

Abstract
Proper cellular localization is required for the function of many proteins. The CaaX prenyltransferases (where CaaX indicates a cysteine followed by two aliphatic amino acids and a variable amino acid) direct the subcellular localization of a large group of proteins by catalyzing the attachment of hydrophobic isoprenoid moieties onto C-terminal CaaX motifs, thus facilitating membrane association. This group of enzymes includes farnesyltransferase (Ftase) and geranylgeranyltransferase-I (Ggtase-1). Classically, the variable (X) amino acid determines whether a protein will be an Ftase or Ggtase-I substrate, with Ggtase-I substrates often containing CaaL motifs. In this study, we identify the gene encoding the β subunit of Ggtase-I (CDC43) and demonstrate that Ggtase-mediated activity is not essential. However, Cryptococcus neoformans CDC43 is important for thermotolerance, morphogenesis, and virulence. We find that Ggtase-I function is required for full membrane localization of Rho10 and the two Cdc42 paralogs (Cdc42 and Cdc420). Interestingly, the related Rac and Ras proteins are not mislocalized in the cdc43Δ mutant even though they contain similar CaaL motifs. Additionally, the membrane localization of each of these GTPases is dependent on the prenylation of the CaaX cysteine. These results indicate that C. neoformans CaaX prenyltransferases may recognize their substrates in a unique manner from existing models of prenyltransferase specificity. It also suggests that the C. neoformans Ftase, which has been shown to be more important for C. neoformans proliferation and viability, may be the primary prenyltransferase for proteins that are typically geranylgeranylated in other species.
INTRODUCTION
In eukaryotic cells, Ras-like GTPases perform vital signaling roles necessary for cell growth, differentiation, and morphogenesis. The majority of their functions are performed at cellular membranes, but their protein sequences alone are not sufficient for membrane interaction. Instead, these proteins must be posttranslationally modified with lipid moieties that facilitate membrane association. Most commonly, these GTPases are modified by the addition of an isoprenoid group in a process known as protein prenylation.
Many members of the Ras-like GTPase family have a characteristic C-terminal CaaX-box motif, which designates them as prenyltransferase substrates. This motif consists of a cysteine followed by two aliphatic amino acids and variable amino acid. A prenyltransferase can bind to this motif and covalently attach an isoprenoid group onto the CaaX-box cysteine. This modification is irreversible and is often essential for stable membrane localization (1–3).
The two prenyltransferase enzymes that catalyze this type of prenylation are farnesyltransferase (Ftase) and geranylgeranyltransferase-I (Ggtase-I). These are structurally similar, dimeric enzyme complexes that share an α subunit and are differentiated by their distinct β subunits. Prenyl pyrophosphates (farnesyl pyrophosphate or geranylgeranyl pyrophosphate) interact with specific β subunits, giving the prenyltransferase donor group specificity. The β subunit typically interacts with certain CaaX-box sequences to allow for target substrate specificity. In general, substrates for Ftase or Ggtase-I enzymes are differentiated by the identity of the variable amino acid (X) of the CaaX box. Although there is overlapping specificity between Ftase and Ggtase-I substrates, CaaX-box proteins terminating in leucine or other hydrophobic amino acids are typically geranylgeranylated, while Ftase substrates can terminate in one of several different amino acids (4–6).
Once prenylated, the C termini of these proteins are further modified in the endoplasmic reticulum prior to plasma membrane localization. First, the terminal aaX amino acids are cleaved by the CaaX-specific protease Rce1 (7–9). The remaining terminal cysteine is then carboxymethylated by the Ste14/Icmt methyltransferase (10, 11). These modifications are important for full membrane association of the prenylated protein but are not absolutely required for protein function.
Because prenylation substrates are involved in a variety of cellular processes, inhibition of this process is often lethal. For this reason, there have been many prenyltransferase inhibitors developed to target cancer cells. More recently, a number of these prenyltransferase inhibitors have been repurposed to inhibit eukaryotic pathogens. Prenyltransferase inhibitors have been shown to be effective against two pathogenic parasites, Trypanosoma brucei and Plasmodium falciparum, in mouse models of infection (12–14). Additionally, prenyltransferases have been shown to be important for fungal pathogen virulence and virulence-associated phenotypes. In the opportunistic pathogen Candida albicans, the shared Ftase and Ggtase-I α subunit is essential (15). In addition, C. albicans Ras1 requires its predicted prenylation site to mediate hyphal growth, a phenotype important for C. albicans virulence (16).
