

Abstract
Cryptosporidiosis, caused by the protozoan parasite Cryptosporidium parvum, can stunt infant growth and can be lethal in immunocompromised individuals. The most widely used drugs for treating cryptosporidiosis are nitazoxanide and paromomycin, although both exhibit limited efficacy. To investigate an alternative approach to therapy, we demonstrate that the clan CA cysteine protease inhibitor N-methyl piperazine-Phe-homoPhe-vinylsulfone phenyl (K11777) inhibits C. parvum growth in mammalian cell lines in a concentration-dependent manner. Further, using the C57BL/6 gamma interferon receptor knockout (IFN-γR-KO) mouse model, which is highly susceptible to C. parvum, oral or intraperitoneal treatment with K11777 for 10 days rescued mice from otherwise lethal infections. Histologic examination of untreated mice showed intestinal inflammation, villous blunting, and abundant intracellular parasite stages. In contrast, K11777-treated mice (210 mg/kg of body weight/day) showed only minimal inflammation and no epithelial changes. Three putative protease targets (termed cryptopains 1 to 3, or CpaCATL-1, -2, and -3) were identified in the C. parvum genome, but only two are transcribed in infected mammals. A homology model predicted that K11777 would bind to cryptopain 1. Recombinant enzymatically active cryptopain 1 was successfully targeted by K11777 in a competition assay with a labeled active-site-directed probe. K11777 exhibited no toxicity in vitro and in vivo, and surviving animals remained free of parasites 3 weeks after treatment. The discovery that a cysteine protease inhibitor provides potent anticryptosporidial activity in an animal model of infection encourages the investigation and development of this biocide class as a new, and urgently needed, chemotherapy for cryptosporidiosis.
INTRODUCTION
Cryptosporidium spp. are unicellular apicomplexan protozoan parasites that infect humans and a wide variety of other vertebrates, including mammals, reptiles, fish, and birds (1). Cryptosporidium parvum and Cryptosporidium hominis, the two species that most commonly cause diarrheal illness in humans, also infect livestock (2, 3). C. parvum infects humans and ruminants, whereas C. hominis utilizes an infectious cycle in humans only. Infection with C. parvum most often occurs following either exposure to contaminated surface water directly or ingestion of contaminated food products (4). The parasite completes its entire life cycle in the small intestine, where it gains access to enterocytes by forced invagination and replicates within cytoplasmic parasitophorous vacuoles (5); the infectious cycle results in the clinical pathophysiological symptoms associated with cryptosporidiosis (2).
C. parvum has historically been considered a pathogen of the developing world; however, it is also found in freshwater throughout the world. C. parvum oocysts are notoriously resistant to conventional water purification efforts, and several large waterborne outbreaks have affected hundreds of thousands of people in the United States, Canada, the United Kingdom, and Japan (6). The scope and impact of these outbreaks has raised the specter of the use of C. parvum as an agent of bioterrorism (7, 8). Indeed, C. parvum is classified as a category B pathogen by the United States National Institutes of Health and the Centers for Disease Control and Prevention (7, 8). More than a billion oocysts can be purified from a single infected calf by simple filtration and centrifugation (9), and the infectious dose for humans is as low as 1 to 5 oocysts (10, 11). Although susceptibility to C. parvum infection appears to be relatively uniform in populations in the developed world, the clinical course of infection varies with age and immunological status. Healthy adults typically develop a mild to moderate diarrheal illness lasting 3 to 4 weeks, followed by complete recovery (11). Infection at an early age is also self-limiting but can result in permanent stunting (12). Infection in immunocompromised individuals, such as transplant recipients or HIV/AIDS patients, can persist indefinitely and may be accompanied by severe, life-threatening diarrhea (13). Chemotherapeutic options for cryptosporidiosis are extremely limited, and they depend on the clinical context. Thus, although paromomycin may be used with HIV-positive subjects infected with C. parvum (14), success with the agent is not guaranteed (15). Likewise, nitazoxanide, which is the only FDA-approved drug for the treatment of cryptosporidiosis in immunocompetent patients older than 1 year (16), is not approved for HIV-infected patients (17). Also, clinical trials demonstrated that there were no differences in mortality or parasitological responses between the patients who received nitazoxanide and placebo (18–20). New drugs are clearly needed.
The clan CA (papain-like) family of cysteine proteases (CPs) is a key family of enzymes for many protozoan parasites, including apicomplexans and kinetoplastids. CPs facilitate cell invasion, nutritive degradation of host proteins, and the modification of parasite proteins during life cycle transitions (21–24). MEROPS (25) lists 20 clan CA proteases in C. parvum, and there is little published experimental characterization of these proteins. Of the cryptopains, only cryptopain 1 has been the subject of work published previously (26). Cryptopains are C. parvum clan CA cathepsin L-like (termed CpaCATL according to an alternate nomenclature [27, 28]) proteases that have been identified in the C. parvum genome (29) and shown to be expressed in the sporozoite stage (26). Cryptopain 1 is annotated in the CryptoDB database (30) as cgd6_4880. The two other cathepsin L-like enzymes, which we designate here cryptopains 2 and 3, are annotated as cgd3_680 and cgd7_2850, respectively. Orthologous cathepsin L-like proteases are validated as promising therapeutic targets in extensive in vitro and in vivo studies with the malaria parasite Plasmodium falciparum (31, 32) and the etiological agent of Chagas' disease, Trypanosoma cruzi (33). In particular, investigations with small-molecule inhibitors targeting parasite clan CA enzymes have shown much promise for their eventual use in the therapy of these and other parasitic diseases (32–36).
One such chemical inhibitor, N-methyl-piperazine-Phe-homoPhe-vinylsulfone phenyl (K11777), is orally bioavailable, has a reasonable safety/toxicity profile (35), and is well-advanced in preclinical development for the therapy of Chagas' disease (37). We were interested, therefore, in understanding whether K11777 could limit or prevent the survival of C. parvum in vitro using several mammalian cell lines and in vivo employing the C57BL/6 gamma interferon receptor knockout (IFN-γR-KO) mouse model, which is highly susceptible to C. parvum. We demonstrate that K11777 arrests the growth of C. parvum in human intestinal cell lines at physiologically achievable concentrations. Further, mice are rescued from an otherwise lethal C. parvum infection by K11777 administered either orally (p.o.) or intraperitoneally (i.p.). Inhibitor competition experiments with an active-site probe of recombinant cryptopain 1, along with homology modeling and docking studies, suggest that K11777 binds to and inhibits this protease target.
MATERIALS AND METHODS
Parasites.
Oocysts of C. parvum (Iowa strain) passaged in newborn calves were purified from feces, as previously described (38). The purified oocysts were stored at 4°C in 2.5% aqueous potassium dichromate until use. All experiments were conducted with oocysts within 6 months of purification.
Test compound.
K11777 (N-methyl piperazine-Phe-homoPhe-vinylsulfone phenyl) was originally a gift from James Palmer, Biota, Inc., Victoria, Australia. A stock solution was prepared in dimethyl sulfoxide (DMSO) at 20 mM. The stock solution was kept at 4°C until it was diluted immediately before use with either UltraCulture medium (Lonza, Walkersville, MD) for in vitro studies or phosphate-buffered saline (PBS) for animal studies. All assays included appropriate DMSO controls (0.01 to 0.5% [vol/vol]). Paromomycin (Sigma, Oakville, ON, Canada) was diluted in water just prior to use.
In vitro infection models.
