

Abstract
We have investigated the mechanism of action of inhibition of the choline kinase of P. falciparum (p.f.-ChoK) by two inhibitors of the human ChoKα, MN58b and RSM-932A, which have previously been shown to be potent antitumoral agents. The efficacy of these inhibitors against p.f.-ChoK is investigated using enzymatic and in vitro assays. While MN58b may enter the choline/phosphocholine binding site, RSM-932A appears to have an altogether novel mechanism of inhibition and is synergistic with respect to both choline and ATP. A model of inhibition for RSM-932A in which this inhibitor traps p.f.-ChoK in a phosphorylated intermediate state blocking phosphate transfer to choline is presented. Importantly, MN58b and RSM-932A have in vitro inhibitory activity in the low nanomolar range and are equally effective against chloroquine-sensitive and chloroquine-resistant strains. RSM-932A and MN58b significantly reduced parasitemia and induced the accumulation of trophozoites and schizonts, blocking intraerythrocytic development and interfering with parasite egress or invasion, suggesting a delay of the parasite maturation stage. The present data provide two new potent structures for the development of antimalarial compounds and validate p.f.-ChoK as an accessible drug target against the parasite.
INTRODUCTION
Choline is the first precursor in the generation of two of the major components of the plasma membrane, phosphatidylcholine (PC) and sphingomyelin (SM). Choline kinase (ChoK) is a cytosolic enzyme expressed in multiple tissues. In the presence of ATP and magnesium, it converts choline (Fig. 1A, panel 4) into phosphorylcholine (PCho), the first step in the biosynthesis of PC, which is also known as the Kennedy pathway (1, 2).
Fig 1.

Structures of the ChoK inhibitors (A) Structures of the ChoK inhibitors HC-3 (panel 1), MN58b (panel 2), RSM-932A (panel 3), and choline (panel 4). (B) Inhibition of p.f.-ChoK activity by human ChoK inhibitors shown by thin-layer chromatography of enzymatic reactions, using E. coli, of recombinant p.f.-ChoK. Reactions were performed in the absence and presence of DMSO (lanes 1 and 2, respectively) and in the presence of 30 μM (each) HC-3 (lanes 3 and 4), MN58b (lanes 5 and 6), and RSM-932A (lane 7). RSM-932A was suspended in DMSO before use. The lower bands represent unconverted choline, and the upper bands correspond to phosphocholine.
Human ChoKα has been shown to be strongly linked to several types of cancer (3–5) and to be oncogenic (6). Our group designed and synthesized ChoKα inhibitors for antitumoral therapy based on Hemicholinium-3 (HC-3), a choline homologue previously described as a ChoK inhibitor (1, 7, 8). Due to its high toxicity in vivo, HC-3 is not a favorable candidate for clinical use (9); however, more-powerful and less toxic compounds were produced (7, 8), followed by an even more efficient second generation (10, 11), one of which, RSM-932A (also designated TCD-717), is currently in phase I clinical trials (http://clinicaltrials.gov/ct2/show/NCT01215864).
ChoK inhibitors can act as effective inhibitors of Plasmodium falciparum choline kinase (p.f.-ChoK) and are effective in in vitro and in vivo assays. The primary sequence of the catalytic site (12) and the tertiary structure (PDB 3FI8; www.pdb.org) of p.f.-ChoK are conserved with respect to other ChoKs. Furthermore, p.f.-ChoK is more highly expressed in growth phases of P. falciparum (12), and inhibition of ChoK affects the parasite's viability in in vitro and mouse models of malaria (13). HC-3 has been shown to inhibit recombinant p.f.-ChoK (14) and to be lethal against the parasite (15). Since it has been validated as a therapeutic target, other groups have found effective inhibitors for p.f.-ChoK (16, 17).
The importance of developing new viable alternatives for current malaria treatments cannot be overstated: while millions are infected and die from the disease yearly, new drug-resistant strains of P. falciparum continuously appear due to selection processes (18). In fact, drug resistance has emerged for artemisinin derivatives, currently the most widely recommended treatment in areas where the disease is endemic (19), underlining the importance of continually searching for new drug therapies and targets.
Here, we have determined that the minimally toxic human ChoKα inhibitors already developed and characterized by our group may be able to function as antimalarial agents. We describe the effects of HC-3 (Fig. 1A, panel 1), the second-generation compound MN58b (Fig. 1A, panel 2), and the third-generation compound RSM-932A (Fig. 1A, panel 3) in enzymatic and in vitro assays. While HC-3 has been already observed in the crystal structures in complex with human ChoK to enter precisely in the same place as phosphocholine (20), and while MN58b, due to structural similarities, may do the same, we show here through enzymatic assays that the mechanism of inhibition of these two inhibitors is not competitive, which suggests a more complex mechanism of action. Importantly, we describe a novel synergistic mechanism of action for RSM-932A. The availability of novel drugs against malaria is important due to the continuous need to overcome resistance to current treatments. Understanding the mechanism of action of drugs under development will help in the design of novel and more effective treatments.
MATERIALS AND METHODS
Enzymatic reactions using Escherichia coli extracts.
The bacterial expression vector containing an N-terminal His-tagged and truncated (amino acids 79 to 439) form of p.f.-ChoK from P. falciparum strain 3D7 was generously provided to us by the Structural Genomics Consortium (www.pdb.org). This vector was expressed in E. coli BL21(λDE3) CodonPlus cells at 37°C. Enzymatic reactions utilizing extracts of recombinant His-tagged p.f.-ChoK expressed in E. coli were performed by placing a 1-μl extract in a reaction mixture containing 0.185 μCi/nmol methyl-[14C]choline, 180 μM choline, 10 mM ATP, 10 mM MgCl2,and 100 mM Tris (pH 8.0) at 37°C for 20 min. The reactions were stopped by placing the mixtures in ice and then at −20°C, defrosted, and resolved by thin-layer chromatography using a Whatman 60A Silica Gel membrane and with a mobile phase consisting of 25 ml 0.9% NaCl, 35 ml methanol, and 2.5 ml 30% NH4OH. Radioactivity was visualized and quantified using a Cyclone Plus Scanner. The IC50 values of ChoKα inhibitors were determined as the concentrations of inhibitor necessary to reach 50% inhibition.
p.f.-ChoK purification.
