

Abstract
The SCAR/WAVE complex drives actin-based protrusion, cell migration, and cell separation during cytokinesis. However, the contribution of the individual complex members to the activity of the whole remains a mystery. This is primarily because complex members depend on one another for stability, which limits the scope for experimental manipulation. Several studies suggest that Abi, a relatively small complex member, connects signaling to SCAR/WAVE complex localization and activation through its polyproline C-terminal tail. We generated a deletion series of the Dictyostelium discoideum Abi to investigate its exact role in regulation of the SCAR complex and identified a minimal fragment that would stabilize the complex. Surprisingly, loss of either the N terminus of Abi or the C-terminal polyproline tail conferred no detectable defect in complex recruitment to the leading edge or the formation of pseudopods. A fragment containing approximately 20% Abi—and none of the sites that couple to known signaling pathways—allowed the SCAR complex to function with normal localization and kinetics. However, expression of N-terminal Abi deletions exacerbated the cytokinesis defect of the Dictyostelium abi mutant, which was earlier shown to be caused by the inappropriate activation of SCAR. This demonstrates, unexpectedly, that Abi does not mediate the SCAR complex's ability to make pseudopods, beyond its role in complex stability. Instead, we propose that Abi has a modulatory role when the SCAR complex is activated through other mechanisms.
INTRODUCTION
Cell movement derives from the extension or protrusion of regions at the front of the cell followed by the coordinated retraction of the rear. When this behavior is coupled to the perception of their situation and surroundings, cells gain the ability to migrate with persistence and direction. For instance, during chemotaxis cells display an ability to detect and move along chemical gradients. Another example is during cytokinesis, where dividing cells establish opposing polarity and migrate apart from each other in a precisely coordinated manner so as to aid in the separation of the newly formed daughter cells (1). Directed migration requires the integrated activities of a multitude of proteins, ranging from sensory receptors and mediators of intracellular signaling to contractile actomyosin filaments, inducers of membrane curvature, and actin nucleators and their activators.
Dictyostelium discoideum has long been the model of choice in the study of eukaryotic chemotaxis, as its movement is very comparable to that of higher eukaryotic cells, such as neutrophils, and it is extremely amenable to genetic manipulation (2). Such traits make Dictyostelium discoideum a powerful model for dissecting the fundamental principles underlying chemotaxis.
In Dictyostelium, as in all motile eukaryotic cells, coordinated cycles of actin polymerization and depolymerization underlie cell motility. As an actin nucleator, the Arp2/Arp3 complex induces actin polymerization that promotes the formation of cellular protrusions (or pseudopodia [3, 4]). This culminates in a dense branched network of actin capable of supporting a growing pseudopod. The Arp2/Arp3 complex alone has a low basal level of activity (3). Several activators of the Arp2/Arp3 complex have now been identified, including the WASP family of proteins. Members of the WASP family include WASP, SCAR (also named WAVE), and the recently discovered WASH (5, 6). Interactions with the Arp2/Arp3 complex are mediated through a highly conserved C-terminal VCA domain, which is capable of promoting Arp2/Arp3 nucleation activity. The N-terminal regions of WASP family proteins are more varied and comprise the regulatory regions that connect the Arp2/Arp3-activating C termini to intracellular signaling events (7).
SCAR has been clearly demonstrated to be important for pseudopod extension and localizes to the extreme front of such protrusions (8). In vivo, SCAR exists as part of a large heteropentameric complex containing PIR121, Nap1, Abi, SCAR, and HSPC300, which are collectively known as the SCAR complex (9). A single, well-conserved homolog of each complex member is found in Dictyostelium discoideum (10). Recently, the crystal structure of the human SCAR/WAVE1 complex has been solved, revealing the complex architecture (11). Sra-1 (also named PIR121) and Nap1 form a platform upon which nestles a trimer composed of the other three complex members. The C-terminal VCA of SCAR/WAVE1 is sequestered within the complex, suggesting a means by which it is inhibited. However, the crystal structure yields little other information as to the purpose of the remaining bulk of the SCAR complex. Attempts to dissect the contributions of the other members to the complex as a whole have been hindered by the inherent instability of the complex to manipulation (12, 13).
As revealed by the crystal structure, SCAR/WAVE1 complex member Abi is situated at the entrance to the PIR121/Nap1 cradle wherein SCAR/WAVE1 is found. It was known that Abi interacted strongly with Nap1, and now that the crystal structure is available, it is notable that of the members of the Abi/SCAR/HSPC300 trimer, Abi is the only one to make any substantial contact with Nap1 (14, 15). The long C-terminal proline-rich tail of Abi had to be removed to aid crystallization, presumably because of its intrinsic disorder. However, it is most probable that it protrudes out from the main body of the complex, where it would be free to interact with other factors, including the extended C terminus of SCAR, following its release from the complex (16).
