

Abstract
Telaprevir is a linear, peptidomimetic small molecule that inhibits hepatitis C virus (HCV) replication by specifically inhibiting the NS3·4A protease. In phase 3 clinical studies, telaprevir in combination with peginterferon and ribavirin (PR) significantly improved sustained virologic response (SVR) rates in genotype 1 chronic HCV-infected patients compared with PR alone. In patients who do not achieve SVR after treatment with telaprevir-based regimens, variants with mutations in the NS3·4A protease region have been observed. Such variants can contribute to drug resistance and limit the efficacy of treatment. To gain a better understanding of the viral resistance profile, we conducted phenotypic characterization of the variants using HCV replicons carrying site-directed mutations. The most frequently observed (significantly enriched) telaprevir-resistant variants, V36A/M, T54A/S, R155K/T, and A156S, conferred lower-level resistance (3- to 25-fold), whereas A156T and V36M+R155K conferred higher-level resistance (>25-fold) to telaprevir. Rarely observed (not significantly enriched) variants included V36I/L and I132V, which did not confer resistance to telaprevir; V36C/G, R155G/I/M/S, V36A+T54A, V36L+R155K, T54S+R155K, and R155T+D168N, which conferred lower-level resistance to telaprevir; and A156F/N/V, V36A+R155K/T, V36M+R155T, V36A/M+A156T, T54A+A156S, T54S+A156S/T, and V36M+T54S+R155K, which conferred higher-level resistance to telaprevir. All telaprevir-resistant variants remained fully sensitive to alpha interferon, ribavirin, and HCV NS5B nucleoside and nonnucleoside polymerase inhibitors. In general, the replication capacity of telaprevir-resistant variants was lower than that of the wild-type replicon.
INTRODUCTION
More than 170 million people worldwide experience chronic hepatitis C virus (HCV) infections, which may lead to severe liver diseases, including fibrosis, cirrhosis, and hepatocellular carcinoma (1–5). Treatment of genotype 1 HCV-infected patients with peginterferon and ribavirin (PR) has a low (34% to 56%) success rate and is associated with substantial adverse events, such as flu-like symptoms, fatigue, depression, and anemia (6, 7), limiting adherence to treatment and impacting treatment outcome in a significant number of patients. In the last decade, the development of new classes of HCV therapy, direct-acting antivirals (DAAs), has been a major focus of drug discovery efforts. Telaprevir, a linear peptidomimetic small molecule, is a specific inhibitor of the HCV NS3·4A protease that is required for polyprotein processing and viral replication (8–10). In phase 3 clinical studies, telaprevir in combination with PR provided significantly improved sustained virologic response (SVR) rates for both treatment-naive and previously treated patients compared with PR alone (11, 12).
The HCV genome exhibits significant genetic heterogeneity, with high sequence diversity both between and within the various genotypes and subtypes (13, 14). The low fidelity of the HCV polymerase, high viral replication rate, and strong selective pressure on the virus result in a unique and diverse viral quasispecies in each patient (15). New HCV populations with every potential substitution, some of which convey various degrees of resistance to DAAs, are likely generated many times each day (14, 16, 17). Thus, it is likely that all patients have DAA-resistant variants prior to treatment. Along with the availability of replication space, the prevalence of a resistant variant in a patient's viral quasispecies is generally determined by its replicative fitness and selective advantage compared with the rest of the viral population (16). Minor populations of preexisting, resistant variants are usually present at levels below the detection limits of current sequencing techniques, as they are less fit than wild-type (WT) virus but have a fitness advantage over WT virus in the presence of a drug and become the dominant viral species (16, 17). Indeed, viral populations with drug resistance substitutions have been shown to emerge in vitro in the presence of DAAs or when patients do not achieve an SVR with DAA treatment (18, 19). During the clinical development of telaprevir, HCV variants associated with treatment failure were identified from extensive viral sequence analyses (11, 12, 20, 21). Variants enriched in the viral population in patients who did not achieve an SVR with a telaprevir-based regimen most commonly had amino acid changes at residues 36, 54, 155, and 156 of the NS3 protease domain (11, 12, 20). Variants V36M, R155K, and V36M+R155K emerged frequently in patients with genotype 1a (G1a) HCV, and V36A, T54A, and A156S/T emerged in patients with genotype 1b (G1b) HCV (22).
Drug resistance is a factor that should be considered in DAA therapies for HCV-infected patients. An understanding of drug resistance is important in optimizing DAA treatment regimens to increase SVR rates and minimize the clinical impact of resistance. In this study, we analyzed the resistance profile of the variants that were observed in clinical studies of telaprevir (22, 23). Using HCV replicons carrying site-directed mutations, we evaluated the in vitro susceptibility of the clinically observed NS3 protease variants to telaprevir, as well as the potential cross-resistance of the variants to other NS3 protease inhibitors, alpha interferon (IFN-α), ribavirin, and NS5B polymerase inhibitors. We also characterized the in vitro replication capacity of these variants.
MATERIALS AND METHODS
Cells and compounds.
Huh-7.5 cells were obtained from Apath LLC (Brooklyn, NY) (24). Huh-7-ET-“cured” cells, a Huh-7 subcell line, were established by curing the Huh-7 cell line containing the subgenomic replicon pFK I389luc-ubi-neo/NS3-3′/5.1 (25).
Cells were maintained in complete Dulbecco's modified Eagle minimal essential medium (cDMEM) composed of DMEM supplemented with 10% fetal bovine serum (FBS), 100 IU/ml penicillin, 100 μg/ml streptomycin, 1× nonessential amino acids (NEAA), and 2 mM glutamine at 37°C in an atmosphere of 5% carbon dioxide and passaged twice weekly to maintain subconfluence.
Linear ketoamide NS3 protease inhibitors telaprevir (8–10) and boceprevir (26), macrocyclic NS3 protease inhibitors ciluprevir (27, 28) and danoprevir (29), nucleoside NS5B polymerase inhibitor NM-107 (30), and nonnucleoside NS5B polymerase inhibitors VX-222 (31), HCV-796 (32), compound 2 (CMPD 2 [33]), and compound 55 (CMPD 55 [34]) were synthesized at Vertex Pharmaceuticals Incorporated (Cambridge, MA). Macrocyclic NS3 protease inhibitors simeprevir (35) and vaniprevir (36) were obtained from Acme Bioscience Inc. (Palo Alto, CA). Nucleoside NS5B polymerase inhibitor mericitabine (37) was obtained from SAI Advantium (Hyderabad, India). All compounds were dissolved in dimethyl sulfoxide (DMSO) to a final concentration of 10 or 20 mM and were stored at −20°C. Human recombinant IFN-α (Calbiochem, La Jolla, CA) and ribavirin (Sigma-Aldrich, St. Louis, MO) were reconstituted in water and stored at −80°C and −20°C, respectively.
Construction of plasmids.
Substitutions of HCV NS3 protease variants were introduced into 4 sets of replicon plasmids using PCR-based site-directed mutagenesis or subcloning to generate variant replicons: (i) in stable cell lines, 50% effective concentration (EC50) values were determined using pBR322-HCV-Neo-mADE, a replicon plasmid that contains adaptive mutations causing 3 amino acid substitutions (E1202G, S2204R, and R2861G) (38) and is derived from the G1b-Con1 strain subgenomic replicon I389neo/NS3-3′/wt (39); (ii) pBR322-HCV-Luc-mADE (40), a G1b-Con1 strain subgenomic replicon that was used to evaluate replication capacity in transient transfections; (iii) pFK I341PiLuc/NS3-3′/ET, a G1b-Con1 strain subgenomic replicon that was kindly provided by R. Bartenschlager (adapted from Lohmann et al. [39]) and was used to determine EC50 and replication capacity in transient transfections; and finally, (iv) 1a-PiLuci-1a6*, a subgenomic replicon that contains the G1a-H77 NS3-3′ sequence with adaptive mutations causing 6 amino acid substitutions (Q1067R, P1496L, V1655I, K1691R, K2040R, and S2204I) and a luciferase gene cassette under the translational control of the encephalomyocarditis virus (EMCV) (internal ribosome entry site [IRES]) and the poliovirus IRES, respectively, and was used to determine the EC50 in transient transfections.
To express the NS3 protease, a cDNA fragment encoding residues 1 (A) to 181 (S) of the NS3 protease domain from a G1a HCV-infected patient enrolled in a phase 1b study was subcloned into pBEV11, which contains a C-terminal hexahistidine tag (38). In each expression construct, NS3 residue L13 (codon: CTC) was replaced with K (codon: AAG) to improve the solubility of the proteins.
