

Abstract
Enterococcus faecalis is a Gram-positive bacterium that is a major cause of hospital-acquired infections, in part due to its intrinsic resistance to cephalosporins. The mechanism that confers intrinsic cephalosporin resistance in enterococci remains incompletely defined. Previously, we have shown that the Ser/Thr protein kinase and phosphatase pair IreK and IreP act antagonistically to regulate cephalosporin resistance in E. faecalis. We hypothesize that IreK senses antibiotic-induced cell wall damage and activates a signaling pathway leading to antibiotic resistance. However, the factors downstream of IreK have not yet been identified. To discover such factors, suppressor mutations that restored resistance to a ΔireK kinase mutant were identified. Mutations were found in IreB, a highly conserved gene of unknown function that is widespread among low-GC Gram-positive bacteria. We show that IreB plays a negative regulatory role in cephalosporin resistance and is an endogenous substrate of both IreK and IreP. IreB is phosphorylated on conserved threonine residues, and mutations at these sites impair cephalosporin resistance. Our results are consistent with a model in which the activity of IreB is modulated by IreK-dependent phosphorylation in a signaling pathway required for cephalosporin resistance and begin to shed light on the function of this previously uncharacterized protein.
INTRODUCTION
Enterococcus faecalis is a Gram-positive bacterium and a ubiquitous member of the gastrointestinal tract (1). E. faecalis is also an opportunistic pathogen and a leading cause of hospital-acquired infections, especially urinary tract infections and endocarditis (2, 3). Like other members of the genus Enterococcus, E. faecalis exhibits intrinsic resistance to a wide array of antimicrobials, including cephalosporins. Cephalosporins are beta-lactam antibiotics that inhibit peptidoglycan synthesis by binding to the active site of penicillin-binding proteins, thereby preventing peptidoglycan cross-linking. Although the underlying mechanisms E. faecalis uses to survive therapeutic concentrations of cephalosporins are still being unraveled, one important contributing factor is a recently described signaling system comprised of a eukaryotic-like serine/threonine kinase (IreK) and phosphatase (IreP) pair that antagonistically regulate cephalosporin resistance (4, 5). Of note, homologs of IreK and IreP (widely referred to as STKs and STPs for serine/threonine kinases or phosphatases, respectively) are encoded in the genomes of nearly all Gram-positive bacteria.
Eukaryotic-like STK/STPs have been reported to play key roles in responding to changes in environmental conditions in Gram-positive bacteria. For example, STK/STPs have been implicated in processes such as stress responses (6), virulence (7), and spore germination (8). It now appears that STK/STPs also perform important functions in basal cellular metabolism such as nucleotide biosynthesis (9), lipid synthesis (10), and cell wall division (11, 12). To delineate the signaling pathways initiated by STK/STPs, considerable effort has been invested in identifying their protein substrates, resulting in a number of reports describing substrates for reversible phosphorylation by IreK/IreP homologs in many Gram-positive bacteria (recently summarized in reference 13). In most cases, these substrates were identified by in vitro assays, and it remains unclear whether many of them are also phosphorylated in vivo.
Previously, we reported that in E. faecalis, IreK and IreP act antagonistically to regulate cephalosporin resistance as mutants lacking ireK exhibit cephalosporin susceptibility, whereas mutants lacking ireP exhibit hyper-resistance (5). We hypothesize that IreK senses cephalosporin-induced cell wall damage, perhaps by the binding of muropeptides (8, 14), and activates a signaling pathway that leads to cephalosporin resistance. However, no endogenous E. faecalis substrates for IreK phosphorylation (other than IreK itself) have yet been identified. In the present study, we have identified a gene (now called ireB) whose product acts to negatively regulate cephalosporin resistance. Biochemical assays show that IreB is an endogenous substrate of both IreK kinase and IreP phosphatase in vitro. IreB is phosphorylated on conserved threonine residues, and mutations at these sites impair cephalosporin resistance in E. faecalis cells. Our results are consistent with a model in which the activity of IreB is modulated by IreK-dependent phosphorylation in a signaling pathway required for cephalosporin resistance and begin to shed light on the function of this previously uncharacterized protein.
MATERIALS AND METHODS
Bacterial strains, growth media, and chemicals.
Bacterial strains and plasmids used in the study are listed in Table 1. Unless otherwise indicated, E. coli strains were grown in lysogeny broth. E. faecalis strains were grown in brain heart infusion (BHI) or half-strength BHI (hBHI) for routine maintenance or in Mueller-Hinton broth (MHB) for susceptibility assays at 37°C. The concentrations of antibiotics were as follows: spectinomycin, 100 μg/ml (E. coli) or 1,000 μg/ml (E. faecalis); kanamycin, 50 μg/ml (E. coli); and chloramphenicol, 10 μg/ml. All antibiotics and chemicals were obtained from Sigma-Aldrich unless stated otherwise.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant description or genotypea | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| Top10 | Routine cloning host | Invitrogen |
| BL21(DE3) | Protein overexpression host | Lab stock |
| E. faecalis | ||
| OG1RF | Wild-type reference strain (MLST 1) | 21 |
| CK119 | OG1RF ΔireK2 | 4 |
| CK121 | OG1RF ΔireP2 | 5 |
| CK164 | OG1RF ΔireB2 | This study |
| CK167 | OG1RF ΔireK2 ireB2 | This study |
| JG4 | Ceftriaxone-resistant suppressor mutant of CK119; carries D50A mutation in IreB | This study |
| T1 (SS498) | Wild-type (MLST 21), CDC reference strain | 22 |
| JL376 | T1 ΔireB2 | This study |
| Plasmids | ||
| pET28b | E. coli expression vector (Knr) | Novagen |
| pJLL20 | pET28b::ireB-His6 | This study |
| pJLL22 | pET28b::ireB T4A T7A-His6 | This study |
| pCLH32 | pET28b::ireB T4A-His6 | This study |
| pCLH33 | pET28b::ireB T7A-His6 | This study |
| pCJK111 | pET28b::ireK-n | 5 |
| pCJK112 | pET28b::ireP | 5 |
| pCJK142 | pET28b::ireK-n K41R | 5 |
| pJRG32 | E. faecalis allelic exchange vector with pheS* counterselectable marker (Emr) | 23 |
| pCJK174 | pJRG32::ΔireB2 (ΔE6-H84) | This study |
| pDL278p23 | E. faecalis expression vector, constitutive p23 promoter (Spr) | 24 |
| pJRG39 | pDL278p23::ireB-strep | This study |
| pJLL14 | pDL278p23::ireB T4A T7A-strep | This study |
| pCI3340 | E. coli/E. faecalis shuttle vector (Cmr) | 25 |
| pCJK187 | pCI3340::p23-ireB-strep | This study |
| pCLH140 | pCI3340::p23-ireB T4V-strep | This study |
| pCLH141 | pCI3340::p23-ireB T7V-strep | This study |
| pCLH142 | pCI3340::p23-ireB T4V T7V-strep | This study |
| pCLH111 | pCI3340::p23-ireB T4I-strep | This study |
| pCLH126 | pCI3340::p23-ireB T7I-strep | This study |
Cmr, chloramphenicol resistance; Emr, erythromycin resistance; Spr, spectinomycin resistance; Knr, kanamycin resistance.
