

Abstract
We designed, synthesized, and identified two novel nonpeptidic human immunodeficiency virus type 1 (HIV-1) protease inhibitors (PIs), GRL-04810 and GRL-05010, containing the structure-based designed privileged cyclic ether-derived nonpeptide P2 ligand, bis-tetrahydrofuranylurethane (bis-THF), and a difluoride moiety, both of which are active against the laboratory strain HIV-1LAI (50% effective concentrations [EC50s], 0.0008 and 0.003 μM, respectively) with minimal cytotoxicity (50% cytotoxic concentrations [CC50s], 17.5 and 37.0 μM, respectively, in CD4+ MT-2 cells). The two compounds were active against multi-PI-resistant clinical HIV-1 variants isolated from patients who had no response to various antiviral regimens. GRL-04810 and GRL-05010 also blocked the infectivity and replication of each of the HIV-1NL4-3 variants selected by up to 5 μM lopinavir (EC50s, 0.03 and 0.03 μM, respectively) and atazanavir (EC50s, 0.02 and 0.04 μM, respectively). Moreover, they were active against darunavir (DRV)-resistant variants (EC50 in 0.03 to 0.034 μM range for GRL-04810 and 0.026 to 0.043 μM for GRL-05010), while DRV had EC50s between 0.02 and 0.174 μM. GRL-04810 had a favorable lipophilicity profile as determined with the partition (log P) and distribution (log D) coefficients of −0.14 and −0.29, respectively. The in vitro blood-brain barrier (BBB) permeability assay revealed that GRL-04810 and GRL-05010 may have a greater advantage in terms of crossing the BBB than the currently available PIs, with apparent penetration indexes of 47.8 × 10−6 and 61.8 × 10−6 cm/s, respectively. The present data demonstrate that GRL-04810 and GRL-05010 exert efficient activity against a wide spectrum of HIV-1 variants in vitro and suggest that two fluorine atoms added to their bis-THF moieties may well enhance their penetration across the BBB.
INTRODUCTION
Combined antiretroviral therapy (cART) has had a major impact on the AIDS epidemic; however, no eradication of human immunodeficiency virus type 1 (HIV-1) appears to be readily possible, in part due to the viral reservoirs remaining in blood and infected tissues. Moreover, we have encountered a number of challenges in bringing about the optimal benefits of the currently available therapeutics of HIV-1 infection and AIDS to individuals receiving cART (1–6). These challenges include (i) drug-related toxicities, (ii) partial restoration of immunologic functions once individuals develop AIDS, (iii) development of various cancers as a consequence of survival prolongation, (iv) immune reconstitution syndrome (IRS) or flare-ups of inflammation in individuals receiving cART, and (v) increased cost of antiviral therapy. Such limitations and flaws of cART are exacerbated by the development of drug-resistant HIV-1 variants (7–11).
One of the sanctuary sites for HIV-1 infection is the central nervous system (CNS) (12). The fact that HIV-1 enters and infects target cells in the CNS represents a significant challenge for the long-term suppression of virus replication and has been linked to the development of several neurological complications (13, 14). Although cART has significantly reduced the incidence of HIV-1-associated dementia, the prevalence of CNS disorders such as HIV-1-associated neurocognitive disorders, or HAND, appears to be increasing as a result of prolonged patient survival and poor antiretroviral drug penetration into the CNS (15–19). Furthermore, subtherapeutic drug concentrations in the CNS may facilitate the development of viral resistance (20). In addition, HIV-1 infection of the CNS may also result in the establishment of a unique viral reservoir, which certain antiretroviral drugs do not have reasonable access to (21–24). Moreover, there is evidence that cART is less effective in lowering virus replication in the CNS than in the blood (21), and unfortunately HIV protease inhibitors (PIs) and several of the nucleoside analogs penetrate only poorly into the CNS (12), allowing early CNS infection to evolve independently over time in the inaccessible brain reservoir (25).
Successful antiviral drugs, in theory, exert their virus-specific effects by interacting with viral receptors, virally encoded enzymes, viral structural components, or viral genes or their transcripts without disturbing cellular metabolism or function. However, at present, no antiretroviral drugs or agents are likely to be completely specific for HIV-1 or to be devoid of toxicity in the therapy of AIDS, which is a critical issue because patients with AIDS and its related diseases will have to receive cART for a long period of time, perhaps for the rest of their lives. Thus, the identification of a new class of antiretroviral drugs which have a unique mechanism(s) of action and produce no or minimal side effects remains an important therapeutic objective.
We have been focusing on the design and synthesis of nonpeptidyl protease inhibitors (PIs) that are capable of overcoming HIV-1 variants resistant to the currently approved PIs. One such anti-HIV-1 agent, darunavir (DRV), containing a structure-based designed privileged nonpeptidic P2 ligand, 3(R),3a(S),6a(R)-bis-tetrahydrofuranylurethane (bis-THF) (26–28), has been clinically used worldwide as a first-line therapeutic for HIV-1-infected individuals. In the present work, we designed and synthesized nonpeptidic HIV-1 protease inhibitors GRL-04810 and GRL-05010, which contain bis-THF and a difluoride moiety, and found that these compounds exert efficacious activity against a wide spectrum of laboratory HIV-1 strains and primary clinical isolates, including multi-PI-resistant variants, with minimal cytotoxicity. We also attempted to elucidate the binding interactions of the two compounds with the wild-type HIV-1 protease by employing molecular modeling. Moreover, we selected HIV-1 variants with GRL-04810 and GRL-05010 by propagating a laboratory wild-type HIV-1NL4-3 in MT-4 cells in the presence of increasing concentrations of GRL-04810 and GRL-05010 and determined amino acid substitutions that emerged under the pressure of these compounds in the protease-encoding region. Finally, in view of the limited penetration of most antiviral drugs into the CNS, we evaluated the partition and distribution coefficients (log P and log D) as well as the apparent permeability coefficient (Papp) for blood-brain barrier (BBB) using an in vitro model, where we were able to demonstrate that GRL-04810 and GRL-05010 had potentially enhanced penetration capabilities across the BBB.
MATERIALS AND METHODS
Cells and viruses.
MT-2 and MT-4 cells were grown in RPMI 1640-based culture medium supplemented with 10% fetal calf serum (FCS; JRH Biosciences, Lenexa, MD), 50 unit/ml penicillin, and 100 μg/ml of kanamycin. The following HIV-1 viruses were employed for the drug susceptibility assay (see below): HIV-1LAI, HIV-1NL4-3, the clinical HIV-1 strain HIV-1ERS104pre isolated from a drug-naive patient with AIDS (29), and six HIV-1 clinical isolates which were originally obtained from patients with AIDS who had received 9 to 11 anti-HIV-1 drugs over the past 32 to 83 months and which were genotypically and phenotypically characterized as multi-PI-resistant HIV-1 variants. All primary HIV-1 strains were passaged once or twice in 3-day-old phytohemagglutinin-activated peripheral blood mononuclear cells (PHA-PBMC), and the culture supernatants were stored at −80°C until use.
Antiviral agents.
Saquinavir (SQV) was kindly provided by Roche Products, Ltd. (Welwyn Garden City, United Kingdom), and Abbott Laboratories (Abbott Park, IL). Amprenavir (APV) was received as a courtesy gift from Glaxo-Wellcome, Research Triangle Park, NC. Lopinavir (LPV) was kindly provided by Japan Energy, Inc., Tokyo, Japan. Atazanavir (ATV) was a contribution from Bristol-Myers Squibb (New York, NY). DRV was synthesized as previously described (30). 3′-Azido-2′,3′-dioxythymidine (AZT) was purchased from Sigma-Aldrich (St. Louis, MO). Indinavir (IDV) was kindly provided by Merck Research Laboratories (Rahway, NJ).
Drug susceptibility assay.
The susceptibility of HIV-1LAI to various drugs was determined as previously described, with minor modifications. Briefly, MT-2 cells (104/ml) were exposed to 100 50% tissue culture infectious doses (TCID50) of HIV-1LAI in the presence or absence of various concentrations of drugs in 96-well microculture plates and incubated at 37°C for 7 days. After 100 μl of the medium was removed from each well, 3-(4,5-dimetylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (10 μl; 7.5 mg/ml in phosphate-buffered saline) was added to each well in the plate, followed by incubation at 37°C for 2 h. After incubation, to dissolve the formazan crystals 100 μl of acidified isopropanol containing 4% (vol/vol) Triton X-100 was added to each well, and the optical density was measured in a kinetic microplate reader (VMax; Molecular Devices, Sunnyvale, CA). All assays were performed in duplicate or triplicate. In some experiments, MT-2 cells were chosen as target cells in the MTT assay since these cells undergo greater HIV-1-elicited cytopathic effects than MT-4 cells. To determine the sensitivity of primary HIV-1 isolates to drugs, phytohemagglutinin-stimulated peripheral blood mononuclear cells (PHA-PBMC) (106/ml) were exposed to 50 TCID50 of each primary HIV-1 isolate and cultured in the presence or absence of various concentrations of drugs in 10-fold serial dilutions in 96-well microculture plates. In determining the drug susceptibility of certain laboratory HIV-1 strains, MT-4 cells were employed as target cells as previously described, with minor modifications. In brief, MT-4 cells (105/ml) were exposed to 100 TCID50 of drug-resistant HIV-1 strains in the presence or absence of various concentrations of drugs and incubated at 37°C. On day 7 of culture, the supernatants were harvested, and the amounts of p24 Gag protein were determined by using a fully automated chemiluminescent enzyme immunoassay system (Lumipulse F; Fujirebio, Inc., Tokyo, Japan). Drug concentrations that suppressed the production of p24 Gag protein by 50% (50% effective concentration [EC50]) were determined by comparison with the p24 production level in drug-free control cell culture. All assays were performed in duplicate or triplicate. PHA-PBMC were derived from a single donor in each independent experiment. Thus, to obtain the data, three different donors were recruited.
