

Abstract
Hydroxymethylnitrofurazone (NFOH) is a trypanocidal prodrug of nitrofurazone (NF), devoid of mutagenic toxicity. The purpose of this work was to study the chemical conversion of NFOH into NF in sodium acetate buffer (pH 1.2 and 7.4) and in human plasma and to determine preclinical pharmacokinetic parameters in rats. At pH 1.2, the NFOH was totally transformed into NF, the parent drug, after 48 h, while at pH 7.4, after the same period, the hydrolysis rate was 20%. In human plasma, 50% of NFOH was hydrolyzed after 24 h. In the investigation of kinetic disposition, the concentration of drug in serum versus time curve was used to calculate the pharmacokinetic parameters after a single-dose regimen. NFOH showed a time to maximum concentration of drug in serum (Tmax) as 1 h, suggesting faster absorption than NF (4 h). The most important results observed were the volume of distribution (V) of NFOH through the tissues, which showed a rate that is 20-fold higher (337.5 liters/kg of body weight) than that of NF (17.64 liters/kg), and the concentration of NF obtained by in vivo metabolism of NFOH, which was about four times lower (maximum concentration of drug in serum [Cmax] = 0.83 μg/ml; area under the concentration-time curve from 0 to 12 h [AUC0–12] = 5.683 μg/ml · h) than observed for administered NF (Cmax = 2.78 μg/ml; AUC0–12 = 54.49 μg/ml · h). These findings can explain the superior activity and lower toxicity of the prodrug NFOH in relation to its parent drug and confirm NFOH as a promising anti-Chagas' disease drug candidate.
INTRODUCTION
American trypanosomiasis (Chagas' disease) is an endemic parasitic disease caused by Trypanosoma cruzi, afflicting much of Mexico, Central America, and South America, where an estimated 100 million people are at risk and 8 to 11 million people are infected, with 14,000 deaths per year. The acute cases are detected in only 1% to 2% of those infected (1, 2). With the increase in immigration to the First World, the disease is spreading to developed countries such as the United States, Europe, and Japan, which should be a concern if it is not controlled or has no treatment available (3). Only two nitro-heterocyclic drugs are available in the worldwide market for its treatment: benznidazole and nifurtimox. Benznidazole is a nitroimidazole derivative active against both the trypomastigote and amastigote forms of T. cruzi. This drug is the first-line drug treatment, with a half-life of 12 h, its elimination is predominantly renal, and it is the only drug on the market in Brazil. Nifurtimox is a nitrofuran compound; its half-life is about 3 h because it is extensively metabolized by the liver. It also presents activity against trypomastigotes and amastigotes. The mechanism of its action is not totally understood (4); however, several derivatives showing potential anti-Chagas' disease activities have been prepared (5, 6, 7).
Both drugs possess negative characteristics such as severe and frequent side effects (vomiting, anorexia, peripheral neuropathy, allergic dermatopathy, mental disturbance, and mutagenicity) and selective sensitivity for different strains of T. cruzi; and the long periods of treatment (8, 9) and lack of active drugs for the treatment of the chronic form of the disease increase the importance of the discovery of new drugs.
New therapeutic drugs have been obtained by using the prodrug design strategy (4, 6, 7, 9, 10, 11, 12, 13). The prodrugs have been an effective way of overcoming absorption, distribution, metabolism, and elimination (ADME) barriers that restrict the application of many chemical compounds as orally administered drugs (14, 15).
Among the studied compounds, hydroxymethylnitrofurazone (NFOH), a prodrug of nitrofurazone (NF) (Fig. 1), was effective when tested in LLC-MK2 cell cultures infected with trypomastigote forms of T. cruzi (10). The results showed higher trypanocidal activity in all stages of development forms of the parasite with low toxicity. Trossini and colleagues (16) demonstrated that NFOH was also able to inhibit 60% of cruzipain activity, compared to 30% inhibition by NF.
Fig 1.
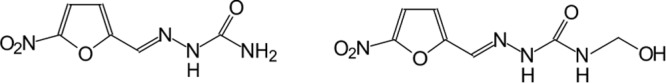
Structures of nitrofurazone (NF) (left) and hydroxymethylnitrofurazone (NFOH) (right).