Determination of replicon EC50.
The method to determine EC50s in stable replicon cells using pBR322-HCV-Neo-mADE has been described previously (38, 41). Briefly, full-length HCV subgenomic replicon RNA was generated from a ScaI-linearized DNA template using a MEGAscript T7 kit (Life Technologies, Grand Island, NY). The resulting RNA was electroporated into Huh-7 cells using a Bio-Rad Gene Pulser II electroporator, and G418 (Geneticin)-resistant HCV replicon cells were selected with 0.5 mg/ml G418–cDMEM for 2 to 3 weeks. The presence of mutations in the replicons was confirmed by sequencing the reverse transcription-PCR (RT-PCR) products amplified from the total cellular RNA. The EC50s of compounds were determined in a 48-h assay using stable replicon cells. Briefly, 1 × 104 HCV replicon cells/well were plated in a tissue-culture-treated 96-well plate in DMEM containing 10% FBS, 2 mM l-glutamine, 1× NEAA, and 0.25 mg/ml G418 and then incubated overnight at 37°C in a humidified incubator with 5% CO2. The next day, the medium was replaced with DMEM containing 2% FBS, 2 mM l-glutamine, 1× NEAA, and serially diluted compound in DMSO (0.5% final concentration). After incubation for 48 h, replicon RNA levels were determined using a QuantiGene Discover assay kit (Panomics, Inc., Fremont, CA) with an HCV-specific primer set.
The EC50s of variant replicons generated with pFK I341PiLuc/NS3-3′/ET or 1a-PiLuci-1a6* were determined in a 96-h assay with transient transfections of Huh-7-ET-cured cells. Briefly, 5 μg of T7 RNA runoff transcripts generated from ScaI-linearized pFK I341PiLuc/NS3-3′/ET or HpaI-linearized 1a-PiLuci-1a6* replicons were electroporated into 4 × 106 Huh-7-ET-cured cells resuspended in Ingenio (Mirus Bio LLC, Madison, WI). Electroporated cells were resuspended in cDMEM and plated in 96-well, tissue-culture-treated plates at 1 × 104 cells in 100 μl. After incubation at 37°C for 24 h, the cultured cells were mixed with 100 μl of medium containing the serially diluted compounds and allowed to grow for 3 days. The cells were lysed with lysis buffer (Promega, Madison, WI), and luciferase activity was measured using a luciferase assay system (Promega, Madison, WI).
The EC50s were calculated from dose-response curves using a 4-parameter curve-fitting method in the Softmax Pro program (Molecular Devices Corp., Sunnyvale, CA). Multiple independent assays were conducted for each viral replicon, and the means and standard deviations (SD) of the replicon EC50s were calculated. The fold change in sensitivity to HCV inhibitors was calculated by dividing the mean EC50 for each variant by the mean EC50 for the WT replicon. Variants were considered to be resistant to the inhibitors if the increase in the EC50 compared with the WT was ≥3-fold because the variation of the EC50s of the assay was <3-fold.
Expression and purification of recombinant NS3 protease.
An HCV NS3 serine protease domain with either a WT sequence or a substitution was expressed in BL21(DE3) Escherichia coli cells (Stratagene, La Jolla, CA) and purified as described previously (38), with minor modifications. Freshly transformed cells were grown at 37°C in a brain heart infusion (BHI) medium (Difco Laboratories, Kansas City, MO) supplemented with 100 μg/ml carbenicillin to reach an optical density of 0.75 at 600 nm, followed by induction with 1 mM isopropyl-1-thio-β-d-galactopyranoside at 24°C for 4 h. Cell paste was resuspended in buffer A (50 mM HEPES [pH 8.0], 300 mM NaCl, 0.1% n-octyl-β-d-glucopyranoside, 5 mM β-mercaptoethanol, and 10% [vol/vol] glycerol) supplemented with 5 mM imidazole and lysed with BugBuster reagent (Novagen, Madison, WI), followed by centrifugation at 16,000 × g for 30 min (41). The lysate was passed over a 2-ml bed of pre-equilibrated Talon affinity resin (Clontech, Palo Alto, CA) at 2 ml/min and washed with 30 column volumes of buffer A plus 5 mM imidazole. The HCV NS3 proteins were eluted in buffer A plus 300 mM imidazole. The eluates were then concentrated and loaded onto a Hi-Load 16/60 Superdex 200 column that had been pre-equilibrated with buffer A. The appropriate fractions of the purified HCV proteins were pooled and stored at −80°C.
Determination of enzyme IC50 or IC50(1 h).
In vitro protease activity was assayed in 96-well microtiter plates (Corning, NY), as described previously (38). Briefly, the NS3 protease was incubated with 5 μM cofactor KK4A (KKGSVVIVGRIVLSGK) (42) in 50 mM HEPES (pH 7.8)–100 mM NaCl–20% glycerol–5 mM dithiothreitol at 25°C for 10 min and at 30°C for 10 min. HCV NS3·4A protease inhibitors, serially diluted in DMSO, were added to the protease mixture. For telaprevir and boceprevir, the mixture was incubated for an additional 60 min at 30°C to derive 1-h 50% inhibitory concentration [IC50(1 h)] values. For ciluprevir or danoprevir, IC50s were obtained without additional preincubation. The reaction was initiated by the addition of 5 μM RET-S1 {Ac-DED(EDANS)EEαAbuψ[COO]ASK(DABCYL)-NH2; Anaspec Inc., San Jose, CA}, an internally quenched fluorogenic depsipeptide substrate (38, 43), and incubated at 30°C. Product release was monitored for 20 min (excitation at 360 nm and emission at 500 nm) in a Tecan SpectraFluorPlus plate reader (Tecan US, Durham, NC). The enzyme assay was performed in a total volume of 100 μl. The protease concentration was chosen such that 10% to 20% of the substrate was turned over during the course of the assay. Prism software (GraphPad Software, Inc., La Jolla, CA) was used to fit a sigmoidal dose-response curve (4-parameter curve fitting method) to the data, with IC50 or IC50(1 h) values derived from each fitted curve.
Determination of replication capacity.
As previously described by Zhou and colleagues, the ScaI-linearized pBR322-Luc-mADE or pFK I341PiLuc/NS3-3′/ET replicons were used to generate T7 RNA runoff transcripts, which were transfected into Huh-7.5 or Huh-7-ET-cured cells by electroporation (41). Transfected cells were plated into duplicated 96-well plates and cultured for 2.5 to 3 h for the first set of plates and 72 h (for pBR322-Luc-mADE) to 96 h (for pFK I341PiLuc/NS3-3′/ET) for the second set of plates. The cells were lysed with lysis buffer and kept frozen at −80°C until assayed for luciferase activity (Promega kit; Promega, Madison, WI). For any given replicon variant, a normalized luciferase signal was calculated by dividing the luciferase signal at 72 or 96 h postelectroporation with that from 2.5 or 3 h postelectroporation of the same replicon variant. The relative replication capacity of the NS3 protease variant is expressed as the percentage of the normalized luciferase signal of the variant replicon compared with that of the WT replicon (as 100%) and that of a HCV polymerase null variant (as 0%).
RESULTS AND DISCUSSION
Susceptibility of NS3 protease variants to telaprevir in HCV replicon cells.
The EC50s of telaprevir against NS3 protease variants were determined in G1b 48-h or 96-h replicon assays, and a subset of the variants was also evaluated using a G1a 96-h replicon assay to assess the consistency of the resistance levels in both genotypes (Table 1). The WT G1b replicon had EC50 (±SD) values of 0.482 (±0.122) μM and 0.269 (±0.096) μM in the 48-h and 96-h assays, respectively, whereas the WT G1a replicon had an EC50 (±SD) of 0.961 (±0.132) μM in the 96-h assay. The NS3 protease variants that were characterized included (i) variants most frequently observed (significantly enriched) in patients who did not achieve SVR in telaprevir-based regimens, including V36A/M, T54A/S, R155K/T, A156S/T, and V36M+R155K (22); (ii) variants rarely observed (not significantly enriched) in patients who did not achieve SVR in telaprevir-based regimens, including V36C/G/I/L, I132V, V151A, R155G/I/M/S, A156F/N/V, V36A+T54A, V36A+R155K/T, V36L+R155K, V36M+R155T, V36A/M+A156T, T54A+A156S, T54S+R155K, T54S+A156S/T, R155T+D168N, and V36M+T54S+R155K (22); and (iii) variants specifically observed for other HCV NS3 protease inhibitors, including Q41R, Q80R, and D168A/N/V, which are resistant to one or more macrocyclic inhibitors such as vaniprevir (36, 44), ciluprevir (38), danoprevir (45), and simeprevir (46), and V55A, R109K, and V170A, which are resistant to linear NS3 protease inhibitors boceprevir (47, 48) and SCH6 (49, 50).