In addition to the above-described structural considerations, it has been reported that Abi directly recruits the SCAR complex to signaling complexes containing its well-known activator, Rac1 (17). Numerous phosphorylation sites have also been identified in Abi, and some of these have been found to be responsive to stimuli, such as epidermal growth factor treatment or serum starvation, in mammalian cells (18). Also, it has been suggested that Abi localizes and activates the complex through its interaction with ABL kinases, from which it originally derived its name (19). Overall, the literature describes Abi to be a fundamental regulator of the SCAR complex.
Previously, our lab reported the phenotype of Dictyostelium cells lacking Abi (abiA-null cells) (20). Unlike knockouts in any of the other complex members, in which SCAR is no longer stable and barely detectable by Western blotting, Dictyostelium abiA-null cells still retain appreciable levels of SCAR protein. It has been demonstrated that cells lacking Abi possess a cytokinesis defect unique to it among the null cells of the SCAR complex members. Since cells devoid of Abi and SCAR (abiA scrA double-null cells) have a phenotype equivalent to that of cells lacking SCAR (scrA-null cells), this defect has been attributed to the residual SCAR that is still present in abiA-null cells and its possible mislocalization or inappropriate activity during cytokinesis.
All of the facts mentioned above strongly imply that Abi is a crucial component of the SCAR complex responsible for its localization and subsequent activation during chemotaxis and cytokinesis. For these reasons, we have undertaken the creation of a comprehensive deletion series of the Dictyostelium Abi gene. The resulting truncated proteins were examined for their ability to stabilize the SCAR complex and to rescue different aspects of the abiA-null phenotype. We have thus identified the minimal functional fragment of Abi required to restore SCAR complex levels and activity in abiA-null cells.
MATERIALS AND METHODS
Cell culture.
Dictyostelium cells were cultured axenically in HL-5 medium (Formedium) at 22°C in petri dishes, and plasmid selection was maintained with 10 μg/ml of G418. For growth curves, 5 × 104 cells were plated in 6-well petri dishes, with cells of an individual well harvested and a cell count performed for each 12-h time point. Alternatively, for growth in shaking culture, flasks were inoculated with 5 × 104 cells/ml and a cell count was again performed every 12 h. Cell counts were performed with a CASY cell counter and analyzer system, model TT (Innovates AG). Doubling times were derived, and statistical significance and two-tailed P values were calculated through the use of unpaired t tests.
Immunoblotting.
For Western blotting, protein lysates were prepared by boiling cells in LDS buffer (Invitrogen) and were separated by electrophoresis on NuPAGE 4 to 12% bis-Tris gels (Invitrogen). Proteins were transferred onto nitrocellulose membranes (Hybond-C-extra; Amersham Biosciences) and then blocked with a solution containing 5% nonfat dried skimmed milk dissolved in Tris-buffered saline. The membranes were then probed with antibodies specific to SCAR, PIR121, or green fluorescent protein (GFP), followed by the use of fluorescence secondary antibodies that were detected using an Odyssey infrared imaging system (LI-COR Biosciences). Quantification of SCAR levels was achieved by normalization of the SCAR band intensity to the PIR121 band intensity for each sample over four blots. For quantification, unpaired, two-tailed Student t tests were used to determine statistical significance.
For native PAGE, lysates were separated by electrophoresis on NativePAGE Novex bis-Tris 4 to 16% gels (Invitrogen) according to the manufacturer's protocol. Proteins were transferred onto polyvinylidene difluoride (PVDF) membranes, blocked, probed with anti-SCAR antibody, and detected by chemiluminescence (Hybond-P PVDF membrane [Amersham], horseradish peroxidase [HRP]-conjugated secondary antibodies and Immobilon Western chemiluminescent HRP substrate [Millipore]).
TIRF microscopy.
The HL-5 medium from confluent plates was replaced with development buffer (10 mM KNaPO4, 2 mM MgCl2, 1 mM CaCl2, pH 6.5), and cells were starved for 7 h. The cells were then harvested from the plates, seeded in glass-bottom dishes (Iwaki), and then slightly compressed under 0.4% agarose slabs. SCAR complex localization and cell morphology were imaged simultaneously using total internal fluorescence (TIRF) microscopy and differential interference contrast (DIC) microscopy, respectively, on a modified Nikon Eclipse TE 2000-U microscope using a ×100 Nikon TIRF objective (numerical aperture [NA], 1.45). Images were recorded every 2 s. Pseudopodia were counted manually with the aid of ImageJ software (National Institutes of Health). Pseudopodia were defined as large convex, semicircular protrusions devoid of granular cytosol. This definition excluded all but the largest runs of blebs. HSPC300-GFP recruitment to these protrusions was then retrospectively determined. Ten cells (collectively amounting to an hour of cell migration) were analyzed for each of the transformants, and statistical significance and two-tailed P values were calculated through the use of unpaired Student t tests.
DAPI-phalloidin staining.