Isolation of ΔireK suppressor mutants.
Independent cultures of the ΔireK mutant (CK119) in BHI were subjected to serial dilutions in BHI and plated on BHI agar supplemented with an inhibitory concentration of ceftriaxone (1 μg/ml; note that, for reasons that are unknown, the MIC of CK119 for ceftriaxone in BHI is lower than the MIC in MHB). After overnight incubation at 37°C, colonies appeared on the ceftriaxone-containing plates at a frequency of ∼5 × 10−6 to 5 × 10−5 per viable CFU. Ceftriaxone-resistant colonies were streaked for purification on BHI plus ceftriaxone and then retested to confirm that they retained enhanced ceftriaxone resistance following growth in the absence of ceftriaxone. Genomic DNA (gDNA) was isolated from the parental ΔireK strain and two suppressor mutants using Qiagen genomic tip 100/G columns, according to the manufacturer's instructions, following an initial incubation with lysozyme (10 mg/ml) and mutanolysin (500 U/ml) at 37°C for 10 min. The gDNA was subjected to next-generation sequencing at the Human and Molecular Genetics Center at the Medical College of Wisconsin.
Whole-genome sequencing.
Genome sequencing was performed by using 454 technology on a Genome Sequencer FLX according to the manufacturer's instructions (Roche). Sequencing yielded ∼20-fold coverage of the genome for the parent CK119 and 15-fold coverage for the suppressor mutant JG4.
Sequences were aligned against the E. faecalis OG1RF (the isogenic parent of CK119) reference genome (NCBI CP002621) to identify single-nucleotide variants using Roche GS Reference Mapper version 2.3. High-confidence variants (differences) between the sequenced and reference genome identified via 454 sequence analysis were confirmed by subsequent Sanger sequencing on PCR amplicons of the corresponding genomic regions derived from the mutants. A single variant was detected in the genome of suppressor mutant JG4 relative to its parent (CK119), giving rise to a D50A substitution in IreB.
Construction of plasmids.
For protein expression and purification of IreB or mutant derivatives, we used the IPTG-inducible pET28b expression vector. Briefly, we encoded substitutions at Thr4 and/or Thr7 within the forward primers designed to amplify the ireB ORF from E. faecalis OG1RF gDNA. Using primer-encoded restriction sites (NcoI/XhoI), the ireB amplicon was digested and ligated into similarly digested pET28b to create IreB with a C-terminal His6 tag provided by the vector.
For two-dimensional immunoblot analysis of Strep-tagged IreB in E. faecalis, we used the pDL278p23 vector encoding the constitutive p23 promoter. ireB was amplified from E. faecalis OG1RF gDNA, using forward primers that encoded the mutations at Thr4 and Thr7 when applicable, and the reverse primer encoded the 8-amino-acid C-terminal Strep-tag (WSHPQFEK). The ireB amplicon was cloned into pDL278p23 using primer-encoded BamHI and SphI restriction sites.
For complementation analysis and antimicrobial susceptibility studies of IreB point mutants, a two-step cloning procedure was used to introduce ireB into the shuttle vector pCI3340. First, ireB was amplified from OG1RF gDNA (with primer-encoded C-terminal Strep-tag) and cloned into pDL278p23 using primer-encoded BamHI and SphI restriction sites. A fragment encoding the p23-ireB-strep was then excised and cloned into pCI3340 using EcoRI and HindIII restriction enzymes (derived from the multiple cloning site of pDL278p23). All plasmid clones were sequenced to verify the absence of any errors.
Construction of mutants lacking ireB.
Modification of the ireB locus in the E. faecalis chromosome was performed using markerless exchange as described previously (15). A derivative of pJRG32 carrying an in-frame deletion allele of ireB (pCJK174) was constructed using a BsaI-based cloning scheme to seamlessly fuse two PCR amplicons flanking EF1202 to generate the in-frame deletion. In an effort to avoid perturbing the expression of adjacent genes, the deletion allele was designed such that the first and last five codons remained (88% of the open reading frame was deleted). This deletion allele was transferred to the native ireB locus in either the wild-type (OG1RF or T1) or ΔireK (CK119) chromosome by allelic exchange, as previously described (15). Deletion mutants were isolated by plating on counterselection medium containing p-Cl-Phe (15) at 30°C for 2 to 3 days. Two such ΔireB mutants isolated completely independently from each other were analyzed and found to exhibit identical phenotypes.
Determination of MICs.
Serial 2-fold dilutions of antibiotics in MHB were prepared in 96-well plates. Overnight cultures of test strains were grown in MHB, normalized by optical density at 600 nm (OD600) values, and inoculated into the wells at a density of ∼105 CFU/ml. Plates were incubated for 24 h at 37°C and 225 rpm. For MICs against daptomycin, CaCl2 was included at 50 mg/liter. MIC values were determined as the lowest antibiotic concentration to inhibit growth as determined by the OD600 measured by a Molecular Devices Spectra Max MS plate reader.
Protein purification.
Proteins were purified from E. coli BL21(DE3) according to the method previously described (5). Of note, IreB proteins were not dialyzed in storage buffer as we observed protein precipitation during dialysis. For all experiments, IreB in the elution buffer after purification (50 mM Tris [pH 8], 300 mM NaCl, 500 mM imidazole) was used directly.
In vitro enzymatic assays. (i) Kinase assays.
Recombinant IreB-His6 (14.2 μM) was incubated alone or in combination with recombinant His6-IreK-n (0.33 μM) in kinase buffer (50 mM Tris [pH 7.5], 25 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 1 mM dithiothreitol [GoldBio], 0.1 mM EDTA). The reaction was initiated by the addition of 2 mM ATP, 1 μCi [γ-32P]ATP, followed by incubation at 37°C for the indicated time. The reactions were stopped with the addition of 5× sodium dodecyl sulfate (SDS) Laemmli sample buffer, boiled for at least 5 min, and subjected to SDS-PAGE. The gels were then fixed, dried, and exposed to a phosphorimager screen for detection of radiolabeled proteins using a Typhoon FLA 7000 phosphorimager.