Table 1.
Replicon EC50 of telaprevir against NS3 protease variantsa
| Variant | EC50 ± SD (μM) | Fold change ± SD |
|---|---|---|
| Frequently observed in telaprevir studies | ||
| G1b replicon (48-h assay) | ||
| WT | 0.482 ± 0.122 | 1.0 ± 0.3 |
| V36A | 3.57 ± 1.05 | 7.4 ± 2.2 |
| V36M | 3.38 ± 0.78 | 7.0 ± 1.6 |
| T54A | 3.04 ± 0.82 | 6.3 ± 1.7 |
| T54S | 2.02 ± 0.19 | 4.2 ± 0.4 |
| R155K | 3.59 ± 0.28 | 7.4 ± 0.6 |
| R155T | 9.60 ± 0.87 | 20 ± 2 |
| A156Sb | 4.65 | 9.6 |
| A156Tc | >30 | >62 |
| V36M + R155K | ∼31 | ∼64 |
| G1a replicon (96-h assay) | ||
| WT | 0.961 ± 0.132 | 1.0 ± 0.1 |
| V36A | 7.25 ± 0.41 | 7.5 ± 0.4 |
| V36M | 6.55 ± 0.56 | 6.8 ± 0.6 |
| R155K | 5.66 ± 1.86 | 5.9 ± 1.9 |
| A156S | 21.5 ± 2.7 | 22 ± 3 |
| V36M + R155K | >25 | >26 |
| Rarely observed in telaprevir studies | ||
| G1b replicon (48-h assay) | ||
| WT | 0.482 ± 0.122 | 1.0 ± 0.3 |
| V36C | 3.78 ± 0.48 | 7.8 ± 1.0 |
| V36G | 5.43 ± 0.21 | 11 ± 0.4 |
| V36L | 1.06 ± 0.20 | 2.2 ± 0.4 |
| R155G | 3.58 ± 0.24 | 7.4 ± 0.5 |
| R155I | 11.6 ± 2.5 | 24 ± 5 |
| R155M | 2.68 ± 0.21 | 5.6 ± 0.4 |
| R155S | 1.97 ± 0.21 | 4.1 ± 0.4 |
| A156F | >30 | >62 |
| A156Vc | >30 | >62 |
| V36A + T54A | 9.73 ± 1.41 | 20 ± 3 |
| V36A + R155K | ∼20 | ∼41 |
| V36A + R155T | >30 | >62 |
| V36L + R155K | 10.3 ± 0.5 | 21 ± 1 |
| V36M + R155T | >30 | >62 |
| V36A + A156T | >30 | >62 |
| V36M + A156T | >30 | >62 |
| T54S + R155K | 10.6 ± 0.2 | 22 ± 0.4 |
| T54A + A156S | >30 | >62 |
| T54S + A156S | >30 | >62 |
| T54S + A156T | >30 | >62 |
| R155T + D168N | 11.3 ± 0.3 | 24 ± 1 |
| V36M + T54S + R155K | >30 | >62 |
| G1b replicon (96-h assay) | ||
| WT | 0.269 ± 0.096 | 1.0 ± 0.4 |
| V36I | 0.0853 ± 0.0059 | 0.3 ± 0.02 |
| V151A | 0.230 ± 0.046 | 0.9 ± 0.2 |
| A156N | >25 | >93 |
| G1a replicon (96-h assay) | ||
| WT | 0.961 ± 0.132 | 1.0 ± 0.1 |
| I132V | 1.68 ± 0.66 | 1.8 ± 0.7 |
| Resistant to other HCV protease inhibitors | ||
| G1b replicon (48-h assay) | ||
| WT | 0.482 ± 0.122 | 1.0 ± 0.3 |
| Q41R | 0.720 ± 0.090 | 1.5 ± 0.2 |
| V55A | 1.03 ± 0.11 | 2.1 ± 0.2 |
| Q80R | 0.250 ± 0.020 | 0.5 ± 0.04 |
| R109K | 0.390 ± 0.110 | 0.8 ± 0.2 |
| D168Ab | 0.193 | 0.4 |
| D168N | 0.306 ± 0.042 | 0.6 ± 0.1 |
| D168Vb | 0.163 | 0.3 |
| V170A | 1.25 ± 0.28 | 2.6 ± 0.6 |
| G1a replicon (96-h assay) | ||
| WT | 0.961 ± 0.132 | 1.0 ± 0.1 |
| D168N | 0.910 ± 0.004 | 0.9 ± 0.004 |
The mean EC50 and SD were derived from 8 to 15 and 3 to 5 individual experiments for the WT and variant replicons, respectively. The mean fold change was determined by dividing the mean EC50 of a given variant replicon by that of the WT replicon. The SD of the fold change was determined by dividing the SD of the EC50 of a given variant replicon by the mean EC50 of the WT replicon. “>” denotes that the EC50 or fold change was greater the value presented; the actual value could not be determined since no significant reduction of HCV RNA level was observed at the maximum concentration of telaprevir used (30 μM for the G1b 48-h assay and 25 μM for the G1a or G1b 96-h assays). “∼” denotes that the EC50 or fold change was estimated from a 40% or greater reduction of the HCV RNA level; the actual value could not be determined due to the reduction of HCV RNA not being sufficient.
Data published previously (38).
Data published previously (40).
The variants that were observed in clinical studies of telaprevir conferred different levels of resistance to telaprevir, with increases in EC50s ranging from 0.3- to >93-fold relative to the WT (Table 1). An increase in the EC50 of <3-fold relative to the WT was considered to be within the assay variation and not reflective of resistance. Variants were categorized as showing either lower-level (EC50 fold change from 3 to 25) or higher-level (EC50 fold change > 25) resistance. As nearly all viral breakthroughs that occur during treatment with telaprevir are associated with variants with higher-level resistance, telaprevir given in combination with PR appears to be sufficient to inhibit variants with lower-level resistance (22).
Frequently observed single variants V36A/M, T54A/S, R155K/T, and A156S showed lower-level resistance to telaprevir, whereas A156T and the double variant V36M+R155K conferred higher-level resistance to telaprevir. Single variants V36C/G and R155G/I/M/S, which were rarely observed, conferred lower-level resistance. However, the rarely observed A156F/N/V variants conferred a higher level of resistance. The rarely observed single variants V36I/L, I132V, and V151A did not change the sensitivity to telaprevir in the replicon system. Rarely observed double and triple variants conferred higher-level resistance, except for V36A+T54A, V36L+R155K, T54S+R155K, and R155T+D168N, which were associated with lower-level resistance to telaprevir.
In general, the fold changes in EC50s relative to the WT replicon were consistent between the G1b 48-h and 96-h replicon assays (data not shown). Similarly, the levels of resistance to telaprevir associated with the major variants V36A/M, R155K, and A156S or V36M+R155K obtained from G1a and G1b replicons were also consistent. The maximum difference in EC50 change between G1a and G1b was 2.3-fold, which was observed for A156S (Table 1).
Variants Q41R, V55A, Q80R, R109K, D168A/N/V, and V170A have been reported to confer resistance to other HCV NS3 protease inhibitors. D168N was also observed in a small number of patients who did not achieve an SVR with telaprevir treatment (22). These variants conferred a less than 3-fold change in the EC50s of telaprevir in G1a or G1b replicons. Thus, these variants are not considered resistant to telaprevir in vitro.
Susceptibility of telaprevir-resistant variants to other HCV NS3 protease inhibitors in HCV replicon cells.
To evaluate whether variants that confer resistance to telaprevir also confer resistance to other HCV NS3 protease inhibitors, WT and NS3 variant replicons (G1b) were tested against boceprevir (26), ciluprevir (27, 28), danoprevir (29), simeprevir (35), and vaniprevir (36) in a 48-h assay (Fig. 1 and Table 2).
Fig 1.