Madin-Darby canine kidney (MDCK) cells (ATCC CCL-34; ATCC, Rockville, MD), the classic tissue culture model for C. parvum, were used and maintained in Dulbecco's modified Eagle's medium (DMEM; Wisent, St. Bruno, Quebec, Canada), as previously described (39). This assay was modified for use with the human intestinal cell lines HCT8 (ATCC CCL-224), I407 (ATCC CCL-6), and CaCo-2 (ATCC HTB-37), which were grown in DMEM, Eagle's minimal essential medium (EMEM; Gibco, Grand Island, NY), and minimal essential medium (MEM) (Wisent), respectively. All media were supplemented with 4 mM l-glutamine, 10 μg/ml gentamicin, 100 U/ml penicillin, 10 μg/ml streptomycin, and 10% fetal bovine serum (FBS). EMEM and DMEM were further supplemented with 1 mM HEPES and nonessential amino acids (Wisent), respectively. All cultures were maintained at 37°C in a humidified, 5% CO2-enriched atmosphere. To assess K11777 activity, cells were then seeded in 2-well coverslip chamber slides (Nalge Nunc International, Naperville, IL) and grown in appropriate media at 37°C in 5% CO2 until nearly confluent. Cell line-specific media were replaced with UltraCulture medium supplemented with 2 mM l-glutamine immediately prior to infection. Oocysts stored in 2.5% potassium dichromate (K2Cr2O7) were washed three times with 0.1 M acetate-NaCl buffer, pH 5.5, and subsequently incubated in 10 mM sodium periodate at 4°C for 20 min. The parasites were then washed three times with PBS, pH 7.4, containing 0.1% bovine serum albumin (BSA) (Sigma). Excystation (release of sporozoites from oocysts) was achieved by incubation in DMEM containing 0.75% sodium taurocholate (Sigma) at 37°C until 50% excystation was determined by microscopy. Each test chamber was inoculated with 1 × 105 excysted C. parvum oocysts. Control slides were either mock inoculated or inoculated in duplicate with 103 to 105 excysted oocysts per chamber. Chamber slides were then cultured for a further 48 h at 37°C in 5% CO2 in the presence of 10 to 100 μM K11777 (6.11 to 61.1 μg/ml), 700 μM paromomycin, or DMSO control with replacement of the UltraCulture medium after 24 h. The slides were processed for immunofluorescent staining by rinsing three times with PBS, fixation for 1 h with Bouin's solution (Sigma), and then decolorization several times with 70% ethanol. The cells were blocked with PBS containing 0.1% BSA for 1 h at 37°C and subsequently labeled with anti-Cryptosporidium monoclonal antibody C3C3 conjugated to Cy3 fluorescent dye (FluoroLink Cy3 reactive dye reagent; Amersham Life Science, Arlington Heights, IL), as previously described (39).
Parasite growth was quantified visually by direct comparison to the series of control slides infected with 103, 104, and 105 parasites. The C. parvum densities in the control slides were assigned scores of 3, 4, and 5, respectively. The relative parasite density observed on test (K11777) and positive-control (paromomycin) slides was estimated in relation to the growth observed on the control slides. Test slides were read in random order and interpreted by an observer masked to the treatment group to avoid potential bias during microscopic examination. Results are reported as the mean values obtained from three independent experiments conducted in duplicate.
Tests for K11777 toxicity in host cells.
Potential inhibition of cell line growth by K11777 was assessed using a modified XTT assay (40). Briefly, the four cell lines were seeded in 96-well flat-bottom plates (BD Falcon, Franklin Lakes, NJ) at 4 × 106 cells/well (in triplicate) and incubated for 24 h and 48 h at 37°C. Culture medium without phenol red was replaced with 100 μl of K11777 diluted in the appropriate medium (without phenol red) to yield inhibitor concentrations of 10 to 100 μM. Paromomycin (700 μM) was also tested. At 24 h and 48 h, 25 μl each of XTT (Sigma) at 250 μg/ml and menadione (Sigma) at 10 μM was added to each well. After 4 h at 37°C, plates were measured at dual wavelengths of 450 and 650 nm using an automated enzyme-linked immunosorbent assay (ELISA) plate reader (EL800 BioTek; Fisher, Nepean, ON, Canada) (40).
Animal model of C. parvum infection.
C57BL/6 IFN-γR-KO mice were purchased from Jackson Laboratories (Bar Harbor, ME). A breeding colony was maintained at the Research Institute of the McGill University Health Center in an isolation room under specific-pathogen-free conditions. Cages, food, water, and bedding were sterilized before use. Between 6 and 8 weeks of age, male and female animals were infected with 1,500 sporulated oocysts suspended in 100 μl PBS using a 20-gauge gavage needle (CDVM; St. Hyacinthe, Quebec, Canada). Mice were divided into 14 groups (8 animals/group). Groups 1, 2, and 3 were infected and treated with K11777 p.o. at 35, 70, and 105 mg/kg of body weight twice a day (BID), respectively. Groups 4, 5, and 6 were infected and treated with K11777 intraperitoneally (i.p.) at 35, 52.5, and 70 mg/kg BID, respectively. Groups 7, 8, and 9 were infected controls, mock treated (p.o.), treated with 0.5% DMSO p.o., and treated with 0.5% DMSO i.p., respectively. Groups 10, 11, 12, 13, and 14 served as uninfected controls, mock treated (p.o.), treated with K11777 p.o. at 105 mg/kg BID, treated with K11777 i.p. at 70 mg/kg BID, treated with 0.5% DMSO p.o., and treated with 0.5% DMSO i.p., respectively. Group 15 was infected and treated with paromomycin p.o. at 125 mg/kg BID. Treatment began 4 days after infection for a period of 10 days. All animals were sacrificed on day 35 after infection. In this model, untreated infected animals are visibly ill by 6 to 7 days after infection, and most either are dead or must be euthanized by 8 to 14 days. To determine whether the mice infected and treated with K11777 subsequently relapse, 4 mice in each of the treated groups were observed for a further 3 months after the endpoint (35 days after infection) of the study. Results were obtained from three independent experiments.
All animal studies were performed in accordance with institutional animal care and use guidelines and were approved by the Animal Care and Use Committee at McGill University. The study was approved by the Research Ethics Committee of the McGill University Health Centre at the Montreal General Hospital.
Flow cytometry for oocyst shedding.
Mouse fecal samples were collected prior to inoculation and every 3 days after infection until the end of the experiment (day 35) and then subjected to discontinuous sucrose gradient centrifugation (38). Briefly, three or four fecal pellets were homogenized at each time point in 2.5% K2Cr2O7 and stored at 4°C until they were processed further. The samples were vortexed and allowed to stand for large debris to settle. Supernatant aliquots were overlaid onto discontinuous sucrose gradients (1.103 and 1.064 M) in microcentrifuge tubes and centrifuged at 1,000 × g for 20 min. The interface between the two sucrose solutions was then collected, washed with saline, and suspended in PBS. The purified stool concentrates were incubated for 30 min at 37°C with 5 μl of an oocyst-specific monoclonal antibody conjugated to fluorescein isothiocyanate (OW50-FITC) and analyzed by flow cytometry, as previously described (41).
Histopathology.
Samples from selected tissues were formalin fixed and paraffin embedded, and 4-μm-thick sections were stained with hematoxylin and eosin for examination by light microscopy.
Expression profile of cryptopains in C. parvum.
To confirm mRNA expression of cryptopain genes, total RNA extracted from oocysts (using the Qiagen RNAeasy kit) was subjected to reverse transcription (RT)-PCR using the Qiagen One Step RT-PCR kit according to the manufacturer's instructions. The respective primer pairs for cryptopains 1, 2, and 3 were as follows: forward, 5′-ATTGTGGGTCATGTTGG-3′, and reverse, 5′-CTCTCCCCACGCTTCACC-3′; forward, 5′-CAGTAAAAGCTTGTAGTGG-3′, and reverse, 5′-TGGTTCTCTATTTAAACAAT-3′; and forward, 5′-ACAAAAGTTGGTACTGCC-3′, and reverse, 5′-ATTCTGATGAAACTTGACGG-3′. These primer pairs generate amplicons of approximately 524, 551, and 575 bp, respectively. The PCR conditions were as follows: step 1, 98°C for 5 min; step 2, 39 cycles of 98°C for 1 min, 55°C for 1 min, and 72°C for 30 s; and step 3, 72°C for 10 min.
Cloning and heterologous expression of cryptopain in P. pastoris.
The putative cryptopain 1 zymogen, devoid of the signal sequence, was cloned into the vector pPICZ alpha A for expression in the yeast Pichia pastoris. The following forward and reverse primers, incorporating XhoI and NotI restrictions sites, respectively, were used as described previously (42): forward, 5′-ATACTCGAGAAAAGAGATTTCGTACCTGGTGATTATGTTGATCCAGCAACTA-3′, and reverse, 5′-TATGCGGCCGCTTATATTGATTGATTAATCACTGGATACAC-3′. Yeast was transformed and induced to express protein as described previously (42).
Active-site competition binding assay using K11777 and the active-site directed probe DCG-04.
P. pastoris clones that secreted processed and active recombinant cryptopain 1 were selected by labeling induction media with the cysteine protease active-site affinity label, 125I-DCG-04 (data not shown). DCG-04 is a covalent probe that binds to the active site of cathepsin L-like cysteine proteases (43). Positive clones containing active cryptopain 1 were incubated at room temperature in the absence or presence of 5 μM K11777 for 15 min in 50 mM sodium acetate (NaOAc), pH 5.5, 250 mM NaCl, 1 mM dithiothreitol (DTT) in a final volume of 100 μl prior to the addition of 1 μl of 125I-DCG04 or 1 μl carrier solvent (acetonitrile). The reaction was continued for 30 min at room temperature before resolving the samples by SDS-PAGE and visualizing the 125I-DCG04-bound recombinant cryptopain 1 by phosphorimaging and fluorography.
