombinants with NS4A (A and C) or NS3P/NS4A (B) of the indicated genotypes(isolates) and with the indicated aa substitutions. ACH-806 at the indicated concentrations was administered at 24 and 48 h postinfection; each concentration was tested in triplicate. Cells were immunostained for HCV NS5A, and single positive cells were counted automatically. Percentages of HCV NS5A-positive cells were calculated by comparison to the mean count for six nontreated cultures. After logarithmic transformation of x values, a sigmoidal concentration-response curve was fitted as described in Materials and Methods. Error bars indicate SEM for 3 replicates. No significant cellular toxicity was observed for the given concentration range; cell viability data are shown in Fig. S3 in the supplemental material. Virus stocks used for infection were as follows. For 2a(JFH1) and original NS4A recombinants without the V1065A substitution, virus stocks are detailed in Table 3, footnote a. 2a(JFH1)V1065A (B), which also had nt change T2656C, coding for the F772S substitution, was a 1st-passage virus stock without further coding ORF mutations. Virus stocks of NS3P/NS4A recombinants (B) are detailed in Table 2 of reference 14. 1a(H77), 1a(TN), and 1b(J4) NS4A recombinants with the V1065A substitution (C) were 1st-passage viruses whose NS3-NS4A sequences were determined. 1a(H77)V1065A had acquired two complete nt changes, encoding A1042T (NS3P) and N1544D (NS3H) substitutions. 1a(TN)V1065A had acquired two nt changes, estimated to be present in 50% of genomes, coding for reversion of the V1065A substitution and coding for the K1643E substitution (NS3H). 1b(J4)V1065A did not acquire nt changes in the NS3-NS4A region.
The A1065V amino acid change (aa 39 of NS3P) was suggested to confer resistance of genotype 1b replicons to ACH-806 (11, 13). To exclude the possibility that the limited efficacy of ACH-806 against the developed J6/JFH1-based NS4A recombinants was caused by the genotype 2a-specific valine at aa position 1065, we constructed 2a(JFH1)V1065A. The V1065A amino acid change was maintained following transfection and 1st passage in Huh7.5 cells. However, compared to 2a(JFH1), ACH-806 did not show increased efficacy against this mutated recombinant (Fig. 4B). In addition, ACH-806 showed only limited efficacy against J6/JFH1-based recombinants with genotype(isolate) 3a(S52)-, 5a(SA13)-, and 6a(HK6a)-specific NS3P/NS4A (14), which all have alanine at NS3P position 1065 (Fig. 4B). Because most inhibitors were designed to target genotype 1 and thus to show optimal efficacy against genotype 1 isolates, we constructed J6/JFH1-based 1a(H77), 1a(TN), and 1b(J4) NS4A recombinants with the V1065A amino acid change. First-passage 1a(H77) and 1b(J4) viruses maintained the V1065A substitution, while the introduced change partially reverted for 1a(TN) (see the legend to Fig. 4 for details). However, these mutants also showed sensitivities to ACH-806 similar to those of the original viruses with genotype 1-specific NS4A and of 2a(JFH1) (Fig. 4C).
The C1042S aa change was also suggested to confer resistance of genotype 1b replicons to ACH-806 (11, 13). J6/JFH1-based NS4A recombinants (Fig. 4A) have an alanine at aa position 1042, while J6/JFH1-based NS3P/NS4A recombinants 3a(S52), 5a(SA13), and 6a(HK6a) (Fig. 4B) have threonine, alanine, and threonine, respectively, at aa position 1042. To test the effects of aa changes at position 1042 on NS4A recombinants, we introduced either the single substitution A1042S or the A1042C substitution in combination with the V1065A substitution in 2a(JFH1), 1a(H77), 1a(TN), and 1b(J4) NS4A recombinants. Introduced substitutions were maintained following transfection and 1st and 2nd viral passages (see the legend to Fig. 5 for details). These changes did, however, not have a major influence on susceptibility to ACH-806 (Fig. 5). Nevertheless, for A1042C/V1065A mutants, we observed a trend toward lower EC50s. Thus, EC50 values for the double mutants were 1.6- to 2.1-fold lower than those of the original recombinants (P < 0.05 for all tested pairs). In conclusion, valine at NS3P position 1065 and alanine at NS3P position 1042, with both positions implicated in resistance to ACH-806, were apparently not the only cause for the relatively limited sensitivities of the developed NS4A recombinants to ACH-806. In addition, serine at NS3P position 1042 did not confer ACH-806 resistance to J6/JFH1-based recombinants with genotype 1a-, 1b-, or 2a-specific NS4A.
Fig 5.

Amino acid substitutions implicated in genotype 1b replicon resistance did not change sensitivity of HCV NS4A recombinants to ACH-806. Cell cultures in 96-well plates were infected with recombinants with NS4A proteins of the indicated genotypes(isolates) and the indicated aa substitutions. ACH-806 at the indicated concentrations was administered at 24 h postinfection; each concentration was tested in triplicate. Cells were immunostained for HCV NS5A, and single positive cells were counted automatically. Percentages of HCV NS5A-positive cells were calculated by comparison to the mean count for six nontreated cultures. After logarithmic transformation of x values, a sigmoidal concentration-response curve was fitted as described in Materials and Methods. Error bars indicate SEM for 3 replicates. No significant cellular toxicity was observed for the given concentration range. Virus stocks used for infection were as follows. For 2a(JFH1) and original NS4A recombinants, virus stocks are detailed in Table 3, footnote a. Recombinants with A1042S or A1042C and V1065A substitutions were 2nd-passage viruses whose NS3-NS4A sequences were determined. 1b(J4)A1042S had acquired one additional coding nt change, estimated to be present in 50% of genomes, coding for the I1061V substitution (NS3P). 1a(TN)A1042C,V1065A had acquired one additional complete coding nt change, encoding the K1398I substitution (NS3H). The other recombinants did not acquire nt changes in the NS3-NS4A region.
Finally, to investigate if the intergenotypic nature of JFH1-based NS4A recombinants had an influence on sensitivity to ACH-806, we carried out treatment experiments with recently developed full-length recombinants (27, 28). For genotype 1a (isolate TN) and genotype 2a (isolate J6), NS4A recombinants as well as full-length recombinants were available. In head-to-head comparisons, no major difference in sensitivity to ACH-806 was recorded for 1a(TN) and 2a(J6) NS4A recombinants versus full-length recombinants (Fig. 6).
