

Abstract
The bacterial cell division protein FtsZ represents a novel antibiotic target that has yet to be exploited clinically. The benzamide PC190723 was among the first FtsZ-targeting compounds to exhibit in vivo efficacy in a murine infection model system. Despite its initial promise, the poor formulation properties of the compound have limited its potential for clinical development. We describe here the development of an N-Mannich base derivative of PC190723 with enhanced drug-like properties and oral in vivo efficacy. The N-Mannich base derivative (TXY436) is ∼100-fold more soluble than PC190723 in an acidic aqueous vehicle (10 mM citrate, pH 2.6) suitable for oral in vivo administration. At physiological pH (7.4), TXY436 acts as a prodrug, converting to PC190723 with a conversion half-life of 18.2 ± 1.6 min. Pharmacokinetic analysis following intravenous administration of TXY436 into mice yielded elimination half-lives of 0.26 and 0.96 h for the TXY436 prodrug and its PC190723 product, respectively. In addition, TXY436 was found to be orally bioavailable and associated with significant extravascular distribution. Using a mouse model of systemic infection with methicillin-sensitive Staphylococcus aureus or methicillin-resistant S. aureus, we show that TXY436 is efficacious in vivo upon oral administration. In contrast, the oral administration of PC190723 was not efficacious. Mammalian cytotoxicity studies of TXY436 using Vero cells revealed an absence of toxicity up to compound concentrations at least 64 times greater than those associated with antistaphylococcal activity. These collective properties make TXY436 a worthy candidate for further investigation as a clinically useful agent for the treatment of staphylococcal infections.
INTRODUCTION
Antibiotics in current clinical use are directed at a limited number of targets (e.g., the cell wall, protein synthesis, and DNA synthesis), many of which are associated with known resistance pathways. As a result, there is a need for new antibiotics with novel mechanisms of action that can help address the growing resistance problem. The bacterial protein FtsZ represents a novel drug target that has yet to be exploited clinically. FtsZ is a widely conserved bacterial cytoskeletal protein that plays a critical role in cell division (1). Using GTP as a cofactor, FtsZ self-assembles into polymers at the bacterial membrane, forming a Z-ring at the midcell site of division (1–5). The Z-ring serves as a scaffold for recruitment of other key protein constituents of the bacterial divisome (1–4). The essential role that FtsZ plays in cell division makes it an appealing new antibiotic target (6–26).
The substituted benzamide derivative PC190723 (see structure in Fig. 1) was among the first FtsZ-targeting agents to be identified (6, 7). It has been shown to bind FtsZ and stimulate the GTP-dependent polymerization of the protein, which, in turn, leads to the formation of stable FtsZ polymers that cannot recapitulate the function of the Z-ring (13, 27). As a result, bacterial cell division is inhibited, eventually leading to cell death. PC190723 is associated with potent and specific activity against Staphylococcus sp., including multidrug-resistant strains of methicillin-resistant Staphylococcus aureus (MRSA) (7, 12). It has also been reported to exhibit in vivo activity in a mouse peritonitis model of S. aureus infection (7).
Fig 1.
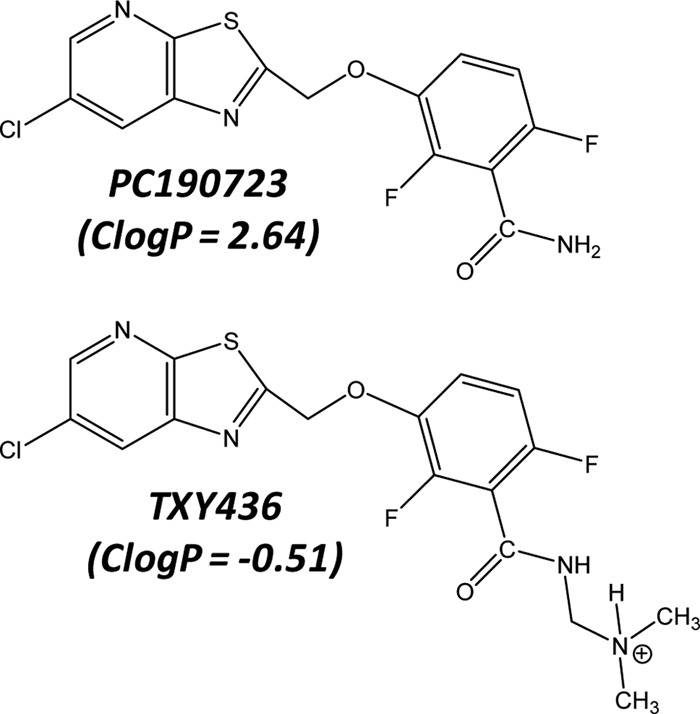
Chemical structures of PC190723 and TXY436. TXY436 is depicted with the Mannich base functionality in its protonated (cationic) form. The indicated ClogP values were calculated using the weighted method (VG = KLOP = PHYS = 1) in the Marvin 5.12 Software Suite (ChemAxon, Ltd.), with Cl− and Na+/K+ concentrations being set at 0.1 mol/dm3.
Although initially promising, the hydrophobic nature of PC190723 (calculated logP [ClogP] = 2.64) makes it difficult to formulate in vehicles suitable for in vivo treatment, a property that has limited the potential clinical utility of the compound. We describe here an N-Mannich base derivative of PC190723 (TXY436; see structure in Fig. 1) that is substantially more polar when protonated under acidic conditions (ClogP = −0.51), and formulates easily in acidic aqueous vehicles suitable for in vivo administration. We show that TXY436 acts as a prodrug at physiological pH, converting to PC190723. Unlike PC190723, the TXY436 prodrug exhibits antistaphylococcal efficacy in vivo after oral administration in a murine model of systemic infection. In addition, TXY436 is minimally toxic to mammalian cells at concentrations up to 64 times those associated with antistaphylococcal activity. These properties make TXY436 an attractive lead for future clinical development.
MATERIALS AND METHODS
Bacterial strains.
S. aureus 8325-4 was a gift from Glenn W. Kaatz (John D. Dingell VA Medical Center, Detroit, MI), S. aureus Mu3 was a gift from George M. Eliopoulos (Beth Israel Deaconess Medical Center, Boston, MA), and Bacillus subtilis FG347 was a gift from Richard Losick (Harvard University, Boston, MA). All other bacterial strains were obtained from the American Type Culture Collection (ATCC).
General chemistry methods and synthesis of PC190723 and TXY436.
All reactions were done under nitrogen atmosphere. Reaction monitoring and follow-up were done using aluminum backed Silica G TLC plates with UV254 (Sorbent Technologies), visualizing with UV light. Flash column chromatography was done on a Combi Flash Rf Teledyne ISCO. The 1H (300 MHz) and 13C (75 MHz) nuclear magnetic resonance (NMR) spectra were acquired on a Varian Unity Inova (300 MHz) multinuclear NMR spectrometer. The data are expressed in parts per million relative to the residual nondeuterated solvent signals, spin multiplicities are given as s (singlet), d (doublet), m (multiplet), and bs (broad singlet), and coupling constants (J) are reported in Hertz (Hz). Melting points were determined using a Mel-Temp II apparatus (Laboratory Devices, Inc.) and are uncorrected.
PC190723 was synthesized as previously described (28). TXY436 was synthesized as follows. In a sealed tube, a mixture of PC190723 (200 mg, 0.56 mmol), formaldehyde (37%, 0.3 ml, 3.7 mmol), and dimethylamine (2 M in tetrahydrofuran [THF], 1.9 ml, 3.70 mmol) in H2O/THF (8/4 ml) was heated at 65°C for 2 h. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate, washed with brine, dried over Na2SO4, and concentrated to afford a brown solid. Subsequent purification by column chromatography afforded the pure product TXY436 as a light brown solid (154 mg, 67% yield): melting point, 152 to 154°C; 1H NMR (CDCl3, 300 MHz); δ: 8.57 (d, J = 2.1 Hz, 1H), 8.24 (d, J = 2.1 Hz, 1H), 7.17 to 7.09 (m, 1H), 6.92-6.86 (m, 1H), 6.20 (bs, 1H), 5.48 (s, 2H), 4.30 (d, J = 6.0 Hz, 2H), 2.37 (s, 6H). HRMS calculated for C17H15ClF2N4O2S (M + H)+ 413.0645. Found 413.0656.
Comparator antibiotics and S. aureus FtsZ.
Vancomycin-HCl, erythromycin, and oxacillin (sodium salt) were obtained from Sigma-Aldrich Co. S. aureus FtsZ was expressed in Escherichia coli and purified as described previously (21).
Compound solubility studies.
The maximum solubilities of PC190723 and TXY436 in 10 mM citrate (pH 2.6) or phosphate-buffered saline (PBS; pH 7.4) were determined at 25°C. In these assays, the compounds were combined with vehicle at weight/volume (wt/vol) ratios above their solubility limit, and the amount of dissolved compound quantified. The wt/vol ratios targeted in the PBS vehicle were 0.2 mg/ml for both compounds, while the wt/vol ratios targeted in the citrate vehicle were 0.2 mg/ml for PC190723 and 2.5 mg/ml for TXY436. Each solubility assessment was conducted in duplicate. All samples were vortexed for 3 min to afford maximal compound dissolution and then centrifuged at 16,000 × g for 1 min to pellet out any unsolubilized compound. The concentration of compound in each of the resulting supernatants was then quantified by using reverse-phase high-performance liquid chromatography (HPLC) as described below and generating standard curves of peak area versus compound concentration (in citrate or PBS vehicle).
Compound stability studies.
