

Abstract
The type II topoisomerases DNA gyrase (GyrA/GyrB) and topoisomerase IV (ParC/ParE) are well-validated targets for antibacterial drug discovery. Because of their structural and functional homology, these enzymes are amenable to dual targeting by a single ligand. In this study, two novel benzothiazole ethyl urea-based small molecules, designated compound A and compound B, were evaluated for their biochemical, antibacterial, and pharmacokinetic properties. The two compounds inhibited the ATPase activity of GyrB and ParE with 50% inhibitory concentrations of <0.1 μg/ml. Prevention of DNA supercoiling by DNA gyrase was also observed. Both compounds potently inhibited the growth of a range of bacterial organisms, including staphylococci, streptococci, enterococci, Clostridium difficile, and selected Gram-negative respiratory pathogens. MIC90s against clinical isolates ranged from 0.015 μg/ml for Streptococcus pneumoniae to 0.25 μg/ml for Staphylococcus aureus. No cross-resistance with common drug resistance phenotypes was observed. In addition, no synergistic or antagonistic interactions between compound A or compound B and other antibiotics, including the topoisomerase inhibitors novobiocin and levofloxacin, were detected in checkerboard experiments. The frequencies of spontaneous resistance for S. aureus were <2.3 × 10−10 with compound A and <5.8 × 10−11 with compound B at concentrations equivalent to 8× the MICs. These values indicate a multitargeting mechanism of action. The pharmacokinetic properties of both compounds were profiled in rats. Following intravenous administration, compound B showed approximately 3-fold improvement over compound A in terms of both clearance and the area under the concentration-time curve. The measured oral bioavailability of compound B was 47.7%.
INTRODUCTION
The significance and impact of antibiotic resistance on human health are widely recognized (1–3). Drug-resistant pathogens that have been identified to be of particular concern include methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant enterococci (VRE), penicillin- and fluoroquinolone-resistant Streptococcus pneumoniae (PRSP and FQSP, respectively), multidrug-resistant Gram-negative bacilli, and extensively drug-resistant (XDR) Mycobacterium tuberculosis (4, 5). The increase in antibiotic resistance has coincided with a decline in the rate of new antibacterial drug discovery (1, 6, 7). Addressing these twin issues involves the continuous discovery and development of new agents that are effective against drug-resistant pathogens. There are several strategies available for the discovery of new antibacterial agents, such as optimizing existing drugs or inhibiting novel targets (8). One approach, which is relevant to this study, is to develop novel compounds with new mechanisms of action against well-established targets.
The bacterial type II topoisomerases DNA gyrase and topoisomerase IV are essential and highly conserved enzymes that function to maintain DNA topology and integrity during replication, recombination, and transcription. DNA gyrase consists of two GyrA and two GyrB subunits in complex, while topoisomerase IV comprises two ParC and two ParE subunits. DNA gyrase and topoisomerase IV are attractive and clinically validated targets for antibacterial therapy (9–11). The quinolone/fluoroquinolone class of antibiotics, an example of which is ciprofloxacin, inhibits GyrA and ParC (12). GyrB is inhibited by the aminocoumarin antibiotics, exemplified by novobiocin (13, 14). There is a high degree of sequence and structural similarity between GyrA and ParC on the one hand and GyrB and ParE on the other. This offers the prospect of multitargeting, also referred to as polypharmacology, in which one ligand simultaneously inhibits two or more targets (15, 16). The compelling advantage of a rational, multitargeting approach in antibacterial design is that the level of spontaneous resistance development will likely be very low, thereby prolonging the potential clinical effectiveness of the therapeutic (17, 18).
Despite the clinical and commercial success of the quinolones and fluoroquinolones, their effectiveness is now limited by the prevalence of target-based resistance. This has prompted the search for new types of compounds with new mechanisms of action against the type II topoisomerases. In recent years, there has been considerable interest in discovering and developing novel inhibitors of both GyrB and ParE to inhibit the ATPase activities of DNA gyrase and topoisomerase IV (16, 18). This effort was stimulated by the elucidation of the crystal structures of GyrB and ParE (19, 20). The aminobenzimidazole class of dual-targeting ATPase inhibitors has been extensively characterized (21–23). Representative compounds from this series demonstrated potent in vitro bactericidal activity against Gram-positive pathogens, very low spontaneous resistance frequencies, and in vivo efficacy in multiple models of infection. Structurally related imidazolopyridine and triazolopyridine analogues with potent biochemical and antibacterial activity have also been described (24, 25). Alternative chemotypes with dual targeting activity have been reported by other workers (26–29; J. Dumas and B. Sherer, 5 March 2009, international patent application WO 2009/02773). Despite the considerable efforts made to develop these novel topoisomerase inhibitors, none have yet progressed into the clinic.
We have synthesized a series of benzothiazole ethyl urea compounds as inhibitors of both DNA gyrase and topoisomerase IV. In the present study, the biochemical, antibacterial, and pharmacokinetic evaluation of two representative compounds, designated compound A and compound B, is described. The chemical structures of the two compounds are shown in Fig. 1. Data on the in vitro activity of the two compounds against bacterial type II topoisomerase enzymes are presented. In addition, their whole-cell potency against drug-susceptible and -resistant bacterial isolates, mode of action, interaction with other antibiotics, propensity for spontaneous resistance development, level of cytotoxicity, and in vivo pharmacokinetic properties are also described.
Fig 1.
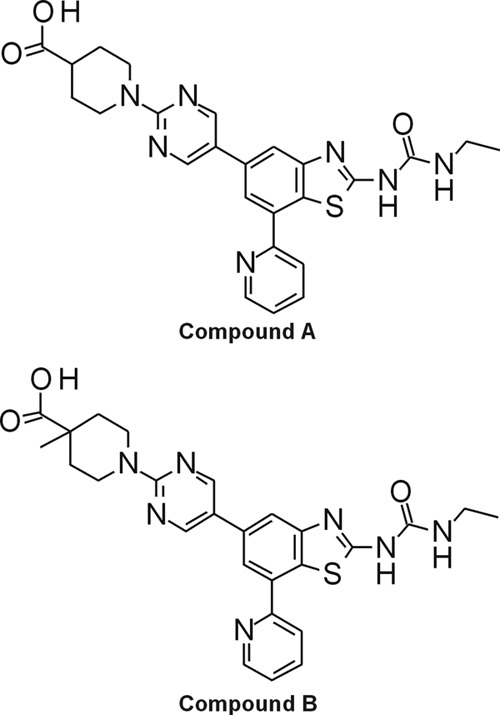
Chemical structures of compound A and compound B.
MATERIALS AND METHODS
Chemicals.
Compound A and compound B were synthesized at BioFocus DPI Ltd. (Saffron Walden, United Kingdom), Jubilant Chemsys Ltd. (Noida, India), and Biota Scientific Management Pty. Ltd. (Melbourne, Australia), as described elsewhere (30). Powder aliquots were dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 16 mg/ml and stored at −20°C. All other chemicals, including commercially available antimicrobials, were purchased from Sigma (Poole, United Kingdom). Microbiological media, agar, and growth supplements were purchased from Oxoid Ltd. (Basingstoke, United Kingdom). Horse serum was purchased from Southern Group Laboratory Ltd. (Corby, United Kingdom).
DNA gyrase and topoisomerase IV ATPase assays.