Fig 6.
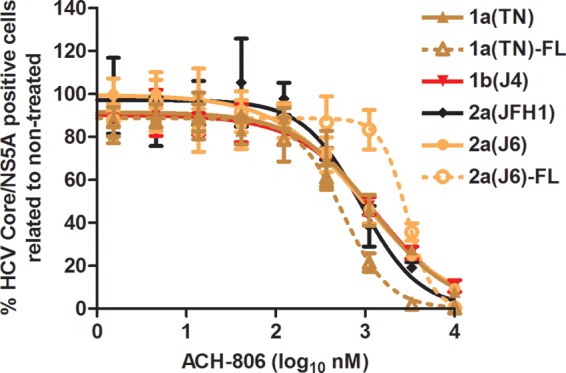
Genotype 1a and 2a HCV NS4A versus full-length recombinants show similar sensitivities to a putative NS4A inhibitor. Cell cultures in 96-well plates were infected with J6/JFH1-based recombinants with genotype(isolate) 1a(TN)-, 1b(J4)-, or 2a(J6)-specific NS4A or with genotype(isolate) 1a(TN) and 2a(J6) full-length recombinants, designated 1a(TN)-FL and 2a(J6)-FL; 2a(JFH1) was included as a control. ACH-806 at the indicated concentrations was administered at 24 h postinfection; each concentration was tested in triplicate. Cells were immunostained for HCV core/NS5A, and single positive cells were counted automatically. Percentages of HCV core/NS5A-positive cells were calculated by comparison to the mean count for six nontreated cultures. After logarithmic transformation of x values, a sigmoidal concentration-response curve was fitted as described in Materials and Methods. Error bars indicate SEM for 3 replicates. No significant cellular toxicity was observed for the given concentration range; cell viability data are shown in Fig. S3 in the supplemental material. Virus stocks used for infection were as follows. For 2a(JFH1) and NS4A recombinants, virus stocks are detailed in Table 3, footnote a. The 1a(TN)-FL virus stock is described in the legend to Fig. 2 in reference 27. The 2a(J6)-FL virus stock is described in the legend to Fig. 3 in reference 28.
NS4A recombinants spread under ACH-806 treatment pressure without acquisition of resistance mutations in the NS3-NS4A region.
Culture under ACH-806 treatment pressure led to the identification of genotype 1b replicon resistance mutations coding for A1065V and C1042S substitutions (11). To further investigate if the NS3-NS4A region was involved in mediating resistance to ACH-806, we aimed at inducing viral escape in long-term cultures treated with ACH-806. We determined that cells could be cultured for several weeks with 1,000 nM ACH-806, while concentrations of 2,000 nM and higher led to cytotoxic effects in this period. ACH-806 at 1,000 nM equals less than 1× EC50 as estimated for the individual NS4A recombinants (Fig. 4 to 7). Treatment of 2a(JFH1), 1a(H77), 1a(TN), and 1b(J4) NS4A recombinants with 1,000 nM ACH-806 delayed virus spread to at least 80% of culture cells by 5 to 11 days (Fig. 7A). By direct sequence analysis of the NS3-NS4A region, the following coding nt changes, all present in an estimated 50% of viral genomes, were identified: A4275G/A in 1a(H77), encoding aa substitution I1312V/I (NS3H); A4275G/A and T5386C/T in 1a(TN), encoding I1312V/I (NS3H) and I1682T/I (NS4A) substitutions, respectively; and G5436A/G in 1b(J4), encoding the E1699K/E substitution (NS4A). For 2a(JFH1), no coding nt changes were identified. We constructed 1a(H77)I1312V, 1a(TN)I1312V, 1a(TN)I1682T, 1a(TN)I1312V,I1682T, and 1b(J4)E1699K recombinants. Following transfection and viral passages in Huh7.5 cells, we found that in 2nd-passage virus stocks, the introduced mutations were maintained, except for in 1b(J4)E1699K, where the introduced change reverted. However, neither 1a(H77)I1312V nor the three 1a(TN) mutants showed resistance to ACH-806 (Fig. 7B).
Fig 7.
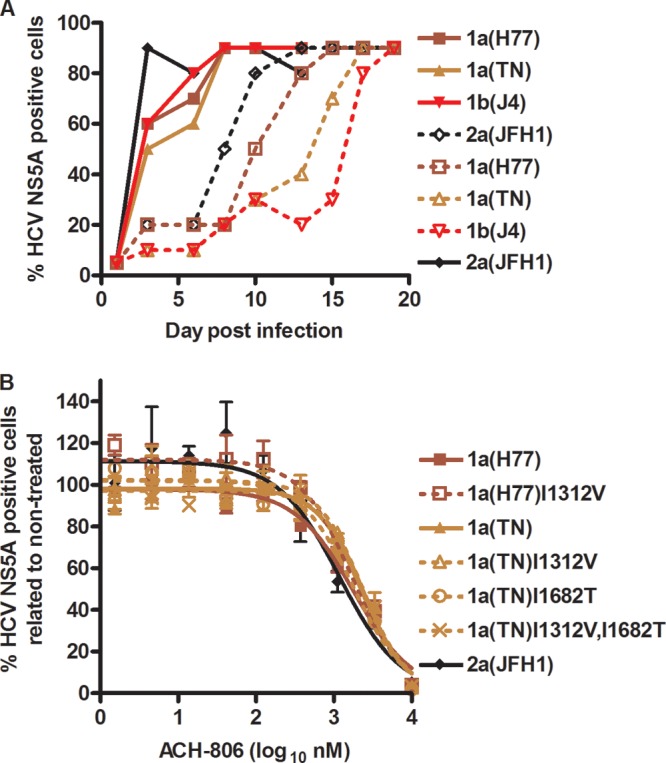
Viral spread under ACH-806 treatment occurred without selection of resistance mutations in the NS3-NS4A region. (A) Cell cultures in 6-well plates were infected at an MOI of 0.05 with the indicated NS4A recombinants and maintained without treatment (filled symbols and lines) or with 1,000 nM ACH-806 (empty symbols and broken lines) until viral spread to the almost-complete cell cultures occurred. Virus stocks used for infection are detailed in Table 3 legend. (B) NS4A recombinants with mutations identified by direct sequencing of the NS3-NS4A region were generated. Second-passage virus stocks of the indicated recombinants had maintained the introduced mutations, as determined by sequencing of the NS3-NS4A region. 1a(TN)I1682T had acquired one additional coding nt change, estimated to be present in 50% of genomes, coding for the T1504P substitution (NS3H). The other recombinants did not acquire nt changes in the NS3-NS4A region. For 2a(JFH1) and original NS4A recombinants, virus stocks are detailed in Table 3, footnote a. For treatments, cell cultures in 96-well plates were infected with the indicated recombinants. ACH-806 at the indicated concentrations was administered at 24 h postinfection; each concentration was tested in triplicate. Cells were immunostained for HCV NS5A, and single positive cells were counted automatically. Percentages of HCV NS5A-positive cells were calculated by comparison to the mean count for six nontreated cultures. After logarithmic transformation of x values, a sigmoidal concentration-response curve was fitted as described in Materials and Methods. Error bars indicate SEM for 3 replicates. No significant cellular toxicity was observed for the given concentration range.