In preclinical genotoxicity studies (Ames mutagenic test using TA102 and TA98 strains of Salmonella enterica serovar Typhimurium in the presence, or not, of S9), NFOH was shown to be four times less toxic than NF (17). These results were confirmed by micronucleus assays that showed NFOH activity similar to that of control (water) as well as by acute-toxicity tests that showed 50% lethal dose (LD50) values higher than 2,000 mg/kg of body weight (rats, per os) versus 556 mg/kg for NF, the parent drug (P. L. Bosquesi, M. Mello, J. L. Santos, and M. C. Chung, unpublished data).
In 2010, NFOH was tested against T. cruzi in a murine model of Chagas' disease and was found to be as effective as benznidazole in keeping direct parasitemia at undetectable levels after 60 days of treatment, and negative PCR results without histopathological lesions after 180 days of finishing the treatments showed that levels of anti-T. cruzi antibodies were very low in mice treated either with NFOH or with benznidazole (11).
The development of prodrugs relies significantly on the knowledge of their pharmacokinetic profile, and at this phase, the pharmacokinetic data from animal studies provide information about their ability to release the drug in biological systems and provide guidance on the level of doses that can be used in humans in future clinical trials (18, 19).
Hence, the present work was conducted with the objectives to study the in vitro hydrolysis of the prodrug NFOH to NF (considering chemical and enzymatic hydrolysis) and to evaluate its pharmacokinetic profile in rats in order to determine the first pharmacokinetic parameters for this new compound. For the accomplishment of these studies, a bioanalytical method for the determination of NF and NFOH in serum was also developed and validated.
MATERIALS AND METHODS
In vitro hydrolysis of prodrug NFOH to NF.
The decomposition of NFOH was studied at 37°C in aqueous sodium acetate buffer solutions (pH 1.2 and 7.4) at a concentration of 15 μg · ml−1. The solution was kept at constant temperature in a water bath, and at appropriate time intervals, samples were analyzed by high-pressure liquid chromatography (HPLC). The analyses were performed on 3 aliquots at each time point. The results were expressed as the average peak area. The same procedure was carried out with human plasma (ex vivo hydrolysis), and the times of the analyses were 0.0, 0.25, 0.50, 1, 2, 4, 8, 12, 24, 48, 72, and 120 h.
Pharmacokinetic profile of NFOH and NF in rats. (i) Animals.
Male Wistar rats (weighing around 250 g) were housed at constant temperature (23 ± 1°C), humidity (55% ± 5%), and light cycle (12/12 h) with food and water ad libitum. The animals were deprived of food 12 h before oral administration of compounds. The experiments were conducted during the light phase.
(ii) Experimental protocol.
Pharmacokinetic studies of NF and NFOH were carried out at an equivalent oral dose of 200 mg/kg in male Wistar rats. This dose was based on recent studies of toxicity in rats (data not published), and the sample size (n = 40 per group) was based on the method published by Liu and Chow (18). NF and NFOH solutions were prepared on the day of administration by dissolving appropriate amounts of drugs directly in dimethyl sulfoxide (DMSO); group I received NF, and group II received NFOH, both by gavage. The animals were killed by decapitation at 0.25, 0.5, 1, 2, 4, 8,12, and 24 h after administration. Five animals were used for each point. The blood samples were centrifuged and submitted to HPLC analysis. All experimental procedures were reviewed and approved by the Research Ethics Committee of the School of Pharmaceutical Science, UNESP, Araraquara, Brazil.
(iii) Pharmacokinetic analysis.
The NFOH and NF kinetic dispositions were evaluated after a single oral administration in rats. The pharmacokinetic parameters were calculated based on the concentration in serum versus time curves. The half-life of elimination (t1/2β) was calculated by the graphic method, and the constant of elimination (β) was calculated by the equation β = 0.693/t1/2β. The area under the curve from 0 to 24 h (AUC0-24) was calculated by the trapezoidal method, and the area under the curve from time zero extrapolated to infinity (AUC0-∞) was calculated by the equation AUC0-∞ = AUC0-24 + Cpn/kel (where Cpn is the last concentration in serum determined and kel is the elimination constant). The AUC0–∞ was used for the calculation of the total clearance (CLTF = dose/AUC0-∞), where F is bioavailability; the apparent distribution volume (V/F) was calculated using the equation V/F = CLT/β).
(iv) Statistical analysis.
Statistical analyses were performed using the software GraphPad Instat, followed by the Sigma-Stat software. The data were analyzed by one-way analysis of variance (ANOVA).