Molecular modeling of the cryptopain-K11777 molecular interaction.
The homology model (“crypto1M”) of cryptopain 1 (CryptoDB identifier [ID], cgd6_4880) was created with the Protein Local Optimization Program (PLOP) (the in-house modeling program written and maintained by the Jacobson laboratory at the University of California, San Francisco [UCSF]) (http://www.jacobsonlab.org/plop_manual/plop_overview.htm), using as the template human cathepsin V complexed with a vinyl sulfone inhibitor (Protein Data Bank [PDB] ID 1FH0) (44). The template structure was chosen from the results of a BLAST search of cgd6_4880 versus the PDB because it had a significant BLAST E value (9e−48) and a reasonable sequence identity to the query (46%). In addition, the crystallography statistics for 1FH0 were high quality (crystal resolution = 1.6 Å; R value = 0.198; Rfree = 0.211), in addition to having a cocrystallized ligand similar to the inhibitor K11777, which would contribute to a more accurate modeling of the active site with the ligand bound. The K11777 ligand was prepared from a SMILES file using Ligprep (Schrodinger LLC, New York, NY). Docking was performed using PRIME docking, a molecular-mechanics energy function-based anchor-and-grow docking program. Structural alignments, structure comparison analyses, and generation of figures were performed using the program Chimera (45). Protein surface rendering and coloring were performed in Chimera.
Multiple-sequence alignment of C1 C. parvum peptidases.
A multiple-sequence alignment (MSA) was generated by inputting all five C1 C. parvum sequences, as well as the PDB IDs for cruzain (2OZ2) and human cathepsin L (1FH0), into PROMALS3D (46) at the PROMALS web server (http://prodata.swmed.edu/promals3d/promals3d.php) using default settings. PROMALS3D incorporates predicted secondary structure, as well as the available three-dimensional (3D) structure(s), in computing an MSA.
Statistical analysis.
Inhibition of parasite growth in vitro and stool oocyst numbers were analyzed by a paired Student's t test. Mantel-Haenszel survival analysis was applied to the animal model (47, 48). Repeated-measures analysis of variance (ANOVA) was used to analyze oocyst excretion. Post hoc least significant difference (LSD) was used to identify the difference between the means of oocyst excretion. A P value of <0.05 was considered significant for all tests.
RESULTS
K11777 eliminates C. parvum infection from cell lines in vitro.
Initial experiments demonstrated that the timing of drug administration relative to infection (i.e., 30 min prior to infection, simultaneous with infection, or up to 24 h postinfection) had no impact on the outcome. At 10 μM, K11777 had no effect on C. parvum growth in any of the cell lines. At 20 μM, 1- to 2-log-unit reductions in parasite numbers were observed in all cell lines (Table 1). At 40 μM, parasite growth was reduced by 3 and 4 log units in MDCK and human intestinal colonic adenocarcinoma (CaCo-2) cell lines, respectively (P < 0.001). Complete elimination of C. parvum was achieved at 40 μM in human ileocecal adenocarcinoma (HCT8) and human fetal enterocyte (I407) cell lines. At 60 μM K11777, complete elimination of C. parvum was observed in all four cell lines. In contrast, 700 μM paromomycin produced only a 1- to 2-log-unit reduction in parasite numbers in all of the cell lines tested.
Table 1.
Effects of K11777 and paromomycin tested against C. parvum in vitro and lack of cytotoxicity to host cells
| Parametera | Scoreb for cell line: |
|||
|---|---|---|---|---|
| MDCK | HCT8 | I407 | CaCo-2 | |
| Drug effect | ||||
| K11777 (μM) | ||||
| 10 | 5 | 5 | 5 | 5 |
| 20 | 4 | 3 | 3 | 3 |
| 40 | 2 | 0 | 0 | 1 |
| 60 | 0 | 0 | 0 | 0 |
| 80 | 0 | 0 | 0 | 0 |
| 100 | 0 | 0 | 0 | 0 |
| Control (105 parasites + monolayer) | 5 | 5 | 5 | 5 |
| 0.5% DMSO + parasites | 5 | 5 | 5 | 5 |
| Paromomycin (700 μM) | 4 | 3 | 3 | 4 |
| Cytotoxicity | ||||
| 60 μM K11777 + monolayer | − | − | − | − |
| 80 μM K11777 + monolayer | + | − | − | − |
| 100 μM K11777 + monolayer | ++ | − | + | − |
| 700 μM paromomycin + monolayer | − | − | − | − |
| 0.5% DMSO + monolayer | − | − | − | − |
For drug effects, cell lines were grown in 2-well coverslip chamber slides in appropriate media at 37°C in 5% CO2. Each test chamber was inoculated with 1 × 105 excysted C. parvum oocysts. Control slides were either mock inoculated or inoculated in duplicate with 103 to 105 excysted oocysts per chamber. Infected cells were washed and incubated in UltraCulture in the presence of various concentrations of K11777, 700 μM paromomycin, or 0.5% DMSO control. The slides were then processed for immunofluorescent staining. For cytotoxicity, uninfected monolayers were incubated in various concentrations of K11777, paromomycin, or 0.5% DMSO.
For drug effects, the score was estimated in a log scale. A score of 5 means growth equivalent to that in control wells infected with 105 sporulated oocysts. At 60 μM K11777, complete inhibition of C. parvum growth was achieved in all cell lines. Results are reported as the mean values obtained from three independent experiments, each conducted in duplicate. For cytotoxicity of K11777 on host cells, scores were as follows: −, no visible toxic effects; +, slight morphological changes/monolayer still intact; ++, 5 to 10% of the monolayer lifted.
K11777 has limited toxicity in vitro.
We next assayed for any direct cytotoxic effects of K11777 on the same cell line in the absence of C. parvum infection (Table 1). As determined by microscopy and the XTT/menadione assay (40), neither K11777 (from 10 to 60 μM) nor DMSO at control concentrations (0.1% and 0.5%) affected the morphologies or growth rates of the four cell lines used. At 80 μM, K11777 produced subtle morphological changes only in MDCK cells (slight rounding and increased refraction in ∼5% of the monolayer) although maintaining >98% of viability. In addition, at 100 μM, 5 to 10% of the MDCK cell monolayer detached with ≥95% of viable cells; however, no effects were observed for the human cell lines.
K11777 depresses or eliminates oocyst shedding from C. parvum-infected mice.
All infected mice began excreting oocysts on day 6 or 7 after infection (2 or 3 days after treatment began). Oocyst numbers increased rapidly in all untreated groups and reached high levels (7,356 to 27,560/100 μl culture medium) in animals surviving 12 to 14 days (Fig. 1A and B). Treatment with K11777 p.o. or i.p. and at all doses decreased oocyst shedding; p.o. treatment was more effective. Differences in oocyst numbers between untreated and treated animals reached significance as early as day 9 for the p.o.-treated groups (9,746 ± 2,079/100 μl for untreated versus 1,305 ± 492/100 μl for treated animals; P < 0.009) and by day 14 for the i.p.-treated groups (20,608 ± 2,332/100 μl for untreated versus 5,365 ± 2,665/100 μl for treated animals; P < 0.007). As observed in the in vitro experiments, as well in the in vivo experiments, the DMSO vehicle did not have any lethal effect on C. parvum. All infected mice from the mock-treated and DMSO-treated groups died or were euthanized by day 14. Overall reductions in the numbers of oocysts shed during the first 14 days were highly significant (P < 0.001). At day 35, this reduction was 98.6 and 79.9% in the p.o.- and i.p.-treated animals, respectively. At day 12, the area under the curve (AUC) was also highly significant, with values of 5,528, 19,940, and 79,162 for the p.o.- and i.p.-treated animals and untreated animals, respectively (P < 0.0001).
Fig 1.