The p.f.-ChoK expression vector described above was expressed in E. coli BL21(λDE3) CodonPlus cells and then induced with 1 mM IPTG (isopropyl-1-thio-d-galactopyranoside) in the presence of 200 μg/ml and 25 μg/ml of ampicillin and chloramphenicol, respectively, overnight at 15°C. The culture was harvested by centrifugation. The pellets were resuspended with 10 ml/liter of cell culture in binding buffer (25 mM Tris [pH 8.8], 100 mM NaCl), 1 mM benzamidine, and 1 mM phenylmethylsulfonyl fluoride (PMSF) and stored at −80°C. Resuspended pellets stored at −80°C were thawed, and prior to lysis each pellet was pretreated with 0.5% CHAPS (3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate hydrate) and 500 units of benzonase and DNase and then taken immediately to be mechanically lysed with a French press at 1,000 lb/in2; and the cell lysate was centrifuged using a Beckman ultracentrifuge at 50,000 rpm in a Beckman 50Ti rotor for 1 h.
The cleared lysate was loaded onto a Hi Trap IMAC HP column (GE Healthcare, USA) charged with Ni+ at 0.5 ml/min and washed (15 column volumes) with binding buffer and then eluted with a gradient using binding buffer supplemented with 500 mM imidazole. Elutions were pooled and then loaded onto a Hi Load Superdex 200 16/60 column (GE Healthcare) preequilibrated with buffer A. The elution volume corresponded to a molecular mass of 34 kDa (compared to an expected size of 45 kDa), confirming, as expected from the crystal structure (www.pdb.org), that the protein was a monomer. Eluted fractions were pooled, concentrated to 100 μM using Amicon Ultra Centrifugal filters with a cutoff of 10 kDa (Millipore, USA), before being frozen in liquid nitrogen and stored at −80°C.
Steady-state enzymatic assays monitored with the pyruvate kinase/lactate dehydrogenase (PK/LDH) reaction.
When a wide range of choline or ATP concentrations was used to determine Michaelis-Menten kinetic values, a negative cooperativity effect was observed as was described for human (21) and yeast (22) ChoKs. In order to compare the substrate kinetic parameters, a range of concentrations in the linear portion of the Lineweaver-Burk plots was selected for analysis and curve fitting into the Michaelis-Menten equation.
These assays were run in 200 μl of a solution containing 100 mM Tris (pH 8.0), 10 mM MgCl2, 75 mM NaCl, 75 mM KCl, 0.66 mM NADH, 0.33 mM phosphoenolpyruvate, 1.1 mM ATP, 10 units lactate dehydrogenase, 5 units pyruvate kinase, and 4.6 nM His-tagged p.f.-ChoK. The assays were carried out at 25°C in UV-Star 96-well plates (Greiner Bio-One, Germany) in a Versamax microplate reader (Molecular Devices, USA), and the linear reduction in NADH levels was observed at A340. The steady-state rate of ADP formation was calculated using an extinction coefficient of 6,200 M−1 cm−1.
Determination of mechanism of action using PK/LDH coupled reactions.
The mechanism of action of the inhibitors with respect to each substrate was determined using a previously described method (23). Briefly, using the IC50 of each inhibitor determined at the Km of choline, the concentration of one substrate was kept constant while initial velocities were measured while varying the second substrate in the presence and absence of inhibitor. The percentage of inhibition (%I) was determined by dividing each initial velocity in the presence of inhibitor with its corresponding initial velocity in the absence of compound. %I was plotted against concentration and fitted using a two-parameter-fit nonlinear regression algorithm of GnuPlot v 4.3 into the equation below, where [S] is the concentration of the varied substrate, [I] is the concentration of inhibitor, Ks is the dissociation constant of the substrate (assumed to be equal to the Km), Ki is the dissociation constant of the inhibitor, and α is a numerical constant that measures the effect the substrate has on the binding of the inhibitor and vice versa.
LC-MS analysis of ATP and hemicholinium.
For the determination of ATP catalysis and HC-3 inhibitor phosphorylation by liquid chromatography-mass spectrometry (LC-MS), 1-ml samples containing 3 mM ATP or 0.5 mM HC-3 plus 3 mM ATP, both in the presence and absence of 3.4 μM p.f.-ChoK in 100 mM Tris (pH 8)–10 mM MgCl2 were incubated overnight and then passed through a centrifugal filter with a cutoff of 10 kDa (Millipore, USA) and stored at −20°C. One hundred microliters of each of these samples was placed into high-pressure liquid chromatography (HPLC) vials, and analyses were performed using an ultrahigh-pressure LC (UHPLC) system (1290 series; Agilent Technologies) coupled to a 6550 ESI-QTOF (Agilent Technologies) operated in positive electrospray ionization (ESI+) mode. Metabolites were separated using an Agilent Zorbax C18 column, 50 by 2.1 mm by 1.8 μm (Agilent). The mobile phase used consisted of A1 (0.1% formic acid in water) and B1 (0.1% formic acid in acetonitrile). The linear gradient elution started at 100% A (time zero to 2 min) and finished at 100% B (14 to 20 min) at a flow rate of 0.4 ml min−1. The injection volume was 2 μl, and the ESI conditions were as follows: gas temperature, 150°C; drying gas, 13 liters min−1; nebulizer, 35 psig (241.32 kPa); sheath gas temperature, 300°C; sheath gas flow, 12 liters min−1; capillary voltage, 3,500 V; nozzle voltage, 500 V; fragmentor, 400 V; and skimmer, 65 V. The instrument was set to acquire data over the m/z range of 100 to 1,000 with an acquisition rate of 2 spectra/second.
Trypsin digestions.
Assays were performed using 0.0375 mg/ml trypsin, 10 μM p.f.-ChoK, 10 mM MgCl2, and 100 mM Tris (pH 8.0) in a total volume of 20 μl incubated at 37°C for 30 min in the case of a dose-response curve or longer in the case of a time course experiment. Resulting digestions were loaded onto a 12% SDS-PAGE and then stained with Coomassie blue. Densitometry was performed on resulting bands using the Quantity One software (Bio-Rad, USA). Percent degradation was determined with respect to a sample load of undigested p.f.-ChoK.