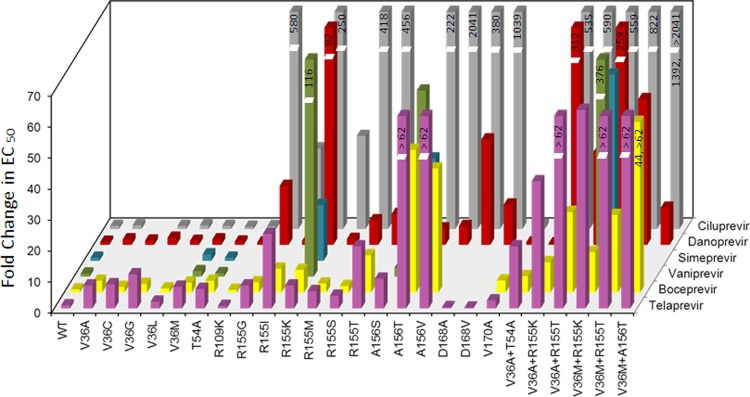
Comparison of the activities of HCV NS3 protease inhibitors on NS3 variant replicons. Fold change in EC50 was determined in the G1b 48-h assay.
Table 2.
Replicon EC50 of other protease inhibitors against NS3 variantsa
| Variant | Boceprevir |
Ciluprevir |
Danoprevir |
Simeprevir |
Vaniprevir |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50(s) (μM) | FC(s) | EC50(s) (μM) | FC(s) | EC50 (μM) | FC | EC50 (μM) | FC | EC50 (μM) | FC | |
| WT | 0.487 | 1.0 | 0.0049 | 1.0 | 0.0049 | 1.0 | 0.0016 | 1.0 | 0.0028 | 1.0 |
| V36A | 1.83 | 3.8 | 0.0058 | 1.2 | 0.0087 | 1.8 | ND | ND | ND | ND |
| V36C | 0.860 | 1.8 | ND | ND | 0.0071 | 1.4 | ND | ND | ND | ND |
| V36G | 1.36 | 2.8 | 0.0048 | 1.0 | 0.012 | 2.3 | ND | ND | ND | ND |
| V36L | 0.610 | 1.3 | 0.0060 | 1.2 | 0.0064 | 1.3 | ND | ND | ND | ND |
| V36M | 1.56 | 3.2 | 0.0049 | 1.0 | 0.0086 | 1.8 | 0.0033 | 2.1 | 0.0050 | 1.8 |
| T54A | 1.85 | 3.8 | 0.0032 | 0.7 | 0.0055 | 1.1 | 0.0020 | 1.0 | 0.0030 | 1.1 |
| R109K | 0.446 | 0.9 | 0.0042 | 0.9 | 0.0037 | 0.8 | ND | ND | ND | ND |
| R155G | 1.59 | 3.3 | 2.8 | 580 | 0.091 | 19 | ND | ND | ND | ND |
| R155I | 3.77 | 7.7 | 0.13 | 26 | 0.0063 | 1.3 | ND | ND | ND | ND |
| R155K | 3.57 | 7.3 | 1.2 | 250 | 0.40 | 82 | 0.030 | 18 | 0.32 | 116 |
| R155M | 1.43 | 2.9 | 0.15 | 30 | 0.0094 | 1.9 | ND | ND | ND | ND |
| R155S | 1.02 | 2.1 | 2.0 | 418 | 0.039 | 7.9 | ND | ND | ND | ND |
| R155T | 5.86 | 12 | 2.2 | 456 | 0.050 | 10 | ND | ND | ND | ND |
| A156S | ND | ND | 0.0070 | 1.4 | ND | ND | 0.00060 | 0.4 | 0.0080 | 2.9 |
| A156T | 22.6 | 46 | 1.1 | 222 | 0.026 | 5.3 | 0.053 | 33 | 0.17 | 60 |
| A156V | 19.3 | 40 | 10 | 2,041 | 0.030 | 6.1 | ND | ND | ND | ND |
| D168A | ND | ND | 1.9 | 380 | 0.17 | 34 | ND | ND | ND | ND |
| D168V | ND | ND | 5.1 | 1,039 | 0.062 | 13 | ND | ND | ND | ND |
| V170A | 1.89 | 3.9 | 0.0070 | 1.4 | 0.0040 | 0.8 | ND | ND | ND | ND |
| V36A + T54A | 2.60 | 5.3 | 0.0022 | 0.5 | 0.0026 | 0.5 | ND | ND | ND | ND |
| V36A + R155K | 4.77 | 9.8 | 2.6 | 535 | 1.5 | 312 | ND | ND | ND | ND |
| V36A + R155T | 12.5 | 26 | 2.9 | 590 | 0.15 | 30 | ND | ND | ND | ND |
| V36M + R155K | 6.48 | 13 | 2.7 | 559 | 1.3 | 259 | 0.097 | 60 | 1.0 | 376 |
| V36M + R155T | 12.2 | 25 | 4.0 | 822 | 0.23 | 47 | ND | ND | ND | ND |
| V36M + A156T | 21.2, >30 | 44, >62 | 6.8, >10 | 1,392, >2,041 | 0.059 | 12 | ND | ND | ND | ND |
The EC50 was the mean of the results of 3 to 5 individual experiments (G1b 48-h assay). FC (fold change) was determined by dividing the mean EC50 of a given variant replicon by that of the WT replicon. “ND” denotes that data were not determined. “>” denotes that the EC50 or FC was greater than the value presented; the actual value could not be determined since no significant reduction of HCV RNA level was observed at the maximum concentration of compound used (30 μM for boceprevir and 10 μM for ciluprevir).
Like telaprevir, boceprevir is a linear ketoamide peptidomimetic NS3 protease inhibitor (26). The EC50 (±SD) value of boceprevir against the WT replicon was 0.487 (±0.130) μM, which was similar to that of telaprevir determined in the same replicon assay. Single amino acid changes at residues 36 (V36A/C/G/L/M), 54 (T54A), and 155 (R155G/I/K/M/S/T) conferred no or lower-level resistance to both boceprevir (EC50 change from 1.3- to 12-fold) and telaprevir (EC50 change from 2.2- to 24-fold) (Table 1). In comparison, A156T/V conferred less resistance to boceprevir (40- to 46-fold) than to telaprevir (>62-fold) (Table 1). Double variants V36A/M+R155K/T and V36A+T54A also conferred less resistance to boceprevir (5.3- to 26-fold) than to telaprevir (20- to >62-fold in G1b) (Table 1). Furthermore, higher-level resistance to both boceprevir and telaprevir (44- to >62-fold) was observed for V36M+A156T.
Macrocyclic NS3 protease inhibitors ciluprevir, danoprevir, simeprevir, and vaniprevir showed comparable levels of potency for the WT replicon, with EC50 (±SD) values ranging from 0.0016 (±0.0001) to 0.0049 (±0.0022) μM. Ciluprevir and danoprevir were fully active against variants with a single amino acid change at residue 36 (V36A/G/L/M) or 54 (T54A), which conferred no or lower-level resistance to telaprevir. Similarly, simprevir and vaniprevir were fully active against V36M and T54A. Substitutions at residue 155 (R155G/I/K/M/S/T), which conferred lower-level resistance to telaprevir, resulted in various levels of resistance to macrocyclic NS3 protease inhibitors. Substitutions with a hydrophobic amino acid (R155I/M) had less impact on the susceptibility to ciluprevir and danoprevir than substitutions at the same position with charged, hydrophilic, or small side-chain residues (R155G/K/S/T). Variant A156S, a variant with lower-level resistance to telaprevir, was sensitive to ciluprevir, simeprevir, and vaniprevir (<3-fold), which is comparable with what was previously observed for danoprevir (4-fold change) (45). Substitution of A156 with T or V, which conferred higher-level resistance to telaprevir, also conferred higher-level resistance to ciluprevir (222- to 2041-fold), simeprevir (33-fold), and vaniprevir (60-fold) but lower-level resistance to danoprevir (5- to 6-fold). Double variants at residues 36 and 155 (V36A/M+R155K/T), which were variants with higher-level resistance to telaprevir, conferred resistance to ciluprevir, danoprevir, simeprevir, and vaniprevir, with a wide range of decreases in susceptibility (30- to 822-fold). A variant with higher-level telaprevir resistance, V36M+A156T, was resistant to ciluprevir (1,392- or >2,041-fold), with much higher resistance than what was imparted by A156T alone (222-fold). However, this double variant conferred less resistance to danoprevir (12-fold); that resistance level was just slightly higher than the resistance imparted by A156T alone (5.3-fold). Double substitution V36A+T54A, which conferred a lower level of resistance to telaprevir, did not confer any resistance to ciluprevir or danoprevir, consistent with the lack of resistance to these macrocyclic inhibitors that was observed for the single variants V36A and T54A.