Generation of PI-resistant HIV-1 variants in vitro.
MT-4 cells (105/ml) were exposed to HIV-1NL4-3 (500 TCID50) and cultured in the presence of various PIs at an initial concentration equal to its EC50. Viral replication was monitored by the determination of the amount of p24 Gag produced by MT-4 cells. The culture supernatants were harvested on day 7 and used to infect fresh MT-4 cells for the next round of culture in the presence of increasing concentrations of each drug. When the virus began to propagate in the presence of the drug, the drug concentration was increased generally 2- to 3-fold. Proviral DNA samples obtained from the lysates of infected cells were subjected to nucleotide sequencing. This drug selection procedure was carried out until the drug concentration reached 5 μM (31–33). In the experiments for selecting drug-resistant variants, MT-4 cells were exploited as target cells since HIV-1 in general replicates at greater levels in MT-4 cells than in MT-2 cells.
Determination of nucleotide sequences.
Molecular cloning and determination of the nucleotide sequences of HIV-1 strains passaged in the presence of anti-HIV-1 agents were performed as described previously (31). In brief, high-molecular-weight DNA was extracted from HIV-1-infected MT-4 cells by using an InstaGene Matrix (Bio-Rad Laboratories, Hercules, CA) and subjected to molecular cloning, followed by sequence determination. The primers used for the first round of PCR with the entire Gag- and protease-encoding regions of the HIV-1 genome were LTR F1 (5′-GAT GCT ACA TAT AAG CAG CTG C-3′) and PR12 (5′-CTC GTG ACA AAT TTC TAC TAA TGC-3′). The first-round PCR mixture consisted of 1 μl of proviral DNA solution, 10 μl of Premix Taq (Ex Taq version; TaKaRa Bio, Inc., Otsu, Japan), and 10 pmol of each of the first PCR primers in a total volume of 20 μl. The PCR conditions used were an initial 3 min at 95°C, followed by 35 cycles of 40 s at 95°C, 20 s at 55°C, and 2 min at 72°C, with a final 10 min of extension at 72°C. The first-round PCR products (1 μl) were used directly in the second round of PCR with primers LTR F2 (5′-GAG ACT CTG GTA ACT AGA GAT C-3′) and Ksma2.1 (5′-CCA TCC CGG GCT TTA ATT TTA CTG GTA C-3′) under the PCR conditions of an initial 3 min at 95°C, followed by 35 cycles of 30 s at 95°C, 20 s at 55°C, and 2 min at 72°C, with a final 10 min of extension at 72°C. The second-round PCR products were purified with spin columns (MicroSpin S-400 HR columns; Amersham Biosciences Corp., Piscataway, NJ), cloned directly, and subjected to sequencing with a model 3130 automated DNA sequencer (Applied Biosystems, Foster City, CA).
Determination of replication kinetics of GRL-04810- and GRL-05010-resistant HIV-1NL4-3 variants and wild-type HIV-1NL4-3.
GRL-04810- and GRL-05010-resistant variants were obtained at passages 18 and 10, respectively, and propagated in fresh MT-4 cells without the drugs for 7 days, and viral stocks were stored at −80°C until use. MT-4 cells (3 × 105) were exposed to the wild-type HIV-1NL4-3 preparation or HIV-1NL4-3 preparations selected with GRL-04810 over 18 passages and GRL-05010 over 10 passages, designated HIV-1GRL-04810RP18 and HIV-1GRL-05010RP10, respectively, containing 10 ng/ml p24, in six-well culture plates for 3 h. Such MT-4 cells were subsequently washed with fresh medium and divided into three fractions, each of which was cultured with or without each compound (final concentration of MT-4 cells, 104/ml; drug concentrations, 0.01 μM and 0.001 μM). The amounts of p24 were measured every 2 days for up to 7 days.
Determination of partition and distribution coefficients of GRL-04810 and GRL-05010 using the shake flask method.
On day −1 of the experimental setting, saturation of 1-octanol [CH3 (CH2)6 CH2 OH] (Nacalai Tesque, Kyoto, Japan) with water and Tris-buffered saline (10× working solution; 20 mM Tris, pH 7.4–0.9% NaCl [Sigma-Aldrich, St. Louis, MO]) took place. Four different flasks were used. One contained 50 ml of water plus 100 ml of 1-octanol, and another flask contained 1-octanol saturated with water by the addition of 50 ml of 1-octanol and 100 ml of water. For the other two flasks, the same ratios and volumes were kept for 1-octanol saturated with Tris buffer and Tris buffer saturated with 1-octanol. The flasks were sealed and placed in a Bioshaker at room temperature for 24 h at 90 rpm. Simultaneously, dilutions of GRL-04810, GRL-05010, and DRV were performed from a 20 mM dimethyl sulfoxide (DMSO) stock to a final concentration of 100 μM using distilled H2O (dH2O), Tris-buffered saline, and 1-octanol as solvents. Successive dilutions were made to obtain concentrations of 10 μM, 1 μM, and 0.1 μM. A standard curve was generated on a light spectrophotometer (DU Series 700; Beckman Coulter, Fullerton, CA) at an absorbance of 230.
On day 1 of the experiment, the lipid and liquid interfaces were separated, and compounds were diluted again from 20 mM DMSO to 100 μM using the 1-octanol, water, and Tris-buffered saline obtained from the shake flask assay. The resulting diluted compounds were then added to separate serum tubes containing equal proportions of 1-octanol and water and of 1-octanol and Tris-buffered saline. The solution was hand shaken for 5 min and then centrifuged at 3,500 rpm and room temperature for 20 min. Finally, the compounds were recovered from the 1-octanol, Tris buffer, and water interfaces and then measured on a light spectrophotometer.
The values for log P and log D were obtained according to the following mathematical formulas: log Poctanol/water = log ([compound]n-octanol/[compound]water) and log Doctanol/water = log ([compound]n-octanol/[compound]ionized + /[compound]neutral), where [compound]n-octanol, [compound]water, [compound]ionized, and [compound]neutral represent the concentrations of compound in n-octanol, water, ionized water, and neutral water, respectively.
Determination of the apparent permeability coefficients of GRL-04810 and GRL-05010 for the blood-brain barrier using a novel in vitro model.
A novel in vitro BBB model (BBB Kit; PharmaCo-Cell, Ltd., Nagasaki, Japan), incorporating a triple culture of rat-derived astrocytes, pericytes, and monkey-derived endothelial cells (34) was used to determine the apparent permeability coefficient (Papp in cm/s) for the BBB of GRL-04810, GRL-05010, AZT, IDV, SQV, LPV, ATV, DRV, caffeine, and sucrose.
The BBB Kit was kept at −80°C until it was thawed on day 0 of the experiments. Nutritional medium was added to both brain and blood sides of the wells. This solution consists of Dulbecco's modified Eagle's medium (DMEM)/F-12 medium with 10% (vol/vol) FCS, 100 μg/ml heparin, 1.5 ng/ml basic fibroblast growth factor (bFGF), 5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml sodium selenite, 500 nM hydrocortisone, and 50 μg/ml gentamicin. Fresh medium was added 3 h after thawing, following the manufacturer's instructions, and 24 h later. The plates were incubated at 37°C until day 4 of the experiment when the condition of astrocytes was checked under a light microscope. Following this, the integrity of the collagen-coated membrane was verified by the measurement of transendothelial electrical resistance (TEER) using an ohmmeter provided by the manufacturer. As TEER increases over time and reaches a peak between days 4 and 6 of the experiment, determinations were done during this period. Membranes were tested individually, and collagen-coated membranes displaying TEER values greater than 150 Ω/cm2 were deemed suitable for the execution of the drug BBB penetration assay. Detailed information regarding the components of the BBB Kit as well as its mechanisms can be seen by accessing the manufacturer's website (PharmaCo-Cell, Nagasaki, Japan).
Once the conditions of cell viability and membrane integrity were met, drug dilutions were performed from 20 mM DMSO stocks of GRL-04810, GRL-05010, AZT, IDV, SQV, LPV, ATV, and DRV, while caffeine and sucrose were used as positive and negative controls, respectively. Standard curves were generated for each compound on a light spectrophotometer as previously described. Each compound (100 μM) was added to the luminal (blood) side of the wells and incubated at 37°C for 30 min, and then the amount of drug that crossed the in vitro BBB was collected and measured under a light spectrophotometer at an absorbance of 230.
Papp was calculated using the following mathematical formula: Papp (cm/s) = (VA/A × [C]luminal) × (Δ[C]abluminal/Δt), where VA is the volume of the abluminal chamber (0.9 cm3), A is the membrane surface area (0.33 cm2), [C]luminal is the initial luminal compound concentration (μM), Δ[C]abluminal is the abluminal compound concentration (μM), and Δt is the time of the experiment (seconds).
Determination of antiviral activities of GRL-04810, GRL-05010, and DRV recovered from the brain side of the BBB assay.
Each drug that successfully crossed the brain interface in the BBB assay described above was harvested; these drugs were designated GRL-04810brain, GRL-05010brain, DRVbrain, AZTbrain, IDVbrain, and SQVbrain. The susceptibility of HIV-1LAI and HIV-1ERS104pre to GRL-04810brain, GRL-05010brain, DRVbrain, AZTbrain, IDVbrain, and SQVbrain was then determined in an MTT assay using MT-2 cells or a p24 assay employing PHA-PBMC as described in the drug susceptibility assay section. The assay was carried out using brain-side stocks of compounds diluted 10-, 100-, 1,000-, and 10,000-fold.