(ii) Staurosporine inhibition of IreK-n kinase activity.
A kinase assay was set up as described above except the IreK-n concentration was reduced to 33 nM. The reaction was split in half. To one aliquot, 0.15 mM staurosporine was added; to the other, an equal volume of vehicle (DMSO) was added. Finally, 0.5 mM ATP, 1 μCi [γ-32P]ATP was added, and the reactions were incubated at 37°C. Samples were taken at the indicated times, and the reactions were stopped with addition of 5× SDS Laemmli sample buffer, boiled for at least 5 min, and subjected to SDS-PAGE and phosphorimaging as described above.
(iii) Phosphatase assays.
Initially, 14.2 μM recombinant IreB and 33 nM IreK-n were incubated with 0.5 mM ATP, 1 μCi [γ-32P]ATP in kinase buffer at 37°C for 60 min to generate phospho-IreB. Subsequently, 0.15 mM staurosporine and 2 μM IreP were added and aliquots of the reaction were taken at the indicated times. The reactions were stopped by the addition of 5× SDS Laemmli sample buffer, boiled for 5 min, and subjected to SDS-PAGE and phosphorimaging as described above.
Phosphoamino acid analysis.
IreB was phosphorylated with radioactive ATP using IreK-n as described above, with slight modifications (1.32 μM IreK-n, 14.2 μM IreB, and 2 mM ATP, 3 μCi [γ-32P]ATP). After 30 min of incubation at 37°C reactions were stopped with the addition of 5× SDS-Laemmli sample buffer, boiled for at least 5 min, and subjected to SDS-PAGE. Proteins were then transferred to Immobilon P membrane (Millipore), and autoradiography was used to identify positions of the radioactive bands on the membrane. Phosphoamino acid analysis was performed as described previously (16) with some modification. Briefly, radioactive IreB bands on membrane were excised and treated with 6 N HCl for 1 h at 110°C. The membrane pieces were removed, and the solution was evaporated in a Speed-Vac to obtain a dried pellet. The pellet was resuspended in 3.5 buffer (5% glacial acetic acid [Fisher] and 0.5% pyrimidine [vol/vol] in water) and split into two aliquots. To one aliquot, phosphoserine and phosphotyrosine standards were added; to the other aliquot, a phosphothreonine standard was added. The samples were then spotted onto a cellulose thin-layer chromatography plate (20 by 20 cm; EMD) and separated by high-voltage electrophoresis at 1,800 V for 35 min. Ninhydrin spray (Acros Organics) was used to visualize phosphoamino acid standards on the cellulose TLC plate, and autoradiography was used to detect radiolabeled amino acids.
Mass spectrometry.
IreK-n was used to phosphorylate IreB in vitro as described above. The protein mixture was separated by SDS-PAGE and stained with Gel-Code Blue (Pierce). The band corresponding to IreB was excised and subjected to tryptic digestion and mass spectrometry at the Proteomics and Mass Spectrometry Facility, University of Massachusetts Medical School to identify the site(s) of phosphorylation.
Two-dimensional gel electrophoresis (2DGE) and immunoblot.
Overnight cultures of E. faecalis strains were diluted 1:50 in hBHI media containing spectinomycin at 37°C. Once the OD600 is 0.6, the cultures were centrifuged and stored overnight at −20°C if necessary. For samples treated with staurosporine: back-diluted overnight cultures were grown until the OD600 reached 0.2, at which point they were treated with 10 μM staurosporine or an equivalent volume of the DMSO solvent, and incubation continued until the OD600 increased to 0.6. Thawed pellets were resuspended in 500 μl of lysis buffer (10 mM Tris [pH 8], 50 mM NaCl, 1× Halt protease inhibitor cocktail [Pierce], 0.5× Halt phosphatase inhibitor cocktail [Pierce], 5 mM EDTA). Soluble protein (50 μg) obtained from cleared lysates after bead-beating was precipitated by using methanol and chloroform as described previously (17) and subjected to two-dimensional electrophoresis as described previously (18). Separated proteins were then transferred to polyvinylidene difluoride membrane for immunoblotting with rabbit anti-strep antibody (Genescript), followed by horseradish peroxidase-conjugated goat anti-rabbit antibody (Invitrogen). Detection was performed using Pierce Super Signal chemiluminescence substrate according to the manufacturer's instructions.
RESULTS AND DISCUSSION
IreB is a negative regulator of IreK-mediated cephalosporin resistance.
Although IreK is required for cephalosporin resistance in E. faecalis, protein targets for phosphorylation by IreK have not been described. To identify genes that act downstream of IreK to mediate cephalosporin resistance, at least a subset of which might be expected to be direct IreK substrates, we isolated single-step cephalosporin-resistant suppressor mutants that restore resistance to ceftriaxone (a broad-spectrum cephalosporin) for the ΔireK mutant. We chose one suppressor mutant (strain JG4) that exhibited enhanced resistance to two different cephalosporins (the MICs for both ceftriaxone and ceftazidime for strain JG4 were 64 μg ml−1) for whole-genome resequencing to identify the mutation(s) responsible for enhanced resistance. Comparison of the suppressor mutant genome to that of the parental ΔireK mutant (which was also resequenced for comparison) revealed a single nucleotide change in EF1202 (OG1RF_10974), resulting in a D50A substitution in the gene product. Targeted sequencing of the EF1202 gene in other independently isolated ceftriaxone-resistant suppressor mutants (which were not subjected to whole-genome sequencing nor studied in depth) identified a EF1202 D50G substitution, as well as two mutants in which the EF1202 protein was truncated prematurely: a 1-bp deletion in the codon for D59 that leads to a frameshift and premature stop, and a mutant in which the codon for E71 was converted to a stop codon. Overall, a sequence variant of some type was found in EF1202 in 9 of 15 independent ceftriaxone-resistant suppressor mutants for which the EF1202 gene locus was sequenced. Thus, the identification of distinct mutations in the EF1202 gene in multiple, independently isolated ΔireK suppressor mutants suggested that the EF1202 gene product participates in IreK-mediated signaling.