Experimental solutions of TXY436 or PC190723 at concentrations of either 10 or 20 μg/ml were prepared from 4-mg/ml dimethyl sulfoxide (DMSO) stock solutions in either 10 mM citrate (pH 2.6) or PBS (pH 7.4) vehicle. The relative amount of TXY436 and PC190723 in each experimental solution was then assessed as a function of time at 25°C using reverse-phase HPLC as described below.
Reverse-phase HPLC.
For all HPLC measurements, a reverse-phase SPHER-100 C18 column (Princeton Chromatography, Inc.) was used on a Shimadzu LC-20AT liquid chromatograph equipped with a Shimadzu SPD-20AV UV/VIS detector (set at 296 nm). The column size was 150 mm by 4.6 mm, with particle and pore sizes of 5 μm and 100 Å, respectively. A 20-μl sample of each experimental solution was injected, and a flow rate of 1 ml/min was applied, along with a gradient of 10 to 90% acetonitrile (containing 0.1% [vol/vol] trifluoroacetic acid) and water in the mobile phase. The total run time was 20 min, with the sampling frequency and response time being 2 Hz and 1 s, respectively. Under these conditions, TXY436 eluted ∼9.8 min after sample injection, and PC190723 eluted ∼11.7 min after sample injection. Peak areas were determined using the Shimadzu EZStart 7.4 SP3 software package.
MIC assays.
MIC assays were conducted in accordance with Clinical and Laboratory Standards Institute (CLSI) guidelines for broth microdilution (29). Briefly, log-phase bacteria were added to 96-well microtiter plates (at 5 × 105 CFU/ml) containing 2-fold serial dilutions of compound or comparator drug in cation-adjusted Mueller-Hinton (CAMH) broth, with each compound concentration being present in duplicate. The final volume in each well was 0.1 ml, and the microtiter plates were incubated aerobically for 24 h at 37°C. Bacterial growth was then monitored by measuring the optical density at 600 nm using a VersaMax plate reader (Molecular Devices, Inc.), with the MIC being defined as the lowest compound concentration at which growth was ≥90% inhibited. The following S. aureus strains were included in these assays: 8325-4 (methicillin-susceptible S. aureus [MSSA]), ATCC 49951 (mucoid MSSA), ATCC 19636 (MSSA), ATCC 29213 (MSSA), ATCC 33591 (MRSA), ATCC 43300 (MRSA), and Mu3 (MRSA). As recommended by CLSI (29), the CAMH broth was supplemented with 2% NaCl in all of the MRSA experiments so as to maintain selection pressure for MRSA. When present, filtered mouse serum (Lampire Biological Laboratories, Inc.) was used at 50% (vol/vol).
MBC assays.
Minimum bactericidal concentration (MBC) assays were conducted in accordance with CLSI guidelines (29). Broth microdilution assays were conducted as described in the preceding section. After the 24-h incubation period, aliquots from the microtiter wells were plated onto tryptic soy agar (TSA). The colonies that grew after 24 h of incubation were counted using an Acolyte colony counter (Synbiosis, Inc.), with MBC being defined as the lowest compound concentration resulting in a ≥3-log reduction in the number of CFU.
Time-kill assays.
Exponentially growing S. aureus 8325-4 bacteria were diluted in CAMH broth to a final count of 105 to 106 CFU/ml. The colony count at time zero was verified by plating serial dilutions of the culture in duplicate on TSA plates. The initial culture was aliquoted into tubes, each containing either a compound or comparator drug at final concentrations ranging from 0× to 8× the MIC. An equivalent volume of DMSO was added to the vehicle control tube. The cultures were then incubated at 37°C with shaking. The CFU/ml in each culture was determined over time by withdrawing samples at time points raging from 2 to 24 h and plating appropriate serial dilutions on to TSA plates. To avoid the possibility of carryover effects in instances when no dilutions were made of the cultures, the samples were centrifuged at 16,000 × g, the supernatant was removed, and the bacterial pellet was resuspended in compound-free medium before plating. All TSA plates were incubated at 37°C, and the CFU/ml at each time point determined by counting colonies after 24 h.
Phase-contrast microscopy.
Log-phase S. aureus 8325-4 cells were cultured in CAMH broth at 37°C for 4 h in the presence of DMSO (solvent control), PC190723 at 3 μg/ml, or TXY436 at 3 μg/ml. A 1-ml sample was withdrawn from each bacterial culture, and centrifuged at 16,000 × g for 3 min at room temperature. The supernatant was removed, and the bacterial pellet was washed with 1 ml of PBS. The final bacterial pellet was then resuspended in 50 μl of PBS, with 5 μl of the resulting bacterial suspension being transferred onto a microscope slide, together with 5 μl of 1% molten agarose (made in PBS). A coverslip was applied, and the slide was visualized with a Zeiss Axioplan 2 microscope equipped with a Plan-Apochromat ×100 objective lens (NA = 1.40). Images were captured with a Zeiss Axiocam HR camera by using the OpenLab software package.
Fluorescence microscopy.
The impact of TXY436 and PC190723 on FtsZ Z-ring formation in B. subtilis FG347 bacteria was assessed in a manner similar to that described previously (30). Specifically, exponentially growing FG347 bacteria were cultured in CAMH broth for 1 h at 37°C in the presence of DMSO (solvent control), PC190723 at 3 μg/ml, or TXY436 at 3 μg/ml. The expression of green fluorescent protein (GFP)-conjugated ZapA was then induced by the addition of xylose to a final concentration of 0.25% (wt/vol), and the cells were incubated for 1 additional hour at 37°C. The bacterial cultures were then treated and visualized as described above, with the additional incorporation of a standard GFP filter set.
Frequency of resistance assay.
The frequency of resistance (FOR) to TXY436 was assayed using a large inoculum approach. In this approach, TSA plates containing TXY436 at concentrations ranging from 4× to 32× the MIC were prepared. A large inoculum of ∼3 × 109 CFU/ml of S. aureus 8325-4 was spread onto each plate. The colony count of the inoculum was verified by plating serial dilutions of the culture onto nonselective TSA plates. All plates were incubated at 37°C overnight and examined after 24 h. Mutational frequency was calculated from the ratio of the number of colonies observed on the selective plates to the total number of plated bacteria (31).
FtsZ polymerization assay.
Polymerization of S. aureus FtsZ was monitored using a microtiter plate-based spectrophotometric assay in which changes in FtsZ polymerization are reflected by corresponding changes in absorbance at 340 nm (A340). PC190723 or TXY436 (at concentrations ranging from 0 to 10 μg/ml) were combined with 5 μM FtsZ in 100 μl of reaction solution. Reaction solutions contained 50 mM Tris-HCl (pH 7.4), 50 mM KCl, and 10 mM magnesium acetate. Reactions were assembled in half-volume, flat-bottom, 96-well microtiter plates and initiated by the addition of 4 mM GTP. Polymerization was continuously monitored at 25°C by measuring A340 in a VersaMax plate reader over a time period of 60 min.
Pharmacokinetic studies.
Pharmacokinetic experiments were conducted by SAI Life Sciences, Ltd. (Pune, India) in accordance with guidelines established by the Indian Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA). Approval by the Institutional Animal Ethics Committee (IAEC) was obtained prior to initiation of the studies. Healthy male BALB/c mice (8 to 12 weeks old and weighing between 25 and 35 g) were obtained from ACTREC (Mumbai, India). Twelve mice were divided into two groups of six mice each, with three mice being housed per cage. Food and water were provided to the mice ad libitum. The first group of mice received a single intravenous (i.v.) dose of TXY436 at 5 mg/kg by tail vein injection. The second group received single peroral (p.o.) dose of TXY436 at 10 mg/kg by gavage. In both cases, the compound was formulated in 10 mM citrate vehicle (pH 2.6) at a concentration of 0.85 mg/ml. Blood samples (∼60 μl) were collected from the retro-orbital plexus at 0.08, 0.25, 0.5, 1, 4, and 8 h after the i.v. dose and 0.25, 0.5, 1, 4, and 8 h after the p.o. dose. The blood samples were collected from a set of three mice at each time point and placed in microcentrifuge tubes containing a 20% K2EDTA solution as an anticoagulant. The blood samples were then centrifuged at 4,000 rpm for 10 min at 4°C to separate the plasma. The plasma samples were stored at −80°C prior to their bioanalysis. Plasma concentrations of TXY436 and PC190723 were quantified by LC-MS/MS, with the lower limit of quantitation (LLOQ) being 20.2 ng/ml for both compounds. The time-dependent plasma concentration data were analyzed by using the sparse sampling mode in the noncompartmental analysis (NCA) tool of the Phoenix WinNonlin version 6.3 software package to yield relevant pharmacokinetic parameters.
In vivo efficacy studies.
Antistaphylococcal efficacy in vivo was assessed in a mouse peritonitis model of systemic infection with S. aureus ATCC 19636 (MSSA) or ATCC 43300 (MRSA). These studies were conducted in full compliance with the standards established by the U.S. National Research Council's Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Rutgers University. Groups of six female Swiss-Webster mice with an average weight of 25 g were infected intraperitoneally with a lethal inoculum of either MSSA (500 μl of saline containing 8 × 106 CFU/ml and 1.5% [wt/vol] porcine mucin [Sigma-Aldrich Co.]) or MRSA (500 μl of saline containing 108 CFU/ml and 5% [wt/vol] porcine mucin).
The mice were fasted overnight prior to their use in the studies. All compound and vehicle administrations were performed p.o. by gavage, with the vehicle being 10 mM citrate (pH 2.6). Both TXY436 and PC190723 were formulated at 2.0 mg/ml, with the resulting TXY436 formulation being a solution and the resulting PC190723 formulation being a suspension.