Substitution R109K did not reduce the susceptibility of the replicon to telaprevir, boceprevir, ciluprevir, or danoprevir. This variant conferred resistance only to inhibitors that have an extension toward the P2′ position, such as SCH6 (49, 50). Amino acid substitutions at position 168 have been associated with resistance to the macrocyclic class of inhibitors but not with resistance to the linear ketoamide class of inhibitors, including telaprevir and boceprevir (46). As is consistent with this observation, variants D168A/V showed resistance to ciluprevir (380- to 1,039-fold) and danoprevir (13- to 34-fold) but not to telaprevir. V170A conferred a 3.9-fold increase in resistance to boceprevir, consistent with the findings of a previous in vitro selection study (47), but did not lead to a change in sensitivity to telaprevir, ciluprevir, or danoprevir.
Susceptibility of NS3 protease variants to telaprevir and other HCV NS3 protease inhibitors in the enzymatic assay.
The enzyme IC50s of telaprevir and the other HCV NS3 protease inhibitors boceprevir, ciluprevir, and danoprevir were derived from the purified NS3 protease of WT and a subset of NS3 variants that were observed in telaprevir clinical studies (Table 3). In general, a good correlation (R2 = 0.66, P < 0.0001) was observed between the resistance levels (increases in IC50 or EC50s over WT) obtained in the enzymatic and G1b replicon assays (Fig. 2).
Table 3.
Enzyme IC50 of telaprevir and other protease inhibitors against NS3 variantsa
| Variant | Telaprevir |
Boceprevir |
Ciluprevir |
Danoprevir |
||||
|---|---|---|---|---|---|---|---|---|
| IC50(1 h) (μM) | FC | IC50(1 h) (μM) | FC | IC50 (μM) | FC | IC50 (μM) | FC | |
| WT | 0.050 | 1.0 | 0.084 | 1.0 | 0.0087 | 1.0 | 0.0073 | 1.0 |
| V36L | 0.099 | 2.0 | ND | ND | ND | ND | ND | ND |
| V36M | 0.27 | 5.4 | 0.37 | 4.4 | 0.010 | 1.2 | 0.0071 | 1.0 |
| R155I | 0.88 | 18 | ND | ND | 0.14 | 16 | ND | ND |
| R155K | 0.51 | 10 | 0.87 | 10 | 0.60, >0.84 | 69, >97 | 0.26 | 35 |
| R155S | 1.2 | 23 | ND | ND | 0.72, >0.84 | 83, >97 | ND | ND |
| R155T | 0.49 | 9.8 | ND | ND | 0.22 | 25 | ND | ND |
| A156S | 2.5 | 50 | 3.1 | 37 | 0.072 | 8.3 | 0.043 | 6.0 |
| A156T | 19 | 380 | 26 | 310 | >0.84 | >97 | 0.028 | 3.8 |
| V36M + R155K | 3.4 | 69 | 3.3 | 39 | 0.69, >0.84 | 79, >97 | 0.48 | 66 |
The IC50(1 h) or IC50 was the mean of the results of 2 to 5 individual experiments. FC (fold change) was determined by dividing the mean IC50(1 h) or IC50 of a given variant by that of WT. “ND” denotes that data were not determined. “>” denotes that the IC50(1 h), IC50, or FC was greater the value presented; the actual value could not be determined since no significant reduction of HCV RNA level was observed at the maximum concentration of compound used (0.84 μM for ciluprevir).
Fig 2.
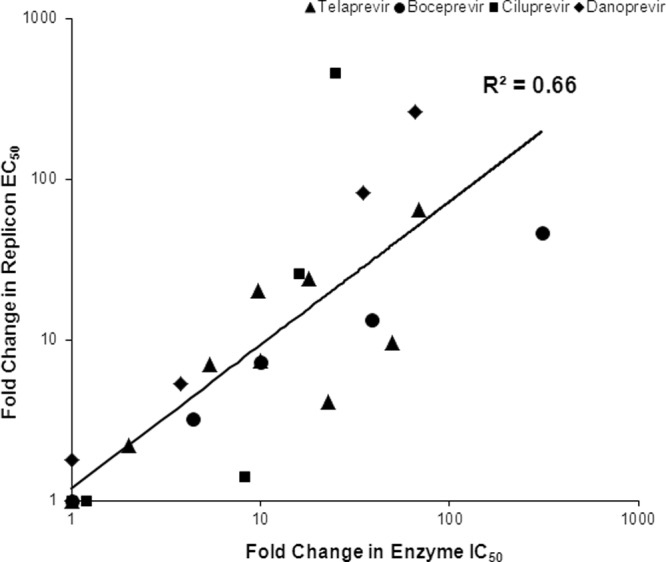
Comparison of fold change of replicon EC50 and enzyme IC50 of NS3 protease inhibitors against NS3 variants. The fold change of replicon EC50 was determined in the G1b 48-h assay. Data for WT (n = 4) and NS3 variants with actual or estimated values (n = 20) (V36L/M, R155I/K/S/T, A156S, and V36M+R155K for telaprevir; V36M, R155K, A156T, and V36M+R155K for boceprevir; V36M, R155I/T, and A156S for ciluprevir; and V36M, R155K, A156T, and V36M+R155K for danoprevir) were included in the comparison. A linear regression was performed on a log-transformed scale, with the resulting coefficient of determination (R2) of 0.66 having a P value < 0.0001.
Susceptibility of telaprevir-resistant variants to IFN-α, ribavirin, and other classes of direct-acting antivirals.
The susceptibility of telaprevir-resistant variants to IFN-α, ribavirin, nucleoside NS5B polymerase inhibitors (mericitabine [37] and NM-107 [30]), and non-nucleoside NS5B polymerase inhibitors (VX-222 [31] binding to the thumb domain, HCV-796 [32] and CMPD 2 [33] binding to the palm domain, and CMPD 55 [34] binding to the finger-loop domain) was evaluated in the G1b 48-h replicon assay (Table 4). The EC50s of IFN-α and ribavirin for replicon cells containing either single variant V36A/M, T54A, or R155K/M/T or double variant V36A/M+R155K/T were comparable to that for WT cells (fold change ranging from 0.3 to 1.3). Similarly, the EC50s of nucleoside or non-nucleoside NS5B polymerase inhibitors against replicon cells containing variant V36A/M, T54A, R155K, A156S/T/V, or V36M+R155K were comparable to that against WT replicon cells (fold change ranging from 0.4 to 2.0). Thus, the telaprevir-resistant variants tested remained fully sensitive to IFN-α, ribavirin, and mechanistically distinct NS5B polymerase inhibitors.
Table 4.
Replicon EC50 of IFN-α, ribavirin, and NS5B polymerase inhibitors against NS3 variantsa
| Variant | IFN-α |
Ribavirin |
Mericitabine |
NM-107 |
VX-222 |
HCV-796 |
CMPD 2 |
CMPD 55 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (U/ml) | FC | EC50 (μM) | FC | EC50 (μM) | FC | EC50 (μM) | FC | EC50 (μM) | FC | EC50 (μM) | FC | EC50 (μM) | FC | EC50 (μM) | FC | |
| WT | 11.6 | 1.0 | 57.8 | 1.0 | 2.06 | 1.0 | 1.34 | 1.0 | 0.0036 | 1.0 | 0.0235 | 1.0 | 0.347 | 1.0 | 0.314 | 1.0 |
| V36A | 10.3 | 0.9 | 43.1 | 0.8 | ND | ND | 2.15 | 1.6 | ND | ND | ND | ND | 0.571 | 1.6 | 0.369 | 1.2 |
| V36M | 11.3 | 1.0 | 32.9 | 0.6 | 1.83 | 0.9 | 1.59 | 1.2 | 0.0037 | 1.0 | 0.0289 | 1.2 | 0.360 | 1.0 | 0.228 | 0.7 |
| T54A | 3.87 | 0.3 | 21.7 | 0.4 | 1.86 | 0.9 | 1.98 | 1.5 | 0.0040 | 1.1 | 0.0274 | 1.2 | 0.564 | 1.6 | 0.337 | 1.1 |
| R155K | 15.2 | 1.3 | 37.2 | 0.6 | 1.45 | 0.7 | 2.37 | 1.8 | 0.0031 | 0.9 | 0.0176 | 0.7 | 0.450 | 1.3 | 0.390 | 1.2 |
| R155M | 4.89 | 0.4 | 38.9 | 0.7 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| R155T | 4.84 | 0.4 | 32.4 | 0.6 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| A156S | ND | ND | ND | ND | 0.850 | 0.4 | ND | ND | 0.0029 | 0.8 | 0.0219 | 0.9 | ND | ND | ND | ND |
| A156T | ND | ND | ND | ND | 1.07 | 0.5 | 1.27 | 0.9 | 0.0029 | 0.8 | 0.0200 | 0.8 | 0.326 | 0.9 | 0.306 | 1.0 |
| A156V | ND | ND | ND | ND | ND | ND | 1.31 | 1.0 | ND | ND | ND | ND | 0.264 | 0.8 | 0.297 | 0.9 |
| V36A + R155K | 6.83 | 0.6 | 35.8 | 0.6 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| V36A + R155T | 3.90 | 0.3 | 41.7 | 0.7 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| V36M + R155K | 10.1 | 0.9 | 40.6 | 0.7 | 2.25 | 1.1 | 2.72 | 2.0 | 0.0040 | 1.1 | 0.0233 | 1.0 | 0.578 | 1.7 | 0.419 | 1.3 |
| V36M + R155T | 3.13 | 0.3 | 36.4 | 0.6 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
The EC50 was the mean of the results of 3 individual experiments (G1b 48-h assay). FC (fold change) was determined by dividing the mean EC50 of a given variant replicon by that of the WT replicon. “ND” denotes that data were not determined.