RESULTS
The study of the kinetic disposition of drugs requires analytical methods with compatible sensitivity and selectivity for determination of their concentration in serum after administration of single or multiple doses. The HPLC method for the determination of NFOH and NF in serum was developed, validated, and applied in the studies of hydrolysis and to evaluate the pharmacokinetic profile of compounds.
Figures 2 and 3 show the results obtained for the in vitro conversion of NFOH to NF at pH 1.2 and 7.4, respectively. Figure 4 shows the results for the in vitro conversion of NFOH to NF* in human plasma at 37°C. NF* represents the NF obtained from NFOH.
Fig 2.
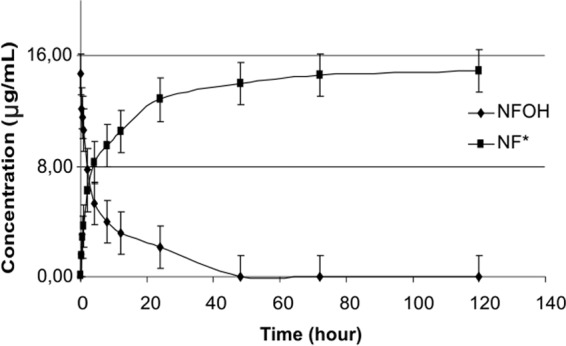
Conversion profile of NFOH to NF* in sodium acetate buffer (pH 1.2) at 37°C.
Fig 3.
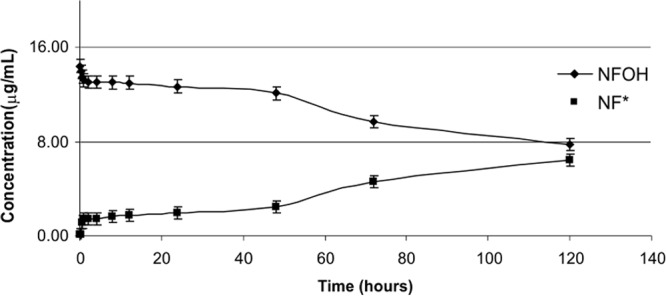
Conversion profile of NFOH to NF* in sodium acetate buffer (pH 7.4) at 37°C.
Fig 4.
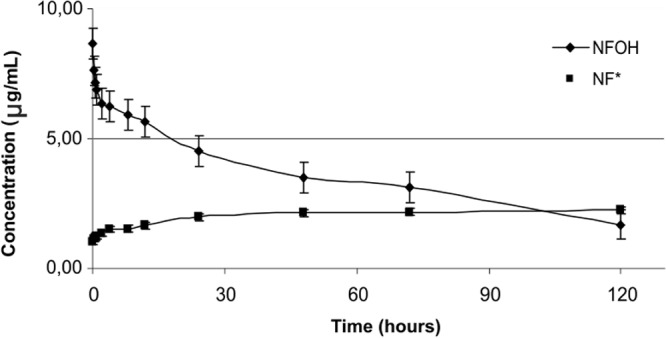
Conversion of NFOH to NF* in human plasma samples (pH 7.4) at 37°C.
In the investigation of kinetic disposition in rats that received NFOH (Fig. 5) and NF (Fig. 6) in a single-dose regimen, the concentration of serum versus time curves obtained were used to calculate the pharmacokinetic parameters.
Fig 5.
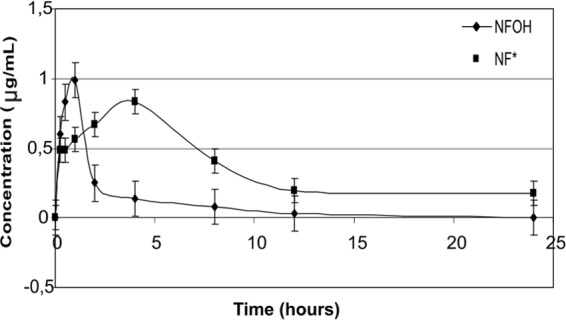
Time-dependent concentrations of NFOH and NF* in plasma, obtained after NFOH administration to male Wistar rats. Data are expressed as means and standard errors of the means (SEM) (n = 50).
Fig 6.