Four experimental groups of MSSA-infected mice were treated as follows: group 1, untreated; group 2, vehicle only; group 3, TXY436 at 128 mg/kg (in four divided doses of 32 mg/kg); and group 4, PC190723 at 128 mg/kg (in four divided doses 32 mg/kg). The first dose of compound was administered 10 min after infection, with subsequent doses being administered at 12-min intervals thereafter. In the MRSA studies, three experimental groups of infected mice were treated as follows: group 1, untreated; group 2, vehicle only; and group 3, TXY436 at 192 mg/kg (in six divided doses of 32 mg/kg). In these studies, the first dose of compound was administered 1 h after infection, with subsequent doses being administered at 12-min intervals thereafter. The dosing volume for all of the p.o. administrations was 16 ml/kg.
An additional group of six infected mice was also included as a positive control group in both the MSSA and the MRSA studies. This group was treated with a single i.v. administration of vancomycin (formulated in saline) at a dose of 16 mg/kg in the MSSA studies and 24 mg/kg in the MRSA studies. The vancomycin was administered 10 min after MSSA infection, while being administered 1 h after MRSA infection. The dosing volume for the i.v. vancomycin control groups was 8 ml/kg.
The body temperatures of all mice were monitored for a period of 5 days after infection. Body temperatures were recorded at the Xiphoid process using a noninvasive infrared thermometer (Braintree Scientific, Inc.). Infected mice with body temperatures of ≤28.9°C were viewed as being unable to recover from the infection (32) and were euthanized.
MTT cytotoxicity assay.
The cytotoxicity of TXY436 versus Vero cells (African green monkey kidney epithelial cells; ATCC) was assessed using a 72-h continuous 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described previously (33).
RESULTS AND DISCUSSION
TXY436 is associated with a 100-fold enhanced solubility relative to PC190723 in an acidic aqueous vehicle (10 mM citrate, pH 2.6) suitable for in vivo drug administration.
We sought to determine whether the N-Mannich base functionality on TXY436 afforded the compound enhanced aqueous solubility relative to PC190723. To this end, we used reverse-phase HPLC to determine the maximal solubilities of TXY436 and PC190723 in two different aqueous vehicles with differing values of pH: 10 mM citrate (pH 2.6) and PBS (pH 7.4). The maximal solubilities derived from these studies are listed in Table 1. The solubility of TXY436 is ∼2.8-fold greater than that of PC190723 in PBS at pH 7.4 (66.8 ± 15.7 μg/ml for TXY436 versus 23.9 ± 4.0 μg/ml for PC190723), while being ∼100-fold greater in citrate at pH 2.6 (2,290 ± 199 μg/ml for TXY436 versus 22.5 ± 1.6 μg/ml for PC190723). Thus, the N-Mannich base functionality affords TXY436 increased aqueous solubility relative to PC190723, with the degree of solubility enhancement being significantly greater at acidic pH (2.6) than at physiological pH (7.4). Importantly, the solubility of TXY436 in the acidic citrate vehicle (2,290 μg/ml) is sufficient in magnitude for in vivo efficacy studies of TXY436 using this vehicle.
Table 1.
Solubilities of TXY436 and PC190723 in 10 mM citrate and PBS at 25°Ca
| Compound | Mean solubility (μg/ml) ± SD |
|
|---|---|---|
| Citrate (pH 2.6) | PBS (pH 7.4) | |
| TXY436 | 2,290 ± 199 | 66.8 ± 15.7 |
| PC190723 | 22.5 ± 1.6 | 23.9 ± 4.0 |
Each solubility value reflects the average of two independent determinations.
TXY436 is chemically stable in the acidic citrate vehicle.
We next sought to assess the chemical stability of TXY436 in the acidic citrate vehicle over time using reverse-phase HPLC. Figure 2 shows the HPLC chromatogram of TXY436 at 10 μg/ml after 24 h of incubation in the acidic citrate vehicle at 25°C, with the corresponding chromatogram of PC190723 at 10 μg/ml also shown for comparative purposes. Under the conditions used in these HPLC measurements (detailed in Materials and Methods), TXY436 elutes at ∼9.8 min after injection, while PC190723 elutes at ∼11.7 min. The shorter retention time of TXY436 relative to PC190723 is consistent with its correspondingly decreased hydrophobicity (ClogP = −0.51 for TXY436 and +2.64 for PC190723). Note that the chromatographic profile of TXY436 remained essentially unchanged, even after 72 h of incubation (data not shown), indicating that TXY436 is chemically stable over this period of time in the acidic citrate vehicle.
Fig 2.
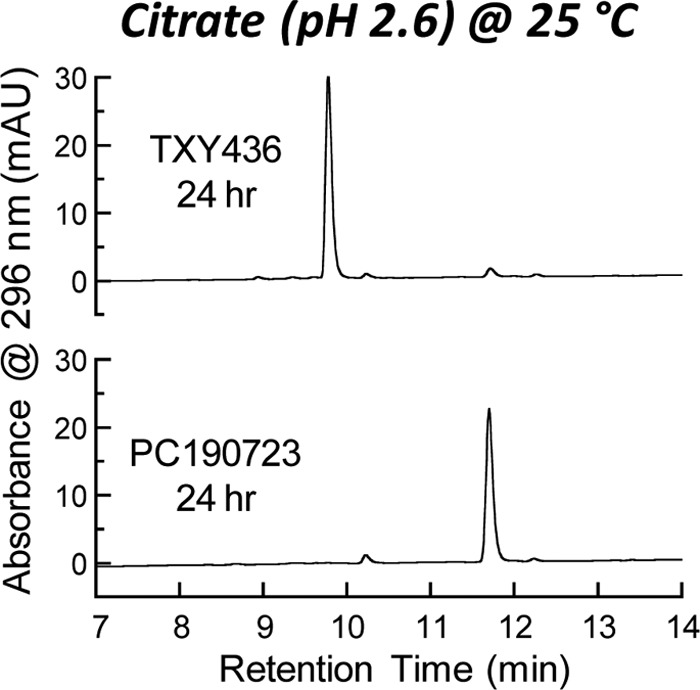
Reverse-phase HPLC chromatograms of TXY436 and PC190723 after incubation for 24 h in 10 mM citrate (pH 2.6) at 25°C. The concentration of each compound was 10 μg/ml.
Although stable at pH 2.6, TXY436 converts to PC190723 at pH 7.4 with a half-life (t1/2) of 18.2 ± 1.6 min at 25°C.
N-Mannich base derivatives of drugs are susceptible to pH-dependent chemical hydrolysis, which has been used as a platform for making prodrugs with improved formulation and delivery properties (34, 35). Rolitetracyclin (transcycline) is an example of a clinically used N-Mannich base prodrug of an antibiotic (tetracycline) (35). One of the by-products of N-Mannich base hydrolysis is formaldehyde (34, 35). Although the release of formaldehyde after N-Mannich base prodrug hydrolysis raised concerns early on regarding potential toxicity, these concerns have been allayed by studies demonstrating that the level of formaldehyde exposure is well below the toxic threshold (36).
If TXY436 were susceptible to pH-dependent chemical hydrolysis like other N-Mannich base derivatives, one of the expected products would be PC190723. We used reverse-phase HPLC to probe for the potential conversion of TXY436 to PC190723 in solution at the physiological pH of 7.4. To this end, we monitored the chemical stability of TXY436 over time in PBS (pH 7.4) at 25°C. Figure 3A shows the HPLC chromatograms of TXY436 at 20 μg/ml after incubation in PBS for various times over a period of 60 min. The chromatogram of PC190723 at 20 μg/ml after 60 min of incubation in PBS is also shown as a reference. Note that over this time period, TXY436 becomes almost fully converted to PC190723. Thus, in essence, TXY436 acts as a stable acid-soluble prodrug that converts to PC190723 under physiological pH conditions. A linear regression analysis of the time-dependent decrease in the peak areas of TXY436 indicates that the TXY436-to-PC109723 conversion follows first-order kinetics, while also yielding a half-life (t1/2) for the conversion of 18.2 ± 1.6 min (Fig. 3B).
Fig 3.
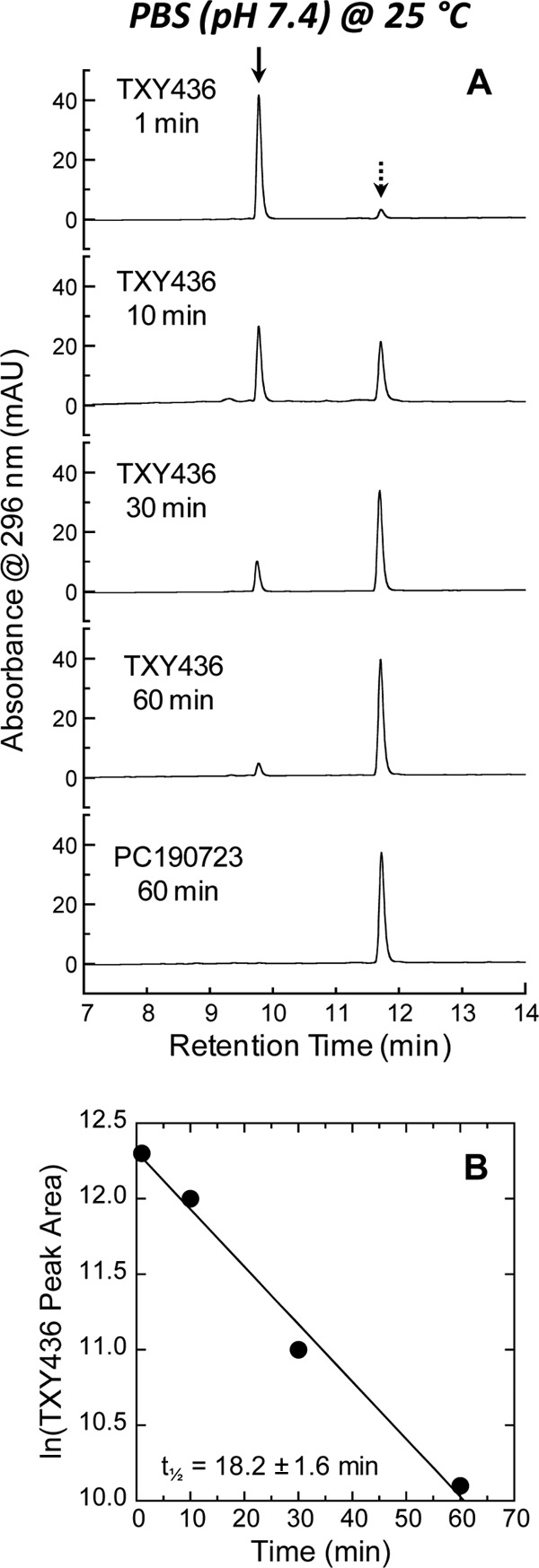
(A) Reverse-phase HPLC chromatograms of TXY436 at 20 μg/ml after the indicated times of incubation in PBS (pH 7.4) at 25°C. For comparative purposes, the corresponding chromatogram of PC190723 at 20 μg/ml after 60 min of incubation in PBS is also presented. The solid arrow indicates the peak corresponding to TXY436, while the dashed arrow indicates the peak corresponding to PC190723. (B) Plot of ln(peak area) versus time for the peak corresponding to TXY436. The indicated half-life (t1/2) for the conversion of TXY436 to PC190723 was determined using the relationship, t1/2 = −0.693/m, where m is the slope derived from the linear least-squares fit of the experimental data points (as indicated by the solid line). The uncertainty in t1/2 reflects the maximal error in m propagated through the above relationship.