Structural interactions of GRL-04810 and GRL-05010 with wild-type HIV-1 protease.
Molecular models of the interactions of GRL-04810 and GRL-05010 with wild-type HIV-1 protease were generated as follows. The coordinates of the structure of a reference compound, GRL-0519, with HIV-1 protease were obtained from the Protein Data Bank (PDB code 3OK9 [http://www.rcsb.org/]). GRL-0519 shares structural similarity with GRL-04810, and its structure was modified to generate the molecular model of interactions of GRL-04810 and HIV-1 protease. The complex was energy minimized using OPLS-2005 force field as implemented in Maestro (version 9.3; Schrödinger, LLC, New York, NY). A model of the interactions of GRL-05010 with HIV-1 protease was obtained in a similar fashion using the crystal structure of darunavir with protease (PDB code 2IEN) as a reference. Visualization, analyses of the models, and figures depicting structural interactions were generated using Maestro.
RESULTS
Antiviral activity of GRL-04810 and GRL-05010 against HIV-1LAI.
We designed and synthesized GRL-04810 and GRL-05010 that contain two fluorine atoms in the bis-THF moiety (Fig. 1) and examined their antiviral activities against a variety of HIV-1 isolates. We found that GRL-04810 and GRL-05010 were highly active in vitro against a wild-type laboratory HIV-1 strain, HIV-1LAI, with EC50s of 0.0008 and 0.003 μM, respectively, as examined using an MTT assay with MT-2 as target cells, while FDA-approved PIs (SQV, LPV, ATV, APV, and DRV) displayed EC50s ranging from 0.005 to 0.03 μM (Table 1). Cytotoxicity was seen for GRL-04810 and GRL-05010 only at high concentrations, with CC50 values of 17.5 and 37.0 μM; the selectivity index proved to be high for GRL-04810 at 21,875, while GRL-05010 scored a moderate selectivity index of 12,333 (Table 1).
Fig 1.

Structures of GRL-04810, GRL-05010, and darunavir. MW, molecular weight.
Table 1.
Antiviral activity of GRL-04810 and GRL-05010 against HIV-1LAI and their cytotoxicitiesa
| Compound | EC50 (μM) | CC50 (μM) | Selectivity indexb |
|---|---|---|---|
| SQV | 0.021 ± 0.001 | 17.7 ± 3.4 | 843 |
| LPV | 0.020 ± 0.001 | 26.8 ± 0.9 | 1,340 |
| ATV | 0.005 ± 0.001 | 28.6 ± 0.9 | 5,720 |
| APV | 0.03 ± 0.001 | >100 | >3,333 |
| DRV | 0.005 ± 0.001 | >100 | >20,000 |
| GRL-04810 | 0.0008 ± 0.0002 | 17.5 ± 0.9 | 21,875 |
| GRL-05010 | 0.003 ± 0.001 | 37.0 ± 0.4 | 12,333 |
MT-2 cells (104/ml) were exposed to 100 TCID50 of HIV-1LAI and cultured in the presence of various concentrations of each PI, and the EC50s were determined by MTT assay. All assays were conducted in duplicate, and the data shown represent mean values derived from the results of three independent experiments.
Each selectivity index denotes a ratio of the CC50 to EC50 against HIV-1LAI.
GRL-04810 and GRL-05010 exert efficacious activity against highly PI-resistant clinical HIV-1 isolates.
In our previous work, we isolated highly multi-PI-resistant primary HIV-1 strains, HIV-1MDR/B (where MDR is multidrug resistant), HIV-1MDR/C, HIV-1MDR/G, HIV-1MDR/TM, HIV-1MDR/JSL, and HIV-1MDR/MM, from patients with AIDS who had failed then-existing anti-HIV regimens after receiving 9 to 11 anti-HIV-1 drugs over 32 to 83 months (35). These primary strains contained 9 to 14 amino acid substitutions in the protease-encoding region that have reportedly been associated with HIV-1 resistance against various PIs (substitutions are identified in the footnote of Table 2). The EC50s of SQV, LPV, ATV, and APV against clinical multidrug-resistant HIV-1 strains were significantly higher than those against a wild-type clinical HIV-1 isolate, HIV-1ERS104pre, as examined in the assay employing PHA-PBMC as target cells and using p24 production inhibition as an endpoint. However, GRL-04810 and GRL-05010 exerted efficient antiviral activities, and their EC50s against those clinical variants were substantially low, varying from 0.002 μM to 0.021 μM (Table 2). GRL-04810 and GRL-05010 proved to be more active against all six multidrug-resistant clinical HIV-1 variants examined than all of the currently available approved PIs examined. The activities of the two compounds were comparable to or greater than the activity of DRV against the variants (Table 2).
Table 2.
Antiviral activity of GRL-04810 and GRL-05010 against multidrug-resistant clinical isolates in PHA-PBMC
| Virus (tropism)a | EC50 (μM)b |
||||||
|---|---|---|---|---|---|---|---|
| SQV | LPV | ATV | APV | DRV | GRL-04810 | GRL-05010 | |
| HIV-1ERS104pre (X4) | 0.0039 ± 0.0001 | 0.033 ± 0.003 | 0.0021 ± 0.0001 | 0.0295 ± 0.0004 | 0.004 ± 0.001 | 0.0023 ± 0.0001 | 0.0027 ± 0.0003 |
| HIV-1MDR/B (X4) | 0.35 (90) ± 0.01 | >1 (>33) | 0.45 (214) ± 0.07 | 0.49 (15) ± 0.05 | 0.021 (5) ± 0.001 | 0.014 (7) ± 0.001 | 0.011 (3) ± 0.001 |
| HIV-1MDR/C (X4) | 0.31 (78) ± 0.02 | >1 (>33) | 0.43 (204) ± 0.01 | 0.21 (7) ± 0.02 | 0.005 (1) ± 0.001 | 0.002 (1) ± 0.001 | 0.002 (1) ± 0.001 |
| HIV-1MDR/G (X4) | 0.039 (10) ± 0.002 | >1 (>33) | 0.042 (19) ± 0.001 | 0.31 (11) ± 0.08 | 0.014 (4) ± 0.009 | 0.004 (2) ± 0.001 | 0.004 (1) ± 0.001 |
| HIV-1MDR/TM (X4) | 0.10 (25) ± 0.04 | >1 (>33) | 0.056 (24) ± 0.007 | 0.328 (12) ± 0.001 | 0.03 (9) ± 0.01 | 0.004 (2) ± 0.001 | 0.004 (2) ± 0.001 |
| HIV-1MDR/JSL (R5) | 0.53 (133) ± 0.01 | >1 (>33) | >1 (>476) | 0.630 (22) ± 0.009 | 0.025 (5) ± 0.002 | 0.021 (10) ± 0.004 | 0.020 (7) ± 0.0002 |
| HIV-1MDR/MM (R5) | 0.11 (27) ± 0.01 | >1 (>33) | 0.081 (38) ± 0.008 | 0.27 (9) ± 0.01 | 0.010 (3) ± 0.001 | 0.002 (1) ± 0.001 | 0.003 (1) ± 0.001 |
The amino acid substitutions identified in the protease-encoding region compared to the consensus type B sequence cited from the Los Alamos database include L63P in HIV-1ERS104pre; L10I, K14R, L33I, M36I, M46I, F53I, K55R, I62V, L63P, A71V, G73S, V82A, L90M, and I93L in HIV-1MDR/B; L10I, I15V, K20R, L24I, M36I, M46L, I54V, I62V, L63P, K70Q, V82A, and L89M in HIV-1MDR/C; L10I, V11I, T12E, I15V, L19I, R41K, M46L, L63P, A71T, V82A, and L90M in HIV-1MDR/G; L10I, K14R, R41K, M46L, I54V, L63P, A71V, V82A, L90M, and I93L in HIV-1MDR/TM; L10I, L24I, I33F, E35D, M36I, N37S, M46L, I54V, R57K, I62V, L63P, A71V, G73S, and V82A in HIV-1MDR/JSL; L10I, K43T, M46L, I54V, L63P, A71V, V82A, L90M, and Q92K in HIV-1MDR/MM. HIV-1ERS104pre served as a source of wild-type HIV-1.
The EC50 values were determined by using PHA-PBMC as target cells, and the inhibition of p24 Gag protein production by each drug was used as an endpoint. The numbers in parentheses represent the fold changes of EC50s for each isolate compared to the EC50s for wild-type HIV-1ERS104pre. All assays were conducted in duplicate or triplicate, and the data shown represent mean values (± 1 standard deviation) derived from the results of three independent experiments. PHA-PBMC were derived from a single donor in each independent experiment.
GRL-04810 and GRL-05010 are effective against PI-selected laboratory HIV-1 variants.
