

Abstract
We recently investigated the pharmacokinetics-pharmacodynamics (PK-PD) of tazobactam in combination with ceftolozane against an isogenic CTX-M-15-producing Escherichia coli triplet set, genetically engineered to transcribe different levels of blaCTX-M-15. The percentage of the dosing interval that tazobactam concentrations remained above a threshold (%Time>threshold) was identified as the PK-PD exposure measure that was most closely associated with efficacy. Moreover, the tazobactam concentration was dependent upon the enzyme transcription level. Given that the aforementioned strains were genetically engineered to transcribe a single β-lactamase enzyme and that clinical isolates typically produce multiple β-lactamase enzymes with various transcription levels, it is likely that the tazobactam threshold concentration is isolate/enzyme dependent. Our first objective was to characterize the relationship between the tazobactam %Time>threshold in combination with ceftolozane and efficacy using clinical isolates in an in vitro PK-PD infection model. Our second objective was to identify a translational relationship that would allow for the comodeling across clinical isolates. The initial challenge panel included four well-characterized β-lactamase-producing E. coli strains with variable enzyme expression and other resistance determinants. As evidenced by r2 values of ranging from 0.90 to 0.99 for each clinical isolate, the observed data were well described by fitted functions describing the relationship between the tazobactam %Time>threshold and change in log10 CFU from baseline; however, the data from the four isolates did not comodel well. The threshold concentration identified for each isolate ranged from 0.5 to 4 mg/liter. We identified an enabling translational relationship for the tazobactam threshold that allowed comodeling of all four clinical isolates, which was the product of the individual isolate's ceftolozane-tazobactam MIC value and 0.5. As evidenced by an r2 value of 0.90, the transformed data were well described by a fitted function describing the relationship between tazobactam %Time>threshold and change in log10 CFU from baseline. Due to these findings, the challenge panel was expanded to include three well-characterized β-lactamase-producing Klebsiella pneumoniae strains with variable enzyme expression and other resistance determinants. The translational relationship for the tazobactam threshold that allowed for the comodeling of the four E. coli isolates performed well for the expanded data set (seven isolates in total; four E. coli and three K. pneumoniae), as evidenced by an r2 value of 0.84. This simple translational relationship is especially useful as it is directly linked to in vitro susceptibility test results, which are used to guide the clinician's choice of drug and dosing regimen.
INTRODUCTION
Tazobactam, a penicillanic acid sulfone β-lactamase inhibitor, has been used in combination with piperacillin to treat infected patients for nearly a quarter-century (1). As a result of the rising clinical concern for β-lactamase-producing Enterobacteriaceae, there has been renewed interest in developing new tazobactam–β-lactam combinations. Ceftolozane-tazobactam is a novel antibacterial with activity against Pseudomonas aeruginosa, including drug-resistant strains, and other common Gram-negative pathogens, including most extended-spectrum β-lactamase (ESBL)-producing Enterobacteriaceae (2).
Until recently, little was known about the pharmacokinetic-pharmacodynamic (PK-PD) underpinnings of β-lactamase inhibitor therapeutics (3, 4, 5). For instance, the PK-PD measure associated with tazobactam efficacy was only recently identified (5). VanScoy et al. utilized an isogenic CTX-M-15-producing Escherichia coli triplet set, genetically engineered to transcribe different levels of blaCTX-M-15, to identify the percentage of the dosing interval that tazobactam concentrations remained above a threshold (%Time>threshold) as the PK-PD efficacy determinant. The results of these studies also demonstrated that the blaCTX-M-15 transcription level had an impact on the tazobactam threshold concentration associated with efficacy. Higher tazobactam threshold concentrations were associated with strains transcribing higher levels of β-lactamase enzyme. Given that the aforementioned strains were genetically engineered to transcribe a single β-lactamase enzyme and that clinical isolates typically produce multiple β-lactamase enzymes with various transcription levels, it is likely that the tazobactam threshold concentration is isolate and enzyme dependent. In such a circumstance, the forecasting of potentially efficacious clinical regimens from preclinical infection model data would be challenging. Thus, it would be useful to identify a translational relationship that would allow for comodeling of the relationship between %Time>threshold and efficacy using data across isolates that produce different β-lactamase enzymes and enzyme combinations with various transcription levels.
The objectives of the PK-PD in vitro infection model studies for ceftolozane-tazobactam described herein were 3-fold. The first objective was to characterize the relationship between tazobactam %Time>threshold and efficacy for each of four well-characterized β-lactamase-producing clinical E. coli isolates. Using data pooled for these four isolates, the second objective was to identify a translational relationship that would allow for comodeling of the relationship between %Time>threshold and efficacy. Upon successful completion of the second objective, the clinical isolate challenge panel was expanded to include three well-characterized β-lactamase-producing clinical Klebsiella pneumoniae isolates in order to evaluate the translational relationship in the context of an expanded ESBL-producing genus data set. Escherichia coli and K. pneumoniae represent those Enterobacteriaceae that dominantly harbor ESBL genes and cause the greatest occurrence of human disease. Such an enabling translational relationship would subsequently allow for the forecasting of effective and noneffective clinical regimens from preclinical models systems based upon in vitro susceptibility test results.