Thermal shift assays.
Assays were performed using a previously described protocol (24). Briefly, 75 μg/ml p.f.-ChoK, a 2,127× dilution of Sypro Orange (Invitrogen, United Kingdom), which was used as the reporter dye, 10 mM MgCl2, and 100 mM Tris (pH 8) in a total volume of 10 μl were plated onto MicroAMP 96-well plates (Applied Biosystems, Life Technologies, United Kingdom). Thermal denaturation using a temperature range of between 25°C and 95°C in steps of 0.5°C was performed in a StepOnePlus real-time PCR system (Applied Biosystems, Life Technologies, United Kingdom) using the ROX emission filter. The resulting data were analyzed using the MATLAB 7.11 program running the ThermoQ curve fitting software, which was used to calculate the melting temperatures (Tm) (24).
Drugs and inhibitors.
Synthesis of ChoK inhibitors has been previously described (25, 26). Chloroquine diphosphate salt was purchased from Sigma-Aldrich. Stock solutions of inhibitors were prepared in 100% dimethyl sulfoxide (DMSO). Chloroquine was dissolved in distilled water at 100 mM. All compounds were stored at −20°C until use. For the drug assays, serial dilutions were made in culture medium and added to 96-well culture plates. Control cultures were treated with equivalent amounts of DMSO diluted in culture medium.
In vitro cultures of Plasmodium falciparum.
Plasmodium falciparum strains Dd2 (clone MRA-150) and 3D7 (clone MRA-102) obtained from the Malaria Research and Reference Reagent Resource Center (MR4) (http://www.mr4.org) were used for this study. Erythrocytes were obtained from type A+ human healthy local donors and collected in tubes with citrate-phosphate-dextrose anticoagulant (Vacuette). The culture medium consisted of standard RPMI 1640 (Sigma-Aldrich) supplemented with 0.5% Albumax I (Gibco), 100 μM hypoxanthine (Sigma-Aldrich), 25 mM HEPES (Sigma-Aldrich), 12.5 μg/ml gentamicin (Sigma-Aldrich), and 25 mM NaHCO3 (Sigma-Aldrich). Each culture was started by mixing uninfected and infected erythrocytes to achieve a 1% hematocrit and incubated in 5% CO2 at 37°C in tissue culture flasks (Iwaki). The progress of growth in the culture was determined by microscopy in thin blood smears stained with Wright's eosin methylene blue solution (Merck), using the freely available Plasmoscore software (27) to monitor the parasitemia. The detailed description of the culture and synchronization methods used has been reported previously (28).
Fluorimetric assays for antimalarial drug activity.
A PicoGreen microfluorimetric DNA-based assay was used to monitor parasite growth inhibition at different drug concentrations (29). PicoGreen (P7589) was purchased from Invitrogen and diluted as indicated by the manufacturer in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.5). Synchronized rings from stock cultures were used to test 2 μM to 0.2 nM serial dilutions of HC-3, MN58b, and RSM-932A in 96-well culture microplates. Thus, 150 μl of parasites at 2% hematocrit and 1% parasitemia were allowed to grow for 48 hours in 5% CO2 at 37°C. The parasites were then centrifuged at 600 × g for 10 min and resuspended in saponin (0.15%, wt/vol, in phosphate-buffered saline [PBS]) to lyse the erythrocytes and release the malaria parasites. To eliminate all traces of hemoglobin, the pellet was washed by the addition of 200 μl of PBS followed by centrifugation at 600 × g. The washing step was repeated twice to ensure complete removal of hemoglobin. Finally, pellets were resuspended in 100 μl of PBS. A 100-μl volume of PicoGreen diluted in TE was added to each well. Plates were incubated for 30 to 60 min in the dark, and the fluorescence intensity was measured at 485-nm excitation and 528-nm emission. Growth inhibition was calculated as previously described (29). The parasite morphology was evaluated by microscopic analysis of thin blood smears stained with Wright's stain. Smears from drug-free cultures were used as a control.
RESULTS
Enzymatic assays using E. coli extracts.
Enzymatic reactions using E. coli extracts expressing recombinant p.f.-ChoK and identical concentrations (30 μM) of each compound showed that while HC-3 and MN58b lightly inhibit the generation of PCho, RSM-932A is a comparatively potent inhibitor (Fig. 1B). A later assay using a range of inhibitor concentrations showed the IC50 of RSM-932A to be 1.5 μM (data not shown).
Steady-state enzymatic reactions and inhibition assays.
Steady-state enzymatic reactions using purified recombinant p.f.-ChoK were performed using the pyruvate kinase/lactate dehydrogrenase (PK/LDH) enzymatic reporter system. The calculated Kms for ATP and choline were 81 ± 13.65 μM and 22 ± 8 μM, respectively. The kcat measured was 338.4 ± 12 min−1. In addition, reactions in the absence of choline showed that p.f.-ChoK had ATPase activity with a Km of 77 ± 9 and a kcat of 82.2 ± 1.8 min−1. This ATPase activity was confirmed by LC/MS analysis of a sample of ATP with and without the p.f.-ChoK enzyme, which showed that 98% of the ATP was consumed relative to a non-enzyme-containing control, while 5.8- and 6.3-fold amounts of ADP and AMP, respectively, were detected relative to the control.
The IC50s of HC-3, MN58b, and RSM-932A were determined to be 250 μM, 106 μM, and 1.75 μM, respectively, in a steady-state reaction in which the concentration of choline was equivalent to its Km. An IC50 of 1.75 for RSM-932A was consistent with the results of enzymatic assays using E. coli extract and was significantly lower than that of HC-3 and MN58b, confirming that it was the more potent inhibitor.
Determination of inhibitor mechanism of action.