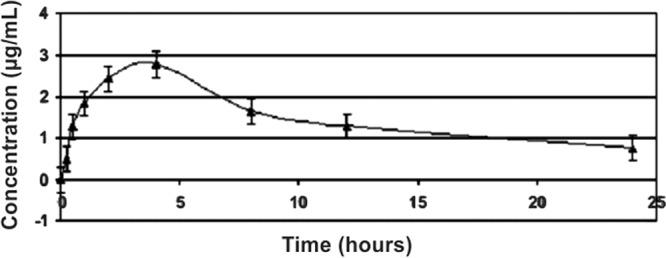
Time-dependent concentration of NF in plasma obtained after NF administration to male Wistar rats. Data are expressed as means and SEM (n = 50).
Figure 7 clearly shows that, after reaching Cmax, the NFOH hydrolysis profile in human plasma (ex vivo) differs from the data obtained in vivo, suggesting the participation of some other systems in the metabolism of NFOH when administered to the animal.
Fig 7.
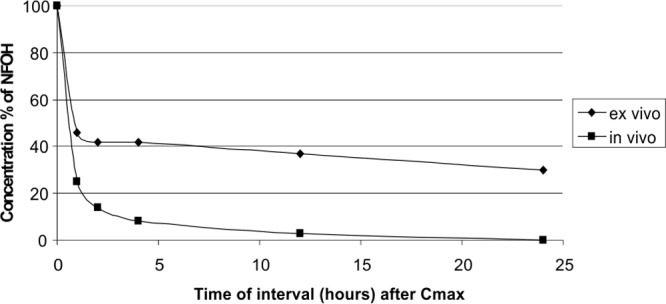
Comparative study of the hydrolysis of NFOH after reaching Cmax in human serum and in vivo.
The pharmacokinetic parameters (Table 1) were calculated as values required for the subsequent evaluation and study of dosage. NFOH reaches Tmax in 1 h compared with 4 h for NF, suggesting faster absorption than NF. The values of Cmax showed that the maximum concentration of NFOH in plasma is 0.99 μg/ml compared to 2.78 μg/ml of NF, but the distribution volume (V) of NFOH is 337.5 liters/kg while that of NF is 17.64 liters/kg.
Table 1.
Pharmacokinetic parameters determined after a single oral dose (200 mg/kg) of NF and NFOH to male Wistar rats (n = 50)a
| Parameter (unit) | NFOH | NF* | NF |
|---|---|---|---|
| Cmax (μg/ml) | 0.99 | 0.83 | 2.78 |
| Tmax (h) | 1.00 | 4.00 | 4.00 |
| kel (h−1) | 0.099 | 0.231 | 0.178 |
| t1/2β (h) | 7.00 | 3.00 | 3.9 |
| CL/F (liters/h · kg) | 33.41 | 3.140 | |
| V/F (liters/kg) | 337.5 | 17.64 | |
| AUC0–12 (μg/ml · h) | 5.683 | 13.36 | 54.49 |
| AUC0-∞ (μg/ml · h) | 5.986 | 14.23 | 63.70 |
| MRT | 10.102 | 5.618 |
Abbreviations: NF, nitrofurazone administered by gavage; NF*, NF obtained by hydrolysis of administered NFOH; Cmax, maximum concentration; Tmax, maximum time; kel, elimination constant; t1/2, half-life; CL/F, clearance/oral bioavailability; V/F, volume of distribution/oral bioavailability; AUC, area under the curve; MRT, mean residence time.
DISCUSSION
According to the literature, N-hydroxymethyl derivatives are hydrolyzed by chemical action and not by enzymatic attack (14, 20, 21). The experiments in this study were performed to verify if NFOH was stable at the pH of stomach and/or blood. The results showed that at pH 1.2, NFOH was totally converted into NF* after 48 h, while at pH 7.4, after the same period, the conversion rate was only 20%, showing chemical stability at pH 7.4. However, when the NFOH experiment was performed in human plasma, it was 50% hydrolyzed into NF* after 24 h; this result could be explained by the action of plasmatic enzymes since in a buffered medium (pH 7.4), 50% degradation occurs only after 120 h, suggesting that the molecule of NFOH has a different behavior from that of other hydroxymethyl compounds.