MATERIALS AND METHODS
Bacteria, antimicrobial, and β-lactamase inhibitor.
Four E. coli and three K. pneumoniae clinical isolates (JMI Laboratories, North Liberty, IA) were utilized in these studies. Isolates were chosen to cover a range ceftolozane/tazobactam MIC values. Ceftolozane and tazobactam were provided by Cubist Pharmaceuticals (Lexington, MA).
Determining transcription levels of intrinsic ampC, efflux pump, and outer membrane protein genes.
The transcription levels of ampC, acrA, ompC, and ompF were determined by comparing the transcription levels of these selected targets to those from the same clinical wild-type E. coli recipient strain used in previous studies (5). For K. pneumoniae, the transcription levels of ampC, acrA, and ompK were determined by comparing the transcription levels of these selected targets to those from a wild-type strain. Total genomic RNA samples were extracted from each challenge isolate using the RNeasy Mini Kit in a fully automated robotic workstation (QIAcube; Qiagen, Valencia, CA), and residual DNA was eliminated with RNase-free DNase (Promega, Madison, WI). Sample quality and quantification of the genomic RNA were assessed using the Agilent 21000 Bioanalyzer with the RNA 6000 Nano kit according to the manufacturer's instructions (Agilent, Santa Clara, CA). Reverse transcription-PCR (RT-PCR) was performed in triplicate using the QuantiTect SYBR green RT-PCR kit (Qiagen, Germantown, MD) in the StepOne Plus instrument (Lie Technologies, Foster City, CA).
Genotypic and phenotypic characterization of β-lactamase content.
Each challenge isolate was screened for plasmidic AmpC (blaCMY-1-41, blaCMY-43-44, blaCMY-49, blaFOX-1-7, blaACC-1-4, blaACT-1-7, blaDHA-1-3, blaLAT-1, blaMIR-1-5, and blaMOM-1-7) extended-spectrum β-lactamase (ESBL) (blaTEM, blaSHV, blaCTX-M, blaGES, blaVEB, blaPER, blaPSE, and blaBEL), and oxacillinases with ESBL spectrum (blaOXA-2, blaOXA-10, and blaOXA-1/30 group, blaOXA-18, and blaOXA-45). β-Lactamase-encoding genes were confirmed by sequencing analysis.
All isolates were subjected to isoelectric focusing (IEF) patterns using standard protocols to rule out the presence of unknown β-lactamase-encoding genes not detected by PCR and sequencing. Crude protein extract preparations were obtained from overnight cultures subjected to cell lysis using BugBuster (Novagen, Darmstadt, Germany). The lysed samples were normalized and loaded into precast IEF gels stained using nitrocefin and a prestained protein marker as a standard. The isoelectric points were determined by linear regression using a combination of preparations from bacteria possessing known β-lactamases.
Determination of hydrolytic activity.
The hydrolytic activity of the pooled β-lactam enzymes produced by each clinical strain was measured by observing the changes in absorbance due to the opening of the nitrocefin β-lactam ring in an Ultrospec 3300 Pro UV/visible spectrophotometer (GE Healthcare Biosciences, Pittsburgh, PA) during a 2-min interval. The degree of hydrolytic activity was calculated using the following formula: hydrolytic activity = [(Δabsorbance/minute)/protein concentration in mg/liter] × −1,000 (factor).
Media and in vitro susceptibility studies.
Susceptibility studies, using cation-adjusted Mueller-Hinton broth (CA-MHB) (BD laboratories, Franklin Lakes, NJ) microdilution methods, were conducted in accordance with Clinical and Laboratory Standards Institute (CLSI) guidelines (6). Isolate susceptibility to ceftolozane was determined for ceftolozane alone as well as in combination with a fixed tazobactam concentration (4 mg/liter) per CLSI guidelines. Minimum bactericidal concentrations (MBCs) were determined in duplicate for each isolate. Briefly, 10 μl was removed from the wells that represented the MIC and 4 concentrations greater than the MIC and was plated onto blood agar plates. To avoid antibacterial carryover, the aliquots were allowed to soak into the agar and then were spread for isolation, thus removing the cells from the drug source. The MBC was defined as the lowest concentration of drug that reduced the initial inoculum by 3 log units. Susceptibility studies were performed in triplicate over a 2-day period. The modal MIC and MBC values are presented and were utilized in all analyses.
PK-PD in vitro model and sample processing.
The one-compartment PK-PD in vitro infection model utilized in these studies has been described previously (7). Briefly, the model consists of a central infection compartment containing growth medium, the challenge isolate, and magnetic stir bars to ensure the homogeneity of the drug(s) within the compartment. The central infection compartment was attached to a stir plate, and the entire unit was placed within a temperature- and humidity-controlled incubator set at 35°C. Drug-free growth medium was pumped into the central infection compartment via a computer-controlled peristaltic pump while growth medium was simultaneously removed through an exit port and captured in a waste container. The challenge isolate was aseptically inoculated directly into the central infection compartment. The peristaltic pump was set at a flow rate which allowed for the simulation of the human free drug concentration-time profile of the drug(s) under study. The test compounds were infused via computer-controlled syringe pumps which allow for the simulation of the desired half-lives, dosing frequencies, and concentrations. Specimens for CFU determination and drug concentration assay were collected from the central infection compartment using a sterile syringe and needle through a rubber septum at predetermined time points.