EF1202 is a small protein (89 amino acids) that is widespread and almost exclusively found among the Firmicutes but has no known function. Sequence analysis shed little light on potential functions of EF1202, since only a domain of unknown function was identified (DUF965, Pfam 06135; E value 1.8 × 10−38). Given that several mutants we identified produced prematurely truncated proteins, we hypothesized that the EF1202 alleles represented loss of function mutations. To confirm that the loss of EF1202 (henceforth called ireB for intrinsic resistance of enterococci, based on studies described below) is responsible for the enhanced ceftriaxone resistance of the ΔireK suppressor mutant, ireB was deleted in the background of the parental ΔireK strain, and antibiotic susceptibility assays were conducted. The susceptibility analyses revealed that the ΔireK ΔireB double mutant exhibits enhanced resistance to ceftriaxone compared to the ΔireK parent (Table 2; see also Fig. S1 in the supplemental material), suggesting that IreB acts to negatively regulate cephalosporin resistance downstream of IreK. Because IreK is required for robust resistance to all expanded- and broad-spectrum cephalosporins tested (4), we examined susceptibility of the double mutant toward other cephalosporins (Table 2), revealing that loss of IreB conferred enhanced resistance to both expanded- and broad-spectrum cephalosporins. Similarly, we found that deletion of ireB in two independent genetic lineages of otherwise wild-type E. faecalis (E. faecalis OG1RF and E. faecalis T1) led to enhanced cephalosporin resistance (Table 2), a finding consistent with the hypothesis that IreB acts as a negative regulator of cephalosporin resistance. Complementation analysis of the ΔireB mutant by expression of plasmid-encoded IreB eliminated the hyper-resistance phenotype, confirming that ireB deletion is indeed responsible (Table 3). In addition, the resistance phenotypes are not due to general changes in growth rate, as the ΔireB and ΔireK ΔireB mutants exhibited identical growth kinetics to the wild type (see Fig. S1 in the supplemental material).
Table 2.
Susceptibility analyses for antibiotics that target various cellular processes
| Drug type or target and drug namea | MICb (μg ml−1) |
|||||
|---|---|---|---|---|---|---|
| OG1RF |
T1 |
|||||
| WT | ΔireB mutant | ΔireK ΔireB mutant | ΔireK mutant | WT | ΔireB mutant | |
| Cephalosporins | ||||||
| Ceftazidime (BS) | 128 | 1,024 | 256 | 4 | 128 | 512 |
| Ceftriaxone (BS) | 128 | 2,048 | 128 | 4 | 64 | 512 |
| Cefuroxime (ES) | 64 | 512 | 256 | 4 | 64 | 256 |
| Cefadroxil (NS) | 64 | 64 | 64 | 32 | 64 | 64 |
| Other cell wall | ||||||
| Ampicillin | 0.5 | 1 | 0.5 | 0.5 | 1 | 1 |
| Vancomycin | 2 | 2 | 2 | 1 | 2 | 2 |
| Daptomycin | 8 | 32 | 16 | 4 | 4 | 4 |
| Protein synthesis | ||||||
| Erythromycin | 2 | 1 | 1 | 2 | 0.5 | 0.5 |
| Chloramphenicol | 4 | 4 | 4 | 4 | 8 | 8 |
| Kanamycin | 128 | 128 | 128 | 128 | 64 | 32 |
| DNA replication | ||||||
| Norfloxacin | 4 | 4 | 4 | 4 | 2 | 4 |
BS, broad spectrum; ES, expanded spectrum; NS, narrow spectrum.
MICs are reported as the medians of three independent experiments performed in MHB broth at 37°C for 24 h. The strain designations in the OG1RF genetic lineage are indicated as follows: the wild type (WT) is strain OG1RF, the ΔireB mutant is strain CK164, the ΔireK ΔireB mutant is strain CK167, and the ΔireK mutant is strain CK119. Strain designations in the T1 genetic lineage are indicated as follows: the wild type is strain T1, and the ΔireB mutant is strain JL376.
Table 3.
Susceptibility analyses for strains expressing IreB mutants
|
E. faecalis strain and plasmid |
Plasmid-borne allele of IreB | Ceftriaxone MIC (μg ml−1)a | |
|---|---|---|---|
| Strain | Plasmid | ||
| ΔireB mutant | pCI3340 | None | 256 |
| pCJK187 | Wild type | 16 | |
| pCLH140 | T4V | 8 | |
| pCLH141 | T7V | 2 | |
| pCLH142 | T4V T7V | 4 | |
| pCLH111 | T4I | 8 | |
| pCLH126 | T7I | 2 | |
| Wild-type strain | pCI3340 | None | 16 |
| ΔireK mutant | pCI3340 | None | 1 |
MICs are reported as the median of three independent experiments performed in MHB broth supplemented with chloramphenicol at 37°C for 24 h. Strain designations are as follows: the wild-type E. faecalis strain is strain OG1RF, the ΔireB mutant is strain CK164, and the ΔireK mutant is strain CK119. All plasmid-encoded IreB proteins contain a C-terminal Strep-tag.
IreK is required for full resistance toward expanded- and broad-spectrum cephalosporins but not toward antibiotics with other cellular targets (4). To determine whether IreB also functions in a cephalosporin-specific pathway, we performed susceptibility tests with noncephalosporin antibiotics that target various cellular functions (Table 2). Wild-type and ΔireB E. faecalis strains exhibit similar levels of susceptibility toward antibiotics that target protein synthesis, DNA replication, and even most noncephalosporin antibiotics that target cell wall biosynthesis, indicating that the activity of IreB is specific to the cephalosporin resistance pathway. Of note, deletion of ireB led to enhanced resistance to daptomycin (thought to be active against the cell membrane) in E. faecalis OG1RF but not in the T1 lineage. The reasons for this discrepancy are not known but may be related to other differences in the genomes of these two lineages, and explanation of this phenomenon will require further investigation. In any case, the subset of antibiotics for which susceptibility of the ΔireB mutant is altered closely parallels that observed for the ΔireK mutant (albeit in the opposite direction; see Table 2), suggesting that IreK and IreB participate in a common biological pathway. Of note, deletion of IreB in an otherwise wild-type E. faecalis strain containing functional IreK also led to enhanced cephalosporin resistance (Table 2), at a higher level than that observed in a mutant lacking both IreB and IreK, indicating that IreK is also able to promote cephalosporin resistance in an IreB-independent manner, likely by phosphorylating other, as-yet-unknown cellular targets.
In addition to reduced cephalosporin resistance, ΔireK strains also exhibit reduced resistance to sodium cholate (4), a detergent found in bile, and to nisin (C. J. Kristich, unpublished data), a lantibiotic that acts in part by binding the lipid II intermediate in peptidoglycan synthesis. To determine whether IreB also influenced these IreK-dependent traits, we sought to determine whether the loss of ireB could enhance resistance to sodium cholate and nisin (see Fig. S2 in the supplemental material). Consistent with the results obtained with the antibiotics above, we observed that the ΔireK ΔireB strains exhibited enhanced resistance to both sodium cholate and nisin compared to the ΔireK strain, further suggesting that IreB acts downstream of IreK to modulate IreK-dependent signaling.
IreB is a substrate of IreK and IreP in vitro.