Doubling times were derived from the growth curves prepared as described above, and cells were seeded at a low density on glass coverslips in a staggered fashion so that the transformants underwent approximately 10 divisions. The coverslips were removed, and cells were then fixed for 5 min with a solution containing 6% (wt/vol) formaldehyde, 15% (vol/vol) picric acid, and 10 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] adjusted to pH 6.5. Cells were then washed with phosphate-buffered saline (PBS) before permeabilization with 70% ethanol for 2 min. The fixed cells were washed with PBS and then stained with 33 nM Texas Red phalloidin (Invitrogen) for 30 min. Coverslips were washed again in PBS and finally mounted on glass slides with antifade reagent containing DAPI (4′,6-diamidino-2-phenylindole; Prolong Gold; Invitrogen). Cells were imaged using an inverted wide-field microscope (IX81; Olympus) and a ×60 objective (NA, 1.42) or a Nikon A1 confocal microscope with a ×60 objective (NA, 1.4). The number of nuclei per cell was counted for >100 cells for each of the transformants, and the cells were then grouped into those with either 1 or ≥2 nuclei. The chi-square test was used for investigation of the differences between the observed frequencies of nuclei per cell. When the expected counts fell below 5, Yates' correction was applied.
RESULTS
Only a small fragment of Abi is required to stabilize the SCAR complex.
From the crystal structure available from the Protein Data Bank (accession number 3P8C; Fig. 1a), it is apparent that Abi2 is incorporated into the mammalian SCAR/WAVE1 complex through the second of its two N-terminal alpha helices (α2; Fig. 1a). This domain was identified in the Dictyostelium Abi by protein alignment (Fig. 1b). We reasoned that this alpha helix alone could be sufficient to fully stabilize the complex in Dictyostelium abiA-null cells, and so we proceeded to incrementally delete all peripheral sequence on either side of it (Fig. 1c). Deletion of the N terminus of Abi resulted in the loss of the anti-Abi epitope, which confounded detection (see Fig. S1 in the supplemental material). We were reluctant to directly tag Abi because the different relative positions of GFP could interfere differentially with Abi's normal activity. However, in order to compare the expression of all our constructs, we additionally N-terminally fused our deletion series to GFP, allowing each member of the series to be detected with anti-GFP. Figure 2a demonstrates that all of the Abi constructs were expressed and there were no gross differences in protein levels. Unless otherwise stated, in all subsequent work we coexpressed our untagged deletion series with HSPC300-GFP, an established marker of the SCAR complex (8). The untagged, truncated Abi proteins were then tested for their ability to stabilize the SCAR complex in vivo when expressed in the abiA-null cells. The SCAR protein level, as assayed by Western blotting, was initially utilized as a readout of complex integrity (Fig. 2a). We have previously shown that SCAR complex member PIR121 is stable in abiA- and scrA-null cells, and so we utilized it as a loading control (12). The domains of Abi that proved dispensable for SCAR stabilization included the first alpha helix (N-terminally truncated Abi [ΔNt Abi]) and—as has been shown previously in mammalian cells (14, 21)—the entire C-terminal polyproline tail (C-terminally truncated Abi1 [ΔCt1 Abi]). The most distal C-terminal Abi2 sequence for which there are crystal structure data available (i.e., immediately before the start of the polyproline tail) was strikingly conserved (Fig. 1b). Following its deletion in combination with the polyproline tail (ΔCt2 Abi), it, too, surprisingly, proved unnecessary for SCAR stabilization (Fig. 2a). Further truncation of the C terminus of Abi yielded fragments that were expressed but did not contribute to SCAR stability (data not shown). Finally, a minimal Abi fragment which had deletions at both termini and consisted of little more than the α2 helix (Abi with a combination of both N-terminal and C-terminal truncations [ΔAbiΔ]) restored SCAR protein levels in abiA-null cells, if only partially. Quantification of SCAR levels in these cells demonstrated that ΔAbiΔ restored SCAR significantly less than wild-type (WT) Abi (unpaired t test, P < 0.05) but restored it to a level significantly more than the basal level found in the null strain expressing HSPC300-GFP alone (unpaired t test, P < 0.001; Fig. 2b). The Abi constructs tagged with GFP also restored SCAR levels in abiA-null cells to a comparable extent (Fig. 2a).
Fig 1.

Design and implementation of Abi deletion series. (a) Illustration of human SCAR/WAVE1 complex derived from the recently resolved crystal structure available from the Protein Data Bank (accession number 3P8C). The sequence corresponding to that deleted in the Dictyostelium Abi is highlighted in red. Red ribbon, the Abi2 polyproline region that is absent from the crystal structure (not to scale). Labels designate different domains/features of Abi2: α1, 1st alpha helix; α2, 2nd alpha helix; CL, conserved loop. (b) Protein alignment between human Abi2 (above), represented in panel a, and Dictyostelium Abi (below) with a color scheme identical to that described for panel a. The sequence corresponding to the C-terminal SH3 domain of human Abi2 was removed to aid alignment, as Dictyostelium Abi does not possess this domain. Asterisks, amino acid identities; underlined sequence, amino acids corresponding to the 2nd alpha helix in panel a. (c) Diagrammatic representation of the Dictyostelium Abi domain structure and the subsequent deletion series undertaken. WT, full-length Abi; ΔNt, N-terminally truncated Abi; ΔCt1 and ΔCt2, C-terminally truncated Abi as shown; ΔAbiΔ, combination of both N-terminally and C-terminally truncated Abi.