A mechanistic analysis was performed for each inhibitor by performing steady-state enzymatic reactions with p.f.-ChoK in which one substrate was varied while the other substrate was fixed both in the absence and in the presence of an inhibitor whose concentration was fixed at its IC50. The percentage of p.f.-ChoK inhibition (%I) due to inhibitor binding was defined as the steady-state initial velocity with inhibitor divided by the initial velocity without inhibitor at a given concentration of the varied substrate. Plotting %I against substrate concentration and fitting the data to the equation described in Materials and Methods, Ki and α values for each inhibitor were derived. In the case of HC-3, when the substrate concentration of either choline (Fig. 2A) or ATP (Fig. 2B) was increased, the %I was reduced, indicating that inhibitor binding was antagonistic to the binding of both ATP and choline (and vice versa). The same result was observed with MN58b (Fig. 2C and D). In contrast, with RSM-932A an increase in either substrate marked a parallel increase in %I (Fig. 2E and F), meaning that inhibitor binding was synergistic to the binding of both ATP and choline. As a measure of the effect of each substrate on inhibitor binding, α values were derived. The α values calculated for HC-3 (αcholine = 2.9 ± 0.3; αATP = 5.2 ± 1) and MN58b (αcholine = 21.3 ± 6.4; αATP = 13.23 ± 2.6) were low; that is, they did not approach infinity. These results show a mixed-inhibition mechanism for these compounds (not competitive). On the other hand, α values derived for RSM-932A (αcholine = 0.48 ± 0.04; αATP = 0.2 ± 0.03) had a value of less than 1, which was consistent with a synergistic mechanism of action. Derived Ki values for HC-3, MN58b, and RSM-932A (with respect to choline) were 204 ± 11.8 μM, 49.2 ± 9.3 μM, and 3.3 ± 0.04 μM, respectively.
Fig 2.

Mechanistic analysis of inhibitors to p.f.-ChoK activity. The percent inhibition of p.f.-ChoK activity caused by each inhibitor (%I) is plotted against the concentration of either choline or ATP in order to perform a mechanistic analysis of inhibitors in relation to each substrate: HC-3 (A, B), MN58b (C, D), and RSM-932A (E, F).
In light of the fact that p.f.-ChoK was observed to have ATPase activity, the mechanistic analysis described above was repeated using ATP in the absence of choline, with the result that ATP alone was shown to be synergistic with RSM-932A (Fig. 3A; αATP = 0.58 ± 0.08; Ki = 2.5 ± 0.2). Here, the calculated αATP was higher than when choline was present (αATP = 0.2 ± 0.03), implying that the ATP synergism with RSM-932A was less pronounced in the absence of choline.
Fig 3.

Mechanistic analysis in the presence of choline or HC-3. (A) Mechanistic analysis between RSM-932A and ATP in the absence of choline. (B) ProQ Diamond staining of an SDS-PAGE of samples of p.f.-ChoK incubated in the absence and presence of ATP. (C) LC/MS results showing the calculated masses of HC-3 (1), phospho-HC-3 (2), and di-phospho-HC-3 (3). (D and E) Mechanistic analysis of RSM-932A with HC-3 as the substrate (D) or of MN58b with HC-3 as the substrate (E).
The observed HC-3 antagonism with ATP was unexpected, as was its lack of competitivity with choline. The crystal structure of human choline kinases α and β in complex with HC-3 and ADP has been solved (20) and shows that the two methyl groups of a symmetrical unit of HC-3 entered precisely into the phosphocholine binding site. With choline kinase β, HC-3 functions as an alternative substrate and is phosphorylated. An attempt to dock ATP onto these structures in the place of ADP shows that HC-3 does not sterically hinder the binding of ATP (20). Given the similarity in tertiary structure between p.f.-ChoK and the human ChoKα, one would expect a completely noncompetitive mechanism with respect to ATP and a completely competitive effect with choline. These results suggest that the structure of p.f.-ChoK is altered when ATP is bound and that the substrates bind to a different conformation than does HC-3.
Inhibition of HC-3 phosphorylation by RSM-932A and MN58b.
Knowing that HC-3 functions as an alternative substrate as well as an inhibitor of human ChoKβ (20), we confirmed by mass spectrometry that with p.f.-ChoK HC-3 was phosphorylated not once as reported with human ChoKβ, but twice (Fig. 3C, peaks labeled 1 to 3), which is logical given the symmetry of the molecule (Fig. 1A, panel 1). This result confirmed that HC-3 enters as expected into the catalytic site and that it functions as an alternative substrate for p.f.-ChoK. As reported previously, the formation of phospho-HC-3 can be monitored at 293.9 nm (20); therefore, we were able to exploit this to perform the same mechanistic analysis as above to determine the relationship between MN58b and RSM-932A on the one hand and HC-3 as a substrate on the other. The Km of HC-3 as a substrate was calculated to be 132 ± 16 μM. The IC50s of MN58b and RSM-932A were 33 μM and 2.51 μM, respectively. The derived αHC-3 values were 4.14 ± 1.11 and 20.3 ± 15.1, indicating a mixed-inhibition model in both cases (Fig. 3D and E). Since an antagonistic (but not a competitive) mechanism of action was observed in both cases, the interpretation of these results was difficult. However, it appears that RSM-932A, MN58b, and HC-3 bind to different enzyme forms, though it is probable that MN58b shares an overlapping binding site with HC-3, due to containing the same two methyl groups used by HC-3 to enter the catalytic site in the same place as phosphocholine (20). It is difficult with the present information to posit whether or not RSM-932A enters the catalytic site; however, it is clear that RSM-932A has a distinct mode of binding, given its synergism with the natural substrates.
Substrate kinetics.
In order to further explore the interaction between the substrates and RSM-932A, an effort was made to determine the order of substrate binding of p.f.-ChoK. Double-reciprocal plots of one substrate at different concentrations of the second substrate as a rule lead to a group of converging lines in the case of a sequential mechanism in which the substrates form a ternary complex before transfer of the phosphate group (such as in the case of brewer's yeast ChoK [22]) and a group of parallel lines in the case of a ping-pong mechanism (30). Parallel lines were observed in double-reciprocal plots of both ATP (Fig. 4A), and choline (Fig. 4B). This result is consistent with the observed ATPase activity, i.e., ATP can be hydrolyzed by p.f.-ChoK in the absence of choline. A ping-pong mechanism presupposes a phosphorylated protein intermediate that mediates the transfer of a phosphate to the product. Consistent with this model, ProQ staining (which is used to detect phosphorylated protein) of an SDS-PAGE containing samples of p.f.-ChoK incubated with ATP showed twice as much fluorescence (when correcting for protein loaded) as the control condition sample (Fig. 3B).
Fig 4.