We also examined GRL-04810 and GRL-05010 against a variety of HIV-1NL4-3 variants selected in vitro with each of four FDA-approved PIs (SQV, LPV, ATV, and APV). Such variants were selected by propagating HIV-1NL4-3 in the presence of increasing concentrations of each PI (up to 5 μM) in MT-4 cells (31); these variants had acquired various PI resistance-associated amino acid substitutions in the protease-encoding region of the viral genomes (substitutions are identified in the footnote of Table 3). These variants were designated HIV-1SQVR5 μM, HIV-1LPVR5 μM, HIV-1ATVR5 μM, and HIV-1APVR5 μM, with the superscript indicating the drug each of the variants was selected against. Each of the variants was highly resistant to the very PI against which the variant was selected and showed significant resistance, with EC50s of >1 μM. GRL-04810 and GRL-05010 were generally as active against HIV-1SQVR5 μM, HIV-1LPVR5 μM, and HIV-1ATVR5 μM as DRV (Table 3) when the absolute EC50s were compared (Table 3). As in the case of DRV, the two compounds were less active against HIV-1APVR5μM, with EC50s of 0.43 and 0.56 μM, respectively, presumably due to their structural resemblance to APV.
Table 3.
Antiviral activity of GRL-04810 and GRL-05010 against laboratory PI-resistant HIV-1 variants and GRL-04810- and GRL-05010-exposed HIV-1 variants
| Virusa | EC50 (μM)b |
||||||
|---|---|---|---|---|---|---|---|
| SQV | LPV | ATV | APV | DRV | GRL-04810 | GRL-05010 | |
| HIV-1NL4-3 | 0.037 ± 0.002 | 0.035 ± 0.005 | 0.0047 ± 0.0001 | 0.081 ± 0.001 | 0.004 ± 0.001 | 0.0005 ± 0.0005 | 0.0037 ± 0.0001 |
| HIV-1SQVR5μM | >1 (>25) | >1 (>25) | >1 (>250) | 0.435 (5) ± 0.001 | 0.04 (10) ± 0.01 | 0.13 (260) ± 0.05 | 0.046 (13) ± 0.001 |
| HIV-1LPVR5μM | 0.025 (1) ± 0.005 | >1 (>25) | 0.033 (8) ± 0.001 | 0.033 (1) ± 0.005 | 0.032 (8) ± 0.001 | 0.03 (66) ± 0.01 | 0.03 (8) ± 0.02 |
| HIV-1ATVR5μM | 0.46 (12) ± 0.02 | >1 (>25) | > 1 (>250) | >1 (>13) | 0.05 (13) ± 0.01 | 0.02 (36) ± 0.01 | 0.04 (12) ± 0.02 |
| HIV-1APVR5μM | 0.09 (2) ± 0.04 | >1 (>25) | 0.66 ± 0.02 | >1 (>13) | 0.51 (128) ± 0.03 | 0.43 (860) ± 0.02 | 0.56 (187) ± 0.03 |
| HIV-1GRL04810RP18 | 0.032 (1) ± 0.005 | 0.37 (11) ± 0.02 | 0.325 (81) ± 0.004 | >1 (>13) | 0.041 (11) ± 0.001 | 0.033 (66) ± 0.003 | 0.039 (13) ± 0.005 |
| HIV-1GRL05010RP10 | 0.036 (1) ± 0.003 | 0.375 (11) ± 0.005 | 0.095 (24) ± 0.035 | >1 (>13) | 0.037 (10) ± 0.003 | 0.029 (58) ± 0.001 | 0.036 (10) ± 0.001 |
The amino acid substitutions identified in the protease-encoding region compared to the wild-type HIV-1NL4-3 include L10F, V32I, M46I, I54M, A71V, and I84V in HIV-1APVR5μM; L23I, E34Q, K43I, M46I, I50L, G51A, L63P, A71V, V82A, and T91A in HIV-1ATVR5μM; L10F, M46I, I54V, and V82A in HIV-1LPVR5μM; and L10I, G48V, I54V, A71V, I84V, and L90M in HIV-1SQVR5μM.
MT-4 cells (105/ml) were exposed to 100 TCID50 of each HIV-1, and the inhibition of p24 Gag protein production by each drug was used as an endpoint. The numbers in parentheses represent the fold changes of EC50s for each isolate compared to the EC50s for wild-type HIV-1NL4-3. All assays were conducted in duplicate or triplicate, and the data shown represent mean values (± 1 standard deviation) derived from the results of two or three independent experiments. In addition, GRL-04810- and GRL-05010-resistant variants selected in vitro were used for antiviral activity determination assays. Time point viruses were harvested at passages 18 and 10, respectively. Washing steps were performed to remove the remaining compounds from the viral stocks, and compound-free viruses were obtained for the experiment. Five commercially available protease inhibitors were used as controls. Absolute values are given plus the fold changes relative to the baseline EC50 for each compound. By passage 18, three amino acid substitutions A28S, L33F, and V82I were identified in HIV-1GRL04810RP18; by passage 10, M46I, I50V, N38K, and M36I were detected in HIV-1GRL05010RP10. Assays were performed in duplicate and the average values (with 1 standard deviation) are shown.
GRL-04810 and GRL-0510 are moderately active against highly DRV-resistant HIV-1 variants.
We also examined the antiviral activity of the two compounds against DRV-resistant variants, which we previously selected out in vitro against DRV (36). These variants were generated using a mixture of eight highly multi-PI-resistant clinical isolates as a starting HIV-1 population and were selected with increasing concentrations of DRV. GRL-04810 and GRL-05010 exhibited slightly decreased activity against variants selected with DRV over 10 and 20 passages (EC50s of 0.03 to 0.034 μM for the former and 0.026 to 0.043 μM for the latter) (see Table S1 in the supplemental material), while DRV was less active against HIV-1DRVRP20 than the two compounds, with an EC50 of 0.174 μM (see Table S1).
In vitro selection of HIV-1 variants resistant to GRL-04810 and GRL-05010.
We attempted to select HIV-1 variants with GRL-04810 and GRL-05010 by propagating a laboratory HIV-1 strain, HIV-1NL4-3, in MT-4 cells in the presence of increasing concentrations of each of the two drugs, as previously described (31). HIV-1NL4-3 was exposed to GRL-04810 with an initial concentration of 0.003 μM and underwent 25 passages when the virus had acquired an ability to replicate in the presence of a 26-fold-increased concentration of GRL-04810 (0.080 μM). A selection assay was also carried out for GRL-05010 starting at 0.003 μM. (Fig. 2), and HIV-1NL4-3 attained an ability to replicate in the presence of 0.037 μM GRL-05010 by passage 15.
Fig 2.
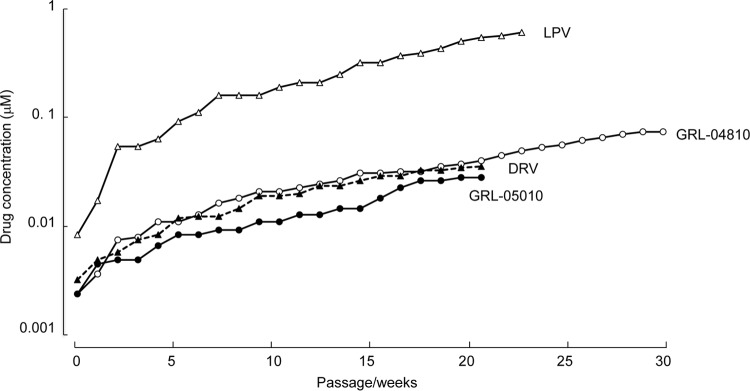
Selection of in vitro variants resistant to GRL-04810 and GRL-05010. MT-4 cells were exposed to the wild-type strain HIV-1NL4-3 and cultured in the presence of increasing concentrations of GRL-04810, GRL-05010, DRV, and LPV. Culture supernatants were collected weekly, and if the amounts of p24 Gag were found to surpass the cutoff value of 200 ng/ml, those supernatants were used for the subsequent passage. Each passage was conducted in a cell-free manner.
Judging from the amounts of p24 Gag protein secreted into the culture medium, the replicative capacity of HIV-1NL4-3 at passages 25 and 15 for GRL-04810 and GRL-05010, respectively, was generally maintained. We compared whether the emergence of resistance-associated amino acid substitutions in GRL-04810- and GRL-05010-exposed HIV-1NL4-3 was delayed in comparison with the emergence of resistant variants against two commercially available FDA-approved PIs (LPV and DRV). The protease-encoding region of proviral DNA isolated from MT-4 cells was cloned and sequenced at passages 5, 10, 15, and 20 during the GRL-04810 selection and at passages 5, 10, and 15 for GRL-05010. HIV-1NL4-3 exposed to GRL-04810 by passage 20 had acquired L33F and V82I in 15 of 22 clones and A28S in 5 clones. HIV-1NL4-3 exposed to GRL-05010 had acquired amino acid substitutions M46I and I50V by passage 15 in all 24 clones examined, while N38K was detected in 8 of 24 clones, and M36I was found in 3 clones (Fig. 3).
Fig 3.

Amino acid sequences of the protease-encoding region of HIV-1 variants selected in the presence of GRL-04810 and GRL-05010. Shown are the amino acid sequences deduced from the nucleotide sequences of the protease-encoding region of proviral DNA isolated from HIV-1NL4-3 variants selected in the presence of GRL-04810, GRL-05010, and DRV at passages 20, 15, and 20, respectively. Identified amino acid substitutions in the protease-encoding region and their frequencies (far-right column) are shown. The amino acid sequence of the wild-type HIV-1NL4-3 protease (pNL4-3 PR) is shown at the top as a reference.
GRL-04810- and GRL-05010-resistant HIV-1NL4-3 variants maintain robust replicative activity.
We determined the replication kinetics of HIV-1GRL-04810RP18, HIV-1GRL-05010RP10, and the wild-type HIV-1NL4-3 using replication kinetics assays as previously described (31). HIV-1NL4-3 replicated well in the presence of 0.001 μM each of the two compounds, but was delayed in starting to replicate or failed to replicate in the presence of 0.01 μM each compound in the 7-day replication kinetics assay (Fig. 4). However, HIV-1GRL-04810RP18 and HIV-1GRL-05010RP10 robustly replicated in the presence of 0.001 and 0.01 μM each of the two compounds (Fig. 4).