The effect of NFOH administered in rats showed that the plasmatic concentrations of NF* observed after the administration of NFOH were significantly lower than the concentrations obtained with direct administration of NF. After calculation, the V of NFOH was about 20-fold higher than the parent drug NF V, an impressive value suggesting that the new compound can cross the tissues to reach the target, while NF remains in the bloodstream. These findings can explain the superiority of the activity of NFOH over that of NF (9, 10). In addition, the active NF* metabolite obtained by in vivo hydrolysis of NFOH showed that the concentration of NF* is about four times lower (Cmax = 0.83 μg/ml; AUC0–12 = 5,683 μg · h/ml) than that of the NF administered orally (Cmax = 2.78 μg/ml; AUC0–12 = 54.49 μg · h/ml). This result can explain the 4-fold-lower toxicity observed by Guido and coworkers (17).
We observed that the simple inclusion of a methylhydroxy group in a molecule of NF may promote the improvement of its ADME profile in vivo. The literature showed that the inhibition of the trypanothione reductase was a possible mechanism of action of nitrofurazone (22, 23, 24); Trossini and coworkers (16) showed cruzipain inhibition and an increase of 30% in affinity for the enzyme. In addition, the most interesting and important point is the simplicity of the NFOH synthesis and its high yield (10), suggesting a very cheap compound for chemical scale-up. All these findings strongly suggested NFOH as a promising anti-Chagas' disease candidate drug.
Conclusions.
Hydrolysis studies carried out at pH 1.2 and 7.4 indicated that NFOH is stable in vitro and is not totally converted in human plasma by plasma enzymes. The in vivo experiments showed different hydrolysis profiles from those obtained in vitro in surprising results. The pharmacokinetics of NFOH showed that this compound is 20-fold more distributed than the parent drug NF. These findings may explain the superior activity against all stages of T. cruzi forms and lower toxicity of NFOH in relation to NF, and unlike what is observed with hydroxymethyl compounds, which are usually unstable, the results showed that NFOH is chemically stable. We recommend NFOH as a promising trypanocidal compound for the continuation of research toward clinical studies.
ACKNOWLEDGMENTS
This study was supported by Brazilian Research Financial Support Programs CAPES, FAPESP, and PADC-FCF-Unesp.
Footnotes
Published ahead of print 30 September 2013
REFERENCES
- 1. Drugs for Neglected Diseases Initiative 2013. Chagas disease. Global view. http://www.dndi.org/diseases-projects/diseases/chagas.html.
- 2. Moncayo A, Silveira AC. 2009. Current epidemiological trends for Chagas disease in Latin America and future challenges in epidemiology, surveillance and health policy. Mem. Inst. Oswaldo Cruz, 104(Suppl 1):17–30 [DOI] [PubMed] [Google Scholar]
- 3. Coura JR, Viñas PA. 2010. Chagas disease: a new worldwide challenge. Nature 465:S6–S7 [DOI] [PubMed] [Google Scholar]
- 4. Cerecetto H, González M. 2011. Antiparasitic prodrug nifurtimox: revisiting its activation mechanism. Future Microbiol. 6:847–850 [DOI] [PubMed] [Google Scholar]
- 5. Bot C, Hall BS, Bashir N, Taylor MC, Helsby NA, Wilkinson SR. 2010. Trypanocidal activity of aziridinyl nitrobenzamide prodrugs. Antimicrob. Agents Chemother. 54:4246–4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bot C, Hall BS, Alvarez G, Di Maio R, González M, Cerecetto H, Wilkinson SR. 2013. Evaluating 5-nitrofurans as trypanocidal agents. Antimicrob. Agents Chemother. 57:1638–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou L, Stewart G, Rideau E, Westwood NJ, Smith TK. 2013. A class of 5-nitro-2-furancarboxylamides with potent trypanocidal activity against Trypanosoma brucei in vitro. J. Med. Chem. 56:796–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wilkinson SR, Bot C, Kelly JM, Hall BS. 2011. Trypanocidal activity of nitroaromatic prodrugs: current treatments and future perspectives. Curr. Top. Med. Chem. 11:2072–2084 [DOI] [PubMed] [Google Scholar]