Enzymatic analysis of ChoK inhibition. (A, B) Determination of the substrate binding order of p.f.-ChoK. Assays distinguishing the ping-pong mechanism from ternary mechanisms were performed by keeping the choline concentration constant while varying the ATP concentration (A) or by keeping the ATP concentration constant while varying that of choline (B). (C) Dead-end inhibitor studies with AMP-PCP versus ATP and choline. The concentration of AMP-PCP was held at 314 mM. (D, E, F) Product inhibition assays using phosphocholine, keeping choline at its Km and saturation levels while varying ATP and plotting the percentage of inhibition (%I) as a function of ATP and keeping the ATP concentration constant at its Km (D) and at saturation (E) while varying the concentration of choline (F).
Dead-end inhibition assays.
A double-reciprocal plot of ATP in the presence of the dead-end inhibitor AMP-PCP (β,γ-methylene adenosine 5′-triphosphate), an analog of ATP, displayed a competitive profile (with an α value approaching infinity) (Fig. 4C). On the other hand, an inhibition assay with respect to choline showed that AMP-PCP was uncompetitive with choline (Fig. 4C). These inhibition patterns are indicative of a ping-pong mechanism (31).
Product inhibition assays.
Double-reciprocal plots of choline with the concentration of ATP fixed at its Km (Fig. 4E) showed a family of lines that met to the left of the y axis (indicative of a mixed-inhibition profile). At saturating concentrations of ATP (Fig. 4F), these lines crossed to the left of the y axis at the x axis, which is consistent with noncompetitivity. A reasonable interpretation of the PCho noncompetitivity with choline is that choline and PCho do not compete because they bind to different enzyme forms.
In order to distinguish between one-site and two-site ping-pong models, we carried out product inhibition assays with PCho. In the case of a one-site ping-pong model, one would expect competitivity between ATP and PCho and noncompetitivity between PCho and choline and that the formation of a ternary complex would be precluded (32). The percentage of inhibition (%I) was plotted against ATP in the presence of different fixed concentrations of PCho with the concentration of choline fixed at its Km and at saturation (Fig. 4D). Both curves showed that an increase in ATP concentration correlates with an increase in %I. These results are indicative of uncompetitivity between PCho and ATP; both can bind simultaneously, which is consistent with two distinct sites for these molecules. The product inhibition results are inconsistent with the one-site model, which means that it can be discarded, leaving only the possibility for a two-site model. The two-site model is consistent with the information from the crystal structures of both P. falciparum (3FI8; www.pdb.org) and human ChoKs (33), which shows that the two substrates do not share a binding site. In fact, the crystal structure of p.f.-ChoK shows that phosphoethanolamine and ADP can simultaneously bind (33). The substrate binding order of p.f.-ChoK therefore appears to follow a nonclassical two-site ping-pong mechanism in which the enzyme first has to be phosphorylated for the reaction to proceed and the substrates have independent binding sites (34).
Protein stability studies.
Time course trypsin degradation experiments showed that while in the presence of AMP-PCP, the degradation of p.f.-ChoK was minimized (Fig. 5A and B) and choline had no effect in either direction (Fig. 5C). Titration of AMP-PCP in the presence of choline showed that choline augmented the protective effect of AMP-PCP (Fig. 5D). This result is consistent with the observed uncompetitivity between AMP-PCP and choline and is indirect evidence of separate binding sites for the two. Meanwhile, the presence of RSM-932A accelerated p.f.-ChoK digestion in a time course experiment (Fig. 5E). RSM-932A caused increased degradation in a dose-dependent manner, and its effect was attenuated in the presence of AMP-PCP (Fig. 5F).
Fig 5.

Trypsin digestions of recombinant ChoK. (A) Representative gel showing a time course experiment in the absence (left of the vertical line) and presence (right of the vertical line) of AMP-PCP, an analog of ATP. (B) Graph of time course experiment in the absence and presence of AMP-PCP. (C) Time course experiment in the presence and absence of choline. (D) AMP-PCP titration in the absence and presence of choline. (E) Time course experiment in the presence of RSM-932A. (F) Titration of RSM-932A in the absence and presence of AMP-PCP.
Thermal shift studies showed that when RSM-932A is added, the melting curve loses cooperativity and the melting temperature is shifted (Fig. 6), while adding AMP-PCP causes a rightward shift in the melting curve. This result agrees with the trypsin degradation results and suggests that RSM-932A destabilizes p.f.-ChoK while ATP stabilizes it.
Fig 6.
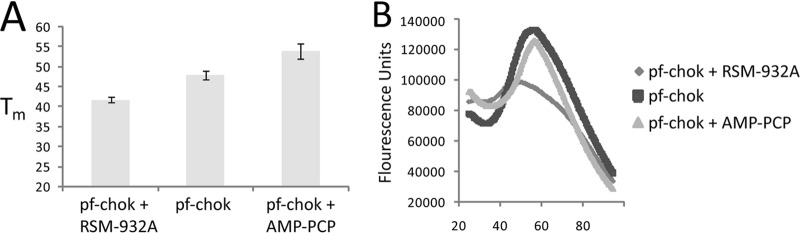
Melting temperature shifts. (A) Melting temperature shifts of p.f.-ChoK by itself in the presence of either RSM-932A or AMP-PCP. (B) Representative fluorescence curves of the same thermal shift assay.
Activity of choline kinase inhibitors on Plasmodium-infected erythrocyte cultures.