Fig 2.

Identification of minimal Abi fragment. (a) Western blot revealing stabilization of SCAR by the untagged/tagged Abi deletion series transformed into an abiA-null strain. (Top) Lysates probed with antibody against PIR121, a stable member of the complex that was used as a loading control; (second panel) lysates probed with anti-SCAR antibody demonstrating stabilization of SCAR by different untagged Abi fragments; (third panel) expression of truncated GFP-tagged Abi constructs confirmed by anti-GFP Western blotting; (bottom) lysates probed with anti-SCAR antibody demonstrating stabilization of SCAR by different GFP-tagged Abi fragments. The dashed line divides blots into those derived from cells that possessed untagged Abi deletion series (above the dashed line) and those that possessed GFP-tagged Abi deletion series (below the dashed line). (b) Quantification of SCAR levels was achieved by normalization of the SCAR band intensity to the PIR121 band intensity for each untagged transformant over four blots. *, significantly different from WT Abi SCAR levels (unpaired t test, P < 0.05); **, significantly different from ΔAbiΔ SCAR levels (unpaired t test, P < 0.0001). Error bars indicate SEMs. (c) Native PAGE demonstrating the stabilization of the entire SCAR complex by the untagged Abi fragments. Intact protein complexes were separated by native PAGE and probed with anti-SCAR antibody following Western blotting. WT, full-length wild-type Abi; ΔNt, N-terminally truncated Abi; ΔCt1 and ΔCt2, C-terminally truncated Abi as shown in Fig. 1c; ΔAbiΔ, combination of both N-terminally and C-terminally truncated Abi; Null, HSPC300-GFP-only vector/empty GFP vector.
The stabilization of the intact complex was confirmed by native gel electrophoresis, as demonstrated in Fig. 2c. Under these conditions Dictyostelium SCAR was found in only one high-molecular-weight complex, the presence of which depended on Abi. Both PIR121 and the GFP-tagged Abi deletion series comigrated with this high-molecular-weight complex, confirming that it was the intact SCAR complex (data not shown; see Fig. S2 in the supplemental material). Neither monomeric nor complexed SCAR was detected in abiA-null strains in native PAGE, which is unsurprising, considering the low sensitivity of the method and the small amount of residual SCAR that is present when abiA is deleted. Also of note was the significant band shift in the intact complexes that had lost the C-terminal polyproline domain of Abi. Since Abi does not contribute a large amount to the total mass of the SCAR complex, it is likely that truncation of the disordered polyproline tail allows more efficient passage of the complex through the gel during electrophoresis.
In summary, we were successfully able to delete 239 of the 332 total amino acids comprising Dictyostelium Abi and still retain SCAR complex integrity. For the first time, we were in a position to interrogate the contribution of an individual complex member to the activity of the whole. With this in mind, we sought to determine if our Abi constructs restored every aspect of the wild-type SCAR complex in abiA-null cells with the aim of identifying functions that could be specifically attributed to Abi.
Abi deletion series rescues the growth defect of abiA-null cells in proportion to SCAR stabilization.
The SCAR complex is required for the optimal growth of Dictyostelium discoideum in liquid medium, presumably due to its involvement in macropinocytosis (22). Therefore, having restored the SCAR protein to wild-type levels, we next sought to determine if the truncated Abi constructs were also capable of rescuing the growth impairment of these cells. As can be seen from Fig. 3a, ΔNt Abi, ΔCt1 Abi, and ΔCt2 Abi all restored growth on substrate to a rate comparable to that of abiA-null cells rescued with full-length Abi. ΔAbiΔ did not significantly improve the growth of abiA-null cells in petri dishes. However, partial rescue was difficult to observe under these conditions, as the defect is relatively small. As shown in Fig. 3b, growth in suspension exacerbated the growth defect of the abiA-null cells and revealed an intermediate phenotype of cells rescued with ΔAbiΔ. Under these conditions, cells rescued with ΔAbiΔ doubled significantly more slowly than cells rescued with WT Abi (by 13.9 ± 3.9 h; unpaired t test, P < 0.05). The severe growth defect of the abiA-null cells in suspension led to uneven growth and made it impractical to calculate a doubling time and perform statistical comparisons. However, it is clear that transformation with ΔAbiΔ consistently improved the growth of abiA-null cells in suspension.
Fig 3.