In a single-cycle production assay, ACH-806 inhibited assembly of infectious intracellular viral particles.
To investigate which step of the viral replication cycle was affected by ACH-806, we carried out studies in S29 cells. These Huh7-derived cells are deficient in the HCV coreceptor CD81, and thus preclude HCV entry but permit studies of effects on viral replication, assembly, and release (32). We first tested NS4A recombinants 1a(TN), 1b(J4), and 2a(J6), as well as 2a(JFH1). For these recombinants, treatment with 4,000 and 8,000 nM ACH-806 for 44 h did not result in major changes of intracellular core levels, indicating that in this system the compound did not affect viral replication/translation (Fig. 8A). However, at 4,000 and 8,000 nM ACH-806, clear decreases in intracellular infectivity titers were observed, suggesting inhibition of viral assembly (Fig. 8B). We found a similar effect on extracellular infectivity titers, not exceeding the effect on intracellular titers, thus suggesting no added effect of ACH-806 on viral release (Fig. 8B). Finally, extracellular core levels decreased for the tested recombinants when 4,000 or 8,000 nM ACH-806 was administered (Fig. 8A).
Fig 8.
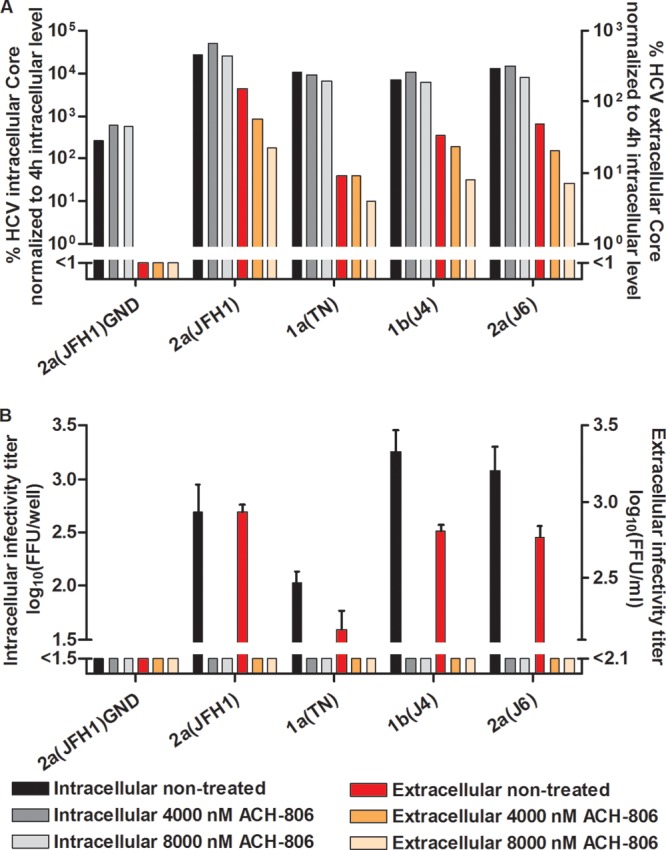
In a single-cycle production assay, ACH-806 affected assembly of infectious intracellular HCV particles for J6/JFH1 recombinants with genotype 1a-, 1b-, and 2a-specific NS4A. CD81-deficient S29 cell cultures were transfected in replicates with HCV RNA transcripts of J6/JFH1-based recombinants with genotype(isolate) 1a(TN)-, 1b(J4)-, or 2a(J6)-specific NS4A. 2a(JFH1) and 2a(JFH1)GND (20) were included as positive and negative controls, respectively. (A) After 4 h, cells of one replicate culture were harvested for determination of intracellular HCV core levels as described in Materials and Methods. The other replicate cultures were treated with the indicated concentrations of ACH-806 or maintained without treatment. At 48 h posttransfection, supernatants and cells of these replicate cultures were harvested; intra- and extracellular core levels were determined and normalized to the 4-h intracellular core levels to normalize for transfection efficiency. Core values are based on single determinations. (B) Cells and supernatants harvested at 48 h posttransfection were also used for determinations of intra- and extracellular infectivity titers in Huh7.5 cells. Infectivity titers are means for 3 replicates with SEM; the lower cutoffs for intracellular and extracellular titers were up to 1.5 log10 FFU/well and 2.1 log10 FFU/ml, respectively, as indicated by the column break. No significant cellular toxicity was observed for the ACH-806 concentrations applied.
To investigate if similar effects were observed at lower ACH-806 concentrations, we tested the effects of 4 to 4,000 nM ACH-806 on 2a(JFH1) as well as 1a(TN), 1b(J4), and 2a(J6) NS4A recombinants (Fig. 9). For all tested recombinants, a pronounced inhibition of viral assembly was observed at 4,000 nM ACH-806, but not at lower concentrations (Fig. 9). Finally, we carried out S29 treatment experiments with 1a(TN) and 2a(J6) full-length recombinants (27, 28) (Fig. 10). Also for these recombinants, we observed a pronounced inhibition of viral assembly at 4,000 nM ACH-806, with decreased intracellular infectivity titers, while viral replication was not affected (Fig. 10).
Fig 9.