Fig 4.

Replication kinetics of GRL-04810- and GRL-05010-resistant HIV-1 variants generated in vitro. GRL-04810- and GRL-05010-resistant viruses were obtained from the selection assay illustrated in Fig. 2. Two time point passages, HIV-1GRL-04810RP18 and HIV-1GRL-05010RP10, were chosen. HIV-1NL4-3 or two resistant viruses were propagated in CD4+ MT-4 cells in the presence of 0.01 μM (▲) and 0.001 μM (■) GRL-04810 and GRL-05010 or in the absence (◆) of the compounds in culture flasks. The amounts of p24 in each culture flask were quantified every 2 days for 1 week. Sup, supernatant.
GRL-04810 and GRL-05010 remain active against the variants selected with GRL-04810 and GRL-05010.
We then attempted to determine the impact of the amino acid substitutions identified in HIV-1GRL-04810RP18 and HIV-1GRL-05010RP10. Five commercially available protease inhibitors (SQV, LPV, ATV, APV, and DRV) were used as controls. GRL-04810 and GRL-05010 generally remained active against HIV-1GRL-04810RP18 and HIV-1GRL-05010RP10, with EC50s ranging from 0.029 μM to 0.039 μM. SQV was active against the two variants HIV-1GRL-04810RP18 and HIV-1GRL-05010RP10, with narrow EC50 ranges of 0.032 to 0.036 μM, while LPV displayed values significantly higher (0.37 and 0.375 μM) than those of GRL-04810 and GRL-05010. ATV was moderately active against the HIV-1GRL-05010RP10 variant (0.095 μM), while the absolute EC50s of GRL-04810 and GRL-05010 were generally lower than the EC50s of ATV against those variants. As for DRV, the values against the variants resistant to GRL-04810 and GRL-05010 were 0.041 and 0.037 μM, respectively. However, APV was not active against any of the selected variants examined (EC50s of >1 μM), presumably due to its structural resemblance to the two PIs used for the selection (Table 3).
GRL-04810 and GRL-05010 show favorable lipophilicity indexes for their partition and distribution coefficients.
The addition of fluorine atoms is generally expected to confer greater lipophilicity on nucleoside analogs and certain compounds (37–39). Thus, we determined the partition (log P) and distribution (log D) coefficients of GRL-04810 and GRL-05010. 1-Octanol, organic alcohol, and water were used for log P determination, while Tris-buffered saline (pH 7.4) and water were utilized for log D determination. Prior to obtaining the actual values, a standard curve was generated as a reference, and drug concentrations attained for each compartment (1-octanol, water, and Tris-buffered saline) were measured on a light spectrophotometer (at an absorbance of 230 nm) as previously described by Rak et al. (40). Of note, GRL-04810 reached the highest concentration in the octanol lipid interface (72 μM) versus 14.09 μM for GRL-05010 and 11.89 μM for DRV (Table 4). GRL-04810 appeared the most lipophilic, with a log D value of −0.29, representing a 4-fold greater log D value than that of DRV (−1.03), since the assumption is that the more negative the log D value is, the less lipophilic the substance is estimated to be (40). GRL-05010 was found to have a log D value (−1.01) comparable to that of DRV.
Table 4.
Partition and distribution coefficients of GRL-04810, GRL-05010, and DRV using the shake flask method
| Compound | Concn (μM) in: |
Log Poctanol/watera | Log Doctanol/Trisa | |||
|---|---|---|---|---|---|---|
| Water | Tris buffer | n-Octanol | n-Octanol | |||
| GRL-04810 | 99.78 | 42.32 | 70.00 | 72.00 | −0.14 | −0.29 |
| GRL-05010 | 94.55 | 49.84 | 18.70 | 14.09 | −0.83 | −1.01 |
| DRV | 81.49 | 48.77 | 15.50 | 11.89 | −0.63 | −1.03 |
The partition (log P) and distribution (log D) coefficients of GRL-04810 and GRL-05010 were determined. DRV was used as a control. n-Octanol, an organic alcohol, and water were used for log P determination, while Tris buffer (pH 7.40) and octanol were utilized for the log D assay. Prior to the retrieval of actual values, a standard curve was generated as a reference. The drug concentrations for each compartment (octanol, water, and Tris buffer) were measured at an absorbance of 230 using a light spectrophotometer. Assays were performed following the OECD guidelines for testing of chemicals (55). Log P and log D values were calculated according to the formulas given in Materials and Methods.
GRL-04810 and GRL-05010 penetrate well across the blood-brain barrier in vitro.
We also attempted to evaluate whether GRL-04810 and GRL-05010 had optimal blood-brain barrier (BBB) apparent permeability coefficients by employing an in vitro model using a triple cell coculture system with rat astrocytes, pericytes, and monkey endothelial cells. This model (BBB Kit) is thought to represent an in vitro BBB model for drug transport assays, permitting adequate cross talk of the cell lines involved and providing a way to test apparent passage of small molecules across the BBB, as previously described by Nakagawa et al. (41). AZT, IDV, SQV, LPV, ATV DRV, GRL-04810, or GRL-05010 was added to the luminal interface (termed the blood side) of microtiter culture wells under the optimal conditions for transendothelial electrical resistance (TEER) determination. The concentrations of each compound that permeated into the abluminal interface (termed the brain side) were determined using a spectrophotometer 30 min after the addition of each drug to the wells. As shown in Table 5, the amounts of caffeine and sucrose, serving as the most and least lipophilic substances, in the abluminal interface were 6.60 and 0.03 μM, respectively. AZT, IDV, SQV, LPV, ATV, and DRV were also used as controls in the assay, giving amounts of 1.50, 2.42, 0.33, 0.94, 1.02, and 0.65 μM, respectively. In contrast, GRL-04810 and GRL-05010 yielded greater concentrations, with 3.16 and 4.08 μM in the abluminal interface of the microtiter culture wells.
Table 5.
Estimation of the apparent blood brain barrier permeability coefficient using an in vitro modela
| Compound | Initial luminal tracer concn (μM) | Final abluminal tracer concn (μM) | Papp (10−6 cm/s) |
|---|---|---|---|
| AZT | 100 | 1.50 ± 0.12 | 22.7 ± 1.9 |
| IDV | 100 | 2.42 ± 0.12 | 36.7 ± 1.7 |
| SQV | 100 | 0.33 ± 0.03 | 4.9 ± 0.4 |
| LPV | 100 | 0.94 ± 0.05 | 14.2 ± 0.7 |
| ATV | 100 | 1.02 ± 0.10 | 15.4 ± 1.4 |
| DRV | 100 | 0.65 ± 0.23 | 9.9 ± 4.2 |
| GRL-04810 | 100 | 3.16 ± 0.48 | 47.8 ± 8.8 |
| GRL-05010 | 100 | 4.08 ± 0.65 | 61.8 ± 12.1 |
| Caffeine | 100 | 6.60 | 100 |
| Sucrose | 100 | 0.03 ± 0.005 | 0.33 ± 0.13 |
In the in vitro model using a triple coculture of rat astrocytes, pericytes, and monkey endothelial cells, AZT, IDV, SQV, LPV, ATV, DRV, GRL-04810, GRL-05010 (all 100 μM), and the positive and negative controls, caffeine and sucrose, respectively, were added to the luminal interface (termed blood side) of duplicate wells. The mathematical formula used for the calculation of Papp is described in Materials and Methods. Results show average values ±1 standard deviation of duplicate determinations.
The apparent permeability coefficient (Papp), referred to as a brain uptake index (BUI), is a way to determine the penetration efficiency of a compound across a BBB model quantitatively and qualitatively (42). The Papp values of GRL-04810 (47.8 × 10−6 cm/s) and GRL-05010 (61.8 × 10−6 cm/s) were significantly greater than the Papp of DRV (9.9 × 10−6 cm/s) and the Papp values of other antiviral drugs tested: AZT (22.7 × 10−6 cm/s), IDV (36.7 × 10−6 cm/s), SQV (4.9 × 10−6 cm/s), LPV (14.2 × 10−6 cm/s), and ATV (15.4 × 10−6 cm/s). Compounds with apparent permeability coefficients greater than 20 × 10−6 cm/s are thought to have reasonably efficient penetration across the BBB; those with values of 10 × 10−6 to 20 × 10−6 cm/s have a moderate degree of penetration, whereas those with values lower than 10 × 10−6 cm/s do not penetrate the BBB well (41).
GRL-04810 and GRL-05010 recovered from the brain interface in the BBB model retain antiviral activity greater than that of DRV.