K11777 eliminates oocyst shedding from C. parvum-infected mice. Animals (8 mice/group) were infected with 1,500 sporulated oocysts and 4 days later were treated with K11777 BID for 10 days p.o. (A) or i.p. (B). The mice were euthanized on day 35 after infection. (A) P.o. treatment at 35 mg/kg (●), 70 mg/kg (■), and 105 mg/kg (▲) and BID treatment with paromomycin p.o. at 125 mg/kg (◆). Infected controls were mock treated (+) and treated with 0.5% DMSO (asterisks). (B) I.p. treatment at 35 mg/kg (▼), 52.5 mg/kg (◆), and 70 mg/kg (○). Infected controls were mock treated (+) and treated with 0.5% DMSO (×). The results are expressed as the mean number of oocysts shed per group ± standard deviations (SD); n = 3 independent experiments. The number of oocysts was determined by flow cytometry.
K11777 rescues mice from an otherwise lethal infection with C. parvum.
As previously reported (39), IFN-γR-KO mice are exquisitely sensitive to C. parvum infection. In preliminary experiments, inocula as low as 10 sporulated oocysts resulted in >50% mortality, whereas inocula in excess of 500 sporulated oocysts were universally 100% lethal.
Four days after infection and for a period of 10 days, K11777 was administered BID to IFN-γR-KO mice either p.o. (Fig. 2A) or i.p. (Fig. 2B) (the data are displayed as Kaplan-Meier survival plots). Mice infected with 1,500 sporulated oocysts of C. parvum and not treated with K11777 were noticeably sick by days 7 to 9 postinfection (e.g., hunched posture, ruffled fur, decreased movement, and weight loss), and all had died or had to be euthanized based on preestablished criteria by day 14; 80% died or were euthanized between days 8 and 11.
Fig 2.

K11777 rescues mice from otherwise lethal infections by C. parvum. Animals (8 mice/group) were infected with 1,500 sporulated oocysts and 4 days later were treated with K11777 BID for 10 days p.o. (A) or i.p. (B). The mice were euthanized on day 35 after infection. (A) P.o. treatment at 35 mg/kg (●), 70 mg/kg (■), and 105 mg/kg (▲) and BID treatment with paromomycin p.o. at 125 mg/kg (◆). Infected controls were mock treated (+) and treated with 0.5% DMSO (asterisks). Uninfected controls were mock treated (□) and treated with 0.5% DMSO p.o. (▽). (B) I.p. treatment at 35 mg/kg (▼), 52.5 mg/kg (◆), and 70 mg/kg (○). Infected controls were mock treated (+) and treated with 0.5% DMSO i.p. (×). Uninfected controls were mock treated (□) and treated with 0.5% DMSO (▽).
For the K11777 treatment groups (either p.o. or i.p.), mice became obviously ill 8 to 10 days postinfection and exhibited the same behavior as the infected, untreated groups. However, for the p.o. administration regimen, there was a dose-dependent increase in survival of mice such that 90% of the mice that received 125 mg/kg K11777 BID survived (P < 0.01). Even at 35 mg/kg BID p.o., over 60% of mice survived the infection. In contrast, only 40% of mice treated with 125 mg/kg of paromomycin p.o. BID survived. For the i.p. administration regimen, dose dependency was less apparent; nevertheless, 75% of the mice survived after treatment with the highest dose of 140 mg/kg/day K11777 (P < 0.01). By day 20 to 25 postinfection (10 to 15 days after completion of therapy), the surviving animals were completely healthy in appearance and behavior, and most had regained their premorbid weight. There was also no gross or behavioral evidence that K11777 or DMSO was toxic at the doses used in the uninfected mice. Mice infected and treated with K11777 and observed for a further 3 months after the endpoint (35 days after infection) of the study were free of oocysts in their stools and intestines. In contrast, the paromomycin-treated mice were positive for oocysts.
K11777 treatment resolves intestinal pathology due to C. parvum infection.
Animals were necropsied, and selected tissues (small intestine, colon, liver, and kidney) were processed for histopathology. Sections of the small intestine from C. parvum-infected mice without K11777 treatment displayed marked acute inflammation, mild villous blunting, and abundance of C. parvum intracellular life cycle stages (Fig. 3A). In contrast, sections from the intestines of mice infected with C. parvum and treated p.o. with 105 mg/kg K11777 BID showed minimal focal inflammation in only 10% of the mice (Fig. 3B) and had small numbers of life cycle stages. The small intestines from uninfected mice treated with K11777 were also normal (Fig. 3C). Sections of liver and kidney were normal in all mouse groups (data not shown). Sections of small intestines from mice infected with C. parvum and treated p.o. with 125 mg/kg paromomycin BID showed focal inflammation and an abundance of C. parvum life cycle stages (Fig. 3D).
Fig 3.

K11777 treatment resolves intestinal pathology due to C. parvum infection. (A and B) Small intestines from C. parvum-infected mice either untreated (A) or treated p.o. with 105 mg/kg K11777 BID as described for Fig. 1 and 2 (B). (C) Small intestine from uninfected mice that were treated p.o. with 105 mg/kg K11777 BID. (D) Small intestine from C. parvum-infected mice that were treated p.o. with 125 mg/kg paromomycin BID. The boxed areas are shown enlarged below (A to C) and to the right (D). Note the marked acute inflammation and abundance of C. parvum (yellow arrows) in panel A. Small intestines were harvested at 14 days (A) and 35 days (B, C, and D) after infection. Transverse sections of the small intestine were stained with hematoxylin and eosin. Magnification, ×400. Scale bars = 24 μm.
Oocysts following in vivo exposure to K11777 are not viable.
We asked whether the small number of sporozoites/oocysts found in the stools and small intestine after exposure to K11777 (Fig. 3B) might be viable. Stools and intestines were harvested, and oocysts were purified as described previously (39). Lack of infectivity was verified by inoculating naive mice with oocysts. These oocysts did not excyst and were therefore incapable of establishing an infection in the mice whereas the oocysts from control infected but untreated mice (Fig. 3A) were able to excyst.
Two cysteine proteases (cryptopains) are transcribed in C. parvum.
The cryptopain 1 sequence (GenBank XM_627814) was used to interrogate the C. parvum genome (http://cryptodb.org/cryptodb/; 30) for homologous clan CA cysteine proteases. Five clan CA family C1 cysteine proteases were identified—two cathepsin C-like sequences (cgd4_2110 and cgd2_3320) and three cathepsin L-like sequences (cgd6_4880, cgd3_680, and cgd7_2850), which we term cryptopains 1, 2, and 3, respectively. Our RT-PCR analysis revealed that cryptopains 1 and 2 are transcribed in the parasite, but not cryptopain 3 (Fig. 4). A previous study showed that cryptopain 1 is also expressed in sporozoites (26). XP_627814, the corresponding protein sequence for GenBank XM_627814, is 100% identical to cgd6_4880 (i.e., cryptopain 1). Peptidyl vinyl sulfones like K11777 were developed in a medicinal chemistry program to inhibit human cathepsins in vivo, including cathepsins B, L, and S, but not cathepsin C (49, 50). Also, K11777 is known to target cysteine proteases in a number of parasitic organisms, including “cruzain,” the cathepsin L-like protease target in T. cruzi (33, 51), and the falcipains of P. falciparum (52). Accordingly, we hypothesized that the cathepsin L-like cryptopain 1 and/or 2 is likely an in vivo target for K11777.
Fig 4.

Cryptopains 1 and 2, but not 3, are transcribed in C. parvum. RNA preparations were DNase treated and reverse transcribed, and the cDNA was subjected to PCR using specific cryptopain primers. Arrows indicate cryptopain.
K11777 inhibits recombinant cryptopain.
Recombinant cryptopain 1 was functionally expressed in the yeast P. pastoris as a secreted soluble protein. The enzyme was active and was processed to the mature catalytic domain, as evidenced by mobility on SDS-PAGE of the enzyme preincubated with the radiolabeled active-site affinity probe, 125I-DCG-04 (43), which irreversibly alkylates the active-site cysteinyl amino acid residue (Fig. 5). The recombinant enzyme could be completely inhibited by K11777, and this inhibition prevented binding of the active-site cysteine by 125I-DCG-04, confirming that cryptopain 1 is a likely target of the inhibitor (Fig. 5). The possibility that cryptopain 2 is also a target for K11777 cannot be excluded; however, attempts to express functional recombinant enzyme for inhibitor analyses were unsuccessful.
Fig 5.