Cryptococcus neoformans is a pathogenic fungus that causes life-threatening disease in immunocompromised individuals. C. neoformans has emerged as a significant global health problem due to rising numbers of immunocompromised patients caused by the HIV pandemic and the increased use of immunosuppressive drugs following organ transplants. This environmental fungus establishes a primary infection in the lungs before disseminating to the brain in immunocompromised patients. Worldwide, over 600,000 deaths are caused by C. neoformans infections each year, primarily as a result of meningoencephalitis (17).
Previous work has documented that protein farnesylation plays an important role in C. neoformans growth, differentiation, and virulence (18). Furthermore, the C. neoformans Ras1 (CnRas1) GTPase, which is a predicted target protein for prenylation, loses all detectable function after disruption of its C-terminal CaaX motif (19). Interestingly, while Ras proteins are typically farnesylated, the C-terminal amino acid sequences of C. neoformans Ras1 and Ras2 proteins suggest that they are Ggtase-I substrates. To date, no studies have documented the role of the Ggtase-I in C. neoformans virulence.
Typical Ggtase-I substrates include Rho family proteins (Rho, Rac, and Cdc42). In many eukaryotes, these and other predicted Ggtase-I substrates play central roles in cell polarity and stress response. In the model yeast Saccharomyces cerevisiae, the Ggtase-I β subunit is essential, likely due to the role of Ggtase-I in modifying Cdc42 and Rho1, both essential proteins in this yeast (20). Many of these predicted Ggtase-I substrates are conserved in C. neoformans; in particular, the Rho family GTPases have been shown to be extremely important in high-temperature growth and virulence. For example, the duplicate C. neoformans Cdc42 and Cdc420 paralogs help to direct septin protein localization and cytokinesis (21). The Rac proteins, Rac1 and Rac2, play a more specialized role in cell polarity, reactive oxygen species localization, and endocytic vesicular trafficking (22–24). C. neoformans Rho proteins are important for cell wall synthesis and integrity (25).
In this study, we define the role of the C. neoformans Ggtase-I by disrupting the Ggtase-I β subunit and show that the Ggtase-I enzyme plays a role in high-temperature growth, virulence, and mating. We also explore several predicted Ggtase-I substrates that reveal potential plasticity in prenyltransferase specificity in C. neoformans.
MATERIALS AND METHODS
Strains, media, and growth conditions.
The strains used in this study are listed in Table 1. All mutants and fluorescent fusion protein-expressing strains were created in the C. neoformans H99 MATα background. Unless otherwise stated, strains were cultured on YPD (yeast extract 1%, peptone 2%, dextrose 2%) (26) plates or in YPD liquid medium. Mating experiments were conducted on MS mating medium (27). The cell wall and membrane stress plates were made by adding indicated concentrations of Congo red, calcofluor white, SDS, or caffeine to YPD medium prior to autoclaving. To create strain KS41 (cdc43Δ MATa), the CH9 (cdc43Δ MATα) strain was crossed with KN99a (MATa), and recombinant spores were isolated by microdissection. To analyze morphogenesis, overnight liquid YPD cultures grown at 30°C with 150 rpm shaking were diluted 1:10 in fresh YPD medium prewarmed to 30°C or 37°C. These cultures were incubated at the indicated temperature (Fig. 1) with shaking (150 rpm) for 18 h prior to imaging.
Table 1.