The TXY436 prodrug retains the antistaphylococcal and FtsZ-targeting activities of the PC190723 product.
For an N-Mannich base prodrug such as TXY436 to be active in vivo, it must maintain the same biological activities in vitro as the product (PC190723) itself. We therefore compared TXY436 and PC190723 with regard to antistaphylococcal activity (as indicated by MIC, MBC, and kinetics of kill), as well as FtsZ-targeting activity (as reflected by the impact on bacterial morphology, FtsZ polymerization, and Z-ring formation).
Antistaphylococcal activity of TXY436.
Our initial studies compared the activities of TXY436 and PC190723 against four different MSSA and three different MRSA strains, with the resulting MICs being summarized in Table 2. The MICs of TXY436 were similar, if not identical, to those of PC190723 against all of the MSSA and MRSA strains examined, with these MICs ranging from 0.5 to 1.0 μg/ml. With regard to the three MRSA strains used in the studies, the activities of TXY436 were comparable to the activity of the prototypical anti-MRSA drug vancomycin (MIC = 2.0 μg/ml), which was included as a comparator antibiotic. Oxacillin (also included as a control agent) was predictably inactive versus the MRSA strains (MIC > 64 μg/ml).
Table 2.
Antibacterial activities of TXY436 and PC190723 against MSSA and MRSAa
| Strain | MIC (μg/ml) |
|||
|---|---|---|---|---|
| TXY436 | PC190723 | Vancomycin | Oxacillin | |
| 8325-4 (MSSA) | 0.5 | 0.5 | 1.0 | 0.125 |
| ATCC 29213 (MSSA) | 1.0 | 1.0 | 1.0 | 0.25 |
| ATCC 19636 (MSSA) | 0.5 | 0.5 | 1.0 | 0.125 |
| ATCC 49951 (MSSA) | 1.0 | 0.5 | 1.0 | 0.25 |
| ATCC 33591 (MRSA) | 0.5 | 0.5 | 2.0 | >64 |
| ATCC 43300 (MRSA) | 0.5 | 0.5 | 2.0 | >64 |
| Mu3 (MRSA) | 0.5 | 0.5 | 2.0 | >64 |
Vancomycin and oxacillin are included as control antistaphylococcal agents. Mu3 is a clinical MRSA isolate also identified as being hetero-GISA (hGISA) (43).
We next evaluated the antistaphylococcal activities of TXY436 and PC190723 with regard to their bactericidal behavior. As a first step toward this goal, we determined the MBC values of TXY436 and PC190723 against two of the MSSA strains (8325-4 and ATCC 19636) and two of the MRSA strains (ATCC 33591 and 43300). The bactericidal antistaphylococcal drug vancomycin and the bacteriostatic drug erythromycin were used as comparator controls in these determinations. In accordance with CLSI standards, an MBC/MIC ratio of 1 to 2 is considered indicative of bactericidal behavior (29). In contrast, an MBC/MIC ratio ≥8 is viewed as being indicative of bacteriostatic behavior. As expected, the control bactericidal agent vancomycin yielded MBC/MIC ratios of 1 to 2 against all of the MSSA and MRSA strains examined (Table 3). Also, as expected, the control bacteriostatic drug erythromycin yielded MBC/MIC ratios of >128 against the two MSSA strains. Both MRSA strains were resistant to erythromycin, precluding MBC determinations against these strains. Significantly, TXY436 and PC190723 exhibited MBC/MIC ratios of 1 to 2 against all of the MSSA and MRSA strains, indicative of bactericidal antistaphylococcal behavior.
Table 3.
Comparison of MICs and MBCs for TXY436 and PC190723 against MSSA and MRSAa
| Strain and compound | MIC (μg/ml) | MBC (μg/ml) | MBC/MIC |
|---|---|---|---|
| 8325-4 (MSSA) | |||
| TXY436 | 0.5 | 1.0 | 2 |
| PC190723 | 0.5 | 1.0 | 2 |
| Vancomycin | 1.0 | 2.0 | 2 |
| Erythromycin | 0.125 | 32 | 256 |
| ATCC 19636 (MSSA) | |||
| TXY436 | 0.5 | 0.5 | 1 |
| PC190723 | 0.5 | 0.5 | 1 |
| Vancomycin | 1.0 | 1.0 | 1 |
| Erythromycin | 0.5 | >64 | >128 |
| ATCC 33591 (MRSA) | |||
| TXY436 | 0.5 | 1.0 | 2 |
| PC190723 | 0.5 | 1.0 | 2 |
| Vancomycin | 2.0 | 4.0 | 2 |
| Erythromycin | >64 | NA | NA |
| ATCC 43300 (MRSA) | |||
| TXY436 | 0.5 | 1.0 | 2 |
| PC190723 | 0.5 | 1.0 | 2 |
| Vancomycin | 2.0 | 4.0 | 2 |
| Erythromycin | >64 | NA | NA |
Vancomycin and erythromycin are included as control bactericidal and bacteriostatic agents, respectively. NA, not applicable.
In addition to assessing the bactericidal activities of TXY436 and PC190723 against S. aureus with regard to MBC/MIC ratios, we also probed for bactericidal activity using a time-kill assay at compound concentrations of 1×, 2×, 4×, and 8× MIC. Figure 4 shows representative time-kill curves, with vancomycin being included as a comparator control agent. Inspection of these time-kill data reveals that both TXY436 and PC109723 produce >3 logs of kill at concentrations of 2× the MIC or greater. Significantly, this observation is consistent with the bactericidal behavior revealed by our MBC/MIC analysis described above. It is also of interest to note that at concentrations of ≥2× the MIC, the killing kinetics of TXY436 and PC190723 are more rapid than those of vancomycin.
Fig 4.
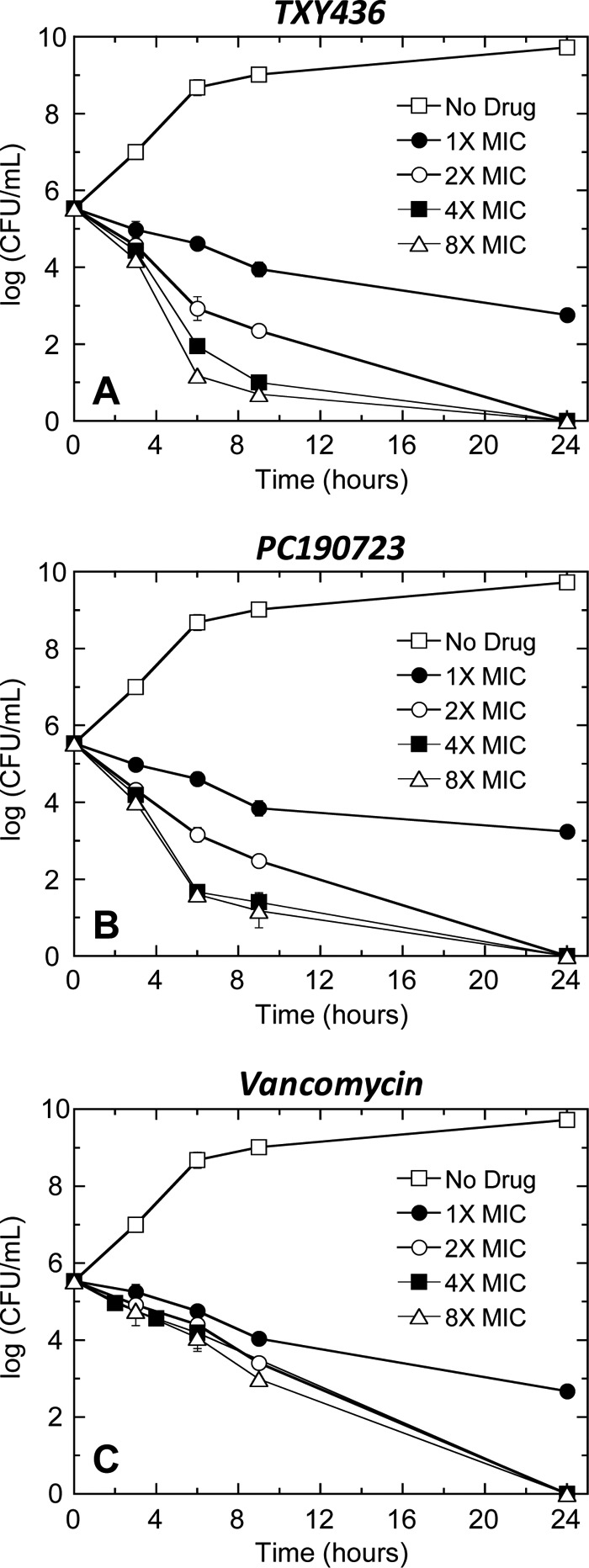
Time-kill curves for S. aureus 8325-4 (MSSA). Each data point reflects the average of two independent measurements, with the error bars reflecting the standard deviation from the mean. Bacteria were treated with DMSO vehicle only (no drug), TXY436 (A), PC190723 (B), or vancomycin (C). All agents were used at concentrations ranging from 1× to 8× the MIC.