In light of the genetic evidence described above linking IreK and IreB function, we hypothesized that IreB might represent a direct substrate for phosphorylation by IreK. To test this, in vitro kinase assays were conducted using recombinant IreB-His6. Two forms of the IreK kinase were used: an N-terminal fragment of IreK containing the wild-type IreK kinase domain (His6-IreK-n), which was previously shown to exhibit kinase activity in vitro (5), or a point mutant of IreK-n bearing a K41R substitution that dramatically reduces kinase activity. We found that IreK-n robustly phosphorylated purified IreB (Fig. 1A), thereby defining IreB as the first identified enterococcal substrate for IreK. In contrast, IreB was only weakly phosphorylated in reactions with the kinase-impaired IreK-n K41R mutant, excluding the possibilities that the robust IreB phosphorylation is due to a contaminating kinase or to IreB-mediated autophosphorylation. In addition, IreB incubated in the absence of any kinase did not become phosphorylated.
Fig 1.
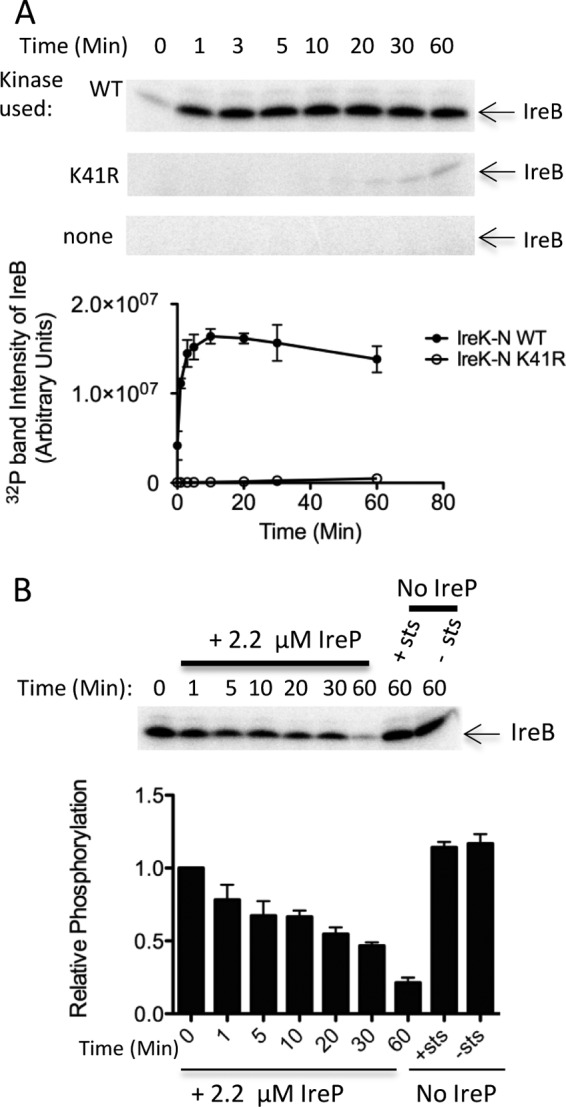
IreB is a substrate of both IreK kinase and IreP phosphatase. (A) Kinase assays were performed to monitor phosphorylation of IreB with [γ-32P]ATP. Reactions contained no IreK kinase (none), wild-type IreK-n kinase (WT), or IreK-n K41R catalytically defective mutant kinase (K41R) and were incubated at 37°C for the indicated times. Radiolabeled proteins were analyzed by SDS-PAGE and phosphorimaging. (B) Prelabeled IreB was incubated in the presence or absence of IreP, as indicated, at 37°C for the indicated times. Staurosporine (sts; to inhibit kinase activity) was included in all reactions except where indicated. Radiolabeled proteins were analyzed by SDS-PAGE and phosphorimaging. Gel images are representative of three independent replicates. Points on the graphs represent the mean and SEM of all three independent replicates.
We previously showed that IreP, encoded immediately upstream of IreK in the E. faecalis genome, is a Ser/Thr protein phosphatase that dephosphorylates IreK and antagonizes IreK activity in the regulation of cephalosporin resistance (5). To test whether IreB is also a substrate of IreP, we performed in vitro phosphatase assays (Fig. 1B). IreK-n and IreB were first incubated with [γ-32P]ATP to phosphorylate IreB, after which IreP phosphatase and staurosporine were added. Staurosporine, a promiscuous kinase inhibitor that is known to inhibit the IreK homolog of Bacillus subtilis (19), also inhibits E. faecalis IreK kinase activity (see Fig. S3 in the supplemental material) and was added to inhibit rephosphorylation of IreB by IreK-n. Addition of IreP resulted in loss of IreK-dependent phosphorylation from IreB, indicating that IreB is a substrate of both IreK kinase and IreP phosphatase in vitro.
IreK-n phosphorylates IreB on threonine residues.
To identify the site(s) of phosphorylation on IreB, we began by performing phosphoamino acid analysis on IreB that had been phosphorylated in vitro by IreK-n. Radiolabeled IreB was separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane. The IreB band was excised, subjected to acid hydrolysis, and analyzed by high-voltage electrophoresis. Radiolabeled amino acids in the hydrolysate comigrated exclusively with the phosphothreonine standard, indicating that IreK-n phosphorylates IreB solely on threonine residues (Fig. 2A). ireB encodes five threonine residues (at positions 4, 7, 23, 26, and 77). Thr7 appears to be highly conserved among IreB homologs in the genomes of most other Firmicutes. In contrast, Thr4 and Thr23 are conserved predominantly in IreB homologs found among the streptococci, and Thr26 and Thr77 are generally not conserved among IreB homologs. To identify which of the five threonine residues encoded in IreB are phosphorylated by IreK-n, mass spectrometry analysis was carried out on IreB previously phosphorylated by IreK-n in vitro. Thr7 of IreB was the only site of phosphorylation identified in tryptic digests of phospho-IreB following phosphopeptide enrichment (data not shown).
Fig 2.