To examine the antimalarial activity of the ChoK inhibitors described above, we assayed the effect of increasing concentrations of these compounds on the in vitro intraerythrocytic life cycle of two P. falciparum strains, one sensitive (3D7) and another resistant (Dd2) to chloroquine. Dose-response curves in human parasite cultures within the 0.2 nM to 2 μM range for MN58b and RSM-932A were obtained (Fig. 7A). HC-3 demonstrated antimalarial activity only at the micromolar range, rendering a hemolytic effect above 50 μM, and consequently it was not possible to estimate a dose-response curve. Compound RSM-932A exhibited a nanomolar range for IC50 in both parasite strains (26.50 ± 5.5 and 36.40 ± 8.6 nM in Dd2 and 3D7, respectively), indicating a remarkable in vitro antimalarial activity (Fig. 7B). MN58b revealed a 10-fold decrease of the IC50 for both strains, 3.50 ± 0.007 nM (Dd2) and 2.60 ± 0.02 nM (3D7), improving considerably the antimalarial activity. The control IC50s obtained with chloroquine in the two strains showed the expected sensitivity (21.6 ± 8.9 nM for 3D7 and 177.6 ± 9.5 nM for Dd2). The comparable IC50s for the two ChoK inhibitors found in chloroquine-resistant and -nonresistant strains suggest that these compounds may affect plasmodial processes different from those targeted by chloroquine, which appears to involve the inhibition of formation of hemozoin. The resistance to chloroquine has been associated to the acquisition of a mutant transporter, PfCRT, which is capable of reducing intracellular exposure to the drug (35). The minor differences observed in the responses of Dd2 and 3D7 to ChoK inhibitors suggest that PfCRT polymorphisms yielding resistance to chloroquine do not affect the activity of these compounds.
Fig 7.

In vitro antiparasite activity of ChoK inhibitors. (A) Dose-response growth curves of RSM-932A (squares), MN58b (triangles), and chloroquine (circles) on human erythrocyte cultures infected with P. falciparum Dd2 and 3D7 strains. (B) Calculated 50% inhibitory concentration (IC50) values of RSM-932A, MN58b, and chloroquine in the two P. falciparum strains. Results are the means of two independent cultures. (C) Dose-dependent effects of ChoK inhibitors on P. falciparum cultures. Synchronized cultures of Dd2 at the mature ring stage were incubated for 48 h with the compounds at the indicated concentrations and compared to an untreated control. Cultures were evaluated by microscopic inspection of thin blood smears stained with Wright's stain. Parasitic stages observed in cultures treated with different concentrations of MN58b (left panel) and RSM-932A (right panel) for 48 h. Bars show the percentages of rings (white), trophozoites (light gray), and schizonts (dark gray) forms observed in cultures treated at the indicated compound concentrations. The percentage of each group accounts for the fraction of cells showing every parasite stage from a total of 1,000 erythrocytes ± standard deviations (SD). Results are representative of 2 experiments. Representative microscopic images of P. falciparum cultures incubated with different concentrations of ChoK inhibitors are shown. Total parasitemia values of the cultures after 48 h of treatment are indicated above.
Generally, antimalarial compounds have intraerythrocytic-stage specificity in their inhibitory effect. For instance, chloroquine kills malaria parasites in its mature stages (36). To gain phenotypic insight into the mode of action of ChoK inhibitors, we analyzed the effects of the increased doses of the compounds on the parasite intraerythrocytic developmental progression by microscopic inspection in thin blood smears. An untreated culture of P. falciparum-infected erythrocytes was monitored as a control.
As shown in Fig. 7C, the untreated 48-h culture reached 10% parasitemia and contained predominantly mature rings/trophozoites, indicating that a complete invasive cycle had taken place, producing new progeny. At identical sampling time (48 h), parasite cultures treated with ChoK inhibitors in the IC50 range accumulated young rings, in parallel with a slight reduction in parasitemia compared to the untreated culture. Concentrations of RSM-932A and MN58b above the IC50s significantly reduced parasitemia and accumulated trophozoites and schizonts from the initial parasite generation, indicating delay of the parasite maturation that did not reach a new invasion stage and induced complete parasite death above 200 nM (Fig. 7C). In contrast, infected cultures treated with chloroquine at the IC50 led to different parasite stage profiles. Cultures treated at 200 nM chloroquine consisted almost exclusively of ring stage parasites, suggesting cell cycle-delayed cells, similarly to the dose-dependent delay recently reported (37). This observation is an additional evidence for different inhibitory mechanisms between ChoK inhibitors and chloroquine.
Noteworthy, our results are also consistent with the effect of another P. falciparum choline kinase inhibitor, HDTAB, which inhibits the enzyme in a dose-dependent manner, exhibiting a very potent antimalarial activity in vitro against P. falciparum and also in vivo against the rodent malaria parasite Plasmodium yoelii strain N67. Its antimalarial action is stage specific, showing the highest inhibitory activity toward trophozoites (13).
DISCUSSION
Substrate kinetic, dead-end inhibitor, and product inhibition assays together with the observed ATPase activity and data indicating a phosphorylated intermediate suggest a nonclassical two-site ping-pong model whereby ATP phosphorylates the enzyme, creating a phosphorylated intermediate that then transfers the phosphate to a choline molecule. Notably, a similar mechanism has very recently been proposed for human ChoK (38).
Inhibitors HC-3 and MN58b both have inhibition profiles that are not competitive with ATP and choline, indicating that they bind to other enzyme forms than those that the substrates bind to. The exact binding sites for MN58b and RSM-932A have yet to be determined, although that of HC-3 can be inferred to enter the catalytic binding site from the fact that it is phosphorylated by p.f.-ChoK. Since MN58b has an antagonistic mechanism of action with respect to both substrates similar to that of HC-3 and, in addition, has two methyl groups in a similar position to that of HC-3, we propose that it also may enter the choline binding site in a manner analogous to HC-3.
RSM-932A, on the other hand, is uncompetitive with choline and with ATP alone, but even more so with ATP in the presence of choline. However, ATP and RSM-932A clearly also do not bind to the same enzyme conformation, judging from degradation assays and melting temperature assays that show that ATP stabilizes while RSM-932A destabilizes the protein. Since RSM-932A inhibits ATPase activity under a model that suggests a phosphate transfer to a protein residue previous to phosphate transfer to choline, this inhibitor could be either blocking ATP hydrolysis or blocking phosphoprotein hydrolysis. In the first case, one would expect an ATP/RSM-932A ternary complex that would either destabilize the protein more than RSM-932A alone or stabilize the protein more than ATP alone, because both molecules would be binding to the same enzyme form in order to fulfill an uncompetitive model. However, we see opposing effects, which suggests that the second case is more likely, i.e., that a complex between a phosphorylated protein and RSM-932A is formed and in this way phosphate transfer to choline is blocked. The noncompetitivity observed between ATP and RSM-932A appears to be not a consequence of ATP binding favoring the binding of this inhibitor but a more indirect mechanism whereby higher concentrations of ATP result in higher levels of phosphoprotein, which more favorably interact with RSM-932A. With this model in mind, and not knowing if a conformation shift occurs before or after phosphate transfer (a conformationally shifted human ChoK is observed when in a complex with phosphocholine [33]), it is possible both that RSM-932A enters the catalytic site and that it binds elsewhere on the protein structure.