In these experiments, initial inocula of 1.0 × 106 CFU/ml of each of the challenge isolates were prepared from a culture grown overnight on Trypticase soy agar enriched with 5% sheep blood (BD, Sparks, MD). Isolates were taken from the overnight cultures and grown to mid-logarithmic phase in a flask of MHB set in a water-shaker bath at 35°C and 125 rotations per minute. The bacterial concentration within the flask of MHB was determined by optical density using a Genesys 30 spectrophotometer (Thermo Scientific, Waltham MA) and a previously confirmed growth curve for each challenge isolate.
Bacteria were then exposed to changing concentrations of ceftolozane and tazobactam simulating human half-lives of 2.5 h for ceftolozane and 1 h for tazobactam. One-milliliter specimens were collected for CFU determination at 0, 2, 4, 8, 12, and 24 h. Each sample was centrifuged, washed, and resuspended with sterile normal saline twice to prevent drug carryover and was then cultured onto Trypticase soy agar enriched with 5% sheep blood. Plated samples were incubated at 35°C for 24 h. One-milliliter specimens for drug assay were collected at 1, 3, 5, 7, 9, and 24 h. These samples were immediately frozen at −80°C until assayed for drug concentration.
Dose-ranging studies.
Dose-ranging studies were conducted using each of the four clinical E. coli isolates to determine the tazobactam %Time>threshold concentration required for cell kill. In these studies, a range of tazobactam doses were administered using a fixed dose of ceftolozane. Each dosing interval was 8 h since this is the dosing schedule that is expected to be used clinically. Both drugs were administered over 1 h, and free drug concentration-time profiles assuming 20% and 30% protein binding for ceftolozane (Cubist; data on file) and tazobactam (Zosyn package insert; Wyeth Pharmaceuticals, Inc., Philadelphia, PA), respectively, were simulated. The ceftolozane dose administered for isolates with MIC values of 0.5 and 1 mg/liter was 1,000 mg, while a 2,000-mg dose was administered for isolates with MIC values of 2 and 4 mg/liter. The tazobactam dose ranged from 125 to 4,000 mg.
Analytical method.
All samples were assayed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Waters, Milford, MA). The standard curve for ceftolozane was linear over a range of 0.1 to 500 mg/liter, and that for tazobactam was quadratic over a range of 0.1 to 100 mg/liter. For the ceftolozane and tazobactam quality control (QC) samples, the deviation of the mean from theoretical values did not exceed ±3.00% and ±4.00%, respectively. The coefficients of variation (CVs) for the ceftolozane and tazobactam QC samples ranged from 2.89% to 4.21% and from 3.22% to 4.20%, respectively.
PK-PD analysis.
Data from the dose-ranging studies were evaluated using Hill-type models and nonlinear least-squares regression. The data were weighted using the inverse of the estimated measurement variance. Relationships between the tazobactam %Time>threshold and change in log10 CFU from baseline at 24 h were evaluated. For each individual isolate, the tazobactam %Time>threshold was identified through an iterative process in which candidate threshold concentrations of 0.05, 0.1, 0.25, 0.5, 1, 2, and 4 mg/liter were evaluated. Threshold value discrimination was based on the evaluation of the dispersion of data along the %Time>threshold axis and optimization of r2 values for the relationship between tazobactam %Time>threshold and change in log10 CFU from baseline at 24 h.
For analyses of pooled data across isolates, two approaches were considered. First, graphical evaluations while viewing an electric telescope (52 Class 530 Series 1080p LCD/HD; Samsung Group, Seoul, South Korea) were used to identify potential tazobactam thresholds, and second, nonlinear regression was considered to explore multivariable models which would predict the tazobactam threshold concentration (dependent variable) as a function of the candidate independent variables. Model discrimination was accomplished according to the corrected Akaike's information criterion (8).
RESULTS
In vitro susceptibility testing.
The in vitro susceptibility test results are presented in Table 1. The MIC values for ceftolozane alone ranged from 128 to 512 mg/liter when tested against the seven selected clinical isolates. As expected, the MIC values of ceftolozane in the presence of 4 mg/liter tazobactam were significantly lower, ranging from 0.5 to 4 mg/liter. The MBC values in the presence of 4 mg/liter tazobactam were identical to the MICs determined under the same conditions for four of the seven isolates and were 2-fold higher for the remaining isolates.
Table 1.
Susceptibility testing results for ceftolozane and ceftolozane combined with tazobactam against an E. coli ATCC control strain and seven clinical isolates
| Species and isolate | Microtiter MIC (mg/liter) |
MBC (mg/liter), ceftolozane-tazobactam (4 mg/liter) | |
|---|---|---|---|
| Ceftolozane alone | Ceftolozane-tazobactam (4 mg/liter) | ||
| E. coli | |||
| ATCC 25922 | 0.5 | 0.5 | 0.5 |
| 4643A | 128 | 0.5 | 0.5 |
| 1801A | 128 | 0.5 | 1 |
| 21711R | 256 | 2 | 4 |
| 13319R | 512 | 4 | 4 |
| K. pneumoniae | |||
| 604C | 256 | 1 | 2 |
| 21904E | 512 | 2 | 2 |
| 4812E | 512 | 4 | 4 |
AmpC, efflux pump, and outer membrane protein genes, hydrolytic activity, and β-lactamase content.