Strains in this study
| Strain | Genotype | Reference or source |
|---|---|---|
| H99 | MATα | 53 |
| KN99 | MATa | 54 |
| CH9 | MATα cdc43::nat | This study |
| KS41 | MATa cdc43Δ::nat | This study |
| KS37 | MATα cdc43Δ::nat CDC43 | This study |
| ERB016 | MATα pHIS-GFP-CDC42-nat | This study |
| KS8 | MATα cdc43Δ::nat pHIS-GFP-CDC42-neo | This study |
| KS13 | MATα pHIS-GFP-cdc42(C190A)a | This study |
| ERB010 | MATα cdc42::nat | 21 |
| ERB011 | MATα cdc42::nat cdc420::neo | 21 |
| ERB013 | MATα cdc42::nat CDC42-neo | 21 |
| KS13 | MATα cdc42::nat cdc42(C190A)-neo | This study |
| KS14 | MATα cdc42::nat cdc42(C190A)-neo | This study |
| KS15 | MATα cdc42::nat cdc42(C190A)-neo | This study |
| ERB053 | MATα rac2::neo pHIS-GFP-RAC2-nat | This study |
| KS49 | MATα rac2::neo pHIS-GFP-rac2(C195A) | This study |
| KS84 | MATα rac2::neo cdc43::nat pHIS-GFP-RAC2-neo | This study |
| CBN55 | MATα ras1::neo ras1(C207A)-nat | 19 |
| KS44 | MATα cdc43Δ::nat pHIS-mCherry-RAS1-neo | This study |
| CBN116 | MATα pHIS-mCherry-Ras1-neo | This study |
| KS126 | MATα pHIS-GFP-RHO1-neo | This study |
| KS129 | MATα cdc43Δ::nat pHIS-GFP-RHO1-neo | This study |
| KS132 | MATα pHIS-GFP-RHO10-neo | This study |
| KS135 | MATα cdc43Δ::nat pHIS-GFP-RHO10-neo | This study |
| KS138 | MATα pHIS-GFP-RHO11-neo | This study |
| KS141 | MATα cdc43Δ::nat pHIS-GFP-RHO11-neo | This study |
The form cdc42(C190A) indicates the C-to-A change at position 190 encoded by cdc42. Similar notation was used for amino acid mutations in ras1 and rac2, as shown.
Fig 1.
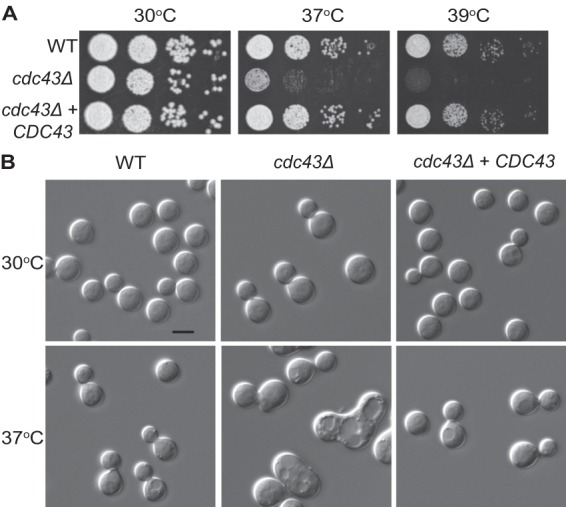
Ggtase-I involvement in thermotolerance and fungal cell morphology. (A) The cdc43Δ mutant has a growth defect at 37°C and 39°C. Five-fold serial dilutions of the indicated strains were spotted onto YPD medium and incubated for 48 h at the indicated temperatures. (B) The cdc43Δ mutant displayed apparent cytokinesis defects when grown at 37°C. Cells were incubated in YPD medium to mid-logarithmic phase at 30°C and shifted to preheated medium at 37°C for 18 h. Cell morphology was assessed by photomicroscopy.
Molecular biology.
Primers used to create each strain are listed in Table 2. The cdc43Δ mutant was created by replacing the entire open reading frame in H99 strain with the dominant nourseothricin (NAT) resistance gene (28). As previously described (29), PCR overlap extension was used to create a cdc43Δ::nat disruption construct, which was integrated into the genome using biolistic transformation as previously described (30). The cdc43Δ::nat construct was created using the following primers (Table 2): CDC43 3′ fragment, AA3014 and AA1996; CDC43 5′ fragment, AA1999 and AA3015; NAT resistance marker, AA1997 and AA1998. Primers AA3014 and AA3045 were used to amplify the final disruption construct. The cdc43Δ mutant was confirmed by Southern blotting. The AA3210 and AA3211 primers were used to create the Southern blot probe.
Table 2.