Replication capacity of telaprevir-resistant variants.
The in vitro replication capacity (fitness) of the HCV replicon containing substitutions at positions 36, 54, 155, and 156 of the HCV NS3 protease was determined in G1b transient replicon systems (Table 5). All variants, except V36A/C/I, had reduced replication capacity compared with the WT replicon. In general, variants that conferred higher-level resistance to telaprevir, such as A156N/T/V and V36M+A156T, tended to have the lowest in vitro replication capacity (9% to 16%), whereas variants that conferred lower-level resistance, such as V36A/C/G/M, T54A/S, R155K, and A156S, tended to be more fit (replication capacity ranging from 59% to 104%). Variant V36I, which was fully susceptible to telaprevir, had a replication capacity even higher than that of the WT. Overall, there appears to be an inverse correlation between the in vitro replication capacity and the resistance level for these NS3 variants (Fig. 3). An exception to this observation is the double variant V36A+T54A, which conferred lower-level resistance but had a lower fitness of 8% ± 2%, much lower than that of either single variant. It is likely that V36A and T54A are more than additive, or synergistic, with regard to their negative impact on NS3 protease structure or activity. This may explain the rare occurrence of this variant in clinical studies.
Table 5.
Relative in vitro replication capacity of NS3 variants
| Variant | Telaprevir replicon EC50 fold changea | Level of resistance to telaprevirb | Replication capacity ± SD (%)c |
|---|---|---|---|
| WT | 1 | None | 100 ± 7 |
| V36I | 0.3 | 146 ± 18 | |
| V36A | 7.4 | Lower | 104 ± 26 |
| V36C | 7.8 | 98 ± 9 | |
| V36G | 11 | 59 ± 8 | |
| V36M | 7.0 | 77 ± 12 | |
| T54A | 6.3 | 65 ± 10 | |
| T54S | 4.2 | 59 ± 3 | |
| R155G | 7.4 | 10 ± 3 | |
| R155I | 24 | 20 ± 7 | |
| R155K | 7.4 | 80 ± 16 | |
| R155M | 5.6 | 18 ± 2 | |
| R155S | 4.1 | 45 ± 8 | |
| R155T | 20 | 39 ± 7 | |
| A156S | 9.6 | 88 ± 8 | |
| V36A + T54A | 20 | 8 ± 2 | |
| A156N | >93 | Higher | 9 ± 4 |
| A156T | >62 | 16 ± 2 | |
| A156V | >62 | 9 ± 2 | |
| V36A + R155K | ∼41 | 57 ± 4 | |
| V36A + R155T | >62 | 23 ± 11 | |
| V36M + R155K | ∼64 | 42 ± 6 | |
| V36M + R155T | >62 | 36 ± 8 | |
| V36M + A156T | >62 | 9 ± 5 |
The fold change of the mean EC50 of the variant replicon compared with that of the WT replicon was determined in G1b 48-h or 96-h assays as presented in Table 1. “>” and “∼” are as described in Table 1.
Lower-level resistance, EC50 change from 3- to 25-fold; higher-level resistance, EC50 change > 25-fold. V36I did not confer resistance to telaprevir (EC50 change < 3-fold).
The mean replication capacity and SD were derived from 31 and 3 to 7 individual experiments for the WT and variant replicons, respectively. Data for V36I, T54S, A156N, and A156S were generated with a pFK I341PiLuc/NS3-3′/ET replicon in Huh-7-ET-cured cells; the rest of the data were generated with a pBR322-Luc-mADE replicon in Huh-7.5 cells.
Fig 3.

Telaprevir EC50 fold change and in vitro replicative fitness of NS3 variants in G1b replicon cells. The fold change in mean EC50 and the SD were derived from Table 1 and plotted in the black bars corresponding to the y axis on the left. The replicative fitness (replication capacity) and the SD were derived from Table 5 and plotted in the gray bars corresponding to the y axis on the right. The upper dashed line represents the maximum change in EC50 (i.e., 93-fold) that could be detected in the 96-h EC50 assay. The lower dashed line represents the maximum change in EC50 (i.e., 62-fold) that could be detected in the 48-h EC50 assay. The fold changes in EC50 above the maximum detection limits are represented with the “>” sign.
Conclusions.
HCV NS3 protease variants observed in clinical studies of telaprevir were characterized phenotypically in both replicon and enzymatic assays. These variants conferred different levels of resistance to telaprevir, ranging from a 0.3-fold to a greater than 93-fold increase in EC50 relative to the WT in replicon cells, and were categorized as variants with either lower-level (3- to 25-fold increase in EC50 from WT) or higher-level (>25-fold increase in EC50) resistance. All telaprevir-resistant variants remained fully sensitive to IFN-α, ribavirin, and HCV NS5B nucleoside and non-nucleoside polymerase inhibitors. In addition, there was a general trend of an inverse relationship between the level of in vitro resistance and replication capacity.
The most frequently observed telaprevir-resistant variants were V36A/M, T54A/S, R155K/T, and A156S, which conferred lower-level resistance to telaprevir; and A156T and V36M+R155K, which conferred higher-level resistance to telaprevir. The levels of resistance to telaprevir conferred by the major variants were consistent both in G1a and G1b replicon assays and in enzymatic assays. Rarely observed variants included V36I/L, I132V, and V151A, which did not confer resistance to telaprevir; V36C/G, R155G/I/M/S, V36A+T54A, V36L+R155K, T54S+R155K, and R155T+D168N, which conferred lower-level resistance to telaprevir; and A156F/N/V, V36A+R155K/T, V36M+R155T, V36A/M+A156T, T54A+A156S, T54S+A156S/T, and V36M+T54S+R155K, which conferred higher-level resistance to telaprevir. Variants resistant to other HCV protease inhibitors (Q41R, V55A, Q80R, R109K, D168A/N/V, and V170A) remained fully sensitive to telaprevir.
In general, variants conferring resistance to telaprevir and boceprevir (covalent, linear, reversible NS3 protease inhibitors) had similar effects on viral sensitivity to the inhibitors. Sensitivity to ciluprevir, danoprevir, simeprevir, and vaniprevir (macrocyclic, reversible, noncovalent NS3 protease inhibitors) was less affected by amino acid substitutions at positions 36, 54, and 156. Substitutions at position 155 showed cross-resistance to all NS3 protease inhibitors.
The in vitro replication capacity of all variants was lower than that of the WT in replicon cells, with the exception of V36A/C/I. Resistant variants that conferred higher-level resistance to telaprevir, such as A156N/T/V and V36M+A156T, generally tended to have the lowest in vitro replication capacity, whereas variants with lower-level resistance to telaprevir, such as V36A/C/G/M, T54A/S, R155K, and A156S, tended to be more fit. These results are consistent with the diminished in vivo fitness estimated from viral load and variant population data in a previous study (21).
Taken together, these results allow a better understanding of the resistant variants observed in patients who do not achieve an SVR with a telaprevir-based regimen. While the variants observed in patients can confer either higher- or lower-level resistance to telaprevir, these variants have a reduced fitness compared with WT virus and are fully sensitive to IFN-α, ribavirin, and other classes of DAAs.
ACKNOWLEDGMENTS
This study was sponsored by Vertex Pharmaceuticals Incorporated.