- 9. Chung MC, Gonçalves MF, Colli W, Ferreira EI, Miranda MTM. 1997. Synthesis and in vitro evaluation of antichagasic dipeptide prodrugs of primaquine. J. Pharm. Sci. 86:1127–1131 [DOI] [PubMed] [Google Scholar]
- 10. Chung MC, Güido RV, Martinelli TF, Gonçalves MF, Polli MC, Botelho KC, Varanda EA, Colli W, Miranda MT, Ferreira EI. 2003. Synthesis and in vitro evaluation of potential antichagasic hydroxymethylnitrofurazone (NFOH-121): a new nitrofurazone prodrug. Bioorg. Med. Chem. 11:4779–4783 [DOI] [PubMed] [Google Scholar]
- 11. Davies C, Marino CR, Sánchez NO, Mora MC, Chung MC, Basombrío MA. 2010. Hydroxymethylnitrofurazone is active in a murine model of Chagas' disease. Antimicrob. Agents Chemother. 54:3584–3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Doyle PS, Chen CK, Johnston JB, Hopkins SD, Leung SS, Jacobson MP, Engel JC, McKerrow JH, Podust LM. 2010. A nonazole CYP51 inhibitor cures Chagas' disease in a mouse model of acute infection. Antimicrob. Agents Chemother. 54:2480–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Engel JC, Ang KK, Chen S, Arkin MR, McKerrow JH, Doyle PS. 2010. Image-based high-throughput drug screening targeting the intracellular stage of Trypanosoma cruzi, the agent of Chagas' disease. Antimicrob. Agents Chemother. 54:3326–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Testa B, Mayer JM. 2003. Hydrolysis in drug and prodrug metabolism: chemistry, biochemistry, and enzymology, p 148–162 VHCA/Wiley-VCH, Zurich, Switzerland [Google Scholar]
- 15. Stuurman FE, Nuijen B, Beijnen JH, Schellens JH. 2013. Oral anticancer drugs: mechanisms of low bioavailability and strategies for improvement. Clin. Pharmacokinet. 52:399–414 [DOI] [PubMed] [Google Scholar]
- 16. Trossini GH, Malvezzi A, Amaral AT, Rangel-Yagui CO, Izidoro MA, Cezari MH, Juliano L, Chin CM, Menezes CM, Ferreira EI. 2010. Trypanosoma cruzi. J. Enzym. Inhib. Med. Chem. 24:1–6 [DOI] [PubMed] [Google Scholar]
- 17. Guido RVC, Ferreira EI, Nassute JC, Varanda EA, Chung MC. 2001. Decreased mutagenic activity of the prodrug NFOH-121 related to nitrofurazone. Rev. Cienc. Farm. 22:319–333 [Google Scholar]
- 18. Liu JP, Chow SC. 2002. Bridging studies in clinical development. J. Biopharm. Stat. 12:359–367 [DOI] [PubMed] [Google Scholar]
- 19. Samara E, Shaw JP, Barriere SL, Wong SL, Worboys P. 2012. Population pharmacokinetics of telavancin in healthy subjects and patients with infections. Antimicrob. Agents Chemother. 56:2067–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Friis GJ, Bundgaard H. 1996. Design and application of prodrugs, p 351–385 In Krogsgaard-Larsen P, Liljefords T, Madsen UA. (ed), A textbook of drug design and development, 2nd ed. Harwood Academic Publishers, Newark, NJ [Google Scholar]
- 21. Bundgaard H, Johansen M. 1980. Pro-drugs as drug delivery systems. VIII. Bioreversible derivatization of hydantoins by N-hydroxymethylation. Int. J. Pharm. 5:67–77 [Google Scholar]
- 22. Henderson GB, Ulrich P, Fairlamb AH, Rosenberg I, Pereira M, Sela M, Cerami A. 1988. Subversive substrates for the enzyme trypanothione disulfide reductase: alternative approach to chemotherapy of Chagas disease. Proc. Natl. Acad. Sci. U. S. A. 85:5374–5378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jockers-Scherübl MC, Schimer RH, Krauth-SiegeL RL. 1989. Trypanothione reductase from Trypanosoma cruzi: catalytic properties of the enzyme and inhibition studies with trypanocidal compounds. Eur. J. Biochem. 180:267–272 [DOI] [PubMed] [Google Scholar]
- 24. Barbosa CF, Okuda ES, Chung MC, Ferreira EI, Cicarelli RM. 2007. Rapid test for the evaluation of the activity of the prodrug hydroxymethylnitrofurazone in the processing of Trypanosoma cruzi messenger RNAs. Braz. J. Med. Biol. Res. 40:33–39 [DOI] [PubMed] [Google Scholar]