PC190723 has been shown to be associated with a frequency of resistance (FOR) in S. aureus of approximately 3 × 10−8 (7, 37). We sought to determine the corresponding FOR to the TXY436 prodrug in S. aureus using the large inoculum approach described in Materials and Methods. These studies yielded an FOR to TXY436 of (2.0 ± 0.7) × 10−8, in agreement with the previously reported value for PC190723.
FtsZ-targeting activity of TXY436.
One of the hallmarks of FtsZ-targeting compounds that disrupt bacterial cell division is the induction of an enlarged morphology in treated bacteria (7, 12, 27, 37). Therefore, in our initial studies probing the FtsZ-targeting activity of TXY436, we determined the impact of the compound on the morphology of S. aureus bacteria using phase-contrast microscopy. Control DMSO-treated S. aureus bacteria are ∼1 μm in diameter, in agreement with previously reported diameters characteristic of S. aureus (38) (Fig. 5A). In contrast, the TXY436-treated bacteria exhibited 2- to 3-fold-enlarged phenotypes, with diameters ranging from 2 to 3 μm (Fig. 5B). PC190723-treated bacteria exhibited a similar behavior (Fig. 5C). Thus, TXY436 is able to induce the type of morphological change in S. aureus that is a hallmark of FtsZ-directed inhibitors of cell division such as PC190723 (6, 7).
Fig 5.
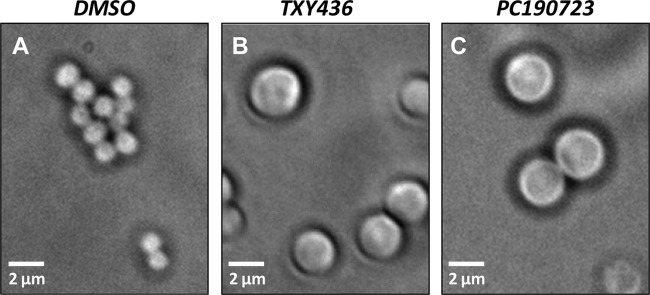
Phase-contrast micrographs of S. aureus 8325-4 bacteria cultured for 4 h in the presence of DMSO (solvent control) (A), TXY436 at 3 μg/ml (B), or PC190723 at 3 μg/ml (C).
Stimulation of FtsZ polymerization dynamics and stabilization of nonfunctional FtsZ polymeric structures are thought to underlie the antibacterial activities of FtsZ-targeting agents such as PC190723 (7, 13, 27, 37, 39). We probed for the propensity of TXY436 to exert these types of effects on the polymerization of purified S. aureus FtsZ in vitro. For this assay, we used a microtiter plate-based spectrophotometric approach in which FtsZ polymerization is detected in solution by a time-dependent increase in solution absorbance at 340 nm (A340). Figure 6A shows the time-dependent A340 profiles of S. aureus FtsZ in the absence or presence of TXY436 at concentrations ranging from 1 to 10 μg/ml. The corresponding results for PC190723 are also included for comparative purposes (Fig. 6B). Note that TXY436 stimulates FtsZ polymerization to a similar extent as PC190723, with the magnitudes of these stimulatory effects also being similarly dependent on compound concentration. Vancomycin was included as a non-FtsZ-targeting control drug in these assays. As expected, vancomycin did not impact the polymerization of S. aureus FtsZ to any significant degree.
Fig 6.
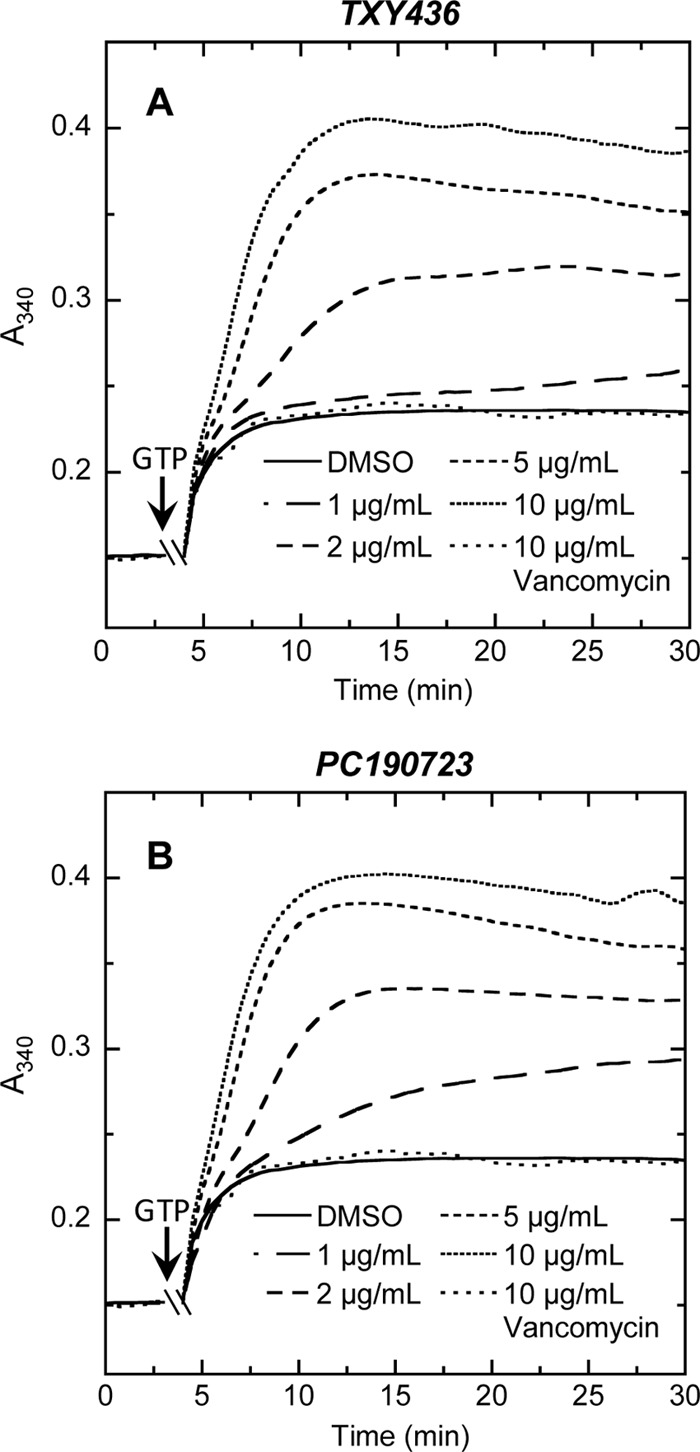
Concentration dependence of the impact of TXY436 (A) or PC190723 (B) on the polymerization of S. aureus FtsZ, as determined by monitoring time-dependent changes in absorbance at 340 nm (A340) at 25°C. The time-dependent A340 polymerization profiles were acquired in the presence of DMSO (solvent control) or the indicated concentrations of compound (ranging from 1 to 10 μg/ml). The polymerization profile acquired in the presence of vancomycin at 10 μg/ml is also included as a negative (non-FtsZ-targeting) control. Experimental conditions for the polymerization studies were 5 μM FtsZ, 50 mM Tris-HCl (pH 7.4), 50 mM KCl, and 10 mM magnesium acetate. Polymerization reactions were initiated by the addition of 4 mM GTP at the times indicated by the arrows.
In addition to investigating the impact of TXY436 on the function of purified FtsZ in vitro, we also sought to examine the impact of the compound on FtsZ function in live bacteria. To this end, we monitored the impact of the compound on the formation of FtsZ Z-rings using a strain of B. subtilis (FG347) that inducibly expresses GFP-tagged ZapA, a known protein marker for FtsZ Z-rings (30). We treated the FG347 bacteria with DMSO (solvent control), TXY436, or PC190723 and examined the bacteria using fluorescence microscopy. In the absence of compound, fluorescent foci corresponding to Z-rings are evident at midcell (highlighted by the arrows in Fig. 7A). In contrast, the TXY436-treated bacteria lack these midcell foci, instead exhibiting punctate fluorescent foci distributed throughout each cell (Fig. 7B). Recall that TXY436 acts as an enhancer of FtsZ polymerization. It is therefore likely that the punctate fluorescent foci distributed throughout the TXY436-treated bacteria reflect multiple nonfunctional FtsZ polymeric structures distinct from Z-rings. This type of behavior is characteristic of agents such as PC190723 that stabilize FtsZ polymers (Fig. 7C) (6, 7). In addition, the TXY436-treated bacteria, like the PC190723-treated bacteria, are significantly more elongated (filamentous) relative to the control DMSO-treated bacteria, a morphological change typical of rod-shaped bacteria in which cell division has been inhibited through disruption of FtsZ function (6, 7, 30, 40–42).