M. Jiang, N. Mani, C. Lin, A. Ardzinski, D. Reagan, D. Bartels, Y. Zhou, O. Nicolas, U. Müh, B. Hanzelka, A. Tigges, R. Rijnbrand, and T. Kieffer are employees of Vertex Pharmaceuticals Incorporated and may own stock or options in that company. M. Nelson and B. G. Rao were employed at Vertex Pharmaceuticals Incorporated at the time this research was conducted and may own or have owned stock or options in that company.
Footnotes
Published ahead of print 7 October 2013
REFERENCES
- 1. Alter HJ, Seeff LB. 2000. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin. Liver Dis. 20:17–35 [DOI] [PubMed] [Google Scholar]
- 2. Ghobrial RM, Steadman R, Gornbein J, Lassman C, Holt CD, Chen P, Farmer DG, Yersiz H, Danino N, Collisson E, Baquarizo A, Han SS, Saab S, Goldstein LI, Donovan JA, Esrason K, Busuttil RW. 2001. A 10-year experience of liver transplantation for hepatitis C: analysis of factors determining outcome in over 500 patients. Ann. Surg. 234:384–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kenny-Walsh E. 2001. The natural history of hepatitis C virus infection. Clin. Liver Dis. 5:969–977 [DOI] [PubMed] [Google Scholar]
- 4. Purcell RH. 1994. Hepatitis C virus: historical perspective and current concepts. FEMS Microbiol. Rev. 14:181–191 [DOI] [PubMed] [Google Scholar]
- 5. Wasley A, Alter MJ. 2000. Epidemiology of hepatitis C: geographic differences and temporal trends. Semin. Liver Dis. 20:1–16 [DOI] [PubMed] [Google Scholar]
- 6. Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Jr, Haussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347:975–982 [DOI] [PubMed] [Google Scholar]
- 7. Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958–965 [DOI] [PubMed] [Google Scholar]
- 8. Kwong AD, Kauffman RS, Hurter P, Mueller P. 2011. Discovery and development of telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat. Biotechnol. 29:993–1003 [DOI] [PubMed] [Google Scholar]
- 9. Lin K, Perni RB, Kwong AD, Lin C. 2006. VX-950, a novel hepatitis C virus (HCV) NS3-4A protease inhibitor, exhibits potent antiviral activities in HCv replicon cells. Antimicrob. Agents Chemother. 50:1813–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perni RB, Almquist SJ, Byrn RA, Chandorkar G, Chaturvedi PR, Courtney LF, Decker CJ, Dinehart K, Gates CA, Harbeson SL, Heiser A, Kalkeri G, Kolaczkowski E, Lin K, Luong YP, Rao BG, Taylor WP, Thomson JA, Tung RD, Wei Y, Kwong AD, Lin C. 2006. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease. Antimicrob. Agents Chemother. 50:899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416 [DOI] [PubMed] [Google Scholar]
- 12. Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, Focaccia R, Younossi Z, Foster GR, Horban A, Ferenci P, Nevens F, Mullhaupt B, Pockros P, Terg R, Shouval D, van Hoek B, Weiland O, Van Heeswijk R, De Meyer S, Luo D, Boogaerts G, Polo R, Picchio G, Beumont M; REALIZE Study Team 2011. Telaprevir for retreatment of HCV infection. N. Engl. J. Med. 364:2417–2428 [DOI] [PubMed] [Google Scholar]
- 13. Ray SC, Thomas DL. 2009. Hepatitis C, p 2157–2187 In Mandell GL, Bennett JE, Dolan R. (ed), Mandell, Douglas, and Bennett's principles and practice of infectious diseases. Churchill Livingstone-Elsevier, Philadelphia, PA [Google Scholar]
- 14. Zeuzem S. 1999. Clinical implications of hepatitis C viral kinetics. J. Hepatol. 31(Suppl 1):61–64 [DOI] [PubMed] [Google Scholar]
- 15. Bukh J, Miller RH, Purcell RH. 1995. Genetic heterogeneity of hepatitis C virus: quasispecies and genotypes. Semin. Liver Dis. 15:41–63 [DOI] [PubMed] [Google Scholar]
- 16. Adiwijaya BS, Herrmann E, Hare B, Kieffer T, Lin C, Kwong AD, Garg V, Randle JC, Sarrazin C, Zeuzem S, Caron PR. 2010. A multi-variant, viral dynamic model of genotype 1 HCV to assess the in vivo evolution of protease-inhibitor resistant variants. PLoS Comput. Biol. 6:e1000745. 10.1371/journal.pcbi.1000745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rong L, Dahari H, Ribeiro RM, Perelson AS. 2010. Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci. Transl. Med. 2:30ra32. 10.1126/scitranslmed.3000544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Halfon P, Sarrazin C. 2012. Future treatment of chronic hepatitis C with direct acting antivirals: is resistance important? Liver Int. 32(Suppl 1):79–87 [DOI] [PubMed] [Google Scholar]
- 19. Sarrazin C, Zeuzem S. 2010. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138:447–462 [DOI] [PubMed] [Google Scholar]
- 20. Hézode C, Forestier N, Dusheiko G, Ferenci P, Pol S, Goeser T, Bronowicki JP, Bourliere M, Gharakhanian S, Bengtsson L, McNair L, George S, Kieffer T, Kwong A, Kauffman RS, Alam J, Pawlotsky JM, Zeuzem S. 2009. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N. Engl. J. Med. 360:1839–1850 [DOI] [PubMed] [Google Scholar]
- 21. Sarrazin C, Kieffer TL, Bartels D, Hanzelka B, Muh U, Welker M, Wincheringer D, Zhou Y, Chu HM, Lin C, Weegink C, Reesink H, Zeuzem S, Kwong AD. 2007. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132:1767–1777 [DOI] [PubMed] [Google Scholar]
- 22. Kieffer TL, De MS, Bartels DJ, Sullivan JC, Zhang EZ, Tigges A, Dierynck I, Spanks J, Dorrian J, Jiang M, Adiwijaya B, Ghys A, Beumont M, Kauffman RS, Adda N, Jacobson IM, Sherman KE, Zeuzem S, Kwong AD, Picchio G. 2012. Hepatitis C viral evolution in genotype 1 treatment-naive and treatment-experienced patients receiving telaprevir-based therapy in clinical trials. PLoS One 7:e34372. 10.1371/journal.pone.0034372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bartels DJ, Sullivan JC, Zhang EZ, Tigges AM, Dorrian JL, De Meyer S, Takemoto D, Dondero E, Kwong AD, Picchio G, Kieffer TL. 2013. Hepatitis C virus variants with decreased sensitivity to direct-acting antivirals (DAAs) were rarely observed in DAA-naive patients prior to treatment. J. Virol. 87:1544–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vrolijk JM, Kaul A, Hansen BE, Lohmann V, Haagmans BL, Schalm SW, Bartenschlager R. 2003. A replicon-based bioassay for the measurement of interferons in patients with chronic hepatitis C. J. Virol. Methods 110:201–209 [DOI] [PubMed] [Google Scholar]
- 26. Malcolm BA, Liu R, Lahser F, Agrawal S, Belanger B, Butkiewicz N, Chase R, Gheyas F, Hart A, Hesk D, Ingravallo P, Jiang C, Kong R, Lu J, Pichardo J, Prongay A, Skelton A, Tong X, Venkatraman S, Xia E, Girijavallabhan V, Njoroge FG. 2006. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob. Agents Chemother. 50:1013–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hinrichsen H, Benhamou Y, Wedemeyer H, Reiser M, Sentjens RE, Calleja JL, Forns X, Erhardt A, Cronlein J, Chaves RL, Yong CL, Nehmiz G, Steinmann GG. 2004. Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology 127:1347–1355 [DOI] [PubMed] [Google Scholar]
- 28. Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, Bos M, Cameron DR, Cartier M, Cordingley MG, Faucher AM, Goudreau N, Kawai SH, Kukolj G, Lagace L, LaPlante SR, Narjes H, Poupart MA, Rancourt J, Sentjens RE, St George R, Simoneau B, Steinmann G, Thibeault D, Tsantrizos YS, Weldon SM, Yong CL, Llinas-Brunet M. 2003. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426:186–189 [DOI] [PubMed] [Google Scholar]