Finally, we found that MN58b and RSM-932A have inhibitory concentrations against the human malaria parasite in the low nanomolar range equally effective against drug-sensitive and drug-resistant strains. Apparently, these compounds block intraerythrocytic development, which may have further effects on parasite egress or invasion. The present data provide two new structures for the development of antimalarial compounds and validate p.f.-ChoK as an accessible drug target of the parasite (17). Therefore, we expect also that these drugs may show potent in vivo antimalaria activity in mice as recently shown for another p.f.-ChoK inhibitor (13) and a choline transporter inhibitor (15).
To summarize, we introduce two new inhibitors for p.f.-ChoK, MN58b and RSM-932A, the second of which has a synergistic mechanism of action and both of which are effective in in vitro assays. The outcome of this study may provide the basis for the development of future inhibitors that are effective in in vivo systems. Moreover, the inhibition model proposed may also provide an idea of how MN58b and RSM-932A interact with the human choline kinase, the system for which they were originally studied and developed.
ACKNOWLEDGMENTS
This work has been supported by Ministerio de Economía y Competitividad, Red Temática de Investigación Cooperativa de Cancer (RTICC, RD12/0036/0019) and SAF2011-29699, and by Comunidad de Madrid (CAM S-2010 BMD-2326). The work of C.M., A.D., and J.M.B. was supported by grant BIO2010-17039 from the Spanish Ministry of Innovation and Science.
We thank Juan Casado for critical reading of the manuscript and for assistance in the mass spectrometry experiments.
Footnotes
Published ahead of print 16 September 2013
REFERENCES
- 1. Lacal JC. 2001. Choline kinase: a novel target for antitumor drugs. IDrugs 4:419–426 [PubMed] [Google Scholar]
- 2. Wittenberg J, Kornberg A. 1953. Choline phosphokinase. J. Biol. Chem. 202:431–444 [PubMed] [Google Scholar]
- 3. Ramirez de Molina A, Rodriguez-Gonzalez A, Gutierrez R, Martinez-Pineiro L, Sanchez J, Bonilla F, Rosell R, Lacal JC. 2002. Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochem. Biophys. Res. Commun. 296:580–583 [DOI] [PubMed] [Google Scholar]
- 4. Ramirez de Molina A, Banez-Coronel M, Gutierrez R, Rodriguez-Gonzalez A, Olmeda D, Megias D, Lacal JC. 2004. Choline kinase activation is a critical requirement for the proliferation of primary human mammary epithelial cells and breast tumor progression. Cancer Res. 64:6732–6739 [DOI] [PubMed] [Google Scholar]
- 5. Ramirez de Molina A, Sarmentero-Estrada J, Belda-Iniesta C, Taron M, Ramirez de Molina V, Cejas P, Skrzypski M, Gallego-Ortega D, de Castro J, Casado E, Garcia-Cabezas MA, Sanchez JJ, Nistal M, Rosell R, Gonzalez-Baron M, Lacal JC. 2007. Expression of choline kinase alpha to predict outcome in patients with early-stage non-small-cell lung cancer: a retrospective study. Lancet Oncol. 8:889–897 [DOI] [PubMed] [Google Scholar]
- 6. Ramirez de Molina A, Gallego-Ortega D, Sarmentero J, Banez-Coronel M, Martin-Cantalejo Y, Lacal JC. 2005. Choline kinase is a novel oncogene that potentiates RhoA-induced carcinogenesis. Cancer Res. 65:5647–5653 [DOI] [PubMed] [Google Scholar]
- 7. Hernandez-Alcoceba R, Fernandez F, Lacal JC. 1999. In vivo antitumor activity of choline kinase inhibitors: a novel target for anticancer drug discovery. Cancer Res. 59:3112–3118 [PubMed] [Google Scholar]
- 8. Rodriguez-Gonzalez A, Ramirez de Molina A, Fernandez F, Ramos MA, Nunez MC, Campos J, Lacal JC. 2003. Inhibition of choline kinase as a specific cytotoxic strategy in oncogene-transformed cells. Oncogene 22:8803–8812 [DOI] [PubMed] [Google Scholar]
- 9. Cannon JG. 1994. Structure-activity aspects of hemicholinium-3 (HC-3) and its analogs and congeners. Med. Res. Rev. 14:505–531 [DOI] [PubMed] [Google Scholar]
- 10. Rodriguez-Gonzalez A, Ramirez de Molina A, Fernandez F, Lacal JC. 2004. Choline kinase inhibition induces the increase in ceramides resulting in a highly specific and selective cytotoxic antitumoral strategy as a potential mechanism of action. Oncogene 23:8247–8259 [DOI] [PubMed] [Google Scholar]
- 11. Rodriguez-Gonzalez A, Ramirez de Molina A, Banez-Coronel M, Megias D, Lacal JC. 2005. Inhibition of choline kinase renders a highly selective cytotoxic effect in tumour cells through a mitochondrial independent mechanism. Int. J. Oncol. 26:999–1008 [PubMed] [Google Scholar]
- 12. Choubey V, Guha M, Maity P, Kumar S, Raghunandan R, Maulik PR, Mitra K, Halder UC, Bandyopadhyay U. 2006. Molecular characterization and localization of Plasmodium falciparum choline kinase. Biochim. Biophys. Acta 1760:1027–1038 [DOI] [PubMed] [Google Scholar]
- 13. Choubey V, Maity P, Guha M, Kumar S, Srivastava K, Puri SK, Bandyopadhyay U. 2007. Inhibition of Plasmodium falciparum choline kinase by hexadecyltrimethylammonium bromide: a possible antimalarial mechanism. Antimicrob. Agents Chemother. 51:696–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alberge B, Gannoun-Zaki L, Bascunana C, Tran van Ba C, Vial H, Cerdan R. 2010. Comparison of the cellular and biochemical properties of Plasmodium falciparum choline and ethanolamine kinases. Biochem. J. 425:149–158 [DOI] [PubMed] [Google Scholar]