Fig 7.
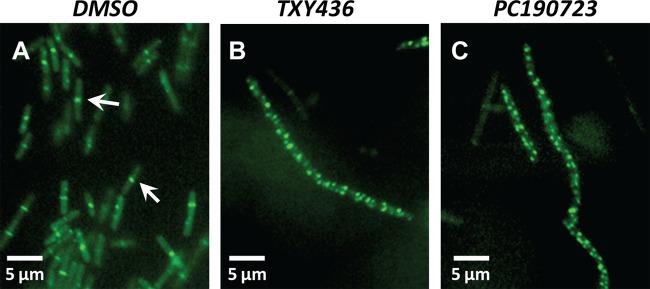
Fluorescence micrographs of B. subtilis FG347 bacteria that express a GFP-tagged, Z-ring marker protein, ZapA. The bacteria were cultured for 1 h in the presence of DMSO (solvent control) (A), TXY436 at 3 μg/ml (B), or PC190723 at 3 μg/ml (C), at which point GFP-ZapA expression was induced by the addition of xylose. The bacteria were incubated for an additional hour at 37°C and then visualized by fluorescence microscopy. The arrows in panel A highlight FtsZ Z-rings at midcell.
Taken together, the MIC/MBC, time-kill, microscopy, and FtsZ polymerization results described in the preceding sections indicate that the TXY436 prodrug maintains all the antistaphylococcal and FtsZ-targeting activities of the PC190723 product itself.
TXY436 retains antistaphylococcal activity in the presence of mouse serum.
An important criterion for a compound with regard to its potential for exhibiting antibacterial efficacy in vivo is for it to retain antibacterial activity in the presence of serum. In recognition of this important property, we assessed the impact of 50% (vol/vol) mouse serum on the activity of TXY436 against S. aureus. As highlighted in Table 4, the presence of mouse serum resulted in a 4-fold reduction in the antistaphylococcal potency of TXY436 (with the MIC increasing from 0.5 μg/ml in the absence of serum to 2.0 μg/ml in the presence of serum). A similar reduction in potency was observed for PC190723 itself. Note that the MIC of TXY436 in the presence of serum is identical to the corresponding MIC for the control drug vancomycin. Thus, TXY436 is still able to retain antistaphylococcal activity in the presence of serum.
Table 4.
Impact of mouse serum on the antistaphylococcal activities of TXY436 and PC190723
| Compound |
S. aureus 8325-4 MIC (μg/ml) |
|
|---|---|---|
| No serum | 50% mouse serum | |
| TXY436 | 0.5 | 2.0 |
| PC190723 | 0.5 | 2.0 |
| Vancomycin | 1.0 | 2.0 |
Pharmacokinetic analysis of the TXY436 prodrug and its PC190723 product after both intravenous and oral administration of TXY436 to mice reveals the prodrug product to be 73% orally bioavailable.
As a prelude to conducting in vivo efficacy experiments, we sought to evaluate the plasma pharmacokinetics of the TXY436 prodrug and its PC190723 product after both i.v. and p.o. administration of TXY436 to mice. In these studies, a single i.v. dose of 5 mg/kg or p.o. dose of 10 mg/kg was administered to mice, and the plasma concentrations of the TXY436 prodrug and the PC190723 product were measured at time points ranging from 0.08 to 8 h using LC-MS. For both the i.v. and the p.o. studies, 10 mM citrate (pH 2.6) was used as the vehicle. The plasma concentration of TXY436 was detectable and quantifiable up to 1 h after i.v. administration and up to 2 h after p.o. administration (Fig. 8A). The plasma concentration of PC190723 was detectable and quantifiable up to 4 h after i.v. administration and up to 8 h after p.o. administration (Fig. 8B).
Fig 8.
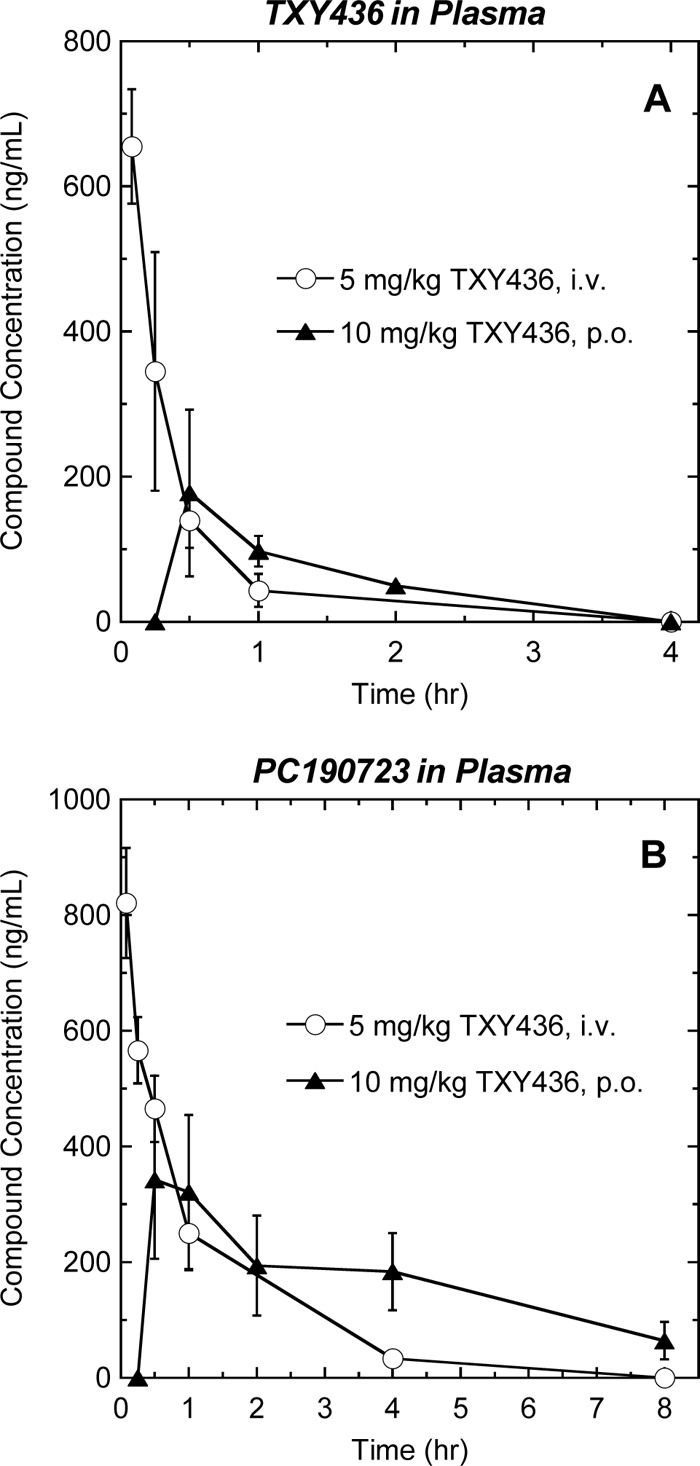
Time-dependent plasma concentrations of TXY436 (A) and PC190723 (B) after a single i.v. administration of TXY436 at 5 mg/kg (○) or a single p.o. administration of TXY436 at 10 mg/kg (▲) to male BALB/c mice.
Pharmacokinetic analyses of the time-dependent plasma concentration profiles for TXY436 and PC190723 with the NCA module of the Phoenix WinNonlin version 6.3 software package yielded the pharmacokinetic parameters listed in Table 5. Analysis of the TXY436 plasma concentrations after i.v. administration yielded an elimination half-life (t1/2) of 0.26 h and a high total body clearance (CL) of 312 ml/min·kg, ∼3.5-fold that of normal liver blood flow in mice (90 ml/min·kg). The volume of distribution at steady state (Vss) was found to be 5.77 liters/kg. Significantly, this value of Vss is ∼8.2-fold greater than that of normal body water (0.7 liters/kg), indicating substantive extravascular distribution of the compound. After p.o. administration of TXY436, the peak plasma concentration was achieved within 30 min (tmax = 0.5 h). A comparison of the areas under the curves up to the last detectable concentrations of TXY436 (AUClast) after both p.o. and i.v. administration yields an absolute oral bioavailability (%F) for the prodrug itself of 33% after normalization for the dose.
Table 5.
Pharmacokinetic parameters of the TXY436 prodrug and the PC190723 product following a single intravenous or peroral administration of TXY436 to male BALB/c micea
| Compound | Route | Dose (mg/kg) | tmax (h) | C0b (ng/liter) | AUClast (ng/liter·h) | t1/2 (h) | CL (ml/min·kg) | Vss (liters/kg) | %Fc |
|---|---|---|---|---|---|---|---|---|---|
| TXY436 | i.v. | 5 | NA | 878 | 252 | 0.26 | 312 | 5.77 | NA |
| p.o. | 10 | 0.50 | 178 | 164 | NA | NA | NA | 33 | |
| PC190723 | i.v. | NA | NA | 978 | 923 | 0.96 | 86.0 | 5.39 | NA |
| p.o. | NA | 0.50 | 343 | 1340 | NA | NA | NA | 73 |
Parameters were calculated by analysis of the plasma concentration versus time plots shown in Fig. 8 using the sparse sampling mode in the noncompartmental analysis (NCA) module of the Phoenix WinNonlin version 6.3 software package. NA, not applicable.; i.v., intravenous; p.o., peroral.
An extrapolated concentration was used for the i.v. group.
The AUClast value was used to calculate the %F value.