- 29. Seiwert SD, Andrews SW, Jiang Y, Serebryany V, Tan H, Kossen K, Rajagopalan PT, Misialek S, Stevens SK, Stoycheva A, Hong J, Lim SR, Qin X, Rieger R, Condroski KR, Zhang H, Do MG, Lemieux C, Hingorani GP, Hartley DP, Josey JA, Pan L, Beigelman L, Blatt LM. 2008. Preclinical characteristics of the hepatitis C virus NS3/4A protease inhibitor ITMN-191 (R7227). Antimicrob. Agents Chemother. 52:4432–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gardelli C, Attenni B, Donghi M, Meppen M, Pacini B, Harper S, Di Marco A, Fiore F, Giuliano C, Pucci V, Laufer R, Gennari N, Marcucci I, Leone JF, Olsen DB, MacCoss M, Rowley M, Narjes F. 2009. Phosphoramidate prodrugs of 2′-C-methylcytidine for therapy of hepatitis C virus infection. J. Med. Chem. 52:5394–5407 [DOI] [PubMed] [Google Scholar]
- 31. Bedard J, Nicolas O, Bilimoria D, L'Heureux L, Fex P, David M, Chan L. 2009. Identification and the characterization of VCH-222, a novel potent and selective non-nucleoside HCV polymerase inhibitor. J. Hepatol. 50(Suppl 1):S340 [Google Scholar]
- 32. Kneteman NM, Howe AY, Gao T, Lewis J, Pevear D, Lund G, Douglas D, Mercer DF, Tyrrell DL, Immermann F, Chaudhary I, Speth J, Villano SA, O'Connell J, Collett M. 2009. HCV796: a selective nonstructural protein 5B polymerase inhibitor with potent anti-hepatitis C virus activity in vitro, in mice with chimeric human livers, and in humans infected with hepatitis C virus. Hepatology 49:745–752 [DOI] [PubMed] [Google Scholar]
- 33. Hutchinson DK, Rosenberg T, Klein LL, Bosse TD, Larson DP, He W, Jiang WW, Kati WM, Kohlbrenner WE, Liu Y, Masse SV, Middleton T, Molla A, Montgomery DA, Beno DW, Stewart KD, Stoll VS, Kempf DJ. 2008. Hepatitis C NS5B polymerase inhibitors: 4,4-dialkyl-1-hydroxy-3-oxo-3,4-dihydronaphthalene-3-yl benzothiadiazine derivatives. Bioorg. Med. Chem. Lett. 18:3887–3890 [DOI] [PubMed] [Google Scholar]
- 34. Harper S, Avolio S, Pacini B, Di Filippo M, Altamura S, Tomei L, Paonessa G, Di Marco S, Carfi A, Giuliano C, Padron J, Bonelli F, Migliaccio G, De Francesco R, Laufer R, Rowley M, Narjes F. 2005. Potent inhibitors of subgenomic hepatitis C virus RNA replication through optimization of indole-N-acetamide allosteric inhibitors of the viral NS5B polymerase. J. Med. Chem. 48:4547–4557 [DOI] [PubMed] [Google Scholar]
- 35. Lin TI, Lenz O, Fanning G, Verbinnen T, Delouvroy F, Scholliers A, Vermeiren K, Rosenquist A, Edlund M, Samuelsson B, Vrang L, de Kock H, Wigerinck P, Raboisson P, Simmen K. 2009. In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor. Antimicrob. Agents Chemother. 53:1377–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liverton NJ, Carroll SS, Dimuzio J, Fandozzi C, Graham DJ, Hazuda D, Holloway MK, Ludmerer SW, McCauley JA, McIntyre CJ, Olsen DB, Rudd MT, Stahlhut M, Vacca JP. 2010. MK-7009, a potent and selective inhibitor of hepatitis C virus NS3/4A protease. Antimicrob. Agents Chemother. 54:305–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ali S, Leveque V, Le Pogam S, Ma H, Philipp F, Inocencio N, Smith M, Alker A, Kang H, Najera I, Klumpp K, Symons J, Cammack N, Jiang WR. 2008. Selected replicon variants with low-level in vitro resistance to the hepatitis C virus NS5B polymerase inhibitor PSI-6130 lack cross-resistance with R1479. Antimicrob. Agents Chemother. 52:4356–4369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin C, Lin K, Luong YP, Rao BG, Wei YY, Brennan DL, Fulghum JR, Hsiao HM, Ma S, Maxwell JP, Cottrell KM, Perni RB, Gates CA, Kwong AD. 2004. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061: structural analysis indicates different resistance mechanisms. J. Biol. Chem. 279:17508–17514 [DOI] [PubMed] [Google Scholar]
- 39. Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 40. Lin C, Gates CA, Rao BG, Brennan DL, Fulghum JR, Luong YP, Frantz JD, Lin K, Ma S, Wei YY, Perni RB, Kwong AD. 2005. In vitro studies of cross-resistance mutations against two hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061. J. Biol. Chem. 280:36784–36791 [DOI] [PubMed] [Google Scholar]
- 41. Zhou Y, Muh U, Hanzelka BL, Bartels DJ, Wei Y, Rao BG, Brennan DL, Tigges AM, Swenson L, Kwong AD, Lin C. 2007. Phenotypic and structural analyses of hepatitis C virus NS3 protease Arg155 variants: sensitivity to telaprevir (VX-950) and interferon alpha. J. Biol. Chem. 282:22619–22628 [DOI] [PubMed] [Google Scholar]
- 42. Landro JA, Raybuck SA, Luong YP, O'Malley ET, Harbeson SL, Morgenstern KA, Rao G, Livingston DJ. 1997. Mechanistic role of an NS4A peptide cofactor with the truncated NS3 protease of hepatitis C virus: elucidation of the NS4A stimulatory effect via kinetic analysis and inhibitor mapping. Biochemistry 36:9340–9348 [DOI] [PubMed] [Google Scholar]
- 43. Taliani M, Bianchi E, Narjes F, Fossatelli M, Urbani A, Steinkuhler C, De Francesco R, Pessi A. 1996. A continuous assay of hepatitis C virus protease based on resonance energy transfer depsipeptide substrates. Anal. Biochem. 240:60–67 [DOI] [PubMed] [Google Scholar]
- 44. Manns MP, Gane E, Rodriguez-Torres M, Stoehr A, Yeh CT, Marcellin P, Wiedmann RT, Hwang PM, Caro L, Barnard RJ, Lee AW. 2012. Vaniprevir with pegylated interferon alpha-2a and ribavirin in treatment-naive patients with chronic hepatitis C: a randomized phase II study. Hepatology 56:884–893 [DOI] [PubMed] [Google Scholar]
- 45. He Y, King MS, Kempf DJ, Lu L, Lim HB, Krishnan P, Kati W, Middleton T, Molla A. 2008. Relative replication capacity and selective advantage profiles of protease inhibitor-resistant hepatitis C virus (HCV) NS3 protease mutants in the HCV genotype 1b replicon system. Antimicrob. Agents Chemother. 52:1101–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lenz O, Verbinnen T, Lin TI, Vijgen L, Cummings MD, Lindberg J, Berke JM, Dehertogh P, Fransen E, Scholliers A, Vermeiren K, Ivens T, Raboisson P, Edlund M, Storm S, Vrang L, de Kock H, Fanning GC, Simmen KA. 2010. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob. Agents Chemother. 54:1878–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Susser S, Welsch C, Wang Y, Zettler M, Domingues FS, Karey U, Hughes E, Ralston R, Tong X, Herrmann E, Zeuzem S, Sarrazin C. 2009. Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology 50:1709–1718 [DOI] [PubMed] [Google Scholar]
- 48. Tong X, Chase R, Skelton A, Chen T, Wright-Minogue J, Malcolm BA. 2006. Identification and analysis of fitness of resistance mutations against the HCV protease inhibitor SCH 503034. Antiviral Res. 70:28–38 [DOI] [PubMed] [Google Scholar]
- 49. Liu R, Abid K, Pichardo J, Pazienza V, Ingravallo P, Kong R, Agrawal S, Bogen S, Saksena A, Cheng KC, Prongay A, Njoroge FG, Baroudy BM, Negro F. 2007. In vitro antiviral activity of SCH446211 (SCH6), a novel inhibitor of the hepatitis C virus NS3 serine protease. J. Antimicrob. Chemother. 59:51–58 [DOI] [PubMed] [Google Scholar]
- 50. Yi M, Tong X, Skelton A, Chase R, Chen T, Prongay A, Bogen SL, Saksena AK, Njoroge FG, Veselenak RL, Pyles RB, Bourne N, Malcolm BA, Lemon SM. 2006. Mutations conferring resistance to SCH6, a novel hepatitis C virus NS3/4A protease inhibitor. Reduced RNA replication fitness and partial rescue by second-site mutations. J. Biol. Chem. 281:8205–8215 [DOI] [PubMed] [Google Scholar]