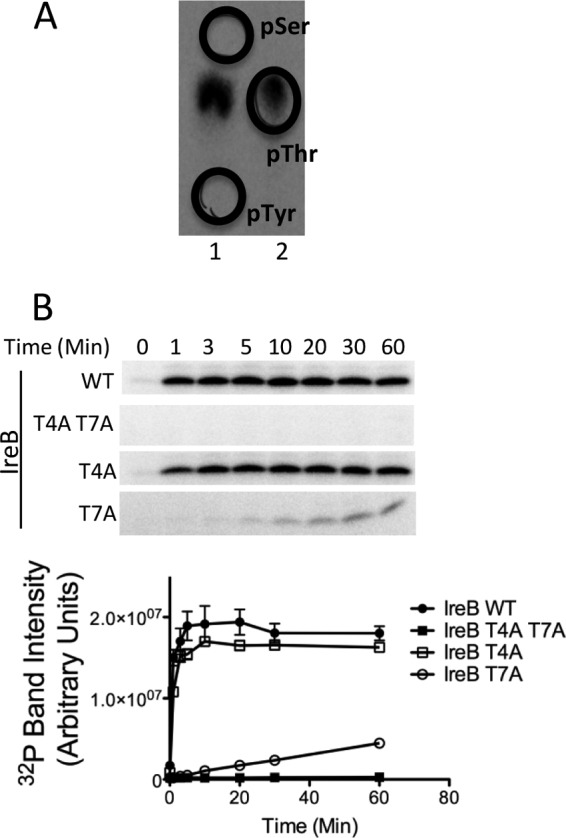
IreK-n phosphorylates IreB on threonine residues. (A) Phosphoamino acid analysis on IreB phosphorylated by IreK-n in vitro. Radiolabeled phospho-IreB protein was subjected to acid hydrolysis and separated by high voltage electrophoresis in 2 adjacent lanes of a cellulose TLC plate. To facilitate unambiguous identification of the phosphorylated amino acid derived from IreB, phosphoamino acid standards were mixed with the IreB hydrolysate and applied in separate lanes (lane 1, phospho-Ser + phospho-Tyr; lane 2, phospho-Thr) and their positions marked (black circles) after staining with ninhydrin. All detectable radiolabeled signal derived from IreB comigrated with the phosphothreonine standard. (B) Kinase assays were performed with 0.33 μM IreK-n and 14.2 μM recombinant hexahistidine-tagged IreB (WT, wild-type IreB; point mutants with substitutions as indicated). Reactions were performed in kinase buffer containing 2 mM ATP, 1 μCi [γ-32P]ATP at 37°C for the indicated times. Radiolabeled proteins were analyzed by SDS-PAGE and phosphorimaging. Gel images are representative of three independent replicates. Points on the graphs represent the mean and SEM of all three independent replicates.
To determine whether IreB T7 is the only site of phosphorylation, we constructed and purified a IreB T7A-His6 mutant and performed kinase assays with IreK-n (Fig. 2B). Phosphorylation of IreB T7A-His6 was substantially diminished compared to wild-type IreB, a finding consistent with the hypothesis that Thr7 is a major site of IreK-n-dependent phosphorylation. However, we observed weak phosphorylation of the IreB T7A mutant, suggesting that other unidentified sites of phosphorylation on IreB existed. A phosphoproteome analysis of group B Streptococcus (GBS) by Silvestroni et al. (20) provided a clue about the identity of the second phosphorylation site: their study reported evidence for phosphorylation of the GBS homolog of IreB (SAK_2030) at sites corresponding to Thr4, as well as Thr7 of E. faecalis IreB. To test whether Thr4 of E. faecalis IreB could be phosphorylated by IreK-n, we constructed and purified an IreB T4A T7A-His6 double mutant and performed kinase assays with IreK-n (Fig. 2). IreK-n kinase was unable to phosphorylate the IreB T4A T7A mutant, confirming that Thr4 and Thr7 represent all sites of IreK-n-dependent phosphorylation, at least in vitro. Analysis of the kinetics of phosphorylation of IreB T4A and IreB T7A mutants (Fig. 2B) revealed that the rate of phosphorylation for IreB T4A was only slightly attenuated compared to that of wild-type IreB, while the rate of IreB T7A phosphorylation was significantly attenuated. These results argue that IreK-n Thr7 represents the primary site of phosphorylation, at least in vitro.
IreB phosphorylation state in vivo is dependent on IreK activity.
To explore the state of IreB phosphorylation in vivo, we introduced plasmids encoding epitope-tagged IreB into E. faecalis strains and performed two-dimensional gel electrophoresis (2DGE). 2DGE, a technique that separates proteins based on both isoelectric point and size, enables interrogation of the posttranslational modification state of a protein because the addition of a phosphoryl group decreases the isoelectric point by 0.3 U. 2DGE analysis of tagged IreB expressed in wild-type E. faecalis showed that IreB is present in two isoforms with distinct isoelectric points (Fig. 3), indicating that IreB is present as a mixed population of modified species. If IreK phosphorylates IreB in vivo, enhancement of IreK activity would be expected to promote a shift in the distribution of IreB species to the more acidic (i.e., phosphorylated) isoform. To test this, we used the E. faecalis ΔireP mutant, in which IreK kinase activity is thought to be unregulated and hyperactivated (5). Expression of tagged IreB in the ΔireP mutant revealed that IreB exists predominantly as one isoform that aligns with the most acidic species of IreB observed in wild-type E. faecalis, a finding consistent with the hypothesis that the majority of IreB is phosphorylated due to the hyperactivated IreK kinase in the ΔireP mutant.
Fig 3.

IreB phosphorylation state in vivo is dependent on IreK activity. Clarified lysates from exponentially growing cells were spiked with an ∼27-kDa Strep-tagged protein standard (to serve as a landmark) and subjected to isoelectric focusing (pH gradient depicted at top) followed by SDS-PAGE and immunoblotting with anti-strep antibody. The protein standard is indicated with an asterisk, and isoforms of IreB-strep are indicated with an arrow in each panel. Strains: WT, OG1RF; ΔireP, CK121. Plasmids: vector, pDL278p23; pIreB strep, pJRG39; pIreB T4A T7A strep, pJLL14.
If IreK were required for phosphorylation of IreB in vivo, IreB would not exist in the most acidic (i.e., phosphorylated) isoform in the absence of IreK. Unfortunately, we were unable to introduce the tagged IreB expression plasmid into E. faecalis strains that lack the IreK kinase, suggesting that increased levels of unphosphorylated IreB in the absence of the IreK kinase are detrimental to normal cell growth. To circumvent this hurdle, we treated wild-type E. faecalis expressing tagged IreB with staurosporine to pharmacologically inhibit IreK activity in vivo and performed 2DGE. We observed a shift in the distribution of IreB isoforms toward the less acidic species compared to that observed in untreated cells (Fig. 3), suggesting that a greater population of IreB molecules exist in a dephosphorylated state after staurosporine treatment, a finding consistent with the hypothesis that IreK phosphorylates IreB in vivo.