Finally, we attempted to examine whether the drug concentrations penetrated into the abluminal interface (brain side) in the BBB Kit as described above had sufficient activity to suppress the replication of HIV-1 in vitro. Each drug that successfully crossed the brain interface in the BBB assay was harvested; these drugs were designated GRL-04810brain, GRL-05010brain, DRVbrain, AZTbrain, IDVbrain, and SQVbrain. The susceptibility of HIV-1LAI and HIV-1ERS104pre to GRL-04810brain, GRL-05010brain, DRVbrain, AZTbrain, IDVbrain, and SQVbrain was then determined in MTT assays using MT-2 cells or in p24 assays employing PHA-PBMC as described in the drug susceptibility assay section. In the assay, we used 10-fold serial dilutions of the original brain side preparation containing GRL-04810 (3.16 μM) (designated GRL-04810brain) and determined antiviral suppression levels. The 10-fold-diluted GRL-04810brain (concentration, 0.316 μM) suppressed HIV-1LAI replication by 94%, whereas 100-fold-diluted GRL-04810brain (concentration, 0.0316 μM) suppressed replication by 45%. The 10-fold-diluted GRL-05010brain (concentration, 0.408 μM) suppressed HIV-1LAI replication by 91%, whereas 100-fold-diluted GRL-05010brain (concentration, 0.0316 μM) suppressed replication by 73%. However, the suppression levels of all of the other preparations examined (DRVbrain, AZTbrain, IDVbrain, and SQVbrain) were less than those of GRL-04810brain and GRL-05010brain (Fig. 5). When PHA-activated PBMC and the treatment-naive clinical isolate HIV-1ERS104pre were used in the drug susceptibility assay, virtually the same antiviral profiles of GRL-04810brain and GRL-05010brain were observed (Fig. 6).
Fig 5.

Antiviral activity of GRL-04810brain, GRL-05010brain, DRVbrain, AZTbrain, IDVbrain and SQVbrain against HIV-1LAI. Compounds recovered from the brain side of the medium in the BBB in vitro assay were challenged with HIV-1LAI, as indicated, in a p24 assay using PHA-PBMC. Dilution ranges were between 10-fold and 10,000-fold, and the percentage of viral inhibition was used as an endpoint. It is assumed that the greater the percentage of inhibition of HIV-1LAI, the greater the concentration of the drug in the brain side medium. Assays were conducted in duplicate, and error bars show standard deviations.
Fig 6.

Antiviral activity of GRL-04810brain, GRL-05010brain, DRVbrain, AZTbrain, IDVbrain and SQVbrain against HIV-1ERS104pre. Compounds recovered from the brain side of the medium in the BBB in vitro assay were challenged with HIV-1ERS104pre, as indicated, in a p24 assay using PHA-PBMC. Dilution ranges were between 10-fold and 10,000-fold, and the percentage of viral inhibition was used as an endpoint. It is assumed that the greater the percentage of inhibition of HIV-1ERS104pre, the greater the concentration of the drug in the brain side medium. Assays were conducted in duplicate, and error bars show standard deviations.
Molecular interactions of GRL-04810 and GRL-05010 with wild-type HIV-1 protease.
We generated molecular models of the interactions of GRL-04810 and GRL-05010 with wild-type HIV-1 protease. A bird's eye view of GRL-04810 bound to protease is shown in Fig. 7A. The interactions of DRV with HIV-1 protease seen in a crystal structure (Fig. 7B) share key interactions with both GRL-04810 and GRL-05010 as follows. Analysis of the model structure revealed that both oxygen atoms present in the bis-THF group of GRL-04810 have polar interactions with the backbone nitrogens of Asp29 and Asp30 in the S2 site of the protease (Fig. 7C). The O-methoxy oxygen has a polar interaction with the backbone NH of Asp30′ in the S2′ site of the protease. GRL-04810 maintains polar interactions with Gly27 and Asp25, as well as with Ile50 and Ile50′, through a bridging water molecule. The oxygens in the bis-THF group of GRL-05010 also exhibit polar interactions with Asp29 and Asp30 in the S2 site of protease (Fig. 7D). Polar interactions with Gly27, the catalytic Asp25, and the bridging water molecule are also seen for GRL-05010. Even though both GRL-04810 and GRL-05010 form polar interactions with Asp30′ in the S2′ site of the protease, there is a subtle difference due to the different chemical moieties present in the P2′ sites of these inhibitors. The O-methoxy oxygen forms a polar contact with the backbone NH of Asp30′, while the NH nitrogen of GRL-05010 forms a polar contact with the backbone carboxyl of Asp30′. The fluoride atoms in the bis-THF group of GRL-04810 and GRL-05010 fill the otherwise empty cavity toward the HIV-1 protease flap.
Fig 7.

Structural interactions of GRL-04810 and GRL-05010 with HIV-1 protease. A model of GRL-04810 bound to protease is shown (A). GRL-04810 is shown in thick sticks, and residues in the protease active site are shown in wire. The following atom colors are used: carbons in gray, oxygens in red, fluorines in green, nitrogens in blue, and sulfurs in yellow. The polar interactions (yellow dotted lines) of DRV, GRL-04810, and GRL-05010, along with other residues in the active site, are shown in panels B, C, and D, respectively. Nonpolar hydrogens are not shown. Figures were generated using Maestro, version 9.3. (Schrödinger, LLC, New York, NY).
DISCUSSION
DRV is an HIV-1 protease inhibitor which was most recently added to the armamentarium of antiretroviral therapeutics. DRV exerts highly effective activity against a wide spectrum of multidrug-resistant HIV-1 variants including multi-PI-resistant variants and has been shown to resist the emergence of DRV-resistant HIV-1 strains in vitro (36, 43) and in the clinical setting (44–46). The mechanisms of the favorable antiretroviral activity and delayed emergence of DRV-resistant strains include the presence of a unique moiety, bis-THF, in the P2′ site and DRV's dual action to block HIV-1 protease's enzymatic activity (28, 47) and protease's dimerization (48). However, the penetration of DRV into the cerebrospinal fluid (CSF) is only moderate, with a ratio of DRV concentration in the CSF to that in peripheral blood of 0.6% (49), probably due to the only moderate penetration of DRV through the blood-brain barrier. The addition of fluorine atoms to nucleoside analogs is known to increase lipophilicity and possibly enhance CSF penetration (37, 39). Thus, in the present study, we newly designed, synthesized, and characterized two PIs, GRL-04810 and GRL-05010, which contain a bis-THF moiety to which a difluoride group is attached.
GRL-04810 and GRL-05010 suppressed various HIV-1 isolates, including multidrug-resistant clinical HIV-1 isolates with reasonably low EC50s and favorable cytotoxicity profiles although, as in the case of DRV, both PIs failed to efficiently block the replication of APV-resistant strains. To our knowledge, the suppression failure of APV-resistant variants has largely been seen in all of the bis-THF-containing PIs thus far published (28, 31–33). The reduced activity of the bis-THF-containing PIs to block the replication of APV-resistant variants is likely due to the structural similarity between the bis-THF-containing PIs, including GRL-04810, GRL-05010, and DRV.
In the present wild-type HIV-1NL4-3 selection experiment with the two PIs, the development of resistance against each compound exhibited different patterns from those of the two control PIs, LPV and DRV. In the case of GRL-05010, by passage 10, two PI resistance-associated amino acid substitutions (M46I and I50V) were identified, while HIV-1NL4-3 exposed to GRL-04810 acquired three amino acid substitutions, A28S, L33F, and V82I, by passage 20 in 5 of 22 clones examined (Fig. 3). The emergence of the A28S substitution has previously been documented as a resistance-conferring amino acid substitution when HIV-1NL4-3 was selected with two bis-THF-containing PIs, TMC-126 (35) and GRL-1398 (32), both of which also contain a paramethoxy group in the P2′ site. Intriguingly, when HIV-1NL4-3 was selected with GRL-0519, which contains the same paramethoxy group in the P2′ site, the A28S substitution never appeared by up to passage 37 (50). GRL-0519 has tris-tetrahydrofuranylurethane (tris-THF) as the P2 ligand and the paramethoxy moiety at the P2′ site, suggesting that the presence of tris-THF prevented the selection of the A28S substitution. Indeed, to our knowledge, even with long-term administration of DRV, HIV-1 containing the A28S substitution has not been documented in the clinical settings (51). Considering that the selection of HIV-1NL4-3 and even a mixture of eight multi-PI-resistant clinical strains with DRV failed to select out the A28S substitution (36) and that only GRL-04810 selection of HIV-1NL4-3 resulted in the emergence of the A28S substitution in the present study, the combination of bis-THF as the P2 ligand and the paramethoxy moiety at the P2′ site seems to be associated with the emergence of the A28S substitution. As illustrated in Fig. 3, it is of note that the emergence of 2 to 3 amino acid substitutions known to be associated with HIV-1's acquisition of PI resistance in the selection assays with the two novel PIs and no emergence of amino acid substitutions in the selection with DRV may possibly pose a disadvantage for GRL-04810 and GRL-05010 with respect to DRV.
Zidovudine (ZDV) is the only AIDS-associated retrovirus (ARV) agent with demonstrated efficacy in the treatment of HIV-1-associated dementia. Considering that HIV-1 causes CNS diseases ranging from HIV-1-associated neurocognitive disorders (HAND) and the milder, but also serious, presentation, HIV-1-associated minor cognitive/motor disorder (MCMD), to the devastating HIV-1-associated encephalopathy, more effective ARV regimens with agents exerting maximal penetration of the BBB are urgently needed. It is well established that the lipophilicity of a molecule is one of the crucial determinants of the molecule's drug-likeness, including the absorption through the digestive tract, penetration of the target cells, oral bioavailability, and penetration through the BBB (37, 39). The shake flask method represents a reasonable way to determine the partition and distribution coefficients of molecules to be tested. The partition (log P) coefficient is an estimate of a compound's overall lipophilicity, which is associated with the compound's solubility, permeability through biological membranes, hepatic clearance, lack of selectivity, and nonspecific toxicity (52). In the present study, the log P determination of GRL-04810 gave a higher concentration in the octanol (lipidic) interface than GRL-05010 and DRV (Table 4). To estimate the actual figures for the ionized form of the drugs (log D) Tris-buffered saline (pH 7.4) must be employed. In both assays the values displayed by both GRL-05010 and DRV were within the acceptable range for the optimal lipophilicity of drug candidates (53, 54).