Analysis of the PC190723 plasma concentrations after i.v. administration of TXY436 yielded an ∼4-fold-longer t1/2 (0.96 h) than that of TXY436 and an ∼3.6-fold-lower CL of 86.0 ml/min-kg, a value that is comparable to normal liver blood flow. The Vss of PC190723 (5.39 liters/kg) was similar to that of TXY436, indicative a similar degree of extravascular distribution. After p.o. administration of TXY436, the peak plasma concentration of PC190723 was achieved within 30 min (tmax = 0.5 h). A comparison of the AUClast values of PC190723 after both p.o. and i.v. administration of TXY436 yields an absolute oral bioavailability for the prodrug product of 73% after normalization for dose. Thus, unlike many clinical MRSA drugs, including vancomycin, daptomycin, and the streptogramins, TXY436 is orally bioavailable.
TXY436 is orally efficacious in vivo against both MSSA and MRSA systemic infections.
Armed with the pharmacokinetic results described above, we next evaluated the oral in vivo efficacy of TXY436 using a mouse peritonitis model of systemic infection with both MSSA and MRSA. In the MSSA studies, four groups of six female Swiss Webster mice were inoculated intraperitoneally with a lethal inoculum of S. aureus ATCC 19636. The four groups of infected mice were treated as follows: group 1, untreated; group 2, treated p.o. with 10 mM citrate vehicle alone; group 3, treated p.o. with TXY436 at 128 mg/kg administered in four divided doses; and group 4, treated p.o. with PC190723 at 128 mg/kg administered as a suspension in four divided doses. All untreated mice died within 1 day of infection. In addition, none of the mice treated with citrate vehicle alone or with PC190723 survived beyond 1 day postinfection (Fig. 9A). Thus, oral administration of PC190723 itself was not efficacious in vivo. It is likely that the lack of oral efficacy associated with PC190723 is related to the compound being administered as a suspension (due to its poor solubility), which may, in turn, limit the oral bioavailability of the compound. In striking contrast to PC190723, oral administration of TXY436 proved to be highly efficacious, with 100% of the TXY436-treated mice surviving the infection.
Fig 9.
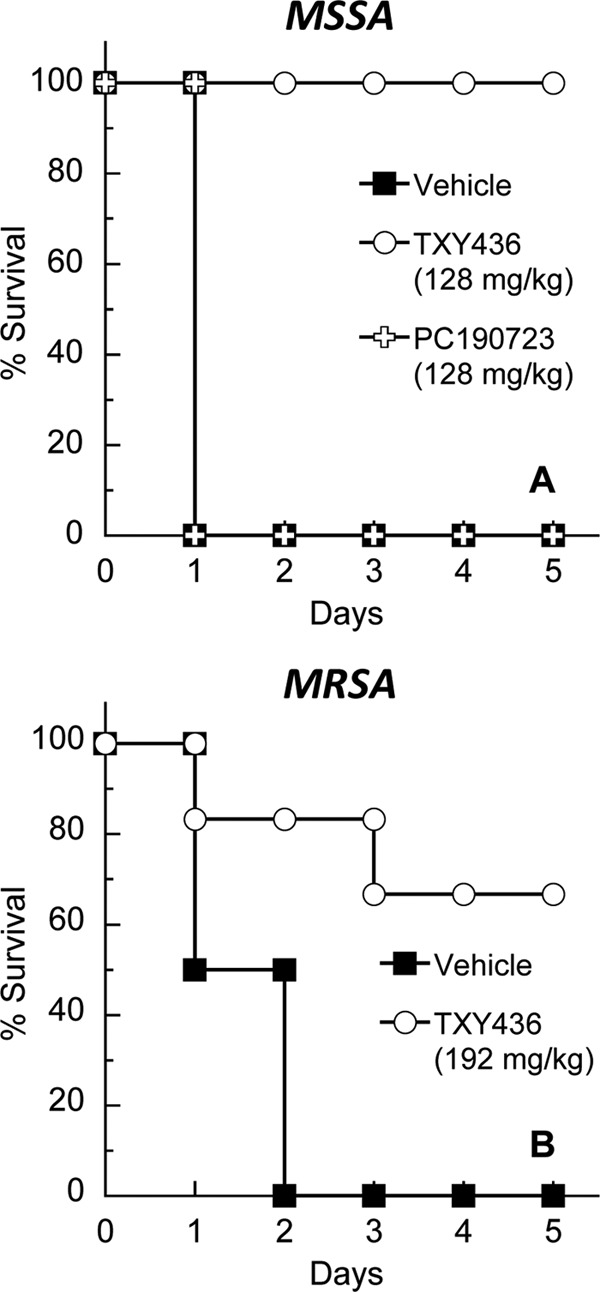
Oral (p.o.) in vivo efficacy of TXY436 in a mouse peritonitis model of systemic infection with MSSA 8325-4 (A) or MRSA ATCC 43300 (B). In the MSSA experiments, TXY436 and PC190723 were administered at 128 mg/kg in four divided doses. In the MRSA experiments, TXY436 was administered at 192 mg/kg in six divided doses. The vehicle was 10 mM citrate (pH 2.6) in all experiments.
In our MRSA studies, three groups of six mice were inoculated intraperitoneally with a lethal inoculum of MRSA ATCC 43300. The three groups of infected mice were treated as follows: group 1, untreated; group 2, treated p.o. with 10 mM citrate vehicle alone; and group 3, treated p.o. with TXY436 at 192 mg/kg administered in six divided doses. All untreated mice died within 2 days of infection. Similarly, none of the mice treated with citrate vehicle alone survived beyond 2 days postinfection (Fig. 9B). However, in the TXY436-treated group, 5 of 6 mice (83%) survived for 3 days postinfection, and 4 of 6 mice (67%) survived the infection entirely. Thus, TXY436 is not only orally efficacious against systemic MSSA infection but also against systemic MRSA infection.
In both the MSSA and the MRSA studies, an additional positive control group of six infected mice was also included. This group was treated i.v. with vancomycin at doses of 16 mg/kg in the MSSA studies and 24 mg/kg in the MRSA studies. As anticipated, all vancomycin-treated mice survived their infections.
TXY436 is minimally toxic to mammalian cells.
An important criterion for the potential clinical utility of an antibacterial agent is minimal toxicity against mammalian cells. In this connection, we used a 3-day tetrazole (MTT)-based assay to assess the cytotoxicity, if any, of TXY436 against Vero African green monkey kidney epithelial cells. TXY436 was found to be minimally toxic to Vero cells, with a 50% inhibitory concentration (IC50) of 64 μg/ml. Significantly, this IC50 is 64× to 128× the antistaphylococcal MICs of TXY436 (which range from 0.5 to 1.0 μg/ml). Albeit derived from a single mammalian cell line, this differential suggests that TXY436 is associated with a significant therapeutic window. Future studies with additional mammalian and human cell lines will be directed toward assessing the general applicability of this therapeutic window.
In conclusion, TXY436 is a prodrug of the FtsZ-targeting benzamide PC190273 with enhanced formulation properties as well as oral efficacy in vivo against both MSSA and MRSA infections. These characteristics, coupled with a minimal potential for toxicity to mammalian cells, make TXY436 a worthy candidate for further development as a clinically useful agent to treat staphylococcal infections.
ACKNOWLEDGMENTS
This study was supported by research agreements between TAXIS Pharmaceuticals, Inc., and both Rutgers Robert Wood Johnson Medical School (D.S.P.) and Rutgers Ernest Mario School of Pharmacy (E.J.L.).
We are indebted to Nancy Connell (Rutgers New Jersey Medical School, Newark, NJ) for assistance with the mammalian cytotoxicity studies. We are also indebted to Glenn W. Kaatz (John D. Dingell VA Medical Center, Detroit, MI) for providing the S. aureus 8325-4 strain and to Richard Losick (Harvard University, Boston, MA) for providing the B. subtilis FG347 strain.