Table 2 shows the β-lactamase content and transcription level of the various resistance genes identified for individual E. coli clinical isolates. All four E. coli isolates produced CTX-M-15, while three of the four clinical isolates produced additional β-lactamases (blaTEM-1 and/or blaOXA-1/30), which are commonly detected among clinical E. coli isolates. Two of the four isolates demonstrated additional resistance determinants, Arab-TolC efflux pump overexpression, or decreased ompC and/or ompF expression. For E. coli, the rate of hydrolytic activity increases in parallel with the number of different β-lactamase genes detected.
Table 2.
Summary of β-lactamase gene content and expression results obtained for the four E. coli clinical isolates
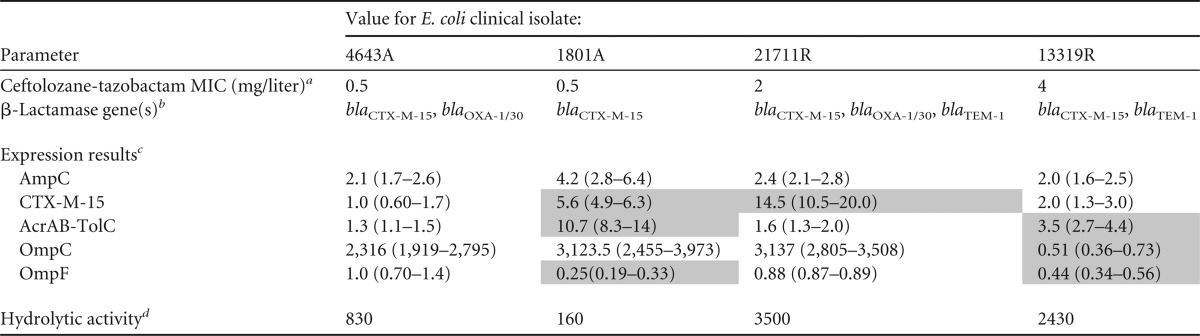
a Modal MIC from triplicate results according to the CLSI specifications (6).
b β-Lactamase gene content was determined by PCR and confirmed by sequencing analysis.
c Quantification of transcriptional levels for the ampC, acrA, ompC, and ompF genes (relative to an rpsL endogenous reference) compared to those obtained from a clinically relevant wild-type ST131 E. coli isolate (ceftolozane-tazobactam MIC, 0.25 mg/liter) (5). Values in parentheses represent the respective relative quantification ± standard deviation. Shading represents expression values that may contribute to the decreased susceptibility to ceftolozane-tazobactam (e.g., overexpression of the AcrAB-TolC efflux pump system or decreased expression of OmpC and/or OmpF porin).
d Hydrolytic activity rates expressed as substrate (nitrocefin) hydrolyzed (Δabsorbance) per minute per milligram of protein.
Table 3 shows the β-lactamase contents and transcription levels of the various resistance genes identified for individual K. pneumoniae clinical isolates. All three K. pneumoniae isolates produced CTX-M-15, while two of the three clinical isolates produced an additional β-lactamase (blaOXA-1). Relative to the wild-type K. pneumoniae strain (918C), there were virtually no differences in AcrAB-TolC efflux pump expression or the level of OmpK expression. For K. pneumoniae, the rate of hydrolytic activity of the clinical isolates was similar to or greater than that for the wild-type strain (918C).
Table 3.
Summary of β-lactamase gene content and expression results obtained for the three K. pneumoniae clinical isolates
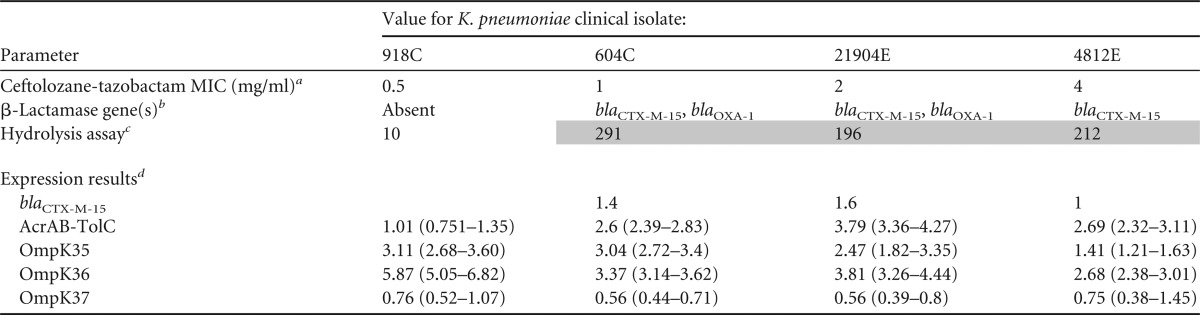
a MICs from triplicate results obtained using frozen-form panels manufactured according to the CLSI specifications (6).
b β-Lactamase gene content determined by PCR and confirmed by sequencing analysis.
c Hydrolytic activity rates expressed as substrate (nitrocefin) hydrolyzed (ΔAbsorbance) per minute per milligram of protein. Shading indicates an impressive difference in hydrolytic activity compared to the result for the wild type.
d Quantification of transcriptional levels for blaCTX-M-15, acrA, ompK35, ompK36, and ompK37 (relative to an rpsL endogenous reference). Results obtained were compared to those for the K. pneumoniae ATCC 13883 strain. Values in parentheses represent the respective relative quantification ± standard deviation.