Cell culture-based models have greatly contributed to the understanding of the physiology, pathology, and pharmacology of the blood-brain barrier (41). Indeed, certain in vitro BBB models have proven to serve as useful tools that permit the estimation of the apparent penetration of molecules into the CNS. GRL-04810 and GRL-05010 gave reasonably favorable indexes, suggesting potentially favorable penetration ability across the BBB compared to DRV and other anti-HIV-1 drugs examined in the present study, including AZT, IDV, SQV, LPV, and ATV.
A well-characterized in vitro BBB cell model can also provide a valuable tool for studying mechanistic aspects of transport as well as biological and pathological processes related to the BBB (42). For any in vitro BBB cell model to be employed successfully, it needs to fulfill a number of criteria, such as reproducible permeability of reference compounds, good screening capacity, the display of complex tight junctions, adequate expression of BBB phenotypic transporters, and transcytotic activity. The BBB model employed in the present study complies with all of these parameters and provides an additional advantage as it incorporates a trilayer of cells consisting of astrocytes, pericytes, and brain endothelial cells, thus increasing its anatomical and physiological reliability. Molecules and compounds that reach Papp values greater than 20 × 10−6 cm/s are deemed to be favorable in terms of relative penetration across the BBB. Conversely, those that display values between 2 × 10−6 cm/s and 10 × 10−6 cm/s are defined as compounds with low BBB penetration. In the present work, both GRL-05010 and GRL-04810 showed the highest values of Papp among the protease inhibitors examined, with values of 47.8 × 10−6 and 61.8 × 10−6 cm/s, respectively (Table 5).
When we asked whether GRL-04810 and GRL-05010 harvested from the brain interface in the BBB model retained antiviral activity, both the recovered GRL-04810 and GRL-05010 (termed GRL-04810brain and GRL-05010brain) significantly more profoundly suppressed the replication of HIV-1LAI and HIV-1ERS104pre than other antiretroviral agents including DRV (Fig. 5 and 6), strongly suggesting that both GRL-04810 and GRL-05010 that crossed into the brain interface in the BBB assay were molecularly intact and exerted robust anti-HIV-1 activity.
The present data suggest that GRL-04810 and GRL-05010 have several advantages: (i) they exert efficient activity against a wide spectrum of drug-resistant HIV-1 variants, presumably due to their interactions with the main chains of the active-site amino acids Asp29 and Asp30, (ii) they have a good lipophilicity profile as expressed by the log D values, and (iii) they have apparently favorable penetration across the BBB. However, their oral bioavailability, pharmacokinetics/pharmacodynamics, and volume of distribution, among other parameters, are yet to be determined in further rigorous preclinical and clinical testing.
Supplementary Material
ACKNOWLEDGMENTS
The present work was supported in part by a grant for global education and a research center aiming at the control of AIDS (Global Center of Excellence supported by Monbu-Kagakusho), the Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan, a grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Re-emerging Infectious Diseases (Renkei Jigyo: number 78, Kumamoto University) of Monbu-Kagakusho (H.M.), the Intramural Research Program of Center for Cancer Research, National Cancer Institute, National Institutes of Health (H.M.), and a grant from the National Institutes of Health (GM53386 to A.K.G.).
Our appreciation also goes to Yasuhiro Koh and Yasushi Tojo for their valuable suggestions during this work.
Footnotes
Published ahead of print 30 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01420-13.
REFERENCES
- 1. Coiras M, Lopez-Huertas M, Perez-Olmeda M, Alcami J. 2009. Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nat. Rev. Microbiol. 7:798–812 [DOI] [PubMed] [Google Scholar]
- 2. De Clercq. 2002. New anti-HIV agents and targets. Med. Res. Rev. 22:531–565 [DOI] [PubMed] [Google Scholar]
- 3. Gorantla S, Poluektova L, Gendelman HE. 2012. Rodent models for HIV-associated neurocognitive disorders. Trends Neurosci. 35:197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lewin SR, Murray J, Solomon A, Wightman F, Cameron PU, Purcell DJ, Zaunders JJ, Grey P, Bloch M, Smith D, Cooper DA, Kelleher AD. 2008. Virological determinants of success after structured treatment interruptions of antiretrovirals in acute HIV-1 infection. J. Acquir. Immune Defic. Syndr. 47:140–147 [PubMed] [Google Scholar]
- 5. Siliciano JD, Siliciano RF. 2004. A long-term latent reservoir for HIV-1: discovery and clinical implications. J. Antimicrob. Chemother. 54:6–9 [DOI] [PubMed] [Google Scholar]
- 6. Simon V, Ho DD. 2003. HIV-1 dynamics in vivo: implications for therapy. Nat. Rev. Microbiol. 1:181–190 [DOI] [PubMed] [Google Scholar]
- 7. Carr 2003. Toxicity of antiretroviral therapy and implications for drug development. Nat. Rev. Drug Discov. 2:624–634 [DOI] [PubMed] [Google Scholar]
- 8. Fumero E, Podzamczer D. 2003. New patterns of HIV-1 resistance during HAART. Clin. Microbiol. Infect. 9:1077–1084 [DOI] [PubMed] [Google Scholar]
- 9. Grabar S, Abraham B, Mahamat A, Del Giudice P, Rosenthal E, Costagliola D. 2006. Differential impact of combination antiretroviral therapy in preventing Kaposi's sarcoma with and without visceral involvement. J. Clin. Oncol. 24:3408–3414 [DOI] [PubMed] [Google Scholar]
- 10. Hirsch HH, Kaufmann G, Sendi P, Battegay M. 2004. Immune reconstitution in HIV-infected patients. Clin. Infect. Dis. 38:1159–1166 [DOI] [PubMed] [Google Scholar]
- 11. Little SJ, Holte S, Routy JP, Daar ES, Markowitz M, Collier AC, Koup RA, Mellors JW, Connick E, Conway B, Kilby M, Wang L, Whitcomb JM, Hellmann NS, Richman DD. 2002. Antiretroviral-drug resistance among patients recently infected with HIV. N. Engl. J. Med. 347:385–394 [DOI] [PubMed] [Google Scholar]
- 12. Enting RH, Hoetelmans R, Lange JM, Burger DM, Beijnen JH, Portegies P. 1998. Antiretroviral drugs and the central nervous system. AIDS 12:1941–1955 [DOI] [PubMed] [Google Scholar]
- 13. Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. 2005. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 111:194–213 [DOI] [PubMed] [Google Scholar]
- 14. McArthur JC, Brew BJ, Nath A. 2005. Neurological complications of HIV infection. Lancet Neurol. 4:543–555 [DOI] [PubMed] [Google Scholar]
- 15. Langford D, Marquie-Beck J, de Almeida S, Lazzaretto D, Letendre S, Grant I, McCutchan JA, Masliah E, Ellis RJ. 2006. Relationship of antiretroviral treatment to postmortem brain tissue viral load in human immunodeficiency virus-infected patients. J. Neurovirol. 12:100–107 [DOI] [PubMed] [Google Scholar]
- 16. Nath A, Sacktor N. 2006. Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Curr. Opin. Neurol. 19:358–361 [DOI] [PubMed] [Google Scholar]
- 17. Potter MC, Figuera-Losada M, Rojas C, Slusher BS. 2013. Targeting the glutamatergic system for the treatment of HIV-associated neurocognitive disorders. J. Neuroimmune Pharmacol. 8:594–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sacktor 2002. The epidemiology of human immunodeficiency virus-associated neurological disease in the era of highly active antiretroviral therapy. J. Neurovirol. 8:115–121 [DOI] [PubMed] [Google Scholar]
- 19. Tan IL, McArthur JC. 2012. HIV-associated neurological disorders: a guide to pharmacotherapy. CNS Drugs 26:123–134 [DOI] [PubMed] [Google Scholar]
- 20. Smit TK, Brew BJ, Tourtellotte W, Morgello S, Gelman BB, Saksena NK. 2004. Independent evolution of human immunodeficiency virus (HIV) drug resistance mutations in diverse areas of the brain in HIV-infected patients, with and without dementia, on antiretroviral treatment. J. Virol. 78:10133–10148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gisolf EH, Van Praaq R, Jurriaans S, Portegies P, Goudsmit J, Danner SA, Lange JM, Prins JM. 2000. Increasing cerebrospinal fluid chemokine concentrations despite undetectable cerebrospinal fluid HIV RNA in HIV-1-infected patients receiving antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 25:426–433 [DOI] [PubMed] [Google Scholar]
- 22. Lipton SA, Brenneman Silverstein DFS, Masliah E, Mucke L. 1995. gp120 and neurotoxicity in vivo. Trends Pharmacol. Sci. 16:122. [DOI] [PubMed] [Google Scholar]
- 23. Neuenburg JK, Brodt HR, Herndier BG, Bickel M, Bacchetti P, Price RW, Grant RM, Schlote W. 2002. HIV-related neuropathology, 1985 to 1999: rising prevalence of HIV encephalopathy in the era of highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 31:171–177 [DOI] [PubMed] [Google Scholar]