Footnotes
Published ahead of print 16 September 2013
REFERENCES
- 1. Bi EF, Lutkenhaus J. 1991. FtsZ ring structure associated with division in Escherichia coli. Nature 354:161–164 [DOI] [PubMed] [Google Scholar]
- 2. Errington J, Daniel RA, Scheffers DJ. 2003. Cytokinesis in bacteria. Microbiol. Mol. Biol. Rev. 67:52–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adams DW, Errington J. 2009. Bacterial cell division: assembly, maintenance, and disassembly of the Z ring. Nat. Rev. Microbiol. 7:642–653 [DOI] [PubMed] [Google Scholar]
- 4. Kirkpatrick CL, Viollier PH. 2011. New(s) to the (Z-)ring. Curr. Opin. Microbiol. 14:691–697 [DOI] [PubMed] [Google Scholar]
- 5. Erickson HP, Anderson DE, Osawa M. 2010. FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol. Mol. Biol. Rev. 74:504–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stokes NR, Sievers J, Barker S, Bennett JM, Brown DR, Collins I, Errington VM, Foulger D, Hall M, Halsey R, Johnson H, Rose V, Thomaides HB, Haydon DJ, Czaplewski LG, Errington J. 2005. Novel inhibitors of bacterial cytokinesis identified by a cell-based antibiotic screening assay. J. Biol. Chem. 280:39709–39715 [DOI] [PubMed] [Google Scholar]
- 7. Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, Baker PJ, Barynin VV, Rice DW, Sedelnikova SE, Heal JR, Sheridan JM, Aiwale ST, Chauhan PK, Srivastava A, Taneja A, Collins I, Errington J, Czaplewski LG. 2008. An inhibitor of FtsZ with potent and selective antistaphylococcal activity. Science 321:1673–1675 [DOI] [PubMed] [Google Scholar]
- 8. Huang Q, Kirikae F, Kirikae T, Pepe A, Amin A, Respicio L, Slayden RA, Tonge PJ, Ojima I. 2006. Targeting FtsZ for antituberculosis drug discovery: noncytotoxic taxanes as novel antituberculosis agents. J. Med. Chem. 49:463–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Czaplewski LG, Collins I, Boyd EA, Brown D, East SP, Gardiner M, Fletcher R, Haydon DJ, Henstock V, Ingram P, Jones C, Noula C, Kennison L, Rockley C, Rose V, Thomaides-Brears HB, Ure R, Whittaker M, Stokes NR. 2009. Antibacterial alkoxybenzamide inhibitors of the essential bacterial cell division protein FtsZ. Bioorg. Med. Chem. Lett. 19:524–527 [DOI] [PubMed] [Google Scholar]
- 10. Schaffner-Barbero C, Martin-Fontecha M, Chacón P, Andreu JM. 2012. Targeting the assembly of bacterial cell division protein FtsZ with small molecules. ACS Chem. Biol. 7:269–277 [DOI] [PubMed] [Google Scholar]
- 11. Ma S. 2012. The development of FtsZ inhibitors as potential antibacterial agents. ChemMedChem 7:1161–1172 [DOI] [PubMed] [Google Scholar]
- 12. Haydon DJ, Bennett JM, Brown D, Collins I, Galbraith G, Lancett P, Macdonald R, Stokes NR, Chauhan PK, Sutariya JK, Nayal N, Srivastava A, Beanland J, Hall R, Henstock V, Noula C, Rockley C, Czaplewski L. 2010. Creating an antibacterial with in vivo efficacy: synthesis and characterization of potent inhibitors of the bacterial cell division protein FtsZ with improved pharmaceutical properties. J. Med. Chem. 53:3927–3936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Andreu JM, Schaffner-Barbero C, Huecas S, Alonso D, Lopez-Rodriguez ML, Ruiz-Avila LB, Núñez-Ramírez R, Llorca O, Martín-Galiano AJ. 2010. The antibacterial cell division inhibitor PC190723 is an FtsZ polymer-stabilizing agent that induces filament assembly and condensation. J. Biol. Chem. 285:14239–14246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lock RL, Harry EJ. 2008. Cell-division inhibitors: new insights for future antibiotics. Nat. Rev. Drug Discov. 7:324–338 [DOI] [PubMed] [Google Scholar]
- 15. Boberek JM, Stach J, Good L. 2010. Genetic evidence for inhibition of bacterial division protein FtsZ by berberine. PLoS One 5:e13745. 10.1371/journal.pone.0013745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Awasthi D, Kumar K, Ojima I. 2011. Therapeutic potential of FtsZ inhibition: a patent perspective. Expert Opin. Ther. Patents 21:657–679 [DOI] [PubMed] [Google Scholar]
- 17. Huang Q, Tonge PJ, Slayden RA, Kirikae T, Ojima I. 2007. FtsZ: a novel target for tuberculosis drug discovery. Curr. Top. Med. Chem. 7:527–543 [DOI] [PubMed] [Google Scholar]
- 18. Kumar K, Awasthi D, Berger WT, Tonge PJ, Slayden RA, Ojima I. 2010. Discovery of anti-TB agents that target the cell-division protein FtsZ. Future Med. Chem. 2:1305–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kumar K, Awasthi D, Lee SY, Zanardi I, Ruzsicska B, Knudson S, Tonge PJ, Slayden RA, Ojima I. 2011. Novel trisubstituted benzimidazoles, targeting Mtb FtsZ, as a new class of antitubercular agents. J. Med. Chem. 54:374–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kapoor S, Panda D. 2009. Targeting FtsZ for antibacterial therapy: a promising avenue. Expert Opin. Ther. Targets 13:1037–1051 [DOI] [PubMed] [Google Scholar]
- 21. Kaul M, Parhi AK, Zhang Y, LaVoie EJ, Tuske S, Arnold E, Kerrigan JE, Pilch DS. 2012. A bactericidal guanidinomethyl biaryl that alters the dynamics of bacterial FtsZ polymerization. J. Med. Chem. 55:10160–10176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kelley C, Zhang Y, Parhi A, Kaul M, Pilch DS, LaVoie EJ. 2012. 3-Phenyl substituted 6,7-dimethoxyisoquinoline derivatives as FtsZ-targeting antibacterial agents. Bioorg. Med. Chem. 20:7012–7029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kelley C, Lu S, Parhi A, Kaul M, Pilch DS, LaVoie EJ. 2013. Antimicrobial activity of various 4- and 5-substituted 1-phenylnaphthalenes. Eur. J. Med. Chem. 60:395–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Parhi A, Lu S, Kelley C, Kaul M, Pilch DS, LaVoie EJ. 2012. Antibacterial activity of substituted dibenzo[a,g]quinolizin-7-ium derivatives. Bioorg. Med. Chem. Lett. 22:6962–6966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y, Giurleo D, Parhi A, Kaul M, Pilch DS, LaVoie EJ. 2013. Substituted 1,6-diphenylnaphthalenes as FtsZ-targeting antibacterial agents. Bioorg. Med. Chem. Lett. 23:2001–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stokes NR, Baker N, Bennett JM, Berry J, Collins I, Czaplewski LG, Logan A, Macdonald R, Macleod L, Peasley H, Mitchell JP, Nayal N, Yadav A, Srivastava A, Haydon DJ. 2013. An improved small-molecule inhibitor of FtsZ with superior in vitro potency, drug-like properties, and in vivo efficacy. Antimicrob. Agents Chemother. 57:317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adams DW, Wu LJ, Czaplewski LG, Errington J. 2011. Multiple effects of benzamide antibiotics on FtsZ function. Mol. Microbiol. 80:68–84 [DOI] [PubMed] [Google Scholar]
- 28. Sorto NA, Olmstead MM, Shaw JT. 2010. Practical synthesis of PC190723, an inhibitor of the bacterial cell division protein FtsZ. J. Org. Chem. 75:7946–7949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. CLSI 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 8th ed. Document M07-A8 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 30. Gueiros-Filho FJ, Losick R. 2002. A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes Dev. 16:2544–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Neill AJ, Cove JH, Chopra I. 2001. Mutation frequencies for resistance to fusidic acid and rifampicin in Staphylococcus aureus. J. Antimicrob. Chemother. 47:647–650 [DOI] [PubMed] [Google Scholar]
- 32. Stiles BG, Campbell YG, Castle RM, Grove SA. 1999. Correlation of temperature and toxicity in murine studies of staphylococcal enterotoxins and toxic shock syndrome toxin 1. Infect. Immun. 67:1521–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ekins S, Reynolds RC, Kim H, Koo M-S, Ekonomidis M, Talaue M, Paget SD, Woolhiser LK, Lenaerts AJ, Bunin BA, Connell N, Freundlich JS. 2013. Bayesian models leveraging bioactivity and cytotoxicity information for drug discovery. Chem. Biol. 20:370–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bundgaard H, Johansen M. 1980. Prodrugs as drug delivery systems. IV. N-Mannich bases as potential novel prodrugs for amides, ureides, amines, and other NH-acidic compounds. J. Pharm. Sci. 69:44–46 [DOI] [PubMed] [Google Scholar]
- 35. Huttunen KM, Rautio J. 2011. Prodrugs: an efficient way to breach delivery and targeting barriers. Curr. Top. Med. Chem. 11:2265–2287 [DOI] [PubMed] [Google Scholar]
- 36. Dhareshwar SS, Stella VJ. 2008. Your prodrug releases formaldehyde: should you be concerned? No! J. Pharm. Sci. 97:4184–4193 [DOI] [PubMed] [Google Scholar]
- 37. Tan CM, Therien AG, Lu J, Lee SH, Caron A, Gill CJ, Lebeau-Jacob C, Benton-Perdomo L, Monteiro JM, Pereira PM, Elsen NL, Wu J, Deschamps K, Petcu M, Wong S, Daigneault E, Kramer S, Liang L, Maxwell E, Claveau D, Vaillancourt J, Skorey K, Tam J, Wang H, Meredith TC, Sillaots S, Wang-Jarantow L, Ramtohul Y, Langlois E, Landry F, Reid JC, Parthasarathy G, Sharma S, Baryshnikova A, Lumb KJ, Pinho MG, Soisson SM, Roemer T. 2012. Restoring methicillin-resistant Staphylococcus aureus susceptibility to β-lactam antibiotics. Sci. Transl. Med. 4:126ra135. [DOI] [PubMed] [Google Scholar]
- 38. Jorge AM, Hoiczyk E, Gomes JP, Pinho MG. 2011. EzrA contributes to the regulation of cell size in Staphylococcus aureus. PLoS One 6:e27542. 10.1371/journal.pone.0027542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaul M, Zhang Y, Parhi AK, LaVoie EJ, Tuske S, Arnold E, Kerrigan JE, Pilch DS. 2013. Enterococcal and streptococcal resistance to PC190723 and related compounds: molecular insights from a FtsZ mutational analysis. Biochimie 95:1880–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haeusser DP, Garza AC, Buscher AZ, Levin PA. 2007. The division inhibitor EzrA contains a seven-residue patch required for maintaining the dynamic nature of the medial FtsZ ring. J. Bacteriol. 189:9001–9010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haeusser DP, Schwartz RL, Smith AM, Oates ME, Levin PA. 2004. EzrA prevents aberrant cell division by modulating assembly of the cytoskeletal protein FtsZ. Mol. Microbiol. 52:801–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ma X, Ehrhardt DW, Margolin W. 1996. Colocalization of cell division proteins FtsZ and FtsA to cytoskeletal structures in living Escherichia coli cells by using green fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 93:12998–13003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hiramatsu K, Aritaka N, Hanaki H, Kawasaki S, Hosoda Y, Hori S, Fukuchi Y, Kobayashi I. 1997. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet 350:1670–1673 [DOI] [PubMed] [Google Scholar]