Pharmacokinetics.
The relationship between the observed and targeted free drug concentrations from all dosing regimens for ceftolozane and tazobactam studied is shown in Fig. 1. As demonstrated by the good agreement between observed and targeted concentrations for ceftolozane (r2 = 0.983) and tazobactam (r2 = 0.988), the drug profiles were well simulated in the PK-PD in vitro model.
Fig 1.

Relationships between the observed and targeted ceftolozane (A) and tazobactam (B) concentrations from all dosing regimens for ceftolozane and tazobactam studied.
Dose-ranging studies.
The results of the dose-ranging studies for each of the seven clinical isolates are shown in Fig. 2. Bacteria in the no-treatment control arms grew well, reaching a bacterial density of greater than 1.0 × 108 CFU/ml by 1 h for each challenge isolate. The monotherapy ceftolozane and tazobactam dosing regimens performed as expected, with little to no cell kill for each dosing regimen and growth reaching greater than 1.0 × 108 CFU/ml by 12 h. For each clinical isolate, the range of tazobactam doses used in combination with a fixed ceftolozane doses provided a full spectrum of drug effect. Low tazobactam exposures resulted in CFU/ml values similar to that of the growth control by 24 h, while the highest tazobactam exposures resulted in reductions of greater than of 2 to 3 log10 CFU relative to baseline at 24 h for three of the four isolates. The intermediate tazobactam exposures resulted in CFU/ml values that were between those observed in the low- and high-exposure study arms.
Fig 2.

Dose-ranging study results for each of the four E. coli (A to D) and three K. pneumoniae (Kp) (E to G) clinical isolates. The effect of each active regimen relative to the no-treatment controls is shown. TOL, ceftolozane; TAZ, tazobactam. Error bars represent the range of data over two studies.
PK-PD analysis.
Relationships between the tazobactam %Time>threshold and change in log10 CFU from baseline at 24 h for the four E. coli clinical isolates evaluated are shown in Fig. 3. As evidenced by r2 values ranging from 0.90 to 0.99 for each clinical isolate, the observed data were well described by fitted functions describing the relationship between tazobactam %Time>threshold and change in log10 CFU from baseline. The threshold concentration identified for each isolate ranged from 0.5 to 4 mg/liter.
Fig 3.
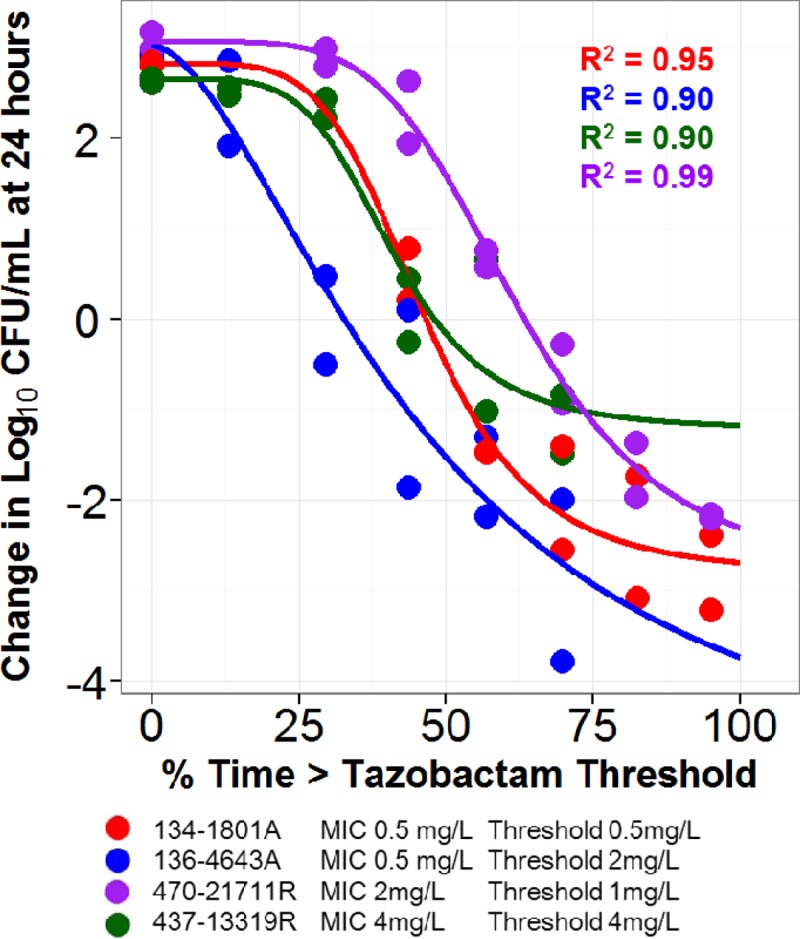
Relationships between tazobactam %Time>threshold and change in log10 CFU from baseline at 24 h for the four E. coli clinical isolates in a PK-PD in vitro infection model. Data points and fitted functions for each isolate are represented by different colors. The threshold for each isolate was identified using an iterative process which allowed for evaluation of dispersion of data along the %Time>threshold axis and optimization of r2 values.