Primers
| Primer | Sequencea |
|---|---|
| AA1510 | 5′-CGGGATCCCGGGAGATTATGCTGCGGATGT-3′ |
| AA1519 | 5′-CGGGATCCCGAGAAGGGGGAGTCTGGAAC-3′ |
| AA1638 | 5′-CGCGGATCCATGCAGACAATCAAGTGTG-3′ |
| AA1928 | 5′-GGGCCCAGATCTATGGCCATGCAGAGTATC-3′ |
| AA1996 | 5′-GCTAGTTTCTACATCTCTTCCGTGTGTGTGGGTGCATGGTGAA-3′ |
| AA1997 | 5′-TTCACCATGCACCCACACACACGGAAGAGATGTAGAAACTAGC-3′ |
| AA1998 | 5′-GGTGATCTCTCTTTGCAGCCTTGCTAGGCTGCGAGGAT-3′ |
| AA1999 | 5′-ATCCTCGCAGCCTAGCAAGGCTGCAAAGAGAGATCACC-3′ |
| AA3014 | 5′-TTCGCGGGAAAGTAAGTCTC-3′ |
| AA3015 | 5′-AAATGTCGGACAGGACAAGG-3′ |
| AA3045 | 5′-TCGGCCTTTCTCCAAAACTA-3′ |
| AA3184 | 5′-GTCTAAAAAGGCCCTCATCCTTTGAAGAC-3′ |
| AA3185 | 5′-GTCTTCAAAGGATGAGGGCCTTTTTAGAC-3′ |
| AA3188 | 5′-GGGCCCGGATCCTATGGGCACAGACAGACAGC-3′ |
| AA3189 | 5′-GGGCCCGGATCCATGGCTCCTCTCGACTCTCC-3′ |
| AA3210 | 5′-ACCAATGCGAGAGGAAGAGA-3′ |
| AA3211 | 5′-TCTGCGTCCTTCAAATACCC-3′ |
| AA3404 | 5′-GGGCCCAGATCTTCAGAGAATCAAAGCCTG-3′ |
| AA3767 | 5′-CCTATAGGATCCCTCAATCCTCATCCACGCCA-3′ |
| AA3768 | 5′-GTTGGAGGATCCTAACGATCCACTCCGGAAAC-3′ |
| AA3769 | 5′-AACGACGGATCCATGTCTGTAAGTGCTGGGAC-3′ |
| AA3770 | 5′-AAGCTCGGATCCAAATTATCACGCGGCAACTC-3′ |
| AA3771 | 5′-CCAAGATCTGCGGCCCACCACCTCTTTTCC-3′ |
| AA3772 | 5′-TATGAAGATCTTTCCGGCGGG-3′ |
H99 DNA sequence is underlined in each primer.
To reconstitute the cdc43Δ mutant, primers AA3189 and AA3188 were used to amplify the CDC43 locus, promoter, and terminator. The PCR product was cloned into the TOPO TA vector pCR2.1 (Invitrogen), digested with BamHI, and ligated into BamHI-digested pJAF1 (29) vector to create pKS1. This plasmid was transformed into the cdc43Δ mutant, and transformants were screened for neomycin resistance and rescued thermotolerance.
To create the green fluorescent protein (GFP)-tagged Cdc42C190A (pKS3) (where Cdc42C190A is Cdc42 with a C-to-A change at position 190) and Rac2C195A (pKS12) fusion proteins, PCR overlap extension was performed to introduce the appropriate base pair changes in addition to adding restriction sites. The primers used to generate the Cdc42C190A point mutations were the following: fragment 1, AA1638 and AA3184 (mutation primer); fragment 2, AA3185 (mutation primer) and AA1519. The full-length product was amplified using AA1638 and AA1519. For GFP-Rac2C195A, the primers AA1928 and AA3404 (mutation primer with BglII restriction site) were used. These point mutants were then cloned into pCN50 (31) containing the neomycin resistance marker, His3 promoter, and GFP.
The GFP-tagged Rho1, Rho10, and Rho11 plasmids were created by amplifying each gene and terminator sequence from H99 genomic DNA and cloning the sequences into the single BamHI site in pCN50. The following primer pairs were used: RHO1, AA3767 and AA3768; RHO10, AA3769 and AA3770; RHO11, AA3771 and AA3772.