Relatively high concentrations of ACH-806 were required for inhibition of viral assembly of genotype 1a, 1b, and 2a recombinants in a single-cycle production assay. CD81-deficient S29 cell cultures were transfected in replicates with HCV RNA transcripts of J6/JFH1-based recombinants with genotype(isolate) 1a(TN)-, 1b(J4)-, or 2a(J6)-specific NS4A. 2a(JFH1) and 2a(JFH1)GND (20) were included as positive and negative controls, respectively. (A) After 4 h, cells of one replicate culture were harvested for determination of intracellular HCV core levels as described in Materials and Methods. The other replicate cultures were treated with the indicated concentrations of ACH-806 or maintained without treatment. At 48 h posttransfection, supernatants and cells of these replicate cultures were harvested; intra- and extracellular core levels were determined and normalized to the 4-h intracellular core levels to normalize for transfection efficiency. Core values are based on single determinations. (B) Cells and supernatants harvested at 48 h posttransfection were also used for determinations of intra- and extracellular infectivity titers in Huh7.5 cells. Infectivity titers are means for 3 replicates with SEM; the lower cutoffs for intracellular and extracellular titers were up to 1.6 log10 FFU/well and 2.1 log10 FFU/ml, respectively, as indicated by the column break. No significant cellular toxicity was observed for the ACH-806 concentrations applied.
Fig 10.
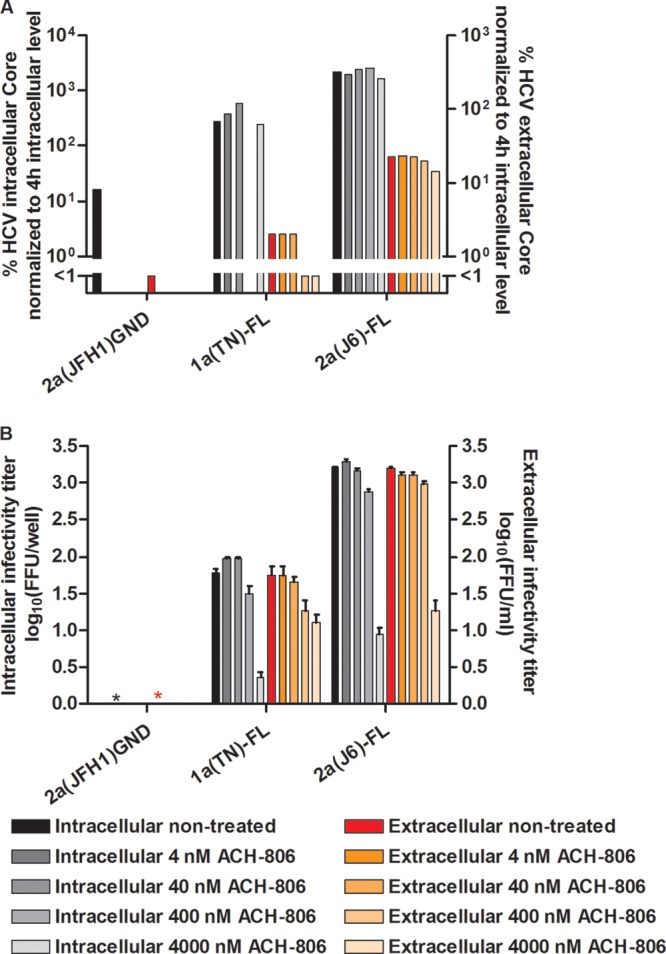
ACH-806 inhibited viral assembly of genotype 1a and 2a full-length recombinants. CD81-deficient S29 cell cultures were transfected in replicates with HCV RNA transcripts of genotype(isolate) 1a(TN) and 2a(J6) full-length recombinants, designated 1a(TN)-FL and 2a(J6)-FL (27, 28). 2a(JFH1)GND (20) was included as a negative control. (A) After 4 h, cells of one replicate culture were harvested for determination of intracellular HCV core levels as described in Materials and Methods. The other replicate cultures were treated with the indicated concentrations of ACH-806 or maintained without treatment. At 48 h posttransfection, supernatants and cells of these replicate cultures were harvested; intra- and extracellular core levels were determined and normalized to the 4-h intracellular core levels to normalize for transfection efficiency. Core values are based on single determinations. For 1a(TN)-FL treated with 400 nM ACH-806, the intracellular core value was not determined. (B) Cells and supernatants harvested at 48 h posttransfection were also used for determinations of intra- and extracellular infectivity titers in Huh7.5 cells. Infectivity titers are means for 3 replicates with SEM. Due to relatively low titers, FFU were counted manually. *, no FFU detected. No significant cellular toxicity was observed for the ACH-806 concentrations applied.
DISCUSSION
We developed efficient J6/JFH1 (2a)-based culture systems expressing NS4A proteins of other genotype (1a, 1b, 4a, 5a, 6a, and 7a) isolates. Efficient growth depended on adaptive mutations, with common mutations found in p7 and NS4A. Criteria for efficacy were relatively fast spread, peak infectivity titers of at least 3.5 log10 FFU/ml, and no obvious dependence on mutations in addition to engineered adaptive mutations. The developed recombinants with NS4A proteins of different genotype isolates did not show major differences in sensitivity to several lead compound protease inhibitors, IFN-alpha2b, or the putative NS4A inhibitor ACH-806. Relatively high doses of ACH-806 were found to inhibit viral assembly but not replication.
To date, efficient cell culture systems recapitulating the complete viral replication cycle have been published for isolates of genotype 1a (27) and genotypes 2a and 2b (19, 28, 32–34). Furthermore, JFH1-based culture-adapted recombinants with various genotype-specific regions were developed (14, 15, 20, 29, 31, 35–45). With the developed recombinants, we efficiently expressed genotype 1a, 1b, 2a, 4a, 5a, 6a, and 7a NS4A in the context of the complete viral replication cycle, producing ∼4 log10 FFU/ml. Expression of genotype 2a, 3a, 5a, and 6a NS4A in the context of the complete viral replication cycle was achieved previously in J6/JFH1-based recombinants with genotype-specific NS3P/NS4A (14, 15). Thus, in comparison to these prior studies (14, 15), expression of genotype 1a, 1b, 4a, and 7a NS4A in the context of the complete viral replication cycle is a novel achievement. Furthermore, this report describes for the first time the expression of genotype 1b, 4a, and 7a NS4A in the context of the complete viral replication cycle in cell culture. Most importantly, the developed panel of recombinants expressing genotype-specific NS4A in a constant backbone will be useful for studies of effects mediated by NS4A, not being influenced by sequence variation of other HCV genome elements.