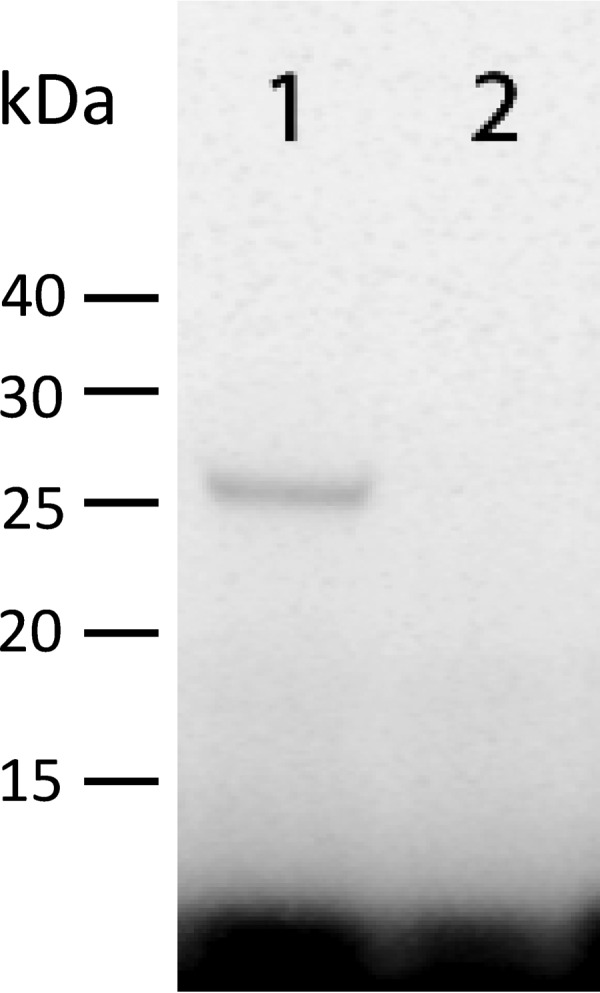
K11777 reacts with the active site of cryptopain 1. Lane 1, recombinant cryptopain 1, expressed in P. pastoris, labeled with the active-site-directed probe 125I-DCG-04 (43), and then subjected to SDS-PAGE and autoradiography; lane 2, same as lane 1 except that recombinant cryptopain was preincubated with 5 μM K11777, which subsequently prevented the reaction of the enzyme with the affinity probe.
K11777 bound to a homology model of cryptopain.
A homology model of cryptopain 1 was produced using human cysteine protease cathepsin V (46% sequence identity), the structure of which has been determined by X-ray crystallography (PDB ID 1FH0) (44). The model crypto1M was structurally aligned with the crystal structure of cruzain (a cathepsin L-like protein of T. cruzi) bound to K11777 (PDB ID 2OZ2) (53). This structural alignment confirmed the superimposition of the active-site catalytic triad (crypto1M/2OZ2 numbering: Cys24/25, His164/162, and Asn186/182) and the Gln18/19 that helps orient the catalytic His (see Fig. S1 in the supplemental material), in addition to an overall agreement between the two sets of coordinates (root mean square deviation [RMSD] = 0.694 Å between 155 alpha carbon pairs). The other C01 C. parvum proteases were not modeled, as they had lower sequence identity (30% to 39%) to the best available templates and would have yielded less reliable models.
K11777 binds covalently to cruzain and crypto1M. In order to examine the binding mode of K11777 to the crypto1M model, we used PRIME docking, a molecular-mechanics energy function-based anchor-and-grow docking program (54) that functions as follows: (i) PRIME docking builds the ligand from a core fragment in an arbitrary conformation; (ii) it then samples the ligand conformations by varying the dihedral angles at 10° resolution, (iii) the conformations are clustered, (iv) a representative conformation from each cluster is energy minimized, and (v) the conformation from the lowest-energy cluster is identified as the docking pose. First, we tested the ability of PRIME docking to reproduce the binding mode of K11777 against the cruzain active site (Fig. 6A). We treated the backbone and side chain atoms of the active-site Cys residue as the core fragment. The protein was held fixed during docking. The heavy-atom RMSD between the predicted and crystal structure pose is 2.0 Å. A similar docking strategy was used to dock K11777 to the crypto1M model (Fig. 6B). We subsequently aligned the K11777-bound crypto1M model to the cruzain structure (2OZ2) and evaluated the heavy-atom RMSD between the inhibitor poses in the cruzain crystal structure and the crypto1M model. The heavy-atom RMSD of the docked pose is 2.3 Å compared to the cruzain crystal structure pose. The S1, S2, and S3 subsites of the enzyme correspond to the homoPhe, Phe, and N-methylpiperazine, respectively, of the inhibitor. In both cruzain and crypto1M, Phe at P2 extends most deeply into the respective S2 subsites. The docked K11777 binds to the same active site in the homology model that K11777 binds to in the cruzain crystal structure, and the orientation is in good general agreement with that of the crystal structure (53).
Fig 6.

(A) Redocking of K11777 to cruzain crystal structure (PDB ID 2OZ2). (B) Docking of K11777 to crypto1M model. The docking results are shown as gray carbon, and crystal structure pose is shown as orange carbon. Nitrogen, oxygen, hydrogen, and sulfur atoms are shown in blue, red, white, and yellow, and binding-pocket residues are shown in a surface representation with the same color scheme.
At the bottom of the S2 pocket in both the model crypto1M and the crystal structure of cruzain is a Glu residue; however, in the corresponding position in human cathepsin L (1FH0), this key amino acid residue is Ala. This residue is highlighted in Fig. S1 in the supplemental material. Although the residue in cryptopain 1 does not aid in binding K11777, it can be an important indicator of specificity in clan CA peptidases and can certainly be critical in binding and stabilizing an incoming P2 Arg (55). Conventionally, in cathepsin L-like peptidases, this is a small hydrophobic residue, and only a few parasites, including P. falciparum (falcipain1), Plasmodium vivax (vivipain), and Toxocara (cathepsin L1), have this quite unusual substitution with a Glu residue. Unlike cathepsin L-like peptidases, it is known that vertebrate cathepsin B-like peptidases all have a Glu in this position (http://merops.sanger.ac.uk; 55).
Figure 7 shows the multiple-sequence alignment of all the family C01 peptidases from C. parvum, as well as cruzain (2OZ2) and human cathepsin L (1FH0) for comparison. As described in Materials and Methods, the MSA incorporated predicted secondary structure and available 3D structures in the alignment calculation. Conservation of active-site residues (Q, C, H, and N) is strict, whereas the residue at the bottom of the S2 binding pocket is variable and occurs in a divergent region, so the identity of the residue should be observed with caution. It is likely that only cryptopain 1 and human cathepsin L have a Glu here (Fig. 7, highlighted with a pink box). The full MSA is shown in Fig. S2 in the supplemental material.
Fig 7.

MSA of family C01 peptidases from C. parvum showing active-site residue regions. All five C01 C. parvum sequences, as well as PDB IDs for cruzain (2OZ2) and human cathepsin L (1FH0), were used as input into PROMALS3D (see Materials and Methods), which incorporates predicted secondary structure and available 3D structure in calculating the MSA. The active-site residues (Q, C, H, and N, in blue boxes) are strictly conserved, but the S2 pocket Glu in cryptopain 1 is in a variable region and appears to be conserved only in cruzain (pink box). Cathepsin L-like sequences (cryptopains) are cgd6 (cryptopain 1), cgd3 (cryptopain 2), and cgd7 (cryptopain 3); cathepsin C-like sequences are cgd2 and cgd4. Higher conservation is shown by higher numbers above the alignments (Conservation). Strictly conserved amino acids are shown as boldface uppercase letters (Consensus_aa). “Consensus_ss” indicates where conserved secondary structure is predicted: e, beta strand (blue letters); h, alpha helix (red letters).
DISCUSSION
Several observations and events in recent history have raised the profile of C. parvum as a serious threat to human health (6, 10). Foremost among these are the impact of cryptosporidial diarrhea in AIDS patients and the permanent stunting of physical growth of children infected at an early age (56). Also, the resistance of C. parvum oocysts to routine water purification, the simplicity of parasite manipulation, the low infectious dose, and the massive disruption caused by waterborne outbreaks (e.g., 1993 in Milwaukee, WI) have resulted in the classification of C. parvum as a category B bioterrorism pathogen (7).
Proteases are critical in cell biology, and those in parasites are implicated in many aspects of the host-parasite interaction (reviewed in reference 24). For example, proteases are essential for host cell invasion, excystation, and intracellular replication by protozoan parasites, including Plasmodium spp. (57), Leishmania mexicana (58) and Toxoplasma gondii (59). Protease activity is also associated with C. parvum excystation and host cell invasion (60–62). Targeting pathogen proteases for therapy has been successful in the control of HIV infection (63) and has spurred the search for novel protease inhibitors to treat other infectious diseases. Indeed, over the last 20 years, small-molecule inhibitors of clan CA cysteine proteases from parasites have been validated as drug leads for amelioration, if not cure, of many protozoan (64) and helminthic (35, 36) diseases.