Figure 4 shows the relationship between tazobactam %Time>threshold and change in log10 CFU from baseline at 24 h based on pooled and transformed data for the four E. coli clinical isolates evaluated. As evidenced by an r2 value of 0.903, the transformed data were well described by a fitted function describing the relationship between tazobactam %Time>threshold and change in log10 CFU from baseline. The enabling translational relationship for the tazobactam threshold that allowed comodeling of all four clinical isolates was the product of the individual isolate's ceftolozane-tazobactam MIC value and 0.5. The parameter estimates (standard errors) for the relationship between tazobactam %Time>threshold and change in log10 CFU from baseline shown in Fig. 4 were as follows: E0, 2.89 (0.215); maximum effect (Emax), 6.45 (1.09); Hill's constant, 4.39 (1.14); and 50% effective concentration (EC50), 68.7 (5.79). The tazobactam %Time>thresholds associated with net bacterial stasis and 1- and 2-log10 CFU reductions in bacteria at 24 h were 65.7, 75.7, and 89.3% of the dosing interval, respectively, regardless of the MIC, number and type of beta-lactamases, or other resistance determinants.
Fig 4.
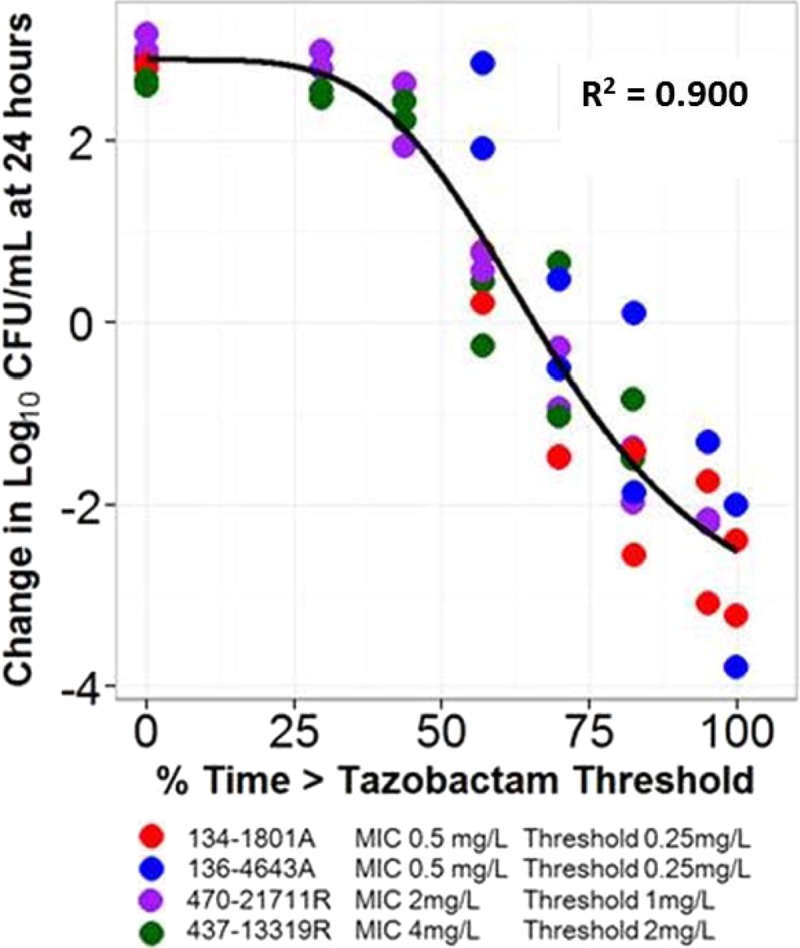
Relationship between tazobactam %Time>threshold and change in log10 CFU from baseline at 24 h for the four E. coli clinical isolates in a PK-PD in vitro infection model. Isolates are represented by different colors. The black line represents the fitted function for the pooled data across isolates. The threshold for each isolate represented the product of the ceftolozane-tazobactam MIC for the individual isolate and 0.5.
Figure 5 shows the relationship between tazobactam %Time>threshold and change in log10 CFU from baseline at 24 h based on pooled and transformed data for the four E. coli and three K. pneumoniae clinical isolates evaluated using the same translational relationship described above. As evidenced by an r2 value of 0.84, the transformed data were well described by a fitted function describing the relationship between tazobactam %Time>threshold and change in log10 CFU from baseline. The parameter estimates (standard errors) for the relationship between tazobactam %Time>threshold and change in log10 CFU from baseline shown in Fig. 5 were as follows: E0, 2.89 (0.212); Emax, 8.09 (2.54); Hill's constant, 3.21 (0.88); and EC50, 79.2 (15.9). The tazobactam %Time>thresholds associated with net bacterial stasis and 1- and 2-log10 CFU reductions in bacteria at 24 h were 65.9, 77.3, and 90.2% of the dosing interval, respectively, regardless of the MIC, number and type of beta-lactamases, or other resistance determinants.
Fig 5.