Several aa substitutions adapting NS4A recombinants were also adaptive for J6/JFH1-based recombinants with genotype-specific NS3P/NS4A (14) or NS5A (43). The key adaptive substitution F772S in p7 also adapted 5a(SA13) and 6a(HK6a) NS3P/NS4A recombinants and was detected during culture adaptation of the 3a(S52) NS3P/NS4 recombinant (14); the F772S substitution further adapted 2a(J6) and 4a(ED43) NS5A recombinants (43) as well as the genotype 2a full-length recombinant J6cc (28). The key adaptive mutation L/V1663A in NS4A was adaptive for the 3a(S52) and 6a(HK6a) NS3P/NS4A recombinants (14, 15). Furthermore, substitution of aa 879 in NS2, adapting the 4a(ED43) NS4A recombinant, was also adaptive for the 3a(S52) NS3P/NS4A recombinant (14). Lastly, the 5a(SA13) NS3P/NS4A recombinant was adapted by the F880S substitution in NS2, adjacent to aa 879, adapting the 4a(ED43) NS4A recombinant, and the I1682L substitution, adjacent to aa 1683, adapting the 5a(SA13) NS4A recombinant (14).
Amino acid positions 772 in p7 and 1663 in NS4A, as well as 879 in NS2, localize to membrane-associated regions of these proteins, possibly indicating the importance of interaction of membrane-associated proteins during the viral replication cycle. F772, conserved for genotypes 1 to 3, localizes to position 26 of p7 in transmembrane domain 1 (TM1), pointing to the outside of the ion channel pore (46). The I879T substitution in putative TM3 of NS2 (8) could substitute for the F772S substitution in the 4a(ED43) NS4A recombinant; also, several aa substitutions adapting NS3P/NS4A recombinants localized to the N-terminal putative TM region of NS2, which is important for virus assembly (8, 47). The p7 and NS2 proteins are apparently not required for replication but for assembly, in interactions with various structural and nonstructural HCV proteins (8, 47–50). Because aa substitutions at position 772 in p7 and N-terminally in NS2 were adaptive for recombinants with genotype-specific NS4A, NS3P/NS4A, and NS5A, these residues might mediate interactions of p7 and NS2 with NS4A and NS5A. However, the fact that amino acid substitution at position 772 also adapted the genotype 2a full-length recombinant J6cc might argue for a role in interaction with cellular factors. Position 1663 is equivalent to position 6 of NS4A in the N-terminal transmembrane alpha-helix, which is important for membrane anchoring, formation of the replication complex, and replication (51). At this position, an alanine is present for most genotype 2a isolates, a leucine for most 3a isolates, and a valine for most other genotype isolates. For 2a(JFH1), exchange of A with V or L led to an ∼1.0- or 2.0-log10 reduction of infectivity titers following transfection; in 2nd-passage viruses, L had reverted to A, while V was maintained. Because 1663A was adaptive for all NS4A and most NS3P/NS4A recombinants, genotype-specific interaction of this residue with other J6 or JFH1 proteins seems to be required. Alternatively, 1663A might be required for efficient cleavage at the intergenotypic NS3H-NS4A junction.
Expression of genotype-specific NS4A sequences in a constant backbone allowed us to address the effect of the NS4A sequence on responses to protease inhibitors and a putative NS4A inhibitor. In treating the developed NS4A recombinants with lead compound protease inhibitors, as previously observed (14), linear inhibitors VX-950 and Sch503034 had lower potencies than macrocyclic inhibitors TMC435350, ITMN-191, and MK-7009 (Fig. 3; Table 3). HCV recombinants or replicating genomes with NS3P/NS4A of different genotype isolates showed differential sensitivity to protease inhibitors (14–16). In contrast, the genotype isolate of the NS3P cofactor NS4A did not have a major influence on sensitivity to the tested inhibitors (Fig. 3; Table 3). Thus, the specific NS4A sequence might not be of major concern for including protease inhibitors in future treatment regimens. The only consistent pattern of statistical significance involved slightly higher sensitivities of 1b(J4) and 6a(HK6a) NS4A recombinants to both linear inhibitors (Fig. 3; Table 3). The small differences in sensitivity observed might be caused by the nature of interaction of NS3P and NS4A (51) and/or the nature of interaction of the specific protease inhibitor with the target. Apparently, genotype-specific NS4A in the tested recombinants did not have a major influence on the response to IFN-alpha2b in vitro (Fig. 3F; Table 3). Thus, other genome regions might be main mediators of genotype-specific sensitivity seen in patients, or factors not present in the applied short-term assay might influence treatment outcomes.
Clinical development of the putative NS4A inhibitor ACH-806 was halted due to nephrotoxicity (11, 13). However, the current lead compound ACH NS4A inhibitor was not available for experiments. As previously described, for ACH-806, we found cell culture toxicity at concentrations above 10,000 nM in short-term treatment assays and above 1,000 nM in long-term treatment assays (see Fig. S3 in the supplemental material) (11). At noncytotoxic concentrations, we found similar efficacies against recombinants with NS4A proteins of various genotype isolates, indicating that antivirals with the same or similar mechanisms of action might show broad genotypic coverage. In the applied assays, ACH-806 showed a lower potency than protease inhibitors. Thus, in contrast to the case with protease inhibitors, after incubation with the highest permissible ACH-806 concentrations for 48 h, HCV antigen-positive cells were still observed in most cases (Fig. 3 to 7). Limited efficacy of ACH-806 was also apparent in the escape experiment, where the highest concentration permissible in long-term treatments led to only a relatively small delay in virus spread (Fig. 7). Furthermore, this viral spread apparently did not depend on resistance mutations in the suggested target regions.