Gyrase B and ParE monomers have only modest ATPase activity which is enhanced in the case of the holoenzymes and stimulated further by DNA. The phosphate released following conversion of ATP into ADP can be detected by the addition of malachite green solution and measured by monitoring the increase in absorbance at 600 nm. Topoisomerases were purchased from Inspiralis Ltd. (Norwich, United Kingdom). For the Escherichia coli DNA gyrase ATPase assay, the final assay composition was 10 nM DNA gyrase (a complex of two GyrA and two GyrB subunits [the A2B2 complex]), 35 mM Tris, pH 7.5, 24 mM KCl, 2 mM MgCl2, 6.5% glycerol, 0.1 mg/ml bovine serum albumin (BSA), 2 mM dithiothreitol (DTT), 1 mM ATP, and 5% DMSO solution containing the compounds. For the E. coli topoisomerase IV ATPase assay, the final assay composition was 10 nM topoisomerase IV (a complex two ParC and two ParE subunits), 40 mM HEPES–KOH, pH 7.6, 100 mM potassium glutamate, 25 mM magnesium acetate, 10 μg/ml single-stranded DNA, 0.2 mg/ml BSA, 10 mM DTT, 0.5 mM ATP, and 5% DMSO solution containing the compounds. The reactions were started by the addition of the ATP, and the reaction mixtures were allowed to incubate at 30°C for 60 min. Reactions were stopped by adding malachite green solution (0.034% malachite green, 10 mM ammonium molybdate, 1 M HCl, 3.4% ethanol, 0.01% Tween 20). Color was allowed to develop for 5 min, and the absorbance at 600 nm was measured spectrophotometrically. The half-maximum (50%) inhibitory concentration (IC50) values were determined from the absorbance readings using no-compound and no-enzyme controls. The values reported are the averages of at least four independent experiments.
DNA gyrase supercoiling assay.
One unit of S. aureus DNA gyrase enzyme (A2B2 complex), supplied by Inspiralis Ltd. (Norwich, United Kingdom), was mixed with 0.5 μg of relaxed pBR322 DNA, also supplied by Inspiralis Ltd., in the presence or absence of compound or novobiocin at the concentrations indicated below. The final assay buffer contained 35 mM Tris HCl, pH 7.5, 24 mM KCl, 2 mM MgCl2, 2 mM DTT, 1.8 mM spermidine, 1 mM ATP, 6.5% (wt/vol) glycerol, and 0.1 mg/ml BSA. Reactions were run for 30 min at 37°C and stopped by the addition of an equal volume of chloroform-isoamyl alcohol (24:1). Gel loading buffer was added, and the samples were briefly centrifuged to separate the phases. Samples (15 μl) from the aqueous phases were resolved by electrophoresis through a Tris-acetate-EDTA (TAE) agarose gel (0.8%, wt/vol). DNA was stained with SYBR Safe DNA gel stain (Life Technologies Ltd., Paisley, United Kingdom) and visualized under UV light.
Bacterial strains.
The bacterial strains used in this study were obtained from the American Type Culture Collection (LGC Promochem, Teddington, United Kingdom) or the Biota Europe Ltd. laboratory collection and were propagated using standard microbiological procedures. Müller-Hinton (MH) agar or MH broth was used for the routine growth of bacterial strains at 37°C in an ambient atmosphere. Brain heart infusion (BHI) agar and broth supplemented with 5% (vol/vol) sterile filtered defibrinated horse serum and NAD, as necessary, were used to culture Haemophilus influenzae, Streptococcus pneumoniae, and Streptococcus pyogenes, H. influenzae, S. pneumoniae, and S. pyogenes were grown at 37°C in an atmosphere containing 5% CO2. Reinforced clostridial medium or reinforced clostridial agar was used to culture Clostridium difficile and Propionibacterium acnes at 37°C under anaerobic conditions. Yeast extract broth or agar supplemented with buffered charcoal yeast extract growth supplement was used to culture Legionella pneumophila at 37°C in an atmosphere containing 5% CO2. Gonococcal broth or agar supplemented with Vitox was used for the growth of Neisseria gonorrhoeae in an atmosphere containing 5% CO2.
Antimicrobial susceptibility testing.
MICs were determined by the broth microdilution or agar dilution method according to the recommendations of the Clinical and Laboratory Standards Institute (31, 32). Twofold variations in the MIC were considered to be within the acceptable reproducibility for the test. Where indicated, MICs were also measured in the presence of 50% horse serum. Testing of panels of clinical isolates of Enterococcus faecalis, Enterococcus faecium, Staphylococcus aureus, Staphylococcus epidermidis, S. pneumoniae, and S. pyogenes was performed by Quotient Bioresearch Ltd. (Fordham, United Kingdom). Antimicrobial combination MICs were determined using the checkerboard method (33). Interpretation of the fractional inhibitory concentration index (FICI) was as described previously (34).
Bactericidal activity assay.
The bactericidal activity of compounds was assessed by the time-kill method according to the recommendations of the Clinical and Laboratory Standards Institute (35). Briefly, a growing culture of S. aureus ATCC 29213 was diluted to approximately 105 CFU/ml in volumes of MH broth containing the concentrations of compounds described below or DMSO (negative control). Cultures were incubated at 37°C with agitation. At the time intervals indicated, 100-μl samples were removed and serially diluted in MH broth, and 100-μl volumes from these dilutions were spread onto MH agar. Cell counts (numbers of CFU/ml) were enumerated after incubating the plates until colonies were readily visible.
Determination of frequency of spontaneous resistance.
Cells of S. aureus ATCC 29213 were grown to late exponential phase (optical density at 600 nm = 0.9 ± 0.1; approximately 109 CFU/ml) and spread onto MH agar containing compound A or compound B at the concentrations indicated below. To determine the number of viable cells in the inoculum, cultures were serially diluted and plated on compound-free MH agar. Plates were incubated at 37°C for 48 h, and the colonies were enumerated. Putative resistant mutants were patched and streaked to single colonies on the same concentration of compound. The frequency of resistance (FoR) was calculated by dividing the number of resistant colonies by the number of CFU in the inoculum. In the case where no resistant colonies were isolated, the FoR was defined to be less than the number of CFU in the inoculum.
Isolation and characterization of first- and second-step compound-resistant mutants.
Spontaneous GyrB G85S and GyrB R144S first-step mutants were generated in S. aureus ATCC 29213 with novobiocin at 8× MIC. The GyrB T173N first-step mutant derived using S. aureus ATCC 29213 was isolated at 8× MIC of a benzothiazole ethyl urea analogue of compound A and compound B. Spontaneous ParE T172A and A53S first-step mutants were derived from S. pneumoniae ATCC 49619 and S. pyogenes ATCC 51339, respectively, at concentrations equivalent to 4× MIC of a different benzothiazole ethyl urea analogue of compound A and compound B. In each case, high-density, late-exponential-phase cultures were inoculated onto compound agar. Putative compound-resistant colonies that arose after incubation at 37°C were patched onto compound agar. The second-step mutants, one with GyrB T173N and GyrB S129T mutations and the other with GyrB T173N and ParE T167N mutations, were subsequently derived from the S. aureus GyrB T173N first-step mutant with other analogues of compound A and compound B following multiple passages. Chromosomal DNA was purified from putative mutants and amplified by PCR using the following oligonucleotides: for S. aureus gyrB, CGATTCAGCATAAAGTACAAACATTTGTC and GCACGAGCAACGATAACAC; for S. aureus parE, GCATTTACGCTGATTTATATAAGAATAACTATTG and GACTGTTTTCGCACTTTTA; for S. pneumoniae gyrB, GCAGCTTATTTTACAGAAGTGG, ATCAGTGATAGAAATTTGAAGACCGC, GGTTTCAGGTGGTCTTCACGGGG, GAAGAACAGTCTGCTAGTTTCC, GCCAATAATATTCATACACATGAAG, CTTGATCTGCACCCGGCTGG, GCAGACTGTTCTTCTAATAACCCTG, and CATTTACTGGAAGATTGTATAGTTC; for S. pneumoniae parE, ATGCGGAAGGTATTGGTCGTT, ATAGTCGCGTCAGGCATAAAAGTA, CCGATGGCGCTGGTCTTCA, AACGGCCGCTAGTCCCTCACGATA, AAGCGAACAGATGAAGCGATTGAG, GGCAGAACCACCGGCAGAGTC, TGACCCCAGCCCAATCTAAG, and CAAAGCCCATAAAATACCAAGTGA; for S. pyogenes gyrB, GTTTTAATACCTTGCTTGTTGACG and ATTAACGCTGTCCCTCAAGATG; and for S. pyogenes parE, GATGAAACTGTCTGGAGGTC and ATGGCGGCTTTCTATTATT. DNA fragments were purified, and full-length gyrB and parE genes were sequenced (GATC Biotech, London, United Kingdom). The amino acid substitutions described were the only ones detected by the sequencing analysis.
Plasma protein binding.