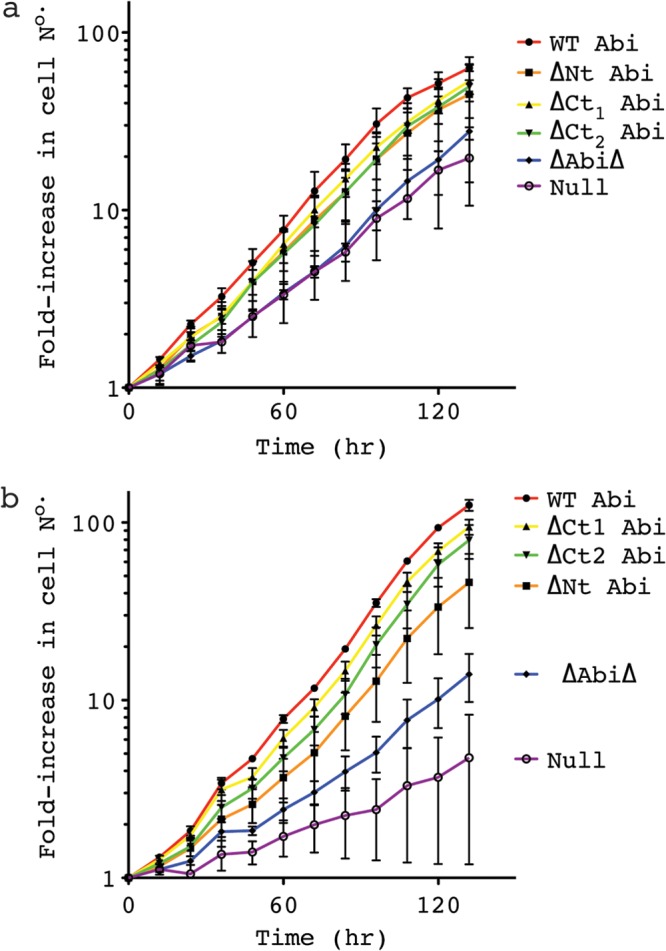
Truncated Abi proteins rescue the growth defect of abiA-null cells. (a, b) Growth curves corresponding to transformants cultured on a petri dish (a) or in shaking suspension (b). A cell count was performed every 12 h, and the fold increase was calculated and plotted. Error bars indicate SEMs. WT, full-length Abi; ΔNt, N-terminally truncated Abi; ΔCt1 and ΔCt2, C-terminally truncated Abi as shown in Fig. 1c; ΔAbiΔ, combination of both N-terminally and C-terminally truncated Abi; Null, HSPC300-GFP-only vector.
We have thus demonstrated that loss of either terminus of Abi has no detrimental effect on cell growth. Furthermore, since ΔAbiΔ partially restored SCAR levels and partially restored growth, the extent to which the growth defect of the abiA-null cells was rescued appeared to simply correlate with the SCAR protein levels in these cells. We infer that Abi contributes little to cell growth beyond stabilization of the SCAR complex. This result implies that even when lacking a large proportion of the Abi sequence, the SCAR complex still retains sufficient functionality to support optimal growth and so argues against a central role for Abi in complex recruitment and activation.
This prompted us to investigate the subcellular localization and dynamics of our mutant SCAR complexes, with particular interest in their ability to drive pseudopod extension.
SCAR complex containing truncated Abi localizes normally in migrating cells.
Coexpression of GFP-tagged complex member HSPC300 alongside the Abi fragments identified as described above in an abiA-null background allowed the localization of the respective mutant complexes to be determined. When visualized by TIRF microscopy, which illuminates only the bottom ∼100 nm of a cell, HSPC300-GFP was seen to localize strongly to the leading edge of migrating wild-type cells, correlating with pseudopod extension. No obvious differences in SCAR complex dynamics were observed between any of the Abi fragments and full-length Abi, which all showed robust concentrations of the complex at the leading edge of randomly migrating cells (Fig. 4a; see Movie S1 in the supplemental material). This was accompanied by the smooth extension of cellular protrusions, as illustrated by the kymographs in Fig. 4b. The HSPC300-GFP recruitment was weaker in cells expressing ΔAbiΔ, and they were also more prone to outbreaks of blebbing. Both observations are consistent with there being reduced levels of SCAR in these cells (23). However, SCAR complex-driven pseudopodia were also clearly evident, although the rate of protrusion was seen to be significantly decreased (unpaired t test, P < 0.05) compared to that of WT Abi-rescued cells (Fig. 4b and c; see Movie S1 in the supplemental material). HSPC300-GFP was essentially completely delocalized in abiA-null cells transformed with HSPC300-GFP alone. These cells almost exclusively moved by means of blebbing (Fig. 4b) and rarely extended anything that could be classified as a pseudopod. This was reflected in a significant reduction in the protrusion rate of ∼30% compared to that of WT Abi-expressing cells (unpaired t test, P < 0.001; Fig. 4b). The few pseudopodia that were generated were devoid of concentrated HSPC300-GFP (Fig. 4c) and were likely driven by WASP, as we have previously shown in SCAR complex-deficient cells, including abiA-null cells (8). All of this implies that not only is the majority of the Abi sequence unnecessary for SCAR complex stabilization but also it is not essential for normal complex localization. Furthermore, since all the transformants were generating the normal diverse range of pseudopodia, which are suppressed in the abiA-null cells, it would appear that the majority of Abi is not required for SCAR complex activation or function either.
Fig 4.