Given the limited efficacy of ACH-806 in the applied assay, calculation of the EC50 was not possible for all treatments done; overall determinable EC50s were in the range of 1,000 to 10,000 nM, thus being several orders of magnitude higher than EC50 values determined in genotype 1 replicon systems (11–13). EC50 values strongly depend on the assay used. However, in the replicon system, long-term treatment also appeared to be more effective, resulting in selection of replicons with aa changes C1042S and A1065V in NS3P, conferring resistance to ACH-806 (11, 13). It seems unlikely that aa residues present at these positions were responsible for the limited efficacy of ACH-806 against NS4A recombinants. This is because 2a(JFH1) and the 1a and 1b NS4A recombinants with V1065A or A1042C/V1065A substitutions showed sensitivities to ACH-806 similar to those of 2a(JFH1) and the original NS4A recombinants (Fig. 4C and 5). Also, genotype 3a, 5a, and 6a NS3P/NS4A recombinants with alanine at aa 1065 and alanine or threonine at aa 1042 showed limited sensitivity to ACH-806 (Fig. 4B). It is notable, however, that the effects of specific aa substitutions might depend on the sequence context (52) as well as on the model system used. Thus, the effects of substitutions at aa 1065 and aa 1042 might be specific to the replicon system. Finally, it needs to be considered that isolated expression of NS4A outside the authentic sequence context might alter NS4A function. However, the limited efficacy of ACH-806 was apparently not due to the intergenotypic nature of the NS4A recombinants, because the 1a(TN) and 2a(J6) NS4A recombinants showed sensitivities to ACH-806 similar to those of full-length recombinants of the same genotype/isolate, in which NS4A was expressed in the authentic sequence context (Fig. 6).
Escape experiments in genotype 1b replicons led to selection of resistance mutations in an NS3P region interacting with NS4A (11). In this study, long-term treatment led to a relatively small delay in virus spread. While for 1a and 1b recombinants several quasispecies aa changes were observed in NS3H and NS4A of recovered viruses, these changes did not result in detectable ACH-806 resistance with the applied short-term assay (Fig. 7B). Due to limited efficacy of ACH-806 in the context of the complete viral replication cycle, the treated recombinants might have spread without selection of resistance mutations. We obtained similar results using protease and NS5A inhibitors, applying drug concentrations of ≤1× EC50 (45; unpublished data). It is less likely, but resistance mutations might localize to genome regions outside NS3-NS4A.
Surprisingly, using a single-cycle production assay (32), we found that ACH-806 did not inhibit viral replication. However, relatively high concentrations of ACH-806 (4,000 and 8,000 nM) showed a clear effect on viral assembly (Fig. 8 to 10); these concentrations also had an antiviral effect in high-throughput treatment assays in Huh7.5 cells (Fig. 4 to 7). Thus, the effect of ACH-806 in Huh7.5 cells might be explained by inhibition of assembly as observed in S29 cells, which, like Huh7.5 cells, are derived from Huh7 cells (32). As in Huh7.5 cells (Fig. 6), ACH-806 had similar effects on 1a(TN) and 2a(J6) full-length versus NS4A recombinants in S29 cells (Fig. 8 to 10). Earlier this year, Yang et al. provided a detailed study on the potential mechanism of action of ACH-806 (13). ACH-806 was found to selectively bind to NS4A, to lead to formation of an NS4A dimer, to reduce NS4A as well as NS3 stability, and to impair NS3-NS4A interaction. These findings could explain a negative effect on assembly, as NS4A and NS3 are involved in regulation of viral assembly (8, 9). However, it is difficult to explain why the potent effect of ACH-806 on viral replication observed in the replicon system, allowing only studies of viral replication, is not observed in the Huh7.5 cell culture system, mimicking the complete viral replication cycle, or in the S29 cell single-cycle production assay, allowing studies of viral replication, assembly, and release. These findings are most likely explained by differences between these experimental systems. Culture systems mimicking the complete viral replication cycle might more accurately reflect the situation in vivo. In line with this, using such systems for studies of antivirals, we so far obtained findings that were in good agreement with clinical observations (14, 27, 28, 43). Although beyond the scope of this study, it would be interesting to carry out studies similar to those carried out by Yang et al. (13) in the context of the complete viral replication cycle.
In conclusion, we developed efficient adapted culture systems with NS4A proteins of important HCV genotypes. To show the applicability of these systems, we studied various antivirals whose efficacy might be influenced by NS4A. These studies revealed that the specific NS4A sequence did not have a major influence on the efficacy of protease inhibitors or IFN-alpha2b in vitro. We also found that the putative NS4A inhibitor ACH-806 had similar, though limited, efficacies against the developed panel of NS4A recombinants. In the context of culture systems allowing the study of various steps of the viral replication cycle, relatively high concentrations of ACH-806 apparently inhibited viral assembly, not replication. The developed systems could be used to study next-generation inhibitors with mechanisms of action similar to that of ACH-806. Inhibitors with greater efficacy in the context of the complete viral replication cycle might allow identification of resistance mutations and might thus contribute to further elucidating the mechanism of action of this compound class. The developed systems could also be used to study the function of NS4A in a genotype-specific manner.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anna Louise Sørensen for technical help, Steen Ladelund for statistical advice, and Jens Ole Nielsen, Ove Andersen, and Kristian Schønning for support (Copenhagen University Hospital, Hvidovre, Denmark). We thank Bodil Nielsen and Karina Jensen for technical support (Aalborg University Hospital). We thank Charles Rice (Rockefeller University), Suzanne Emerson, and Robert Purcell (both from the National Institutes of Health) for reagents and CTL Europe GmbH for customized software.
This work was supported by grants from Copenhagen University Hospital, Hvidovre (J.M.G., S.B.N.S., and T.K.H.S.), the Region H Foundation (J.M.G. and J.B.), The Lundbeck Foundation (J.M.G., T.K.H.S., and J.B.), The Novo Nordisk Foundation (J.M.G. and J.B.), The Danish Council for Independent Research, Medical Science (J.B.), The A. P. Møller and Chastine Mc-Kinney Møller Foundation (J.M.G., T.K.H.S., and J.B.), and The Danish Cancer Society (J.M.G. and J.B.), by a master's stipend from The Danish Cancer Society (S.B.J.), and by Ph.D. stipends from the Faculty of Health Sciences, University of Copenhagen (S.B.N.S., T.K.H.S., and T.B.J.).
Footnotes
Published ahead of print 23 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01176-13.