Protein binding was measured using an ultracentrifugation method with undiluted rat plasma (Sera Laboratories International Ltd., Haywards Heath, United Kingdom). Compounds were tested at 10 μM in a final DMSO concentration of 1% (vol/vol) and allowed to equilibrate at 37°C for 60 min. Duplicate samples were then centrifuged at 250,000 × g at 37°C for 260 min in a Sorvall MTX150 microultracentrifuge fitted with a S80-AT2 rotor (Thermo Scientific, Basingstoke, United Kingdom). Samples were extracted using acetonitrile and centrifugation. Supernatants were analyzed by liquid chromatography-mass spectrometry (LC-MS) to determine the free compound concentration by comparison with the concentration in samples that had not been centrifuged.
Human topoisomerase II relaxation assay.
Testing of compounds in the human topoisomerase II assay was performed by Inspiralis Ltd. (Norwich, United Kingdom). Compounds were prepared in DMSO and added to the reaction mixtures to a final compound concentration of 10 μM and a DMSO concentration of 10% (vol/vol). Reaction mixes comprised 0.5 μg supercoiled plasmid DNA (pBR322) and reaction buffer (50 mM Tris HCl at a pH of 7.5, 125 mM NaCl, 10 mM MgCl2, 5 mM DTT, 0.5 mM EDTA, 0.1 mg/ml BSA, 1 mM ATP). One unit of human topoisomerase II was added to all reaction mixtures, with 1 U defined to be the amount of enzyme required to just fully relax the substrate. The control compounds were levofloxacin, novobiocin, and m-amsacrine (AMSA). Following incubation at 37°C for 30 min, each reaction was stopped by the addition of chloroform-isoamyl alcohol (26:1) and stop dye, before the reaction mixture was loaded on a 1% TAE gel and run at 85 V for 90 min. Once the gels had been stained with ethidium bromide and processed with gel-scanning software, the level (percent) of inhibition (INH) was determined by measuring the amount of supercoiled plasmid DNA as a percentage of that for the no-enzyme negative control.
HepG2 cytotoxicity assay.
Human cell viability was assessed by measuring the levels of ATP using a CellTiter-Glo kit (Promega United Kingdom, Southampton, United Kingdom), according to the manufacturer's instructions. Cells were seeded at 5,000 cells/well in Eagle's minimal essential medium (EMEM) supplemented with 1% fetal bovine serum (FBS) and left to settle overnight. Compound dilutions were prepared in DMSO and added to cells to a DMSO concentration of 1% (vol/vol). The positive-control compound was chlorpromazine. Cells were incubated with compound at 37°C in 5% CO2 for 48 h. The CellTiter-Glo reagent was added, and the luminescence was read on a Tecan plate reader.
Pharmacokinetics.
Pharmacokinetic experiments were performed by GVK Biosciences Pvt. Ltd. (Hyderabad, India) in compliance with the Regulations of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India. Groups of healthy adult male Sprague-Dawley rats received compounds by intravenous (i.v.) administration as a single bolus through the tail vein at a dose level of 3 or 10 mg/kg of body weight or by oral (p.o.) administration via an oral gavage needle at a dose level of 3 mg/kg. In all cases the dose volume was 5 ml/kg. Compounds were formulated in 10% dimethyl acetamide (DMA), 40% tetraethylene glycol (TEG), and 10% 2-hydroxypropyl-β-cyclodextrin (HPβCD) in water (final concentrations). At the time points indicated below, samples of blood were collected for analysis. Three animals were used for each time point. Plasma concentrations of compounds were measured by LC-MS/MS, and pharmacokinetic parameters were calculated using WinNonlin software.
RESULTS
Inhibition of DNA gyrase and topoisomerase IV in vitro activity.
The compounds were tested for their ability to inhibit the ATPase function of DNA gyrase and topoisomerase IV in in vitro biochemical assays. Purified, recombinant enzymes from Escherichia coli were used. Novobiocin, a competitive inhibitor of the ATPase activity of the bacterial type II topoisomerases (36), was included as a control. Compound A and compound B potently inhibited the turnover of ATP by both the DNA gyrase and topoisomerase IV enzymes, with mean IC50s ranging from 0.0033 μg/ml to 0.046 μg/ml (Table 1). On a molar basis, the compounds inhibited the topoisomerase enzymes to a greater extent than novobiocin, with IC50s being between 3- and 11-fold lower than those of novobiocin. The effect of the compounds on the in vitro function of DNA gyrase was also measured using a gel-based DNA supercoiling assay. Relaxed plasmid DNA (pBR322) incubated with S. aureus DNA gyrase was converted to supercoiled DNA that migrated more rapidly through an agarose gel (Fig. 2). Inclusion of 1.0 μg/ml novobiocin or 1.0 μg/ml compound A in the reaction mixture prevented the conversion of the relaxed DNA to the supercoiled form (Fig. 2). Thus, compounds A and B are potent inhibitors of gyrase and topoisomerase in enzyme assays.
Table 1.
In vitro activity of compound A, compound B, and novobiocin against the ATPase function of the E. coli DNA gyrase and topoisomerase IV enzymes
| Compound | IC50 (μg/ml)a |
|
|---|---|---|
| DNA gyrase | Topoisomerase IV | |
| A | 0.0039 ± 0.0017 (0.0077 ± 0.0034) | 0.046 ± 0.030 (0.091 ± 0.060) |
| B | 0.0033 ± 0.0021 (0.0064 ± 0.0041) | 0.024 ± 0.012 (0.046 ± 0.023) |
| Novobiocin | 0.045 ± 0.027 (0.073 ± 0.044) | 0.18 ± 0.059 (0.29 ± 0.096) |
Data in parentheses are IC50s in μM.
Fig 2.
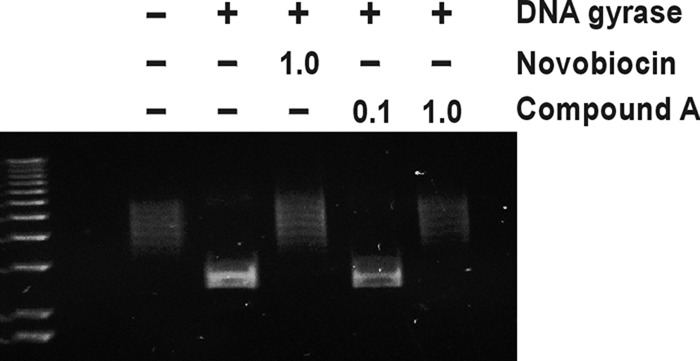
Effect of compound A on DNA supercoiling by DNA gyrase. Relaxed pBR322 DNA was incubated with or without S. aureus DNA gyrase enzyme and in the absence or presence of 1.0 μg/ml novobiocin (equivalent to 1.6 μM) or 0.1 or 1.0 μg/ml of compound A (equivalent to 0.2 or 2.0 μM, respectively). At the end of the assay, samples were resolved by agarose gel electrophoresis along with a DNA molecular weight marker (leftmost lane). The upper ladders of topoisomers represent relaxed (nonsupercoiled) DNA, while lower, tighter bands are supercoiled DNA.
Antibacterial profiles of compound A and compound B.
The scope of the antibacterial activity of the two compounds was determined by assaying their MICs against strains of clinically important Gram-positive and Gram-negative bacterial pathogens. The results are listed in Table 2. Data for linezolid and novobiocin are also included for comparison. Both compound A and compound B potently inhibited the growth of all Gram-positive species tested (MICs ≤ 0.12 μg/ml). The two compounds displayed similar levels of potency against these organisms. The MICs of the compounds increased up to 16-fold in mutant strains of S. aureus, S. pneumoniae, and S. pyogenes carrying spontaneous first-step mutations encoding single-point amino acid substitutions in either GyrB (T173N) or ParE (T172A or A53S) (Table 3). These mutants, which were derived from the respective type strains, had been previously isolated against analogues of compound A and compound B (see Materials and Methods). The S. aureus gyrB mutant (encoding the T173N substitution) and the S. pneumoniae parE mutant (encoding the T172A substitution) have been isolated and characterized with the aminobenzimidazole chemotype of GyrB/ParE inhibitors (22). The compound susceptibility data for the mutants support the in vitro biochemical data presented in Table 1, indicating that compound A and compound B target the type II topoisomerase enzymes.
Table 2.