SCAR complexes containing truncated Abi proteins localize normally in migrating cells. (a) Localization of HSPC300-GFP in abiA-null cells expressing different Abi fragments. (Top row) DIC images of migrating cells coexpressing Abi-truncated constructs and the SCAR complex marker HSPC300-GFP; (middle row) TIRF images of the same cells revealing localization of mutant SCAR complexes; (bottom row) merged images of the upper two rows. (b) Representative kymographs highlighting smooth progression of advancing protrusions in cells expressing Abi truncations. Null cells present with a ragged slope indicative of blebbing. (c) All truncated Abi proteins rescue the suppressed pseudopod formation of abiA-null cells. *, significantly reduced rate of pseudopod formation compared to that for WT Abi-rescued cells (unpaired t test, P < 0.05); **, significantly reduced rate of pseudopod formation compared to that for ΔAbiΔ-rescued cells (unpaired t test, P < 0.001). The percentages in the box on the right indicate the proportion of pseudopodia with robust HSPC300-GFP localization ± SD. WT Abi, full-length Abi; ΔNt, N-terminally truncated Abi; ΔCt2, C-terminally truncated Abi; ΔAbiΔ, combination of both N-terminally and C-terminally truncated Abi; Null, HSPC300-GFP-only vector.
Deletion of the α1 helix alone confers a multinucleate phenotype.
Our lab has previously shown that Dictyostelium abiA-null cells have a tendency to become multinucleate due to a failure in cytokinesis (20). Many Dictyostelium mutants accumulate multinucleate cells during growth in shaking culture but can resolve such cells when introduced to a substrate by myosin-independent cytokinesis or by the mitosis-independent process of traction-mediated cytofission (24). However, even in the presence of a substrate, abiA-null cells often become multinucleate. The abiA constructs generated here were tested to determine which deletions could rescue this phenotype. Doubling times were derived from the growth curves shown in Fig. 3a. Cells were plated at a low density and cultured for 10 divisions. Following this, the cells were then fixed and stained with DAPI to visualize the nuclei and with Texas Red phalloidin for actin so as to clearly define the cell boundaries (Fig. 5a). The number of nuclei per cell was then counted, and the proportion of multinucleate cells for each of the transformants is shown in Fig. 5b. Cells with ≥2 nuclei were rarely seen in abiA-null cells rescued with full-length Abi or Abi lacking the C-terminal polyproline tail. The vast majority of these cells were mononucleate, with only a small population of cells having ≥2 nuclei (12 and 7% of the total, respectively). As has been previously documented, the abiA-null cells expressing only HSPC300-GFP accumulated multinucleate cells, with a significant increase in the number of cells with ≥2 nuclei compared to that for the WT Abi controls (26%; chi-squared test, P < 0.0001) being detected. Furthermore, there were also more severely multinucleate cells present, including some with as many as 10 nuclei per cell (Fig. 5b). Surprisingly, deletion of the N-terminal α1 helix resulted in a significant increase in the number of cells with ≥2 nuclei (42%) even compared to the number of abiA-null cells (chi-square test, P < 0.0001). This was also true of ΔAbiΔ-expressing cells (39% of cells with ≥2 nuclei). Again, this proved to be a significant increase compared to the results for both the WT Abi controls and the abiA-null cells (chi-square test, P < 0.0001 and P < 0.01, respectively), although there was no statistically significant difference compared to the results for the ΔNt Abi-expressing cells (chi-square test, P = 0.36). The different severity in the phenotype observed between the abiA-null cells and the ΔAbiΔ- and ΔNt Abi-expressing cells correlates with the increasing SCAR levels in these cells (Fig. 2a and b) and, therefore, presumably, an increasing amount of deregulated SCAR complex.
Fig 5.
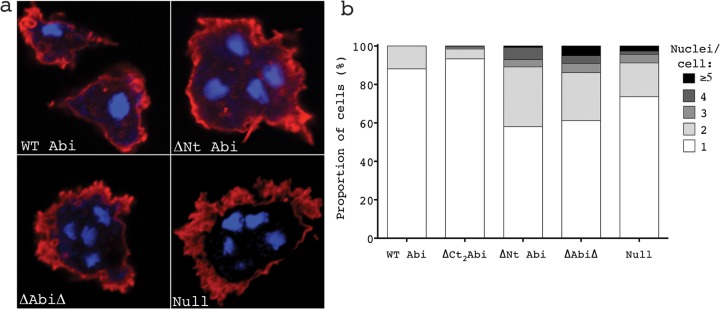
Deletion of the 1st alpha helix is sufficient to reproduce and to exacerbate the multinucleate phenotype of abiA-null cells. Cells were cultured for the time taken for 10 divisions on glass coverslips before fixation and stained with Texas Red phalloidin and DAPI. The number and extent of multinucleate cells were then counted. (a) Examples of normal and multinucleate cells found in abiA-null cells expressing WT Abi, ΔNt Abi, ΔAbiΔ, and the HSPC300-GFP vector only (Null). (b) Percentages of cells with 1, 2, 3, 4, or ≥5 nuclei cells rescued with the different Abi constructs. WT Abi, full-length Abi; ΔNt, N-terminally truncated Abi; ΔCt2, C-terminally truncated Abi; ΔAbiΔ, combination of both N-terminally and C-terminally truncated Abi; Null, HSPC300-GFP-only vector.