To determine whether phosphorylation of IreB at Thr4 and Thr7 occurs in vivo, we expressed tagged IreB T4A T7A in wild-type E. faecalis cells and performed 2DGE. The distribution of IreB isoforms shifted toward the less acidic species (Fig. 3), which is consistent with the loss of phosphorylated IreB and indicates that phosphorylation of Thr4 or Thr7 occurs in E. faecalis cells. Intriguingly, two isoforms with distinct isoelectric points were still observed for IreB T4A T7A, suggesting that IreB is subject to an additional modification in vivo at an as-yet-unidentified site.
Mutations at sites of phosphorylation in IreB impair cephalosporin resistance in E. faecalis.
Because IreB acts a negative regulator of cephalosporin resistance and is a substrate of the IreK kinase, we hypothesized that phosphorylation of IreB would influence its activity in the cephalosporin resistance pathway. To test whether IreB phosphorylation plays a role in regulation of cephalosporin resistance, we expressed IreB T4A T7A in the ΔireB mutant and assessed cephalosporin resistance by determining MIC values. Surprisingly, ΔireB strains expressing IreB T4A T7A exhibited similar levels of ceftriaxone resistance to the ΔireB mutant expressing wild-type IreB (data not shown). However, substitutions at Thr4 or Thr7 of IreB with nonphosphorylatable amino acids valine and isoleucine, which bear greater structural similarity to threonine than does alanine, resulted in reduced cephalosporin resistance (Table 3). For example, expression of IreB T7V led to an 8-fold reduction in resistance toward ceftriaxone compared to the wild-type control. Similarly, expression of the IreB T4V T7V double mutant led to a 4-fold reduction in resistance, and expression of the IreB T4V mutant also resulted in modestly reduced ceftriaxone resistance. Expression of IreB mutants carrying isoleucine substitutions at either Thr4 or Thr7 also resulted in changes in susceptibility that were identical to those observed with the valine single mutants (Table 3). Thus, the reduced ceftriaxone resistance exhibited by strains expressing the threonine-to-valine or -isoleucine IreB point mutants suggests that phosphorylation is required for normal IreB function, and further argues that the nonphosphorylatable valine and isoleucine IreB point mutants are locked in an inhibitory state that prevents activation of cephalosporin resistance pathways in E. faecalis. The observations that the Ala substitutions at Thr4 and Thr7 did not yield a similar phenotype remain unexplained; we propose that the Ala mutants are inherently impaired in their ability to inhibit cephalosporin resistance due to structural differences in the amino acid side chains (Ala lacks a methyl group found in the side chain of Thr; Val and Ile possess an equivalent methyl group), independent of any effect of phosphorylation. For example, the IreB Ala mutants might not be able to bind to an interacting partner as efficiently due to the loss of the methyl group.
If phosphorylation of IreB at T4 and/or T7 relieves its inhibitory state and enables activation of a cephalosporin resistance pathway, we predicted that an IreB mutant carrying phosphomimetic mutations would lead to constitutive and high-level resistance comparable to that of the ΔireB mutant. Although not universally successful, a general strategy to mimic the effect of phosphorylation in some phosphoproteins is to introduce glutamate substitutions at the sites of phosphorylation, thereby introducing a negatively charged amino acid side chain to mimic the negatively charged phosphoryl group. Therefore, we constructed an IreB phosphomimetic mutant containing glutamate at Thr4 and Thr7. However, expression of IreB T4E T7E in ΔireB E. faecalis resulted in wild-type levels of ceftriaxone resistance (data not shown), suggesting that glutamate substitutions at Thr4 and Thr7 do not behave as phosphomimetics in IreB.
It remains unclear how, at the molecular level, IreB acts to inhibit cephalosporin resistance. We speculate that IreB may participate in protein-protein interactions with one or more as-yet-unknown factors that promote resistance, thereby sequestering those factors and preventing their activity. In this model, IreK-mediated phosphorylation of IreB at Thr4 and/or Thr7 results in the release of IreB from the unknown factor X, enabling factor X to actively promote cephalosporin resistance (Fig. 4). Although phosphorylation at either Thr4 or Thr7 of IreB appears to modulate its influence on cephalosporin resistance (Table 3), the relationship between phosphorylation events at these sites remains unclear. Future studies will be required to determine whether phosphorylation at one site on IreB influences phosphorylation at the other, whether the different phosphorylated isoforms exhibit distinct biological properties, and whether the effect of phosphorylation at Thr4 and Thr7 is additive, partially redundant, or independent. Our in vitro phosphorylation analyses (Fig. 2) suggest that Thr7 represents the primary site of phosphorylation. Consistent with that hypothesis, in vivo susceptibility studies (Table 3) indicate that phosphorylation at IreB T7 plays a more important role than phosphorylation at Thr4 in relieving the inhibitory activity of IreB. Because Thr7 is conserved in nearly all homologs of IreB found in GenBank, the critical functional role of Thr7 may be widely shared among many Gram-positive bacteria.
Fig 4.
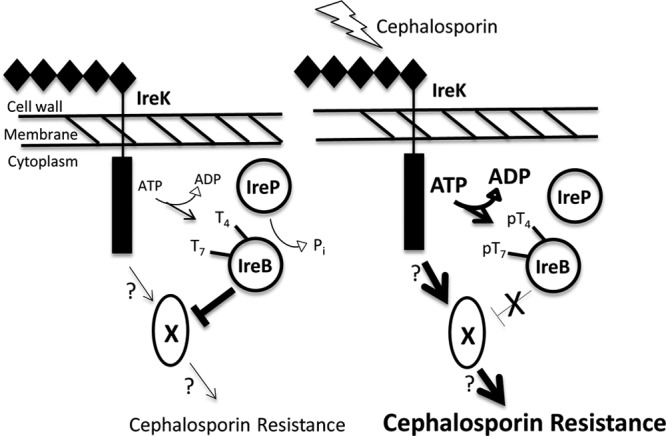
Model for IreB function in IreK-mediated cephalosporin resistance in E. faecalis. (Left) In the absence of cell-wall stressors such as cephalosporins, the kinase activity of IreK is low, and the IreB population is maintained in a relatively unphosphorylated state by IreP-mediated dephosphorylation. Unphosphorylated IreB inhibits the function of an as-yet-unknown resistance factor (X) to restrict the activity of the signaling pathway and reduce phenotypic cephalosporin resistance. (Right) During cephalosporin-induced cell wall stress, kinase activity of IreK is enhanced (bold arrows), resulting in phosphorylation of IreB and relief of its inhibitory activity against X, thereby enabling active signaling and enhanced cephalosporin resistance (boldface). Mutants lacking IreB entirely will possess a completely uninhibited factor X and therefore exhibit elevated levels of cephalosporin resistance, as observed.