Antibacterial activities of compound A, compound B, linezolid, and novobiocin
| Organism and strain | MIC (μg/ml) |
|||
|---|---|---|---|---|
| Linezolid | Novobiocin | Compound A | Compound B | |
| Acinetobacter baumannii ATCC 19606 | 64 | 8 | >64 | >64 |
| Clostridium difficile ATCC 9689 | 1 | 2 | 0.03 | 0.015 |
| Enterococcus faecalis ATCC 29212 | 2 | 4 | 0.015 | 0.03 |
| Escherichia coli ATCC 25922 | >64 | 64 | >64 | >64 |
| E. coli N43 (acrA) | 16 | 8 | 4 | 2 |
| Haemophilus influenzae ATCC 49247 | 4 | 0.5 | 2 | 2 |
| Klebsiella pneumoniae ATCC 13882 | 32 | 8 | >64 | >64 |
| Legionella pneumophila ATCC 33152 | 4 | 0.06 | 0.25 | 0.25 |
| Moraxella catarrhalis ATCC 25240 | 4 | 0.25 | 0.12 | 0.12 |
| Neisseria gonorrhoeae ATCC 49226 | 4 | 1 | 0.12 | 0.12 |
| Propionibacterium acnes ATCC 6919 | 0.5 | 2 | 0.015 | 0.03 |
| Pseudomonas aeruginosa ATCC 27853 | >64 | >64 | >64 | >64 |
| Salmonella enterica serovar Typhimurium ATCC 19585 | >64 | >64 | >64 | >64 |
| Staphylococcus aureus ATCC 29213 | 2 | 0.12 | 0.12 | 0.12 |
| S. aureus ATCC 29213 + 50% serum | 4 | 8 | 2 | 2 |
| Staphylococcus epidermidis ATCC 12228 | 1 | 0.06 | 0.03 | 0.03 |
| Streptococcus pneumoniae ATCC 49619 | 1 | 0.5 | 0.008 | 0.008 |
| Streptococcus pyogenes ATCC 51339 | 1 | 1 | 0.06 | 0.06 |
Table 3.
Potencies of compound A, compound B, linezolid, and novobiocin against topoisomerase mutant strains of S. aureus, S. pneumoniae, and S. pyogenes
| Organism and strain | MIC (μg/ml) |
|||
|---|---|---|---|---|
| Linezolid | Novobiocin | Compound A | Compound B | |
| Staphylococcus aureus ATCC 29213 | 2 | 0.12 | 0.12 | 0.12 |
| S. aureus GyrB G85S | 2 | 8 | 0.06 | 0.06 |
| S. aureus GyrB R144S | 2 | 8 | 0.06 | 0.06 |
| S. aureus GyrB T173N | 2 | 0.25 | 0.25 | 0.25 |
| S. aureus GyrB T173N, GyrB S129T | 2 | 1 | 0.25 | 0.12 |
| S. aureus GyrB T173N, ParE T167N | 2 | 1 | 16 | 4 |
| Streptococcus pneumoniae ATCC 49619 | 1 | 0.5 | 0.008 | 0.008 |
| S. pneumoniae ParE T172A | 1 | 0.5 | 0.12 | 0.12 |
| Streptococcus pyogenes ATCC 51339 | 1 | 1 | 0.06 | 0.06 |
| S. pyogenes ParE A53S | 1 | 1 | 0.5 | 0.5 |
Variable levels of potency against the Gram-negative species were observed. Both compounds had activity against the Gram-negative respiratory bacterial pathogens Legionella pneumophila, Moraxella catarrhalis, and Haemophilus influenzae. Neisseria gonorrhoeae was also susceptible to the two inhibitors. In contrast, the growth of wild-type Acinetobacter baumannii, E. coli, Pseudomonas aeruginosa, and Salmonella enterica serovar Typhimurium was not inhibited by either compound. The compound A and compound B MICs for the E. coli pump mutant strain N43 were 4 μg/ml and 2 μg/ml, respectively (Table 2), indicating that the lack of activity against the wild-type E. coli strain is due to export mediated by the AcrAB-TolC multidrug efflux transport system.
To determine whether the compounds were lethal, the compounds were tested at 1× and 8× MICs in a time-kill assay with S. aureus ATCC 29213. The results from representative experiments are shown in Fig. 3. Both compounds produced comparable time-kill curves at each concentration, causing a >99% reduction in the number of CFU within 24 h. This indicates a bactericidal mode of action (35) and has been seen previously with novobiocin (37). The bactericidal activity was concentration independent, as evidenced by the similarity of the profiles at the two concentrations. At the higher concentration, the rate of killing with compound A and compound B was less rapid than that measured with levofloxacin, which caused a >99% reduction in the number of CFU within 2 h. This observation is consistent with a mechanism of action of the benzothiazole ethyl ureas against the topoisomerase enzymes different from that of the quinolone class of antibiotics.
Fig 3.
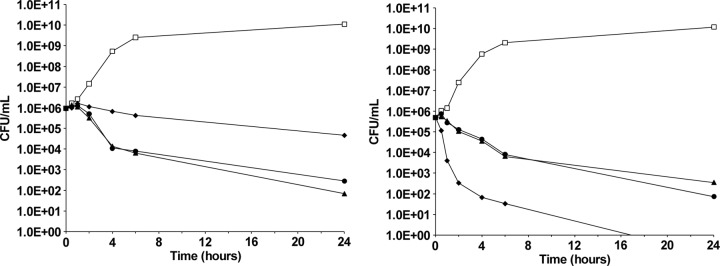
Time-kill profiles of compound A and compound B at 1× MIC (left) and 8× MIC (right). Cells of S. aureus ATCC 29213 were cultured in the presence of compound A (solid triangles), compound B (solid circles), levofloxacin (solid diamonds), or DMSO (negative control; open squares) for 24 h. Samples were taken at the time intervals indicated for the determination of viable cell numbers (numbers of CFU/ml) by serial dilution.
The two compounds were then tested against panels of clinical isolates of six major Gram-positive pathogens (Table 4). The compounds potently inhibited the growth of all 66 isolates tested. MIC90 values for both compounds ranged from 0.015 μg/ml for S. pneumoniae to 0.25 μg/ml for S. aureus, whereas the MIC90 values for the two organisms were 2 μg/ml and 4 μg/ml, respectively, for linezolid and >16 μg/ml and 16 μg/ml, respectively, for levofloxacin. Importantly, compound A and compound B showed narrow MIC ranges within the sets of isolates. The compounds retained excellent potency against characterized drug-resistant strains, including VRE, MRSA, vancomycin-resistant S. aureus, linezolid-nonsusceptible S. aureus, daptomycin-nonsusceptible S. aureus, methicillin-resistant S. epidermidis, PRSP, and FQSP strains, indicating that there is no cross-resistance caused by the mechanisms responsible for conferring resistance to these antibiotics (Table 5).
Table 4.
Antibacterial activities of compound A, compound B, linezolid, and levofloxacin against panels of clinical isolates of six major Gram-positive pathogens
| Organism (no. of isolates) | Compound | MIC (μg/ml) |
|
|---|---|---|---|
| Range | 90% | ||
| Enterococcus faecalis (10) | A | 0.015–0.03 | 0.03 |
| B | 0.015–0.06 | 0.06 | |
| Linezolid | 1–2 | 1 | |
| Levofloxacin | 1–>16 | >16 | |
| Enterococcus faecium (12) | A | 0.06–0.25 | 0.12 |
| B | 0.06–0.25 | 0.12 | |
| Linezolid | 1–4 | 2 | |
| Levofloxacin | 0.5–>16 | >16 | |
| Staphylococcus aureus (13) | A | 0.12–0.5 | 0.25 |
| B | 0.06–0.25 | 0.25 | |
| Linezolid | 1–16 | 4 | |
| Levofloxacin | 0.12–>16 | 16 | |
| Staphylococcus epidermidis (10) | A | 0.03–0.12 | 0.06 |
| B | ≤0.004–0.12 | 0.06 | |
| Linezolid | 0.5–8 | 1 | |
| Levofloxacin | 0.12–>16 | >16 | |
| Streptococcus pneumoniae (11) | A | ≤0.008–0.03 | 0.015 |
| B | ≤0.008–0.03 | 0.015 | |
| Linezolid | 0.5–2 | 2 | |
| Levofloxacin | 0.5–>16 | >16 | |
| Streptococcus pyogenes (10) | A | 0.06–0.12 | 0.12 |
| B | 0.12–0.12 | 0.12 | |
| Linezolid | 1–1 | 1 | |
| Levofloxacin | 0.25–1 | 0.5 | |
Table 5.