To create the pKS4 plasmid, which contains the Cdc42C190A under its endogenous promoter, PCR overlap extension was performed, and the resulting fragment was cloned into the BamHI site in pJAF1 (29). Primers used were the following: fragment 1, AA1510 and AA3184; fragment 2, AA3185 and AA1519. The full-length product was amplified using AA1510 and AA1519.
Microscopy.
Morphology and mating images were obtained using differential interference microscopy (DIC); fluorescent mating images were captured using a Zeiss Axio Imager A1 fluorescence microscope equipped with an AxioCam MRM digital camera. The high-resolution fluorescent images were captured using a DeltaVision Elite deconvolution microscope equipped with a Coolsnap HQ2 high resolution charge-coupled-device (CCD) camera.
For images of mating structures, samples were fixed with 70% ethanol and permeabilized with 1% Triton X-100. The cell wall was visualized using calcofluor white, and nuclei were stained with SYTOX green (S7020; Molecular Probes).
Animal and macrophage experiments.
As previously described (32), J774.1 murine macrophage-like cells were used to assess survival within macrophages. J774.1 cells (1 × 105 cells/well) were plated in a 96-well plate and incubated for 18 h. J774.1 cells were then activated for 1 h in 10 nM phorbol myristate acetate (PMA) diluted in Dulbecco's modified Eagles medium (DMEM). C. neoformans cells were washed two times with phosphate-buffered saline (PBS) and opsonized for 1 h at 37°C in DMEM containing 1 μg/ml anti-glucuronoxylomannan (GXM) monoclonal antibody (MAb) 18b7 (33, 34). The PMA was removed from the J774.1 cells, and 1 × 105 opsonized C. neoformans cells were added to each well (multiplicity of infection [MOI] of 1). After 1 h of coincubation, the nonengulfed/adherent C. neoformans cells were removed by washing three times with 200 μl of PBS. DMEM was then added to each well, and cells were incubated for 24 h. To release phagocytosed C. neoformans cells, sterile distilled H2O (dH2O) was added to each well, and the macrophages were lysed by vigorous pipetting. Wells were washed two more times with sterile dH2O and combined. Quantitative culturing was used to assess the number of viable C. neoformans cells.
Virulence was tested using the inhalation model of infection described in Cox et al. (35). Briefly, 10 female A/Jcr mice were anesthetized with 140 μl of 12 mg/ml ketamine HCl and 1 mg/ml xylazine in PBS. Each mouse was then intranasally inoculated with 1 × 105 cells in 25 μl of PBS. The mice were monitored and sacrificed based on predetermined symptoms that predict imminent death. Groups were compared using the log rank test (JMP software; SAS Institute, Cary, NC). All studies were performed in compliance with Duke University institutional guidelines for animal experimentation.
RESULTS
Identification of the C. neoformans geranylgeranyltransferase-I.
In S. cerevisiae and C. albicans, the Ggtase-I β subunit is encoded by CDC43. One potential Cdc43 ortholog (CNAG_02756) was identified in the C. neoformans genome by reciprocal BLAST searches using the S. cerevisiae and C. albicans Cdc43 protein sequences. The C. neoformans CDC43 (CnCDC43) gene encodes a 259-amino-acid protein with 24% and 32.5% sequence similarity to the S. cerevisiae Cdc43 (ScCdc43) and C. albicans Cdc43 (CaCdc43) proteins, respectively.
Cryptococcus neoformans geranylgeranyltransferase-I is involved in high-temperature growth and morphogenesis.
To elucidate the role of the C. neoformans Ggtase-I, we created a C. neoformans cdc43Δ mutant by replacing the entire open reading frame (ORF) with a drug resistance marker. The CDC43 locus was completely deleted in several isolates, and Southern blot analysis showed that each isolate had a single integration event removing the entire CDC43 coding region. Therefore, unlike S. cerevisiae, the C. neoformans cdc43 gene is not essential.
Ggtase-I activity is required for proper cell proliferation and differentiation in other eukaryotes, especially under conditions of cell stress (36–39). Therefore, we tested the cdc43Δ mutant for growth defects under high-temperature stress, which is one of the major cell stresses this organism encounters during infection. While the C. neoformans cdc43Δ mutant grew equally as well as the wild type at 30°C, it displayed a marked growth defect at 37°C and a more severe defect at 39°C (Fig. 1A).