K11777 is a clan CA selective inhibitor now in late-stage preclinical investigation for treatment of Chagas' disease (37). K11777 targets the resident cathepsin L-like cysteine protease activity in T. cruzi, termed cruzain. The compound has reasonable oral bioavailability and no major toxicity concerns in rodents, dogs, and primates (35). In the current study, K11777 was evaluated in vitro and in animals with severe immune deficiency (mimicking cryptosporidiosis in AIDS patients). We demonstrate that K11777 eliminates C. parvum infection from cell lines in vitro and rescues highly susceptible IFN-γR-KO mice from a uniformly lethal challenge. Consistent with the known mechanism of action of K11777 for clan CA proteases, this potent anticryptosporidial activity is hypothesized to be the result of K11777 inhibiting the C. parvum cathepsin L-like cryptopain enzymes. Significantly, K11777, both in vitro and in vivo, was more effective than the current anticryptosporidial drug, paromomycin.
The in vitro efficacy of K11777 against C. parvum observed here is consistent with its activity against other parasites, including T. cruzi (33) and P. falciparum (32). The present observations are also consistent with the conserved nature of the target cathepsin L-like proteases among protozoa (55). Using both the “classic” MDCK model and three human intestinal cell line models, we demonstrated complete inhibition of C. parvum growth with K11777 concentrations of ≥60 μM. It is significant that when Armson et al. used MDCK cells to screen 71 compounds for activity against C. parvum (65), the positive control, paromomycin, achieved only 75 to 85% inhibition of growth at a concentration (3.2 mM) 53 times that of the present compound. Studies in our human cell line models (HCT8, I407, and CaCo-2) suggest that complete inhibition of C. parvum growth can be achieved with K11777 concentrations as low as 20 μM when the drug is applied twice (at time zero and after 24 h). However, the inferences regarding the therapeutic potential that can be drawn from tissue culture models of C. parvum are limited by the nature of those models. To date, it has not been possible to achieve the full replicative cycling of C. parvum in vitro, i.e., parasites invade and replicate in a direct, linear fashion through sexual stages, but efficient production of oocysts does not occur. Likewise, no significant “recycling” of asexual stages is observed and no “autoinfective” cycle has been demonstrated, where thin-walled oocysts release sporozoites and “restart” the life cycle. Nonetheless, these in vitro models can be informative about compounds that interfere with intracellular growth, a key stage in the amplification of parasite numbers, and the in vitro data obtained for K11777 encouraged us to pursue efficacy testing of the compound in a mammalian model of C. parvum infection.
Although several small-animal models have been described for C. parvum, including dexamethasone-treated (66) and SCID (67) mice, we opted for IFN-γR-KO mice for the simplicity of their manipulation and the clarity of the outcome (68). These mice are highly sensitive to C. parvum. As few as 10 oocysts reliably infect a large proportion of animals (69). Once symptoms develop, disease progresses inexorably to death in 9 to 14 days. We used 1,500 sporulated oocysts in our studies, achieving a 100% infection rate, as evidenced by symptoms and shedding of oocysts. Almost 80% of the infected control mice in our studies died or had to be euthanized between days 8 and 11.
Four days after establishing infections, mice were treated BID with K11777 for 10 days. During and after treatment with K11777, oocyst shedding gradually decreased to very low levels or disappeared by day 35 postinfection. The overall reductions in oocyst numbers during treatment were more pronounced in the animals treated p.o. (mean, 27.8 ± 15.4 oocysts/day during the last 2 weeks of the experiment) than in the mice that were treated i.p. (mean, 2,507.8 ± 735.6 oocysts/day; P < 0.01). Given that the location of C. parvum replication is in parasitophorous vacuoles at the apices of intestinal enterocytes, it is perhaps not surprising that the p.o. route was more efficient than the i.p. route. This may also explain why relatively low doses of K11777 (35 mg/kg or 70 mg/kg BID), given p.o., had a significant impact on C. parvum infection in vivo (Fig. 2). In contrast to previous studies using immunosuppressed rats treated with azithromycin (70) or SCID mice treated with maduramicin (67), we did not observe recrudescent cryptosporidial infection upon cessation of K11777 treatment. For a small number of mice monitored for up to 3 months after cessation of therapy, there was no evidence of infection.
We observed no significant side effects attributable to K11777 at the highest BID doses used in our studies, namely, 105 mg/kg/day p.o. and 70 mg/kg/day i.p. K11777 is well tolerated when given to C3H mice at doses as high as 667 mg/kg/day p.o. and 222 mg/kg/day i.p. (33). Also, rats treated with K11777 showed no toxic effects at plasma concentrations up to 76 μM in males and 118 μM in females (35). Our own observations of significant inhibition (90 to 99%) of C. parvum growth in vitro with K11777 at 10 to 20 μM and efficacy in vivo following 35- to 70-mg/kg BID dosing regimens suggest that the therapeutic index for the agent is acceptable. The higher therapeutic index of K11777 may be due, in part, to the active uptake of the inhibitor by the parasite, decreased functional redundancy (paralogs and orthologs) in the parasite's protease repertoire relative to the mammalian host (71, 72), and higher concentrations (millimolar) of cysteine proteases in host cell organelles (64, 73, 74).
Because K11777 targets clan CA cathepsin-like cysteine proteases, the genome of C. parvum was mined for similar enzymes. Three cathepsin L-like proteases, termed cryptopains 1, 2, and 3, were identified. Only cryptopains 1 and 2 were actively transcribed in the infectious stage of the parasite, and accordingly, they are likely targets of K11777 in vivo. Support for this hypothesis comes first from structural homology modeling of cryptopain 1 and computer docking of K11777 in the model of cryptopain 1 (crypto1M), which demonstrated the similarity of the binding site to that of the cathepsin L-like protease cruzain (53). Second, binding by the radiolabeled clan CA active-site-directed probe, 125I-DCG-04 (43), in the active site of recombinant cryptopain 1 could be blocked by K11777.
In conclusion, we demonstrated that the vinyl sulfone cysteine protease inhibitor K11777, which is currently in late stage preclinical studies for the treatment of Chagas' disease, displays potent anticryptosporidial activity. The inhibitor arrests C. parvum infection in human gastrointestinal cell lines and rescues otherwise lethal infections in a murine model of C. parvum infection at concentrations/doses comparable to or lower than those of the current drug therapy, paromomycin. The toxicity of K11777 seems to be low. Homology modeling, ligand docking, and experimental results using recombinant protease provide evidence that cryptopain 1 is a target of K11777 and thus represents one feasible mechanism for the rescue of C. parvum infection in mice using the drug. However, this does not exclude the possibility that other clan CA family C1 cysteine proteases in C. parvum may be additional targets of the compound, and further validation would be required to show that cryptopain(s) is indeed the target(s) of K11777 in vivo. Other small-molecule inhibitors of cysteine proteases should also be evaluated as potential drug leads for treatment of cryptosporidiosis, given the pressing need for new drugs.
Supplementary Material
ACKNOWLEDGMENTS
We thank Christopher McClendon of UCSF for help with use of the Maestro programs.
The study was supported by the Sandler Foundation, the Foundation of the Montreal General Hospital, and the Research Institute of the McGill University Health Centre. Additional support was provided by the PhRMA Foundation (a Postdoctoral Fellowship in Informatics to S.T.M.).
Footnotes
Published ahead of print 23 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00734-13.