Although we do not yet understand the molecular details of the inhibitory activity of IreB, the observation that we were unable to introduce an expression vector encoding IreB into an E. faecalis strain lacking the IreK kinase suggests that overexpression of unphosphorylated IreB either impairs growth or is overtly toxic to the cells. We hope to exploit this property in future genetic screens designed to identify the molecular target(s) of IreB. Finally, because putative homologs of IreB are conserved in most low GC Gram-positive bacteria, further investigation of the important role of IreB in bacterial physiology and stress responses promises to provide a better understanding of the biology of other harmful and opportunistic pathogens.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jaime Little and Jamie Genthe for the construction of plasmids and strains, Mike Gilmore for providing E. faecalis T1, Thomas Zahrt for assistance with isoelectric focusing, and Matthew Greseth and Paula Traktman for assistance with phosphoamino acid analysis.
This study was supported by grant AI081692 from the National Institute of Allergy and Infectious Disease (NIAID).
The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the National Institutes of Health.
Footnotes
Published ahead of print 30 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01472-13.
REFERENCES
- 1. Tannock GW, Cook G. 2002. Enterococci as members of the intestinal microflora of humans, p 101–132 In The enterococci: pathogenesis, molecular biology, and antibiotic resistance. ASM Press, Washington, DC [Google Scholar]
- 2. Richards MJ, Edwards JR, Culver DH, Gaynes RP. 2000. Nosocomial infections in combined medical-surgical intensive care units in the United States. Infect. Control Hosp. Epidemiol. 21:510–515 [DOI] [PubMed] [Google Scholar]
- 3. Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK, National Healthcare Safety Network Team, Participating National Healthcare Safety Network Facilities 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect. Control Hosp. Epidemiol. 29:996–1011 [DOI] [PubMed] [Google Scholar]
- 4. Kristich CJ, Wells CL, Dunny GM. 2007. A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc. Natl. Acad. Sci. U. S. A. 104:3508–3513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kristich CJ, Little JL, Hall CL, Hoff JS. 2011. Reciprocal regulation of cephalosporin resistance in Enterococcus faecalis. mBio 2:e00199–11. 10.1128/mBio.00199-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hussain H, Branny P, Allan E. 2006. A eukaryotic-type serine/threonine protein kinase is required for biofilm formation, genetic competence, and acid resistance in Streptococcus mutans. J. Bacteriol. 188:1628–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burnside K, Lembo A, de Los Reyes M, Iliuk A, Binhtran N-T, Connelly JE, Lin W-J, Schmidt BZ, Richardson AR, Fang FC, Tao WA, Rajagopal L. 2010. Regulation of hemolysin expression and virulence of Staphylococcus aureus by a serine/threonine kinase and phosphatase. PLoS One 5:e11071. 10.1371/journal.pone.0011071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shah IM, Laaberki M-H, Popham DL, Dworkin J. 2008. A eukaryotic-like Ser/Thr kinase signals bacteria to exit dormancy in response to peptidoglycan fragments. Cell 135:486–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rajagopal L, Vo A, Silvestroni A, Rubens CE. 2005. Regulation of purine biosynthesis by a eukaryotic-type kinase in Streptococcus agalactiae. Mol. Microbiol. 56:1329–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Veyron-Churlet R, Zanella-Cléon I, Cohen-Gonsaud M, Molle V, Kremer L. 2010. Phosphorylation of the Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein reductase MabA regulates mycolic acid biosynthesis. J. Biol. Chem. 285:12714–12725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beltramini AM, Mukhopadhyay CD, Pancholi V. 2009. Modulation of cell wall structure and antimicrobial susceptibility by a Staphylococcus aureus eukaryote-like serine/threonine kinase and phosphatase. Infect. Immun. 77:1406–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Banu LD, Conrads G, Rehrauer H, Hussain H, Allan E, van der Ploeg JR. 2010. The Streptococcus mutans serine/threonine kinase, PknB, regulates competence development, bacteriocin production, and cell wall metabolism. Infect. Immun. 78:2209–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pereira SFF, Goss L, Dworkin J. 2011. Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol. Mol. Biol. Rev. 75:192–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Squeglia F, Marchetti R, Ruggiero A, Lanzetta R, Marasco D, Dworkin J, Petoukhov M, Molinaro A, Berisio R, Silipo A. 2011. Chemical basis of peptidoglycan discrimination by PrkC, a key kinase involved in bacterial resuscitation from dormancy. J. Am. Chem. Soc. 133:20676–20679 [DOI] [PubMed] [Google Scholar]
- 15. Kristich CJ, Chandler JR, Dunny GM. 2007. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 57:131–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Traktman P, Liu K, DeMasi J, Rollins R, Jesty S, Unger B. 2000. Elucidating the essential role of the A14 phosphoprotein in vaccinia virus morphogenesis: construction and characterization of a tetracycline-inducible recombinant. J. Virol. 74:3682–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wessel D, Flügge UI. 1984. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138:141–143 [DOI] [PubMed] [Google Scholar]
- 18. Zahrt TC, Wozniak C, Jones D, Trevett A. 2003. Functional analysis of the Mycobacterium tuberculosis MprAB two-component signal transduction system. Infect. Immun. 71:6962–6970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shah IM, Dworkin J. 2010. Induction and regulation of a secreted peptidoglycan hydrolase by a membrane Ser/Thr kinase that detects muropeptides. Mol. Microbiol. 75:1232–1243 [DOI] [PubMed] [Google Scholar]
- 20. Silvestroni A, Jewell KA, Lin W-J, Connelly JE, Ivancic MM, Tao WA, Rajagopal L. 2009. Identification of serine/threonine kinase substrates in the human pathogen group B streptococcus. J. Proteome Res. 8:2563–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dunny GM, Brown BL, Clewell DB. 1978. Induced cell aggregation and mating in Streptococcus faecalis: evidence for a bacterial sex pheromone. Proc. Natl. Acad. Sci. U. S. A. 75:3479–3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maekawa S, Yoshioka M, Kumamoto Y. 1992. Proposal of a new scheme for the serological typing of Enterococcus faecalis strains. Microbiol. Immunol. 36:671–681 [DOI] [PubMed] [Google Scholar]
- 23. Vesić D, Kristich CJ. 2012. MurAA is required for intrinsic cephalosporin resistance of Enterococcus faecalis. Antimicrob. Agents Chemother. 56:2443–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Y, Staddon JH, Dunny GM. 2007. Specificity determinants of conjugative DNA processing in the Enterococcus faecalis plasmid pCF10 and the Lactococcus lactis plasmid pRS01. Mol. Microbiol. 63:1549–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hayes F, Daly C, Fitzgerald GF. 1990. Identification of the minimal replicon of Lactococcus lactis subsp. lactis UC317 plasmid pCI305. Appl. Environ. Microbiol. 56:202–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.