REFERENCES
- 1. Gottwein JM, Bukh J. 2008. Cutting the gordian knot—development and biological relevance of hepatitis C virus cell culture systems. Adv. Virus Res. 71:51–133 [DOI] [PubMed] [Google Scholar]
- 2. Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin IT, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A. 2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42:962–973 [DOI] [PubMed] [Google Scholar]
- 3. Antaki N, Craxi A, Kamal S, Moucari R, Van der Merwe S, Haffar S, Gadano A, Zein N, Lai CL, Pawlotsky JM, Heathcote EJ, Dusheiko G, Marcellin P. 2010. The neglected hepatitis C virus genotypes 4, 5 and 6: an international consensus report. Liver Int. 30:342–355 [DOI] [PubMed] [Google Scholar]
- 4. Dustin LB, Rice CM. 2007. Flying under the radar: the immunobiology of hepatitis C. Annu. Rev. Immunol. 25:71–99 [DOI] [PubMed] [Google Scholar]
- 5. Beran RK, Lindenbach BD, Pyle AM. 2009. The NS4A protein of hepatitis C virus promotes RNA-coupled ATP hydrolysis by the NS3 helicase. J. Virol. 83:3268–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lindenbach BD, Pragai BM, Montserret R, Beran RK, Pyle AM, Penin F, Rice CM. 2007. The C terminus of hepatitis C virus NS4A encodes an electrostatic switch that regulates NS5A hyperphosphorylation and viral replication. J. Virol. 81:8905–8918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shiryaev SA, Chernov AV, Shiryaeva TN, Aleshin AE, Strongin AY. 2011. The acidic sequence of the NS4A cofactor regulates ATP hydrolysis by the HCV NS3 helicase. Arch. Virol. 156:313–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phan T, Beran RK, Peters C, Lorenz IC, Lindenbach BD. 2009. Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 83:8379–8395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stapleford KA, Lindenbach BD. 2011. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 85:1706–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sarrazin C, Hezode C, Zeuzem S, Pawlotsky JM. 2012. Antiviral strategies in hepatitis C virus infection. J. Hepatol. 56(Suppl 1):S88–S100 [DOI] [PubMed] [Google Scholar]
- 11. Yang W, Zhao Y, Fabrycki J, Hou X, Nie X, Sanchez A, Phadke A, Deshpande M, Agarwal A, Huang M. 2008. Selection of replicon variants resistant to ACH-806, a novel hepatitis C virus inhibitor with no cross-resistance to NS3 protease and NS5B polymerase inhibitors. Antimicrob. Agents Chemother. 52:2043–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wyles DL, Kaihara KA, Schooley RT. 2008. Synergy of a hepatitis C virus (HCV) NS4A antagonist in combination with HCV protease and polymerase inhibitors. Antimicrob. Agents Chemother. 52:1862–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang W, Sun Y, Hou X, Zhao Y, Fabrycki J, Chen D, Wang X, Agarwal A, Phadke A, Deshpande M, Huang M. 2013. ACH-806, an NS4A antagonist, inhibits hepatitis C virus replication by altering the composition of viral replication complexes. Antimicrob. Agents Chemother. 57:3168–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gottwein JM, Scheel TK, Jensen TB, Ghanem L, Bukh J. 2011. Differential efficacy of protease inhibitors against HCV genotypes 2a, 3a, 5a, and 6a NS3/4A protease recombinant viruses. Gastroenterology 141:1067–1079 [DOI] [PubMed] [Google Scholar]
- 15. Imhof I, Simmonds P. 2010. Development of an intergenotypic hepatitis C virus (HCV) cell culture method to assess antiviral susceptibilities and resistance development of HCV NS3 protease genes from HCV genotypes 1 to 6. J. Virol. 84:4597–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Imhof I, Simmonds P. 2011. Genotype differences in susceptibility and resistance development of hepatitis C virus to protease inhibitors telaprevir (VX-950) and danoprevir (ITMN-191). Hepatology 53:1090–1099 [DOI] [PubMed] [Google Scholar]
- 17. Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. 2006. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. U. S. A. 103:2310–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pietschmann T, Zayas M, Meuleman P, Long G, Appel N, Koutsoudakis G, Kallis S, Leroux-Roels G, Lohmann V, Bartenschlager R. 2009. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog. 5:e1000475. 10.1371/journal.ppat.1000475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 21. Yanagi M, Purcell RH, Emerson SU, Bukh J. 1997. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc. Natl. Acad. Sci. U. S. A. 94:8738–8743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sakai A, Takikawa S, Thimme R, Meunier JC, Spangenberg HC, Govindarajan S, Farci P, Emerson SU, Chisari FV, Purcell RH, Bukh J. 2007. In vivo study of the HC-TN strain of hepatitis C virus recovered from a patient with fulminant hepatitis: RNA transcripts of a molecular clone (pHC-TN) are infectious in chimpanzees but not in Huh7.5 cells. J. Virol. 81:7208–7219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yanagi M, St Claire M, Shapiro M, Emerson SU, Purcell RH, Bukh J. 1998. Transcripts of a chimeric cDNA clone of hepatitis C virus genotype 1b are infectious in vivo. Virology 244:161–172 [DOI] [PubMed] [Google Scholar]
- 24. Yanagi M, Purcell RH, Emerson SU, Bukh J. 1999. Hepatitis C virus: an infectious molecular clone of a second major genotype (2a) and lack of viability of intertypic 1a and 2a chimeras. Virology 262:250–263 [DOI] [PubMed] [Google Scholar]
- 25. Gottwein JM, Scheel TK, Callendret B, Li YP, Eccleston HB, Engle RE, Govindarajan S, Satterfield W, Purcell RH, Walker CM, Bukh J. 2010. Novel infectious cDNA clones of hepatitis C virus genotype 3a (strain S52) and 4a (strain ED43): genetic analyses and in vivo pathogenesis studies. J. Virol. 84:5277–5293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bukh J, Meuleman P, Tellier R, Engle RE, Feinstone SM, Eder G, Satterfield WC, Govindarajan S, Krawczynski K, Miller RH, Leroux-Roels G, Purcell RH. 2010. Challenge pools of hepatitis C virus genotypes 1–6 prototype strains: replication fitness and pathogenicity in chimpanzees and human liver-chimeric mouse models. J. Infect. Dis. 201:1381–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li YP, Ramirez S, Jensen SB, Purcell RH, Gottwein JM, Bukh J. 2012. Highly efficient full-length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc. Natl. Acad. Sci. U. S. A. 109:19757–19762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li YP, Ramirez S, Gottwein JM, Scheel TK, Mikkelsen L, Purcell RH, Bukh J. 2012. Robust full-length hepatitis C virus genotype 2a and 2b infectious cultures using mutations identified by a systematic approach applicable to patient strains. Proc. Natl. Acad. Sci. U. S. A. 109:E1101–E1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gottwein JM, Scheel TK, Hoegh AM, Lademann JB, Eugen-Olsen J, Lisby G, Bukh J. 2007. Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology 133:1614–1626 [DOI] [PubMed] [Google Scholar]