Relationship between tazobactam %Time>threshold and change in log10 CFU from baseline at 24 h for the four E. coli and three K. pneumoniae clinical isolates in a PK-PD in vitro infection model. Isolates are represented by different colors. The black line represents the fitted function for the pooled data across isolates. The threshold for each isolate represented the product of the ceftolozane-tazobactam MIC for the individual isolate and 0.5.
DISCUSSION
There were two objectives of the studies described here. The first objective was to characterize the relationship between the tazobactam %Time>threshold and efficacy for each of the four well-characterized β-lactamase-producing clinical E. coli isolates studied. Subsequently, the second objective was to identify a translational relationship that would allow for the comodeling of the relationship between %Time>threshold and efficacy using pooled data for these isolates. Upon successful completion of the second objective, the clinical isolate challenge panel was expanded to include three well-characterized β-lactamase-producing clinical K. pneumoniae isolates in order to evaluate the translational relationship in the context of an expanded ESBL-producing genus data set.
We successfully described a relationship between tazobactam exposure and change in bacterial density for each study isolate. As previously demonstrated using genetically engineered strains (5), the joint actions of ceftolozane and tazobactam were well-described by %Time>threshold. However, in the current study, the threshold identified for each individual isolate did not allow for the identification of a unifying exposure-response relationship. That is, although we were able to fit exposure-response models to each of the four E. coli isolates individually, we were unable to fit an integrated model to the pooled data (Fig. 3). This is not unexpected, as there was considerable variability in the number of resistance determinants among the four clinical isolates. For example, the E. coli isolate with the lowest MIC (JMI 4643A; MIC, 0.5 mg/liter) possessed an acquired resistance determinant, β-lactamase (blaCTX-M-15 and blaoxa-1/30), while the isolate with the highest MIC (JMI 13319R; 4 mg/liter) had acquired β-lactamase (blaCTX-M-15 and blaTEM-1) possibly associated with up- and downregulated drug efflux (AcrAB-TolC) and porin genes (ompC and ompF).
For an antimicrobial agent, an integrated exposure-response model allows for the forecasting of effective and noneffective clinical dosing regimens from preclinical model systems based upon in vitro susceptibility test results. Given that the four clinical isolates studied have differing %Time>threshold requirements for a given amount of bactericidal activity, forecasting effective clinical dosing regimens would be challenging. Moreover, supporting in vitro susceptibility test interpretive criteria from a PK-PD perspective would be fraught with difficulty. For this reason, we sought to develop a translational relationship that would allow for the comodeling of exposure-response relationships across isolates.
We successfully identified a surprisingly simple translational relationship that allowed for the integration of the individual exposure-response relationships using pooled data for each of the four study isolates. The enabling translational relationship that led to a high coefficient of determination (r2 = 0.900) for the fitted function between tazobactam %Time>threshold and change in log10 CFU from baseline was that for which the tazobactam threshold was the product of each individual isolate's MIC to ceftolozane in the presence of tazobactam (4 mg/liter) and 0.5 (Fig. 4). One important benefit of this simple translation relationship is that it is directly linked to in vitro susceptibility test results, which guide the clinician's choice of drug and dosing regimen.
Subsequently, we expanded the clinical isolate challenge panel to include three well-characterized β-lactamase-producing clinical K. pneumoniae isolates and confirmed the translational relationship in a second genus (Fig. 5). As E. coli and K. pneumoniae represent those Enterobacteriaceae that dominantly harbor ESBL genes and cause the greatest occurrence of human disease, we felt the confirmation of the translational relationship using data for these two genera was of critical importance. It is worth noting that this translational relationship was identified despite the fact that this group of isolates displayed different resistance mechanisms (e.g., up- and downregulated drug efflux and porin expression). Thus, the identification of PK-PD relationships for tazobactam, based on the unifying translational relationship for %Time>tazobactam threshold and using data from this genetically diverse set of isolates, implies that β-lactamase and not the other resistance mechanisms represents the dominant resistance determinant.
One likely explanation for the success of this simple enabling translational relationship is directly related to the ceftolozane-tazobactam susceptibility test itself. It should be remembered that the fixed 4-mg/liter tazobactam concentration utilized in the static susceptibility test method is functionally equivalent to exposing the bacterial inoculum to a continuous infusion of drug and that the test result itself represents net bacterial stasis over a 24-h test duration. In reality, β-lactam antimicrobial agents do not require 100% Time>MIC to achieve net bacterial stasis in dynamic preclinical model systems. Using a neutropenic murine thigh infection model, Craig and Andes demonstrated that for ceftolozane-tazobactam against Enterobacteriaceae elaborating extended-spectrum β-lactamases, 31.1% (±4.9%) Time>MIC was required for net bacterial stasis over a 24-h experiment (9).
There are several important caveats regarding the implications of the identified simple translational relationship. The relationship was identified based upon studies involving four E. coli and three K. pneumoniae β-lactamase-producing clinical isolates and one β-lactamase inhibitor, tazobactam. Thus, we do not know if the identified translational relationship will apply across β-lactam–β-lactamase inhibitor combinations. Additional studies are warranted in this regard.