Antibacterial activities of compound A, compound B, linezolid, and levofloxacin against drug-resistant strains
| Organism | Phenotype | MIC (μg/ml) |
|||
|---|---|---|---|---|---|
| Linezolid | Levofloxacin | Compound A | Compound B | ||
| E. faecalis | Vancomycin resistant | 1 | >16 | 0.03 | 0.03 |
| E. faecium | Vancomycin resistant | 2 | >16 | 0.06 | 0.06 |
| S. aureus | Methicillin resistant | 1 | 0.12 | 0.25 | 0.25 |
| S. aureus | Vancomycin resistant | 1 | 16 | 0.5 | 0.25 |
| S. aureus | Linezolid nonsusceptible | 16 | 16 | 0.25 | 0.25 |
| S. aureus | Tigecycline resistant | 1 | 8 | 0.12 | 0.12 |
| S. aureus | Daptomycin nonsusceptible | 1 | >16 | 0.12 | 0.12 |
| S. aureus | Fluoroquinolone resistant | 1 | 16 | 0.25 | 0.25 |
| S. epidermidis | Methicillin resistant | 0.5 | 16 | 0.06 | 0.015 |
| S. epidermidis | Linezolid nonsusceptible | 8 | 8 | 0.06 | 0.12 |
| S. epidermidis | Quinupristin-dalfopristin resistant | 0.5 | 8 | 0.06 | 0.06 |
| S. epidermidis | Daptomycin nonsusceptible | 0.5 | >16 | 0.06 | ≤0.004 |
| S. pneumoniae | Penicillin resistant | 1 | 0.5 | 0.015 | 0.015 |
| S. pneumoniae | Macrolide resistant | 0.5 | 0.5 | 0.015 | 0.015 |
| S. pneumoniae | Linezolid resistant | 2 | 1 | 0.015 | 0.015 |
| S. pneumoniae | Fluoroquinolone resistant | 1 | >16 | 0.015 | 0.015 |
| S. pyogenes | Macrolide resistant | 1 | 0.25 | 0.12 | 0.12 |
Interaction of compound A and compound B with other topoisomerase and nontopoisomerase inhibitors.
To evaluate the potential for interactions between the compounds and other antibacterial agents, checkerboard MIC experiments were performed using S. aureus strain ATCC 29213. The antibacterial activities of the two compounds were tested in the presence of a range of concentrations of five antibiotics comprising the known type II topoisomerase inhibitors, levofloxacin and novobiocin, as well as agents with diverse mechanisms of action. The occurrence of synergism or antagonism was assessed by calculating the fractional inhibitory concentration index (FICI), as described previously (33). Synergy is defined as a FICI of ≤0.5, while antagonism is evidenced by a FICI of >4.0 (34). The results from these experiments are displayed in Table 6. Compound A and compound B had no effect on the activity of the β-lactam antibiotic oxacillin, the glycopeptide vancomycin, or the protein synthesis inhibitor linezolid. In each case, the measured FICIs were between 1.0 and 2.0. No interactions were also observed between the two compounds and levofloxacin, which targets the GyrA and ParC subunits, or novobiocin, which preferentially binds to GyrB. On the basis of these results, it would be predicted that compound A or compound B could be used in combination with the antibiotics tested without a loss of activity due to antagonism; however, no synergism would be expected.
Table 6.
Interaction of compound A and compound B with other topoisomerase and nontopoisomerase inhibitors determined by the checkerboard assay with S. aureus ATCC 29213
| Compound | Antibiotic | FICI | Interaction |
|---|---|---|---|
| A | Novobiocin | 2.0 | None |
| Levofloxacin | 0.75 | None | |
| Linezolid | 2.0 | None | |
| Vancomycin | 1.0 | None | |
| Oxacillin | 2.0 | None | |
| B | Novobiocin | 1.0 | None |
| Levofloxacin | 0.75 | None | |
| Linezolid | 1.0 | None | |
| Vancomycin | 1.1 | None | |
| Oxacillin | 2.0 | None |
Spontaneous frequency of resistance to compound A and compound B.
Single-enzyme-target inhibitors have spontaneous frequencies of resistance (FoRs) in the range of 10−6 to 10−9 (38). A dual-targeting intracellular inhibitor of GyrB and ParE would be expected to show a lower FoR. To test this hypothesis, the in vitro FoR against S. aureus strain ATCC 29213 was measured at high multiples of the MICs of the compounds. At concentrations equivalent to 4- and 8-fold the MICs of compound A (agar MIC = 0.12 μg/ml) and compound B (agar MIC = 0.06 μg/ml), no spontaneous mutants were isolated, giving measured FoRs on the order of ≤10−10 (Table 7). This magnitude of FoR provides a further piece of experimental evidence in support of a multitargeting mechanism of action for compound A and compound B. By comparison, the equivalent FoRs with novobiocin, which preferentially targets GyrB in S. aureus, were measured as 5.8 × 10−8 (4× MIC) and 3.2 × 10−8 (8× MIC). These values for novobiocin are in line with published data (21, 39). The data also indicate that the concentration required to stop the isolation of first-step mutants, i.e., the mutant prevention concentration (MPC), is ≤0.5 μg/ml for compound A and ≤0.25 μg/ml for compound B. With S. pyogenes ATCC 51339, the frequencies at 8× MIC were <7.1 × 10−11 for compound A and <2.2 × 10−10 for compound B, giving an MPC of ≤0.5 μg/ml for both compounds with this species. For the fluoroquinolone ciprofloxacin, a concentration equivalent to 16-fold the agar MIC was necessary to achieve a multitargeting FoR of <7.4 × 10−11 in S. aureus (Table 7). This translates to an MPC of 4 μg/ml for this antibiotic with S. aureus.
Table 7.
Measured frequencies of spontaneous resistance of compound A, compound B, and selected antibiotics in S. aureus strain ATCC 29213
| Compound | MIC multiple | FoR |
|---|---|---|
| A | 4× | <2.3 × 10−10 |
| 8× | <2.3 × 10−10 | |
| B | 4× | <4.5 × 10−10 |
| 8× | <5.8 × 10−11 | |
| Novobiocin | 4× | 5.8 × 10−8 |
| 8× | 3.2 × 10−8 | |
| Ciprofloxacin | 4× | 1.1 × 10−8 |
| 8× | 7.5 × 10−9 | |
| 16× | <7.4 × 10−11 | |
| Linezolid | 4× | <7.4 × 10−11 |
| 8× | <5.3 × 10−11 |
Toxicity of compounds A and B.
The potential for toxicity of the compounds was investigated by determining the specificity of the compounds for bacterial topoisomerase II and measuring the cytotoxic effect against the HepG2 human liver cell line. The compounds were tested for their ability to inhibit the activity of human topoisomerase II in the DNA relaxation assay. At 10 μM, the control inhibitor (AMSA) resulted in 36.7% inhibition of human topoisomerase II, whereas novobiocin, ciprofloxacin, and both compounds A and B resulted in less than 1% inhibition at the same concentration. This represents a >100-fold window to the IC50s observed in the E. coli DNA gyrase and topoisomerase IV ATPase assays and a >5-fold window to the inhibition of DNA supercoiling by S. aureus DNA gyrase in the supercoiling assay. In the HepG2 cytotoxicity assay, no inhibition was seen for either compound A or compound B up to the maximum concentration tested of 64 μg/ml. For both compounds, this represents a >500-fold window to the S. aureus ATCC 29213 MIC of 0.12 μg/ml.
Pharmacokinetic properties of compound A and compound B.