- 15. Ancelin ML, Vial HJ. 1986. Quaternary ammonium compounds efficiently inhibit Plasmodium falciparum growth in vitro by impairment of choline transport. Antimicrob. Agents Chemother. 29:814–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dardonville C, Fernandez-Fernandez C, Gibbons SL, Jagerovic N, Nieto L, Ryan G, Kaiser M, Brun R. 2009. Antiprotozoal activity of 1-phenethyl-4-aminopiperidine derivatives. Antimicrob. Agents Chemother. 53:3815–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crowther GJ, Napuli AJ, Gilligan JH, Gagaring K, Borboa R, Francek C, Chen Z, Dagostino EF, Stockmyer JB, Wang Y, Rodenbough PP, Castaneda LJ, Leibly DJ, Bhandari J, Gelb MH, Brinker A, Engels IH, Taylor J, Chatterjee AK, Fantauzzi P, Glynne RJ, Van Voorhis WC, Kuhen KL. 2011. Identification of inhibitors for putative malaria drug targets among novel antimalarial compounds. Mol. Biochem. Parasitol. 175:21–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guantai E, Chibale K. 2010. Chloroquine resistance: proposed mechanisms and countermeasures. Curr. Drug Deliv. 7:312–323 [DOI] [PubMed] [Google Scholar]
- 19. Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361:455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hong BS, Allali-Hassani A, Tempel W, Finerty PJ, Jr, Mackenzie F, Dimov S, Vedadi M, Park HW. 2010. Crystal structures of human choline kinase isoforms in complex with hemicholinium-3: single amino acid near the active site influences inhibitor sensitivity. J. Biol. Chem. 285:16330–16340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Infante JP, Kinsella JE. 1976. Choline kinase kinetic studies: dual role of Mg2+ in the sequential ordered mechanism at low reactant concentrations. Regulatory mechanisms. Int. J. Biochem. 7:483–496 [Google Scholar]
- 22. Brostrom MA, Browning ET. 1973. Choline kinase from brewers' yeast. Partial purification, properties, and kinetic mechanism. J. Biol. Chem. 248:2364–2371 [PubMed] [Google Scholar]
- 23. Lai CJ, Wu JC. 2003. A simple kinetic method for rapid mechanistic analysis of reversible enzyme inhibitors. Assay Drug Dev. Technol. 1:527–535 [DOI] [PubMed] [Google Scholar]
- 24. Phillips K, de la Pena AH. 2011. The combined use of the Thermofluor assay and ThermoQ analytical software for the determination of protein stability and buffer optimization as an aid in protein crystallization. Curr. Protoc. Mol. Biol. Chapter 10:Unit10.28. 10.1002/0471142727.mb1028s94 [DOI] [PubMed] [Google Scholar]
- 25. Conejo-Garcia A, Banez-Coronel M, Sanchez-Martin RM, Rodriguez-Gonzalez A, Ramos MA, Ramirez de Molina A, Espinosa A, Gallo MA, Campos JM, Lacal JC. 2004. Influence of the linker in bispyridium compounds on the inhibition of human choline kinase. J. Med. Chem. 47:5433–5440 [DOI] [PubMed] [Google Scholar]
- 26. Sanchez-Martin R, Campos JM, Conejo-Garcia A, Cruz-Lopez O, Banez-Coronel M, Rodriguez-Gonzalez A, Gallo MA, Lacal JC, Espinosa A. 2005. Symmetrical bis-quinolinium compounds: new human choline kinase inhibitors with antiproliferative activity against the HT-29 cell line. J. Med. Chem. 48:3354–3363 [DOI] [PubMed] [Google Scholar]
- 27. Proudfoot O, Drew N, Scholzen A, Xiang S, Plebanski M. 2008. Investigation of a novel approach to scoring Giemsa-stained malaria-infected thin blood films. Malar. J. 7:62. 10.1186/1475-2875-7-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Radfar A, Mendez D, Moneriz C, Linares M, Marin-Garcia P, Puyet A, Diez A, Bautista JM. 2009. Synchronous culture of Plasmodium falciparum at high parasitemia levels. Nat. Protoc. 4:1899–1915 [DOI] [PubMed] [Google Scholar]
- 29. Moneriz C, Marin-Garcia P, Bautista JM, Diez A, Puyet A. 2009. Haemoglobin interference and increased sensitivity of fluorimetric assays for quantification of low-parasitaemia Plasmodium infected erythrocytes. Malar. J. 8:279. 10.1186/1475-2875-8-279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frey PA, Hegeman AD. 2007. Enzymatic reaction mechanisms. Oxford University Press, Oxford, England [Google Scholar]
- 31. Purich DL. 1983. Contemporary enzyme kinetics and mechanism. Academic Press, New York, NY [Google Scholar]
- 32. Cleland WW. 1977. Determining the chemical mechanisms of enzyme-catalyzed reactions by kinetic studies. Adv. Enzymol. Relat. Areas Mol. Biol. 45:273–387 [DOI] [PubMed] [Google Scholar]
- 33. Malito E, Sekulic N, Too WC, Konrad M, Lavie A. 2006. Elucidation of human choline kinase crystal structures in complex with the products ADP or phosphocholine. J. Mol. Biol. 364:136–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Northrop DB. 1969. Transcarboxylase. VI. Kinetic analysis of the reaction mechanism. J. Biol. Chem. 244:5808–5819 [PubMed] [Google Scholar]
- 35. Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naude B, Deitsch KW, Su XZ, Wootton JC, Roepe PD, Wellems TE. 2000. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 6:861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peters W. 1987. Chemotherapy and drug resistance in malaria, 2nd ed. Academic Press, London, United Kingdom [Google Scholar]
- 37. Veiga MI, Ferreira PE, Schmidt BA, Ribacke U, Bjorkman A, Tichopad A, Gil JP. 2010. Antimalarial exposure delays Plasmodium falciparum intra-erythrocytic cycle and drives drug transporter genes expression. PLoS One 5:e12408. 10.1371/journal.pone.0012408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hudson CS, Knegtel RM, Brown K, Charlton PA, Pollard JR. 2013. Kinetic and mechanistic characterisation of choline kinase-alpha. Biochim. Biophys. Acta 1834:1107–1116 [DOI] [PubMed] [Google Scholar]