These data are consistent with our lab's previous conclusion that it is the remaining SCAR in the abiA-null cells that is responsible for its multinuclearity. The data presented here identify the α1 helix of Abi as being important for the regulation of the SCAR complex during cytokinesis.
DISCUSSION
Abi is considered a key component of the SCAR complex that is thought to control its activation by acting as a target for phosphorylation and through its interaction with various regulatory proteins, such as receptor tyrosine kinases. However, here we deleted the majority of the Abi sequence, including all the conserved phosphorylation sites identified, and still observed robust localization of the SCAR complex at the leading edge of migrating cells. The loss of Abi in its entirety resulted in a loss of complex integrity, and so we cannot rule out the possibility that the remaining alpha helix required for complex stability is involved in activation. However, structural considerations suggest that this is unlikely, as the minimal Abi fragment that still stabilized the complex consisted of little more than a single alpha helix that incorporated Abi into the complex. Furthermore, on the basis of the model that emerged from the resolution of the crystal structure, this helix would be oriented away from the plasma membrane, where activation is presumed to take place. Most surprisingly, deletion of the entire C-terminal polyproline tail of Abi, which accounts for almost half of the entire sequence, yielded no detectable phenotype. Instead, it was deletion of the first 44 amino acids of Abi that was sufficient to reproduce and apparently exacerbate the multinucleate defect of the abiA-null cells.
Dictyostelium possesses multiple robust mechanisms to ensure successful cell division. These include the substrate-independent action of myosin at the cleavage furrow of a dividing cell, which acts to pinch the cell in two. This is coupled to adhesion-dependent directed migration, which acts to drive the newly forming daughter cells apart. These two modes of cytokinesis almost certainly work together in concert to ensure efficient cell division; however, they are also sufficient to compensate in the absence of one or the other. As a final safeguard, multinucleate cells on a substrate can randomly tear themselves apart independently of mitosis by traction-mediated cytofission. We have previously shown that these two substrate-dependent means of cell division require the SCAR complex (1). The scar-null cells themselves have a low level of multinuclearity, as myosin is sufficient to drive efficient cytokinesis. The fact that the abiA-null cells become multinucleate has been attributed to a misregulation or localization of their residual SCAR protein, with the resulting aberrant activity disrupting cell division. Despite restoring SCAR to wild-type levels, expression of the N-terminally deleted Abi merely aggravated the cytokinesis defect, supporting our previous conclusions. It is notable that the multinucleate phenotype documented here is not strong compared with the phenotype derived from mutations that specifically block cytokinesis. Given that the cells were cultured for 10 divisions, the vast majority of even the most severely affected ΔNt Abi- or ΔAbiΔ-expressing cells were no more than binucleate. This is well below what the particularly robust Dictyostelium can tolerate and possibly explains why ΔNt Abi confers no significant adverse effect on growth. By stabilizing the SCAR complex with our N-terminally truncated Abi proteins, we were also likely restoring traction-mediated cytofission in these cells, which works to counteract any specific defect in cytokinesis.
From all this we conclude that Abi is not required for recruitment or activation of the SCAR complex even during cell migration and cytokinesis. Instead, we suggest here that Abi's role is to modulate the activity of the SCAR complex and act as a signal integrator. During events such as cytokinesis, where the SCAR complex appears to be hyperactivated, the small imbalance in activity conferred by the absence of the α1 helix of Abi would have the greatest effect and could possibly accumulate to ultimately contest and disrupt normal cell division (1). As to how exactly the α1 helix of Abi is regulating SCAR complex activity and what it is interacting with remain unknown. Nevertheless, this work has identified one functional domain on what still remains a mostly impenetrable complex.
There exists strong conservation between the Dictyostelium SCAR complex members and those found in higher eukaryotes, in terms of both sequence and function. However, in higher eukaryotes, Abi is evidently more complicated, possessing an SH3 domain and interactors not present in Dictyostelium. These presumably accommodate the increasingly complex layers of regulation required to control SCAR activity in multicellular organisms. As a modulator of SCAR complex activity, Abi would appear to be a natural target through which to exert these additional layers of regulation. This observation, together with the findings presented in this paper, lead us to propose that Abi's role in all motile cells is as a modulator. Abi is unlikely to directly mediate either SCAR complex recruitment to the membrane or activation in any cell type, including cells of higher animals. Instead, it most likely tunes the activity of the SCAR complex during or after activation.
Supplementary Material
ACKNOWLEDGMENTS
We are thankful for the funding provided by Cancer Research UK.
We also thank Margaret O'Prey of the Beatson Advanced Imaging Resource for technical help with the microscopy and Douwe Veltman for providing various plasmids. Finally, we voice our appreciation for the Dictyostelium community resource, dictybase.org.
Footnotes
Published ahead of print 13 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00116-13.