The pharmacokinetic profiles of compound A and compound B were determined in the rat by measuring the total concentration in plasma by LC-MS/MS following a single intravenous (i.v.) administration of the compounds. The full set of calculated pharmacokinetic parameters from these experiments is shown in Table 8. For animals receiving a 10-mg/kg dose of compound A, the area under the curve (AUC) was 16.6 μg·h/ml, the clearance (CL) was 9.4 ml/min/kg, and the half-life (t1/2) was 210 min. For animals receiving a 3-mg/kg i.v. dose of compound B, the AUC was 17.4 μg·h/ml, the CL was 2.8 ml/min/kg, and the t1/2 was 104 min. In vitro, the levels of protein binding of compounds A and compound B were similar, with the fraction unbound for both compounds determined to be 0.007 using rat plasma. The MIC in the presence of 50% horse serum was also the same for both compounds, with a shift of 16-fold compared to that in serum-free medium (Table 2). The total plasma concentrations of the two compounds scaled to a 3-mg/kg i.v. dose are shown in Fig. 4. The graph in Fig. 4 reveals that a markedly greater AUC and reduced clearance were observed with compound B. The in vivo pharmacokinetics of compound B were also assessed in rats following a single oral (p.o.) administration of 3 mg/kg (Fig. 4). By this route, the AUC was 8.3 μg·h/ml (Table 8). The oral bioavailability following p.o. administration of compound B was calculated to be 47.7%.
Table 8.
In vivo pharmacokinetic parameters of compound A and compound B following intravenous or oral administration in ratsa
| Compound (dose) | C0 (μg/ml) | Cmax (μg/ml) | Tmax (min) | AUC (μg·h/ml) | t1/2 (min) | CL (ml/min/kg) | V (liters/kg) | Bioavailability (%) |
|---|---|---|---|---|---|---|---|---|
| A (10 mg/kg i.v.) | 53.6 | 16.6 | 210 | 9.4 | 2.6 | |||
| B (3 mg/kg i.v.) | 9.8 | 17.4 | 104 | 2.8 | 0.4 | |||
| B (3 mg/kg p.o.) | 1.8 | 120 | 8.3 | 47.7 |
C0, extrapolated concentration in plasma at 0 min; Cmax, maximum concentration in plasma; Tmax, time to maximum concentration in plasma; V, volume of distribution.
Fig 4.
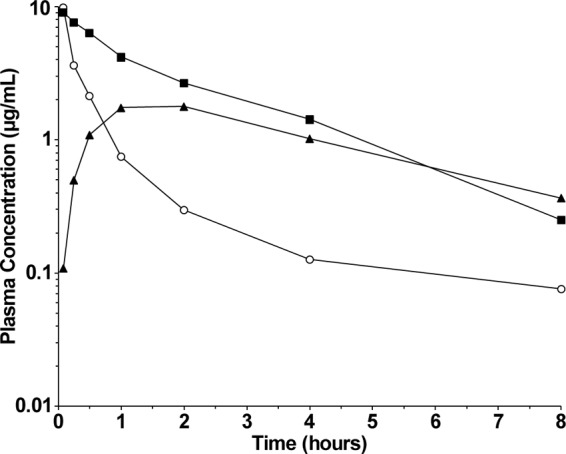
In vivo pharmacokinetic profiles of compound A and compound B in rats. The plasma concentrations of compound A (open circles) scaled to a 3-mg/kg dose are shown following a single i.v. administration of 10 mg/kg. The plasma concentrations of compound B following a single i.v. (solid squares) or p.o. (solid triangles) administration of 3 mg/kg are shown.
DISCUSSION
The objective of this study was to evaluate the in vitro and in vivo properties of substituted benzothiazole ethyl urea-based molecules, as exemplified by compound A and compound B. The mechanism of action and time-kill profiles of the compounds were comparable to those reported for the coumarin class of antibiotics (39). Molecular docking studies predict that the putative binding site is similar to that in the aminobenzimidazole class of type II topoisomerase inhibitors (23). Like novobiocin, both compounds inhibited the ATPase function of DNA gyrase (GyrB) and topoisomerase IV (ParE), with IC50s of <0.1 μg/ml (Table 1). On a molar basis, the compounds inhibited the topoisomerase enzymes to a greater extent than novobiocin, with IC50s being between 3- and 11-fold lower than those of novobiocin. Like novobiocin, compound A prevented the supercoiling of relaxed DNA in a gel-based supercoiling assay (Fig. 2).
In terms of their intracellular action, the activity of compound A and compound B is different from that of novobiocin. In S. pneumoniae and S. pyogenes, the first-step amino acid substitutions were in ParE (T172A or A53S), indicating that ParE is the primary target in these species. The MICs against the two mutants were 0.5 μg/ml or lower, a ≤16-fold increase in the MIC compared to that for the wild type (Table 3). In contrast, in the case of S. aureus, the first-step amino acid substitutions were in GyrB (T173N), indicating that GyrB is the primary target of compounds A and B. The MICs against the S. aureus first-step mutant GyrB (T173N) and the second-step mutant strain that harbors two mutations in GyrB (T173N and S129T) were only slightly raised, with a ≤2-fold increase compared to the MIC for the wild type (Table 3). Consistent with a multitargeting mechanism of action, in the event of one or more mutations in the primary target (GyrB), the compounds are still able to exert their antibacterial effect by inhibiting ParE. Multitargeting by compounds A and B is also supported by the marked decrease in susceptibility to the two compounds (a >32-fold increase in the MIC) that was observed when both target enzymes were mutated, as in the second-step S. aureus GyrB (T173N) ParE (T167N) double mutant (Table 3). This situation contrasts with that for novobiocin, which preferentially targets either GyrB or ParE inside the cell (13, 14, 20). A primary mutation event in response to novobiocin, for example, at residue G85 or R144 of GyrB in S. aureus, drastically raises the MIC of novobiocin by 64-fold (Table 3) (39). The dual-targeting mechanism of action of the compounds is further evidenced by the spontaneous FoR data (Table 7). The FoR in S. aureus for both compounds, at ≤10−10, is below the range expected for a single-enzyme-target inhibitor and is at least 2 orders of magnitude less than the FoR to novobiocin. Taken together, these biochemical and microbiological data support a dual-targeting mechanism of action in which the compounds are able simultaneously to inhibit both type II topoisomerase enzyme complexes.
As expected, the compounds did not show synergy or antagonism with antibiotics that do not target the bacterial type II topoisomerases, i.e., linezolid, vancomycin, and oxacillin (Table 6). Interestingly, compound A and compound B also did not display any apparent in vitro interaction with levofloxacin or novobiocin, both of which target the type II topoisomerases. Additivity and synergism between the coumarin drugs (novobiocin or coumermycin) and quinolones has been reported in in vitro assays with S. aureus, Enterococcus spp., and E. coli (40–43). These divergent observations further support the inference that the mechanism of action and molecular interaction between compound A or compound B and GyrB/ParE are distinct from the interaction between novobiocin and GyrB/ParE. Consistent with this conclusion, the GyrB G85S and GyrB R144S amino acid substitutions that arise in the presence of novobiocin and have a substantial impact on its MIC (Table 3) (39) showed no effect on the potency of compound A and compound B (Table 3).
The compounds demonstrated potent antibacterial activity against a broad range of Gram-positive bacteria, with MICs ranging from 0.008 to 0.12 μg/ml (Table 2). This included sets of representative clinical isolates of the major Gram-positive pathogens (Table 4). Potency was unaffected by a wide spectrum of drug resistance phenotypes among representative Gram-positive isolates (Table 5). This result is consistent with the novel mechanism of action of the compounds. It also demonstrates a lack of cross-resistance between other resistance mechanisms, including those for other bacterial type II topoisomerase inhibitors and the benzothiazole ethyl urea inhibitors of GyrB and ParE. The compounds had mixed activity against Gram-negative organisms. Certain species, such as Moraxella catarrhalis and Neisseria gonorrhoeae, were as susceptible to the compounds as their counterpart Gram-positive bacteria. In contrast, the majority of the Gram-negative bacterial species tested were not susceptible to inhibition by the compounds, despite the ability of the compounds to inhibit potently the ATPase function of the purified E. coli enzymes. The susceptibility of the E. coli N43 strain (MIC = 4 μg/ml), which lacks a functional AcrA protein (44), indicates a role for efflux rather than target-based factors in the apparent lack of susceptibility of some of the Gram-negative organisms. AcrA forms one component of the tripartite AcrA-AcrB-TolC multidrug efflux pump (45). Efflux pumps of the resistance-nodulation-division (RND) superfamily of transporters, which includes the AcrA-AcrB-TolC complex, are known to play a major role in multidrug resistance in Gram-negative bacteria, such as E. coli and P. aeruginosa (46).