In conclusion, we identified relationships between the tazobactam %Time>threshold and efficacy for each of the seven clinical Enterobacteriaceae isolates studied, representing two genera. Moreover, we identified a translational relationship that would allow for the comodeling of the relationship between %Time>threshold and efficacy using data pooled for these seven isolates. The enabling translational relationship was that for which the tazobactam threshold was the product of the individual MIC of ceftolozane in the presence of tazobactam (4 mg/liter) and 0.5. This simple translational relationship is especially useful as it is directly linked to in vitro susceptibility test results, which are used to guide the clinician's choice of drug and dosing regimen.
ACKNOWLEDGMENTS
We thank Kim A. Charpentier from ICPD (Latham, NY, USA) and Lalitagauri M. Deshpande from JMI Laboratories (North Liberty, IA, USA) for manuscript assistance and technical support.
This study was sponsored by Cubist Pharmaceuticals, Inc., Lexington, MA. The Institute for Clinical Pharmacodynamics (B.V., J.M., C.C.B., O.O.O., S.M.B., A.F., and P.G.A.) has received research support from Achaogen, Astellas, AstraZeneca, Basilea Pharmaceutica, Bayer HealthCare, Bristol-Meyers Squibb, Cempra Pharmaceuticals, Cerexa, Cubist Pharmaceuticals, Durata Pharmaceuticals, Fedora Pharmaceuticals, Forest Research Institute, Furiex Pharmaceuticals, GlaxoSmithKline, Meiji Seika Pharma, Nabriva Therapeutics, Nimbus, Pfizer, PolyMedix, Rib-X, Roche Bioscience, Rock Therapeutics, Tetraphase Pharmaceuticals, and The Medicines Company. JMI Laboratories, Inc. (R.E.M. and R.N.J.) has received research and educational grants in 2009 to 2012 from the American Proficiency Institute (API), Anacor, Astellas, AstraZeneca, Bayer, Cempra, Cerexa, Contrafect, Cubist, Daiichi, Dipexium, Enanta, Furiex, GlaxoSmithKline, Johnson & Johnson (Ortho McNeil), LegoChem Biosciences Inc., Meiji Seika Kaisha, Merck, Nabriva, Novartis, Pfizer (Wyeth), Rempex, Rib-X Pharmaceuticals, Seachaid, Shionogi, The Medicines Co., Theravance, and ThermoFisher. Some JMI employees are advisors/consultants for Astellas, Cubist, Pfizer, Cempra, Cerexa-Forest, J&J, and Theravance. With regard to speaker bureaus and stock options, there are none to declare. L.V.F. and J.N.S. are both employees and stockholders of Cubist Pharmaceuticals.
Footnotes
Published ahead of print 16 September 2013
REFERENCES
- 1. Gould IM, Ansari A, Harvey G, Douglas JG, Smith CC, Reid TM. 1991. Piperacillin/tazobactam in the treatment of serious acute soft tissue infection. Drugs Exp. Clin. 17:187–190 [PubMed] [Google Scholar]
- 2. Chandorkar G, Huntington J, Parsons T, Gotfried M, Rodvold KA, Umeh O. 2012. Intrapulmonary penetration of ceftolozane/tazobactam and piperacillin/tazobactam in healthy adult subjects. J. Antimicrob. Chemother. 67:2463–2469 [DOI] [PubMed] [Google Scholar]
- 3. Strayer AH, Gilbert DH, Pivarnik P, Medeiros AA, Zinner SH, Dudley MN. 1994. Pharmacodynamics of piperacillin alone and in combination with tazobactam against piperacillin-resistant and -susceptible organisms in an in vitro model of infection. Antimicrob. Agents Chemother. 38:2351–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Louie A, Castanheira M, Liu W, Grasso C, Jones RN, Williams G, Critchley I, Thye D, Brown D, Vanscoy B, Kulawy R, Drusano GL. 2012. Pharmacodynamics of β-lactamase inhibition by NXL104 in combination with ceftaroline: examining organisms with multiple types of β-lactamases. Antimicrob. Agents Chemother. 56:258–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. VanScoy B, Mendes RE, Nicasio AM, Castanheira M, Bulik CC, Okusanya OO, Bhavnani SM, Forrest A, Jones RN, Friedrich LV, Steenbergen JN, Ambrose PG. 2013. Pharmacokinetics-pharmacodynamics of tazobactam in combination with ceftolozane in an in vitro infection model. Antimicrob. Agents Chemother. 57:2809–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clinical and Laboratory Standards Institute 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved standard, 9th ed. CLSI document M07-A9. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 7. Garrison MW, Vance-Bryan K, Larson TA, Toscano JP, Rotschafer JC. 1990. Assessment of effects of protein binding on daptomycin and vancomycin killing of Staphylococcus aureus by using an in vitro pharmacodynamic model. Antimicrob. Agents Chemother. 34:1925–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Akaike HA. 1979. Bayesian extension of the minimum AIC procedure of autoregressive model fitting. Biometrika 66:237–242 [Google Scholar]
- 9. Craig WA, Andes DR. 2013. In vivo activity of ceftolozane, a new cephalosporin, with and without tazobactam against Pseudomonas aeruginosa and Enterobacteriaceace, including strains with extended-spectrum β-lactamases, in the thighs of neutropenic mice. Antimicrob. Agents Chemother. 57:1577–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]