REFERENCES
- 1. Fayer R, Santin M, Macarisin D. 2010. Cryptosporidium ubiquitum n. sp. in animals and humans. Vet. Parasitol. 172:23–32 [DOI] [PubMed] [Google Scholar]
- 2. O'Donoghue PJ. 1995. Cryptosporidium and cryptosporidiosis in man and animals. Int. J. Parasitol. 25:139–195 [DOI] [PubMed] [Google Scholar]
- 3. Tzipori S, Widmer G. 2008. A hundred-year retrospective on cryptosporidiosis. Trends Parasitol. 24:184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Juranek DD. 1995. Cryptosporidiosis: sources of infection and guidelines for prevention. Clin. Infect. Dis. 21(Suppl. 1):S57–S61 [DOI] [PubMed] [Google Scholar]
- 5. Fayer R. 2008. Cryptosporidium and cryptosporidiosis, p 1–42 In Fayer R, Xiao L. (ed), General biology. CRC Press, Boca Raton, FL [Google Scholar]
- 6. Smith HV, Rose JB. 1998. Waterborne cryptosporidiosis: current status. Parasitol. Today 14:14–22 [DOI] [PubMed] [Google Scholar]
- 7. Rotz LD, Khan AS, Lillibridge SR, Ostroff SM, Hughes JM. 2002. Public health assessment of potential biological terrorism agents. Emerg. Infect. Dis. 8:225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moran GJ. 2002. Threats in bioterrorism. II. CDC category B and C agents. Emerg. Med. Clin. North Am. 20:311–330 [DOI] [PubMed] [Google Scholar]
- 9. Arrowood MJ, Donaldson K. 1996. Improved purification methods for calf-derived Cryptosporidium parvum oocysts using discontinuous sucrose and cesium chloride gradients. J. Eukaryot. Microbiol. 43:89S. [DOI] [PubMed] [Google Scholar]
- 10. Guerrant RL. 1997. Cryptosporidiosis: an emerging, highly infectious threat. Emerg. Infect. Dis. 3:51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DuPont HL, Chappell CL, Sterling CR, Okhuysen PC, Rose JB, Jakubowski W. 1995. The infectivity of Cryptosporidium parvum in healthy volunteers. N. Engl. J. Med. 332:855–859 [DOI] [PubMed] [Google Scholar]
- 12. Janoff EN, Mead PS, Mead JR, Echeverria P, Bodhidatta L, Bhaibulaya M, Sterling CR, Taylor DN. 1990. Endemic Cryptosporidium and Giardia lamblia infections in a Thai orphanage. Am. J. Trop. Med. Hyg. 43:248–256 [DOI] [PubMed] [Google Scholar]
- 13. Griffiths JK. 1998. Human cryptosporidiosis: epidemiology, transmission, clinical disease, treatment, and diagnosis. Adv. Parasitol. 40:37–85 [DOI] [PubMed] [Google Scholar]
- 14. Mead JR. 2002. Cryptosporidiosis and the challenges of chemotherapy. Drug Resist. Updat. 5:47–57 [DOI] [PubMed] [Google Scholar]
- 15. Hewitt RG, Yiannoutsos CT, Higgs ES, Carey JT, Geiseler PJ, Soave R, Rosenberg R, Vazquez GJ, Wheat LJ, Fass RJ, Antoninievic Z, Walawander AL, Flanigan TP, Bender JF. 2000. Paromomycin: no more effective than placebo for treatment of cryptosporidiosis in patients with advanced human immunodeficiency virus infection. AIDS Clinical Trial Group. Clin. Infect. Dis. 31:1084–1092 [DOI] [PubMed] [Google Scholar]
- 16. Davies AP, Chalmers RM. 2009. Cryptosporidiosis. BMJ 339:b4168. [DOI] [PubMed] [Google Scholar]
- 17. Capparelli EV, Syed SS. 2008. Nitazoxanide treatment of Cryptosporidium parvum in human immunodeficiency virus-infected children. Pediatr. Infect. Dis. J. 27:1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Amadi B, Mwiya M, Musuku J, Watuka A, Sianongo S, Ayoub A, Kelly P. 2002. Effect of nitazoxanide on morbidity and mortality in Zambian children with cryptosporidiosis: a randomised controlled trial. Lancet 360:1375–1380 [DOI] [PubMed] [Google Scholar]
- 19. Zulu I, Kelly P, Njobvu L, Sianongo S, Kaonga K, McDonald V, Farthing M, Pollok R. 2005. Nitazoxanide for persistent diarrhoea in Zambian acquired immune deficiency syndrome patients: a randomized-controlled trial. Aliment. Pharmacol. Ther. 21:757–763 [DOI] [PubMed] [Google Scholar]
- 20. Amadi B, Mwiya M, Sianongo S, Payne L, Watuka A, Katubulushi M, Kelly P. 2009. High dose prolonged treatment with nitazoxanide is not effective for cryptosporidiosis in HIV positive Zambian children: a randomised controlled trial. BMC Infect. Dis. 9:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McKerrow JH, Sun E, Rosenthal PJ, Bouvier J. 1993. The proteases and pathogenicity of parasitic protozoa. Annu. Rev. Microbiol. 47:821–853 [DOI] [PubMed] [Google Scholar]
- 22. Koussis K, Withers-Martinez C, Yeoh S, Child M, Hackett F, Knuepfer E, Juliano L, Woehlbier U, Bujard H, Blackman MJ. 2009. A multifunctional serine protease primes the malaria parasite for red blood cell invasion. EMBO J. 28:725–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Teo CF, Zhou XW, Bogyo M, Carruthers VB. 2007. Cysteine protease inhibitors block Toxoplasma gondii microneme secretion and cell invasion. Antimicrob. Agents Chemother. 51:679–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McKerrow JH, Caffrey C, Kelly B, Loke P, Sajid M. 2006. Proteases in parasitic diseases. Annu. Rev. Pathol. 1:497–536 [DOI] [PubMed] [Google Scholar]
- 25. Rawlings ND, Barrett AJ, Bateman A. 2012. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40:D343–D350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Na BK, Kang JM, Cheun HI, Cho SH, Moon SU, Kim TS, Sohn WM. 2009. Cryptopain-1, a cysteine protease of Cryptosporidium parvum, does not require the pro-domain for folding. Parasitology 136:149–157 [DOI] [PubMed] [Google Scholar]
- 27. Clayton C, Adams M, Almeida R, Baltz T, Barrett M, Bastien P, Belli S, Beverley S, Biteau N, Blackwell J, Blaineau C, Boshart M, Bringaud F, Cross G, Cruz A, Degrave W, Donelson J, El-Sayed N, Fu G, Ersfeld K, Gibson W, Gull K, Ivens A, Kelly J, Vanhamme L. 1998. Genetic nomenclature for Trypanosoma and Leishmania. Mol. Biochem. Parasitol. 97:221–224 [DOI] [PubMed] [Google Scholar]
- 28. Caffrey CR, Steverding D. 2009. Kinetoplastid papain-like cysteine peptidases. Mol. Biochem. Parasitol. 167:12–19 [DOI] [PubMed] [Google Scholar]
- 29. Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, Buck GA, Xu P, Bankier AT, Dear PH, Konfortov BA, Spriggs HF, Iyer L, Anantharaman V, Aravind L, Kapur V. 2004. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science 304:441–445 [DOI] [PubMed] [Google Scholar]
- 30. Heiges M, Wang H, Robinson E, Aurrecoechea C, Gao X, Kaluskar N, Rhodes P, Wang S, He CZ, Su Y, Miller J, Kraemer E, Kissinger JC. 2006. CryptoDB: a Cryptosporidium bioinformatics resource update. Nucleic Acids Res. 34:D419–D422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rosenthal PJ, McKerrow JH, Rasnick D, Leech JH. 1989. Plasmodium falciparum: inhibitors of lysosomal cysteine proteinases inhibit a trophozoite proteinase and block parasite development. Mol. Biochem. Parasitol. 35:177–183 [DOI] [PubMed] [Google Scholar]
- 32. Rosenthal PJ, Wollish WS, Palmer JT, Rasnick D. 1991. Antimalarial effects of peptide inhibitors of a Plasmodium falciparum cysteine proteinase. J. Clin. Invest. 88:1467–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Engel JC, Doyle PS, Hsieh I, McKerrow JH. 1998. Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J. Exp. Med. 188:725–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ang KK, Ratnam J, Gut J, Legac J, Hansell E, Mackey ZB, Skrzypczynska KM, Debnath A, Engel JC, Rosenthal PJ, McKerrow JH, Arkin MR, Renslo AR. 2011. Mining a cathepsin inhibitor library for new antiparasitic drug leads. PLoS Negl. Trop. Dis. 5:e1023. 10.1371/journal.pntd.0001023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abdulla MH, Lim KC, Sajid M, McKerrow JH, Caffrey CR. 2007. Schistosomiasis mansoni: novel chemotherapy using a cysteine protease inhibitor. PLoS Med. 4:e14. 10.1371/journal.pmed.0040014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vermeire JJ, Lantz LD, Caffrey CR. 2012. Cure of hookworm infection with a cysteine protease inhibitor. PLoS Negl. Trop. Dis. 6:e1680. 10.1371/journal.pntd.0001680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sajid M, Robertson SA, Brinen LS, McKerrow JH. 2011. Cruzain: the path from target validation to the clinic. Adv. Exp. Med. Biol. 712:100–115 [DOI] [PubMed] [Google Scholar]