- 24. Schrager LK, D'Souza MP. 1998. Cellular and anatomical reservoirs of HIV-1 in patients receiving potent antiretroviral combination therapy. JAMA 280:67–71 [DOI] [PubMed] [Google Scholar]
- 25. Kaul M, Garden G, Lipton SA. 2001. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410:988–994 [DOI] [PubMed] [Google Scholar]
- 26. Ghosh AK, Krishnan K, Walters DE, Cho W, Cho H, Koo Y, Trevino J, Holland L, Buthod J. 1998. Potent HIV protease inhibitors incorporating high-affinity P2-ligands and (R)-(hydroxyethylamino)sulfonamide isostere. Bioorg. Med. Chem. Lett. 8:687–690 [DOI] [PubMed] [Google Scholar]
- 27. Ghosh AK, Cho W, Walters DE, Krishnan K, Hussain KA, Koo Y, Cho H, Rudall C, Holland L, Buthod J. 1998. Structure based design: novel spirocyclic ethers as nonpeptidal P2-ligands for HIV protease inhibitors. Bioorg. Med. Chem. Lett. 8:979–982 [DOI] [PubMed] [Google Scholar]
- 28. Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. 2003. Novel bis-tetrahydro-furanylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 47:3123–3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shirasaka T, Chokekijchai S, Yamada A, Gosselin G, Imbach JL, Mitsuya H. 1995. Comparative analysis of anti-human immunodeficiency virus type 1 activities of dideoxynucleoside analogs in resting and activated peripheral blood mononuclear cells. Antimicrob. Agents Chemother. 39:2555–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ghosh AK, Leshchenko S, Noetzel M. 2004. Stereoselective photochemical 1,3-dioxolane addition to 5-alkoxymethyl-2(5H)-furanone: synthesis of bis-tetrahydrofuranyl ligand for HIV protease inhibitor UIC-94017 (TMC-114). J. Org. Chem. 69:7822–7829 [DOI] [PubMed] [Google Scholar]
- 31. Amano M, Koh Y, Das D, Li J, Leschenko S, Wang YF, Boross PI, Weber IT, Ghosh AK, Mitsuya H. 2007. A novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI), GRL-98065, is potent against multiple-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 51:2143–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ide K, Aoki M, Amano M, Koh Y, Yedidi RS, Das D, Leschenko S, Chapsal B, Ghosh AK, Mitsuya H. 2011. Novel HIV-1 protease inhibitors (PIs) containing a bicyclic P2 functional moiety, tetrahydropyrano-tetrahydrofuran, that are potent against multi-PI-resistant HIV-1 variants. Antimicrob. Agents Chemother. 55:1717–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tojo Y, Koh Y, Amano M, Aoki M, Das D, Kulkarni S, Anderson DD, Ghosh AK, Mitsuya H. 2010. Novel protease inhibitors (PIs) containing macrocyclic components and 3(R),3a(S),6a(R)-bis-tetrahydrofuranylurethane that are potent against multi-PI-resistant HIV-1 variants in vitro. Antimicrob. Agents Chemother. 54:3460–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakagawa S, Deli MA, Kawaguchi H, Shimizudani T, Shimono T, Kittel A, Tanaka K, Niwa M. 2009. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem. Int. 54:253–263 [DOI] [PubMed] [Google Scholar]
- 35. Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW, Mitsuya H. 2002. A potent human immunodeficiency virus type 1 protease inhibitor, UIC-94003 (TMC-126), and selection of a novel (A28S) mutation in the protease active site. J. Virol. 76:1349–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koh Y, Amano M, Towata T, Danish M, Leshchenko-Yashchuk S, Das D, Nakayama M, Tojo Y, Ghosh AK, Mitsuya H. 2010. In vitro selection of highly darunavir-resistant and replication-competent HIV-1 variants by using a mixture of clinical HIV-1 isolates resistant to multiple conventional protease inhibitors. J. Virol. 84:11961–11969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bright TV, Dalton F, Elder VL, Murphy CD, O'Connor NK, Sandford G. 2013. A convenient medical-microbial method for developing fluorinated pharmaceuticals. Org. Biomol. Chem. 11:1135–1142 [DOI] [PubMed] [Google Scholar]
- 38. Karppi J, Akerman S, Akerman K, Sundell A, Nyyssonen K, Penttila I. 2007. Adsorption of drugs onto apH responsive poly(N,N dimethyl aminoethyl methacrylate) grafted anion-exchange membrane in vitro. Int. J. Pharm. 338:7–14 [DOI] [PubMed] [Google Scholar]
- 39. Liu P, Sharon A, Chu CK. 2008. Fluorinated nucleosides: synthesis and biological implication. J. Fluor. Chem. 129:743–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rak J, Dejlová B, Lampová H, Kaplánek R, Matĕjíček P, Cígler P, Král V. 2013. On the solubility and lipophilicity of metallacarborane pharmacophores. Mol. Pharm. 10:1751–1759 [DOI] [PubMed] [Google Scholar]
- 41. Nakagawa S, Deli MA, Nakao S, Honda M, Hayashi K, Nakaoke R, Kataoka Y, Niwa M. 2007. Pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells. Cell. Mol. Neurobiol. 27:687–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, Fenart L. 2007. Modelling of the blood brain barrier in drug discovery and development. Nat. Rev. Drug Discov. 6:650–661 [DOI] [PubMed] [Google Scholar]
- 43. de Bethune MP, Hertogs K. 2006. Screening and selecting for optimized antiretroviral drugs: rising to the challenge of drug resistance. Curr. Med. Res. Opin. 22:2603–2612 [DOI] [PubMed] [Google Scholar]
- 44. Geretti AM, Arribas J, Lathouwers E, Foster GM, Yakoob R, Kinloch S, Hill A, van Delft Y, Moecklinghoff C. 2013. Dynamics of cellular HIV-1 DNA levels over 144 weeks of darunavir/ritonavir monotherapy versus triple therapy in the MONET trial. HIV Clin. Trials 14:45–50 [DOI] [PubMed] [Google Scholar]
- 45. Orkin C, DeJesus E, Khanlou H, Stoehr A, Supparatpinyo K, Lathouwers E, Lefebvre E, Opsomer M, Van de Casteele T, Tomaka F. 2013. Final 192-week efficacy and safety of once-daily darunavir/ritonavir compared to lopinavir/ritonavir in HIV-1-infected treatment-naive patients in the ARTEMIS trial. HIV Med. 14:49–59 [DOI] [PubMed] [Google Scholar]
- 46. Banhegyi D, Katlama C, da Cunha CA, Schneider S, Rachlis A, Workman C, De Meyer S, Vandevoorde A, Van De Casteele T, Tomaka F. 2012. Week 96 efficacy, virology and safety of darunavir/r versus lopinavir/r in treatment-experienced patients in TITAN. Curr. HIV Res. 10:171–181 [DOI] [PubMed] [Google Scholar]
- 47. Ghosh AK, Xu CX, Rao KV, Baldridge A, Agniswamy J, Wang YF, Weber IT, Aoki M, Miguel SG, Amano M, Mitsuya H. 2010. Probing multidrug-resistance and protein-ligand interactions with oxatricyclic designed ligands in HIV-1 protease inhibitors. ChemMedChem 5:1850–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koh Y, Matsumi S, Das D, Amano M, Davis DA, Leschenko S, Baldridge A, Shioda T, Yarchoan R, Ghosh AK, Mitsuya H. 2007. Potent inhibition of HIV-1 replication by novel non-peptidyl small molecule inhibitors of protease dimerization. J. Biol. Chem. 282:28709–28720 [DOI] [PubMed] [Google Scholar]
- 49. Calcagno A, Yilmaz A, Cusato J, Simiele M, Bertucci R, Siccardi M, Marinaro L, D'Avolio A, Di Perri G, Bonora S. 2012. Determinants of darunavir cerebrospinal fluid concentrations: impact of once-daily dosing and pharmacogenetics. AIDS 26:1529–1533 [DOI] [PubMed] [Google Scholar]
- 50. Amano M, Tojo Y, Salcedo-Gomez PM, Campbell JR, Das D, Aoki M, Xu CX, Rao KV, Ghosh AK, Mitsuya H. 2013. GRL-0519, a novel oxatricyclic ligand-containing nonpeptidic HIV-1 protease inhibitor (PI), potently suppresses replication of a wide spectrum of multi-PI-resistant variants in vitro. Antimicrob. Agents Chemother. 57:2036–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mitsuya Y, Varghese V, Wang C, Liu TF, Holmes SP, Jayakumar P, Gharizadeh B, Ronaghi M, Klein D, Fessel WJ, Shafer RW. 2008. Minority human immunodeficiency virus type 1 variants in antiretroviral-naive persons with reverse transcriptase codon 215 revertant mutations. J. Virol. 82:10747–10755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hughes JD, Blagg J, Price DA, Bailey S, Decrescenzo GA, Devraj RV, Ellsworth E, Fobian YM, Gibbs ME, Gilles RW, Greene N, Huang E, Krieger-Burke T, Loesel J, Wager T, Whiteley L, Zhang Y. 2008. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 18:4872–4875 [DOI] [PubMed] [Google Scholar]
- 53. Ryckmans T, Edwards MP, Horne VA, Correia AM, Owen DR, Thompson LR, Tran I, Tutt MF, Young T. 2009. Rapid assessment of a novel series of selective CB2 agonists using parallel synthesis protocols: a lipophilic efficiency (LipE) analysis. Bioorg. Med. Chem. Lett. 19:4406–4409 [DOI] [PubMed] [Google Scholar]
- 54. Waring MJ. 2009. Defining optimum lipophilicity and molecular weight ranges for drug candidates—molecular weight dependent lower log D limits based on permeability. Bioorg. Med. Chem. Lett. 19:2844–2851 [DOI] [PubMed] [Google Scholar]
- 55. Organisation for Economic Cooperation and Development 1995. OECD guidelines for the testing of chemicals, section 1: physical-chemical properties. Test no. 107. Organisation for Economic Cooperation and Development, Paris, France. 10.1787/20745753 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.