REFERENCES
- 1. King JS, Veltman DM, Georgiou M, Baum B, Insall RH. 2010. SCAR/WAVE is activated at mitosis and drives myosin-independent cytokinesis. J. Cell Sci. 123:2246–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andrew N, Insall RH. 2007. Chemotaxis in shallow gradients is mediated independently of PtdIns 3-kinase by biased choices between random protrusions. Nat. Cell Biol. 9:193–200 [DOI] [PubMed] [Google Scholar]
- 3. Mullins RD, Heuser JA, Pollard TD. 1998. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci. U. S. A. 95:6181–6186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Svitkina TM, Borisy GG. 1999. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 145:1009–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Machesky LM, Insall RH. 1998. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr. Biol. 8:1347–1356 [DOI] [PubMed] [Google Scholar]
- 6. Linardopoulou EV, Parghi SS, Friedman C, Osborn GE, Parkhurst SM, Trask BJ. 2007. Human subtelomeric WASH genes encode a new subclass of the WASP family. PLoS Genet. 3:e237. 10.1371/journal.pgen.0030237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pollitt AY, Insall RH. 2009. WASP and SCAR/WAVE proteins: the drivers of actin assembly. J. Cell Sci. 122:2575–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Veltman DM, King JS, Machesky LM, Insall RH. 2012. SCAR knockouts in Dictyostelium: WASP assumes SCAR's position and upstream regulators in pseudopods. J. Cell Biol. 198:501–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. 2002. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 418:790–793 [DOI] [PubMed] [Google Scholar]
- 10. Caracino D, Jones C, Compton M, Saxe CL., III 2007. The N-terminus of Dictyostelium Scar interacts with Abi and HSPC300 and is essential for proper regulation and function. Mol. Biol. Cell 18:1609–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Z, Borek D, Padrick SB, Gomez TS, Metlagel Z, Ismail AM, Umetani J, Billadeau DD, Otwinowski Z, Rosen MK. 2010. Structure and control of the actin regulatory WAVE complex. Nature 468:533–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ibarra N, Blagg SL, Vazquez F, Insall RH. 2006. Nap1 regulates Dictyostelium cell motility and adhesion through SCAR-dependent and -independent pathways. Curr. Biol. 16:717–722 [DOI] [PubMed] [Google Scholar]
- 13. Kunda P, Craig G, Dominguez V, Baum B. 2003. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr. Biol. 13:1867–1875 [DOI] [PubMed] [Google Scholar]
- 14. Innocenti M, Zucconi A, Disanza A, Frittoli E, Areces LB, Steffen A, Stradal TE, Di Fiore PP, Carlier MF, Scita G. 2004. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat. Cell Biol. 6:319–327 [DOI] [PubMed] [Google Scholar]
- 15. Gautreau A, Ho HY, Li J, Steen H, Gygi SP, Kirschner MW. 2004. Purification and architecture of the ubiquitous Wave complex. Proc. Natl. Acad. Sci. U. S. A. 101:4379–4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davidson AJ, Insall RH. 2011. Actin-based motility: WAVE regulatory complex structure reopens old SCARs. Curr. Biol. 21:R66–R68. 10.1016/j.cub.2010.12.001 [DOI] [PubMed] [Google Scholar]
- 17. Innocenti M, Frittoli E, Ponzanelli I, Falck JR, Brachmann SM, Di Fiore PP, Scita G. 2003. Phosphoinositide 3-kinase activates Rac by entering in a complex with Eps8, Abi1, and Sos-1. J. Cell Biol. 160:17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lebensohn AM, Kirschner MW. 2009. Activation of the WAVE complex by coincident signals controls actin assembly. Mol. Cell 36:512–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leng Y, Zhang J, Badour K, Arpaia E, Freeman S, Cheung P, Siu M, Siminovitch K. 2005. Abelson-interactor-1 promotes WAVE2 membrane translocation and Abelson-mediated tyrosine phosphorylation required for WAVE2 activation. Proc. Natl. Acad. Sci. U. S. A. 102:1098–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pollitt AY, Insall RH. 2008. Abi mutants in Dictyostelium reveal specific roles for the SCAR/WAVE complex in cytokinesis. Curr. Biol. 18:203–210 [DOI] [PubMed] [Google Scholar]
- 21. Stradal T, Courtney KD, Rottner K, Hahne P, Small JV, Pendergast AM. 2001. The Abl interactor proteins localize to sites of actin polymerization at the tips of lamellipodia and filopodia. Curr. Biol. 11:891–895 [DOI] [PubMed] [Google Scholar]
- 22. Seastone DJ, Harris E, Temesvari LA, Bear JE, Saxe CL, Cardelli J. 2001. The WASp-like protein scar regulates macropinocytosis, phagocytosis and endosomal membrane flow in Dictyostelium. J. Cell Sci. 114:2673–2683 [DOI] [PubMed] [Google Scholar]
- 23. Ura S, Pollitt AY, Veltman DM, Morrice NA, Machesky LM, Insall RH. 2012. Pseudopod growth and evolution during cell movement is controlled through SCAR/WAVE dephosphorylation. Curr. Biol. 22:553–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nagasaki A, de Hostos EL, Uyeda TQ. 2002. Genetic and morphological evidence for two parallel pathways of cell-cycle-coupled cytokinesis in Dictyostelium. J. Cell Sci. 115:2241–2251 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.