- 38. Arrowood MJ, Sterling CR. 1987. Isolation of Cryptosporidium oocysts and sporozoites using discontinuous sucrose and isopycnic Percoll gradients. J. Parasitol. 73:314–319 [PubMed] [Google Scholar]
- 39. Von Oettingen J, Nath-Chowdhury M, Ward BJ, Rodloff AC, Arrowood MJ, Ndao M. 2008. High-yield amplification of Cryptosporidium parvum in interferon gamma receptor knockout mice. Parasitology 135:1151–1156 [DOI] [PubMed] [Google Scholar]
- 40. Roehm NW, Rodgers GH, Hatfield SM, Glasebrook AL. 1991. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J. Immunol. Methods 142:257–265 [DOI] [PubMed] [Google Scholar]
- 41. Arrowood MJ, Hurd MR, Mead JR. 1995. A new method for evaluating experimental cryptosporidial parasite loads using immunofluorescent flow cytometry. J. Parasitol. 81:404–409 [PubMed] [Google Scholar]
- 42. Caffrey CR, Hansell E, Lucas KD, Brinen LS, Alvarez HA, Cheng J, Gwaltney SL, Roush WR, Stierhof YD, Bogyo M, Steverding D, McKerrow JH. 2001. Active site mapping, biochemical properties and subcellular localization of rhodesain, the major cysteine protease of Trypanosoma brucei rhodesiense. Mol. Biochem. Parasitol. 118:61–73 [DOI] [PubMed] [Google Scholar]
- 43. Greenbaum D, Baruch A, Hayrapetian L, Darula Z, Burlingame A, Medzihradszky KF, Bogyo M. 2002. Chemical approaches for functionally probing the proteome. Mol. Cell. Proteomics 1:60–68 [DOI] [PubMed] [Google Scholar]
- 44. Somoza JR, Zhan H, Bowman KK, Yu L, Mortara KD, Palmer JT, Clark JM, McGrath ME. 2000. Crystal structure of human cathepsin V. Biochemistry 39:12543–12551 [DOI] [PubMed] [Google Scholar]
- 45. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 46. Pei J, Kim BH, Grishin NV. 2008. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36:2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mantel N, Haenszel W. 1959. Statistical aspects of the analysis of data from retrospective studies of disease. J. Natl. Cancer Inst. 22:719–748 [PubMed] [Google Scholar]
- 48. Mantel N. 1966. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother. Rep. 50:163–170 [PubMed] [Google Scholar]
- 49. Bromme D, Klaus JL, Okamoto K, Rasnick D, Palmer JT. 1996. Peptidyl vinyl sulphones: a new class of potent and selective cysteine protease inhibitors: S2P2 specificity of human cathepsin O2 in comparison with cathepsins S and L. Biochem. J. 315:85–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Palmer JT, Rasnick D, Klaus JL, Bromme D. 1995. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J. Med. Chem. 38:3193–3196 [DOI] [PubMed] [Google Scholar]
- 51. Engel JC, Doyle PS, Palmer J, Hsieh I, Bainton DF, McKerrow JH. 1998. Cysteine protease inhibitors alter Golgi complex ultrastructure and function in Trypanosoma cruzi. J. Cell Sci. 111:597–606 [DOI] [PubMed] [Google Scholar]
- 52. Schulz F, Gelhaus C, Degel B, Vicik R, Heppner S, Breuning A, Leippe M, Gut J, Rosenthal PJ, Schirmeister T. 2007. Screening of protease inhibitors as antiplasmodial agents. Part I: Aziridines and epoxides. ChemMedChem. 2:1214–1224 [DOI] [PubMed] [Google Scholar]
- 53. Kerr ID, Lee JH, Farady CJ, Marion R, Rickert M, Sajid M, Pandey KC, Caffrey CR, Legac J, Hansell E, McKerrow JH, Craik CS, Rosenthal PJ, Brinen LS. 2009. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J. Biol. Chem. 284:25697–25703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rapp C, Kalyanaraman C, Schiffmiller A, Schoenbrun EL, Jacobson MP. 2011. A molecular mechanics approach to modeling protein-ligand interactions: relative binding affinities in congeneric series. J. Chem. Inf. Model. 51:2082–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sajid M, McKerrow JH. 2002. Cysteine proteases of parasitic organisms. Mol. Biochem. Parasitol. 120:1–21 [DOI] [PubMed] [Google Scholar]
- 56. Chalmers RM, Davies AP. 2010. Minireview: clinical cryptosporidiosis. Exp. Parasitol. 124:138–146 [DOI] [PubMed] [Google Scholar]
- 57. Rosenthal PJ. 1998. Proteases of malaria parasites: new targets for chemotherapy. Emerg. Infect. Dis. 4:49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Frame MJ, Mottram JC, Coombs GH. 2000. Analysis of the roles of cysteine proteinases of Leishmania mexicana in the host-parasite interaction. Parasitology 121:367–377 [DOI] [PubMed] [Google Scholar]
- 59. Shaw MK, Roos DS, Tilney LG. 2002. Cysteine and serine protease inhibitors block intracellular development and disrupt the secretory pathway of Toxoplasma gondii. Microbes Infect. 4:119–132 [DOI] [PubMed] [Google Scholar]
- 60. Okhuysen PC, Chappell CL, Kettner C, Sterling CR. 1996. Cryptosporidium parvum metalloaminopeptidase inhibitors prevent in vitro excystation. Antimicrob. Agents Chemother. 40:2781–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nesterenko MV, Tilley M, Upton SJ. 1995. A metallo-dependent cysteine proteinase of Cryptosporidium parvum associated with the surface of sporozoites. Microbios 83:77–88 [PubMed] [Google Scholar]
- 62. Forney JR, Yang S, Healey MC. 1996. Protease activity associated with excystation of Cryptosporidium parvum oocysts. J. Parasitol. 82:889–892 [PubMed] [Google Scholar]
- 63. Pozio E, Morales MA. 2005. The impact of HIV-protease inhibitors on opportunistic parasites. Trends Parasitol. 21:58–63 [DOI] [PubMed] [Google Scholar]
- 64. McKerrow JH, Rosenthal PJ, Swenerton R, Doyle P. 2008. Development of protease inhibitors for protozoan infections. Curr. Opin. Infect. Dis. 21:668–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Armson A, Meloni BP, Reynoldson JA, Thompson RC. 1999. Assessment of drugs against Cryptosporidium parvum using a simple in vitro screening method. FEMS Microbiol. Lett. 178:227–233 [DOI] [PubMed] [Google Scholar]
- 66. Yang S, Healey MC. 1993. The immunosuppressive effects of dexamethasone administered in drinking water to C57BL/6N mice infected with Cryptosporidium parvum. J. Parasitol. 79:626–630 [PubMed] [Google Scholar]
- 67. Mead JR, You X, Pharr JE, Belenkaya Y, Arrowood MJ, Fallon MT, Schinazi RF. 1995. Evaluation of maduramicin and alborixin in a SCID mouse model of chronic cryptosporidiosis. Antimicrob. Agents Chemother. 39:854–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. You X, Mead JR. 1998. Characterization of experimental Cryptosporidium parvum infection in IFN-gamma knockout mice. Parasitology 117:525–531 [DOI] [PubMed] [Google Scholar]
- 69. Griffiths JK, Theodos C, Paris M, Tzipori S. 1998. The gamma interferon gene knockout mouse: a highly sensitive model for evaluation of therapeutic agents against Cryptosporidium parvum. J. Clin. Microbiol. 36:2503–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rehg JE. 1991. Activity of azithromycin against cryptosporidia in immunosuppressed rats. J. Infect. Dis. 163:1293–1296 [DOI] [PubMed] [Google Scholar]
- 71. McKerrow JH, Engel JC, Caffrey CR. 1999. Cysteine protease inhibitors as chemotherapy for parasitic infections. Bioorg. Med. Chem. 7:639–644 [DOI] [PubMed] [Google Scholar]
- 72. Renslo AR, McKerrow JH. 2006. Drug discovery and development for neglected parasitic diseases. Nat. Chem. Biol. 2:701–710 [DOI] [PubMed] [Google Scholar]
- 73. Xing R, Addington AK, Mason RW. 1998. Quantification of cathepsins B and L in cells. Biochem. J. 332:499–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Steverding D, Sexton DW, Wang X, Gehrke SS, Wagner GK, Caffrey CR. 2012. Trypanosoma brucei: chemical evidence that cathepsin L is essential for survival and a relevant drug target. Int. J. Parasitol. 42:481–488 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.