The mode of action of compound A and compound B was bactericidal with a time-dependent, concentration-independent kill profile (Fig. 3). A bactericidal mode of action against S. pyogenes, M. catarrhalis, and H. influenzae was also observed (data not shown). A bactericidal mode of action may be advantageous in an antibacterial agent for the treatment of severe Gram-positive bacterial infections (47). Novobiocin, the chemical structure of which is distinct from that of the benzothiazole ethyl urea ligands, had a broadly similar profile in the time-kill experiment (data not shown). Arathoon and coworkers reported a slow, bactericidal mode of action for novobiocin in time-kill studies with clinical isolates of MRSA (37). The combination of these published data with our own results suggests a common mode of action for diverse GyrB and/or ParE inhibitors. At concentrations above the MICs, the rate of killing by GyrB/ParE inhibitors is relatively slow compared with the rapid drop in CFU counts observed with levofloxacin, which binds to the topoisomerase-DNA complex close to the active site in GyrA/ParC (Fig. 3). This indicates that, at higher relative concentrations, interfering with the ATPase function of bacterial type II topoisomerases by novobiocin, compound A, or compound B may be less catastrophic for the cells than poisoning by the formation of enzyme-DNA-quinolone complexes (11, 48, 49).
The compounds showed specificity for bacterial topoisomerases, with no inhibition of human topoisomerase II being observed. Furthermore, no cytotoxicity against HepG2 cells was seen for either compound A or compound B.
Preliminary pharmacokinetic studies in rats indicated that compound B generally showed superior pharmacokinetic parameters to compound A (Table 8). Scaled to an identical dose of 3 mg/kg, the AUC of compound B was approximately 3-fold greater than that of compound A (Fig. 4). Both compounds were similarly highly bound to plasma proteins (>99%). The reduced clearance and higher AUC of compound B compared with those of compound A are likely due to the α substitution to the carboxylate moiety off the piperidine ring of the molecule. This conclusion is supported by pharmacokinetic data for other α-substituted carboxylate analogues that also display enhanced pharmacokinetics relative to compound A (30).
In summary, we report the biological profiles of two representative molecules of the benzothiazole ethyl urea chemotype. These compounds are potent inhibitors of the bacterial type II topoisomerases with activity against drug-susceptible and drug-resistant bacteria. The multitargeting nature of the inhibitors translates into a very low spontaneous FoR. The pharmacokinetic properties of both compounds have also been described. Further investigation of this class of compounds as potential antibacterial agents is under way and will be reported elsewhere in due course.
ACKNOWLEDGMENTS
We thank Steve Ruston and Simon Tucker for their encouragement during the execution of this work.
Footnotes
Published ahead of print 16 September 2013
REFERENCES
- 1. Infectious Diseases Society of America 2004. Bad bugs, no drugs: as antibiotic discovery stagnates, a public health crisis brews. Infectious Diseases Society of America, Alexandria, VA [Google Scholar]
- 2. Spellberg B, Guidos R, Gilbert D, Bradley J, Bucher HW, Scheld WM, Bartlett JG, Edwards J., Jr 2008. The epidemic of antibiotic-resistant infections: a call to arms for the medical community from the Infectious Diseases Society of America. Clin. Infect. Dis. 46:155–164 [DOI] [PubMed] [Google Scholar]
- 3. Bush K, Courvalin P, Dantas G, Davies J, Eisenstein B, Huovinen P, Jacoby GA, Kishony R, Kreiswirth BN, Kutter E, Lerner SA, Levy S, Lewis K, Lomovskaya O, Miller JH, Mobashery S, Piddock LJV, Projan S, Thomas CM, Tomasz A, Tulkens PM, Walsh TR, Watson JD, Witkowski J, Witte W, Wright G, Yeh P, Zgurskaya HI. 2011. Tackling antibiotic resistance. Nat. Rev. Microbiol. 9:894–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rice LB. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 197:1079–1081 [DOI] [PubMed] [Google Scholar]
- 5. Fischbach MA, Walsh CT. 2009. Antibiotics for emerging pathogens. Science 325:1089–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6:29–40 [DOI] [PubMed] [Google Scholar]
- 7. Jabes D. 2011. The antibiotic R&D pipeline: an update. Curr. Opin. Microbiol. 14:564–569 [DOI] [PubMed] [Google Scholar]
- 8. Brötz-Oesterhelt H, Sass P. 2010. Postgenomic strategies in antibacterial drug discovery. Future Microbiol. 5:1553–1579 [DOI] [PubMed] [Google Scholar]
- 9. Maxwell A. 1997. DNA gyrase as a drug target. Trends Microbiol. 5:102–109 [DOI] [PubMed] [Google Scholar]
- 10. Pommier Y, Leo E, Zhang HL, Marchand C. 2010. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 17:421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Collin F, Karkare S, Maxwell A. 2011. Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl. Microbiol. Biotechnol. 92:479–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Drlica K, Hiasa H, Kerns R, Malik M, Mustaev A, Zhao X. 2009. Quinolones: action and resistance updated. Curr. Top. Med. Chem. 9:981–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lewis RJ, Singh OMP, Smith CV, Skarzynski T, Maxwell A, Wonacott AJ, Wigley DB. 1996. The nature of inhibition of DNA gyrase by the coumarins and the cyclothialidines revealed by X-ray crystallography. EMBO J. 15:1412–1420 [PMC free article] [PubMed] [Google Scholar]
- 14. Stieger M, Angehrn Wohlgensinger B, Gmünder H. 1996. GyrB mutations in Staphylococcus aureus strains resistant to cyclothialidine, coumermycin, and novobiocin. Antimicrob. Agents Chemother. 40:1060–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Silver LL. 2012. Polypharmacology as an emerging trend in antibacterial discovery, p 167–202 In Peters JU. (ed), Polypharmacology in drug discovery. John Wiley & Sons, Inc, Hoboken, NJ [Google Scholar]
- 16. East SP, Czaplewski LG, Haydon DJ. 2012. Case study 10: ethyl urea inhibitors of the bacterial type II topoisomerases DNA gyrase (GyrB) and topoisomerase IV (ParE), p 335–352 In Morphy RJ, Harris CJ. (ed), Designing multi-target drugs. Royal Society of Chemistry, Cambridge, United Kingdom [Google Scholar]
- 17. Silver LL. 2011. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 24:74–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. East SP, Silver LL. 2013. Multitarget ligands in antibacterial research: progress and opportunities. Expert Opin. Drug Discov. 8:143–156 [DOI] [PubMed] [Google Scholar]
- 19. Brino L, Urzhumstev A, Mousli M, Bronner C, Mitschler A, Oudet P, Moras D. 2000. Dimerization of Escherichia coli DNA-gyrase B provides a structural mechanism for activating the ATPase catalytic center. J. Biol. Chem. 275:9468–9475 [DOI] [PubMed] [Google Scholar]