- 30. Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gottwein JM, Scheel TK, Jensen TB, Lademann JB, Prentoe JC, Knudsen ML, Hoegh AM, Bukh J. 2009. Development and characterization of hepatitis C virus genotype 1–7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49:364–377 [DOI] [PubMed] [Google Scholar]
- 32. Russell RS, Meunier JC, Takikawa S, Faulk K, Engle RE, Bukh J, Purcell RH, Emerson SU. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 105:4370–4375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Date T, Kato T, Kato J, Takahashi H, Morikawa K, Akazawa D, Murayama A, Tanaka-Kaneko K, Sata T, Tanaka Y, Mizokami M, Wakita T. 2012. Novel cell culture-adapted genotype 2a hepatitis C virus infectious clone. J. Virol. 86:10805–10820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ramirez S, Li YP, Jensen SB, Pedersen J, Gottwein JM, Bukh J. 2013. Highly efficient infectious cell culture of three HCV genotype 2b strains and sensitivity to lead protease, NS5A, and polymerase inhibitors. Hepatology 10.1002/hep.26660 [DOI] [PubMed] [Google Scholar]
- 35. Li YP, Ramirez S, Gottwein JM, Bukh J. 2011. Non-genotype-specific role of the hepatitis C virus 5′ untranslated region in virus production and in inhibition by interferon. Virology 421:222–234 [DOI] [PubMed] [Google Scholar]
- 36. Li YP, Gottwein JM, Scheel TK, Jensen TB, Bukh J. 2011. MicroRNA-122 antagonism against hepatitis C virus genotypes 1–6 and reduced efficacy by host RNA insertion or mutations in the HCV 5′ UTR. Proc. Natl. Acad. Sci. U. S. A. 108:4991–4996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408–7413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yi M, Lemon SM. 2004. Adaptive mutations producing efficient replication of genotype 1a hepatitis C virus RNA in normal Huh7 cells. J. Virol. 78:7904–7915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Scheel TK, Gottwein JM, Jensen TB, Prentoe JC, Hoegh AM, Alter HJ, Eugen-Olsen J, Bukh J. 2008. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc. Natl. Acad. Sci. U. S. A. 105:997–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jensen TB, Gottwein JM, Scheel TK, Hoegh AM, Eugen-Olsen J, Bukh J. 2008. Highly efficient JFH1-based cell-culture system for hepatitis C virus genotype 5a: failure of homologous neutralizing-antibody treatment to control infection. J. Infect. Dis. 198:1756–1765 [DOI] [PubMed] [Google Scholar]
- 41. Scheel TK, Gottwein JM, Carlsen TH, Li YP, Jensen TB, Spengler U, Weis N, Bukh J. 2011. Efficient culture adaptation of hepatitis C virus recombinants with genotype-specific core-NS2 by using previously identified mutations. J. Virol. 85:2891–2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gottwein JM, Jensen TB, Mathiesen CK, Meuleman P, Serre SB, Lademann JB, Ghanem L, Scheel TK, Leroux-Roels G, Bukh J. 2011. Development and application of hepatitis C reporter viruses with genotype 1 to 7 core-nonstructural protein 2 (NS2) expressing fluorescent proteins or luciferase in modified JFH1 NS5A. J. Virol. 85:8913–8928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Scheel TK, Gottwein JM, Mikkelsen LS, Jensen TB, Bukh J. 2011. Recombinant HCV variants with NS5A from genotypes 1–7 have different sensitivities to an NS5A inhibitor but not interferon-alpha. Gastroenterology 140:1032–1042 [DOI] [PubMed] [Google Scholar]
- 44. Murayama A, Kato T, Akazawa D, Sugiyama N, Date T, Masaki T, Nakamoto S, Tanaka Y, Mizokami M, Yokosuka O, Nomoto A, Wakita T. 2012. Production of infectious chimeric hepatitis C virus genotype 2b harboring minimal regions of JFH-1. J. Virol. 86:2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gottwein JM, Jensen SB, Li YP, Ghanem L, Scheel TK, Serre SB, Mikkelsen L, Bukh J. 2013. Combination treatment with hepatitis C virus protease and NS5A inhibitors is effective against recombinant genotype 1a, 2a, and 3a viruses. Antimicrob. Agents Chemother. 57:1291–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Patargias G, Zitzmann N, Dwek R, Fischer WB. 2006. Protein-protein interactions: modeling the hepatitis C virus ion channel p7. J. Med. Chem. 49:648–655 [DOI] [PubMed] [Google Scholar]
- 47. Jirasko V, Montserret R, Lee JY, Gouttenoire J, Moradpour D, Penin F, Bartenschlager R. 2010. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog. 6:e1001233. 10.1371/journal.ppat.1001233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T. 2007. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 3:e103. 10.1371/journal.ppat.0030103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. 2007. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 81:8374–8383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ma Y, Anantpadma M, Timpe JM, Shanmugam S, Singh SM, Lemon SM, Yi M. 2011. Hepatitis C virus NS2 protein serves as a scaffold for virus assembly by interacting with both structural and nonstructural proteins. J. Virol. 85:86–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brass V, Berke JM, Montserret R, Blum HE, Penin F, Moradpour D. 2008. Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3-4A complex. Proc. Natl. Acad. Sci. U. S. A. 105:14545–14550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sun JH, O'Boyle IDR, Zhang Y, Wang C, Nower P, Valera L, Roberts S, Nettles RE, Fridell RA, Gao M. 2012. Impact of a baseline polymorphism on the emergence of resistance to the hepatitis C virus nonstructural protein 5A replication complex inhibitor, BMS-790052. Hepatology 55:1692–1699 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.