- 20. Bellon S, Parsons JD, Wei Y, Hayakawa K, Swenson LL, Charifson PS, Lippke JA, Alsape R, Gross CH. 2004. Crystal structures of Escherichia coli topoisomerase IV ParE subunit (24 and 43 kilodaltons): a single residue dictates differences in novobiocin potency against topoisomerase IV and DNA gyrase. Antimicrob. Agents Chemother. 48:1856–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mani N, Gross CH, Parsons JD, Hanzelka B, Müh U, Mullin S, Liao Y, Grillot A-L, Stamos D, Charifson PS, Grossman TH. 2006. In vitro characterization of the antibacterial spectrum of novel bacterial type II topoisomerase inhibitors of the aminobenzimidazole class. Antimicrob. Agents Chemother. 50:1228–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grossman TH, Bartels DJ, Mullin S, Gross CH, Parsons JD, Liao Y, Grillot A-L, Stamos D, Olson ER, Charifson PS, Mani N. 2007. Dual targeting of GyrB and ParE by a novel aminobenzimidazole class of antibacterial compounds. Antimicrob. Agents Chemother. 51:657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Charifson PS, Grillot A-L, Grossman TH, Parsons JD, Badia M, Bellon S, Deininger DD, Drumm JE, Gross CH, LeTiran A, Liao Y, Mani N, Nicolau DO, Perola E, Ronkin A, Shannon D, Swenson LL, Tang Q, Tessier PR, Tian S-K, Trudeau M, Wang T, Wei Y, Zhang H, Stamos D. 2008. Novel dual-targeting benzimidazole urea inhibitors of DNA gyrase and topoisomerase IV possessing potent antibacterial activity: intelligent design and evolution through the judicious use of structure-guided design and structure-activity relationships. J. Med. Chem. 51:5243–5263 [DOI] [PubMed] [Google Scholar]
- 24. East SP, White CB, Barker O, Barker S, Bennett J, Brown D, Boyd EA, Brennan C, Chowdhury C, Collins I, Convers-Reignier E, Dymock BW, Fletcher R, Haydon DJ, Gardiner M, Hatcher S, Ingram P, Lancett P, Mortenson P, Papadopoulos K, Smee C, Thomaides-Brears HB, Tye H, Workman J, Czaplewski LG. 2009. DNA gyrase (GyrB)/topoisomerase IV (ParE) inhibitors: synthesis and antibacterial activity. Bioorg. Med. Chem. Lett. 19:894–899 [DOI] [PubMed] [Google Scholar]
- 25. Starr JT, Sciotti RJ, Hanna DL, Huband MD, Mullins LM, Cai H, Gage JW, Lockard M, Rauckhorst MR, Owen RM, Lall MS, Tomilo M, Chen H, McCurdy SP, Barbachyn MR. 2009. 5-(2-Pyrimidinyl)-imidazo[1,2-a]pyridines are antibacterial agents targeting the ATPase domains of DNA gyrase and topoisomerase IV. Bioorg. Med. Chem. Lett. 19:5302–5306 [DOI] [PubMed] [Google Scholar]
- 26. Tanitame A, Oyamada Y, Ofuji K, Fukimoto M, Iwai N, Hiyama Y, Suzuki K, Ito H, Terauchi H, Kawasaki M, Nagai K, Wachi M, Yamagishi J-i. 2004. Synthesis and antibacterial activity of a novel series of potent DNA gyrase inhibitors. Pyrazole derivatives. J. Med. Chem. 47:3693–3696 [DOI] [PubMed] [Google Scholar]
- 27. Manchester JI, Dussault DD, Rose JA, Boriack-Sjodin A, Uria-Nickelsen M, Ioannidis G, Bist S, Fleming P, Hull KG. 2012. Discovery of a novel azaindole class of antibacterial agents targeting the ATPase domains of DNA gyrase and topoisomerase IV. Bioorg. Med. Chem. Lett. 22:5150–5156 [DOI] [PubMed] [Google Scholar]
- 28. Tari LW, Trzossa M, Bensen DC, Li X, Chen Z, Lam T, Zhang J, Creighton CJ, Cunningham ML, Kwan B, Stidham M, Shaw KJ, Lightstone FC, Wong SE, Nguyen TB, Nix J, Finn J. 2013. Pyrrolopyrimidine inhibitors of DNA gyrase B (GyrB) and topoisomerase IV (ParE), part I: structure guided discovery and optimization of dual targeting agents with potent, broad-spectrum enzymatic activity. Bioorg. Med. Chem. Lett. 23:1529–1536 [DOI] [PubMed] [Google Scholar]
- 29. Trzoss M, Bensen DC, Li X, Chen Z, Lam T, Zhang J, Creighton CJ, Cunningham ML, Kwan B, Stidham M, Nelson K, Brown-Driver V, Castellano A, Shaw KJ, Lightstone FC, Wong SE, Nguyen TB, Finn J, Tari LW. 2013. Pyrrolopyrimidine inhibitors of DNA gyrase B (GyrB) and topoisomerase IV (ParE), part II: development of inhibitors with broad spectrum, Gram-negative antibacterial activity. Bioorg. Med. Chem. Lett. 23:1537–1543 [DOI] [PubMed] [Google Scholar]
- 30. Palmer JT, Lunniss CJ, Offermann DA, Axford LC, Blair M, Mitchell D, Palmer N, Steele C, Atherall J, Watson D, Haydon D, Czaplewski L, Davies D, Collins I, Tyndall EM, Andrau L, Pitt GRW. April 2012. International patent WO 2012/045124 Biota Europe Ltd, Begbroke, United Kingdom
- 31. Clinical and Laboratory Standards Institute 2006. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 7th ed. CLSI document M7-A7 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 32. Clinical and Laboratory Standards Institute 2007. Methods for antimicrobial susceptibility testing of anaerobic bacteria; approved standard, 7th ed. CLSI document M11-A7 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 33. Pillai SK, Moellering RC, Jr, Eliopoulos GM. 2005. Antimicrobial combinations, p 365–440 In Lorian V. (ed), Antibiotics in laboratory medicine. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 34. Odds FC. 2003. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 52:1. [DOI] [PubMed] [Google Scholar]
- 35. Clinical and Laboratory Standards Institute 1999. Methods for determining bactericidal activity of antimicrobial agents; approved guideline. CLSI document M26-A Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 36. Sugino A, Higgins NP, Brown PO, Peebles CL, Cozzarelli NR. 1978. Energy coupling in DNA gyrase and the mechanism of action of novobiocin. Proc. Natl. Acad. Sci. U. S. A. 75:4838–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arathoon EG, Hamilton JR, Hench CE, Stevens DA. 1990. Efficacy of short courses of oral novobiocin-rifampin in eradicating carrier state of methicillin-resistant Staphylococcus aureus and in in vitro killing studies of clinical isolates. Antimicrob. Agents Chemother. 34:1655–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O'Neill AJ, Chopra I. 2004. Preclinical evaluation of novel antibacterial agents by microbiological and molecular techniques. Expert Opin. Investig. Drugs 13:1045–1063 [DOI] [PubMed] [Google Scholar]
- 39. Vickers AA, O'Neill AJ, Chopra I. 2007. Emergence and maintenance of resistance to fluoroquinolones and coumarins in Staphylococcus aureus: predictions from in vitro studies. J. Antimicrob. Chemother. 60:269–273 [DOI] [PubMed] [Google Scholar]
- 40. Neu HC, Chin N-X, Labthavikul P. 1984. Antibacterial activity of coumermycin alone and in combination with other antibiotics. Antimicrob. Agents Chemother. 25:687–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. French P, Venuti E, Fraimow HS. 1993. In vitro activity of novobiocin against multiresistant strains of Enterococcus faecium. Antimicrob. Agents Chemother. 37:2736–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schick DG, Canawati HN, Montgomerie JZ. 1997. In vitro activity of the combination of trovafloxacin and other antibiotics against enterococci. Diagn. Microbiol. Infect. Dis. 29:233–239 [DOI] [PubMed] [Google Scholar]
- 43. Chao L. 1978. An unusual interaction between the target of nalidixic acid and novobiocin. Nature 271:385–386 [DOI] [PubMed] [Google Scholar]
- 44. Ma D, Cook DN, Albert M, Pon NG, Nikaido H, Hearst JE. 1993. Molecular cloning and characterization of acrA and acrE genes of Escherichia coli. J. Bacteriol. 175:6299–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Symmons MF, Bokma E, Koranakis V, Hughes C, Koranakis V. 2009. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc. Natl. Acad. Sci. U. S. A. 106:7173–7178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nikaido H. 2009. Multidrug resistance in bacteria. Annu. Rev. Biochem. 78:119–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pankey GA, Sabath LD. 2004. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive infections. Clin. Infect. Dis. 38:864–870 [DOI] [PubMed] [Google Scholar]
- 48. Kreuzer KN, Cozzarelli NR. 1979. Escherichia coli mutants thermosensitive for deoxyribonucleic acid gyrase subunit A: effects on deoxyribonucleic acid replication, transcription, and bacteriophage growth. J. Bacteriol. 140:424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Drlica K, Zhao X. 1997. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 61:377–392 [DOI] [PMC free article] [PubMed] [Google Scholar]