Temperature sensitivity often results from dysregulation of the cell cycle at high temperature, leading to specific morphological defects in C. neoformans cells (22, 40–42). Consistent with this hypothesis, there were notable morphological changes in the cdc43Δ cells compared to wild-type cells when they were incubated at 37°C. At this elevated temperature, the majority of the cdc43Δ cells displayed wide bud necks and chains of multiple budding cells, indicative of a cytokinesis defect. As expected, the morphology of the cdc43Δ mutant was indistinguishable from that of the wild type when it was grown at 30°C. Importantly, both the temperature sensitivity and the morphological defects of the cdc43Δ mutant were completely rescued by the reintroduction of the wild-type CDC43 allele in the cdc43Δ CDC43 complemented strain.
C. neoformans Ggtase-I is required for virulence.
Because C. neoformans must be able to grow at high temperatures to cause disease, we hypothesized that the temperature-sensitive cdc43Δ mutant would have a defect in virulence. C. neoformans is a facultative intracellular pathogen, and the ability to replicate within macrophages is strongly correlated with the ability to cause disease (43). Therefore, we tested the cdc43Δ strain by coculturing J774A.1 murine macrophages with opsonized C. neoformans cells at a multiplicity of infection of 1:1 for 1 h. At this point, unengulfed cells were removed by gentle washing. After 24 h, the macrophages were lysed, and the intracellular fungal replication/survival rate was determined by quantitative culture. The cdc43Δ mutant had a significant growth/survival defect in macrophages, with an output/input viable cell ratio that was 41% lower than that of wild-type C. neoformans (Fig. 2A). This indicates that Cdc43 and therefore Ggtase-I activity are required for full growth in association with macrophages.
Fig 2.
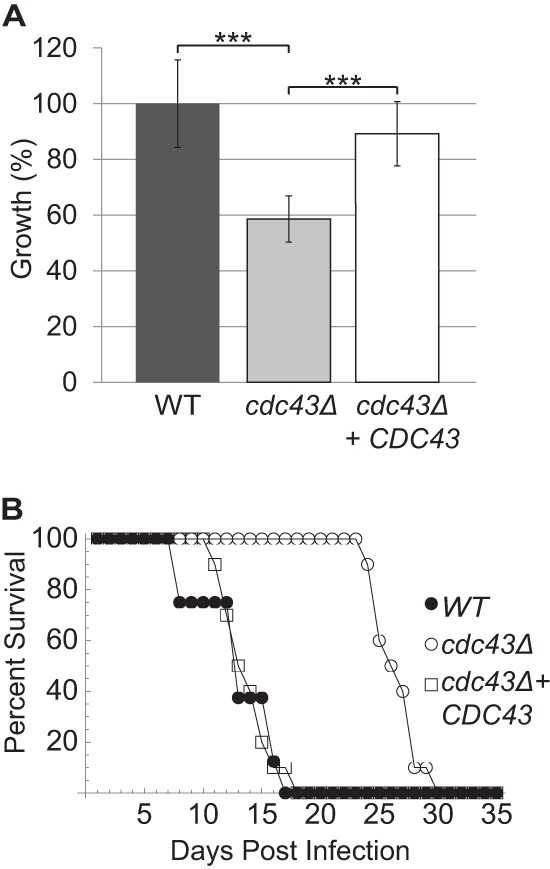
Effect of cdc43Δ mutation on virulence. (A) The cdc43Δ mutant has a relative growth defect compared to wild-type and reconstituted strains in murine macrophages. C. neoformans cells were opsonized with anticapsular antibody (18B7) and coincubated with J774.1 murine macrophage-like cells that had been activated in PMA. After 1 h, unphagocytosed Cryptococcus cells were washed away, and phagocytosed cells were coincubated with macrophages for 24 h. Macrophages were lysed, and surviving fungal cells were quantitatively cultured. The viable output/input cell number was calculated for each strain and normalized to that of the wild type. ***, P < 0.001. (B) The cdc43Δ mutant has a delayed virulence in mice. Ten A/J mice were intranasally infected with 1 × 105 cells for each strain and monitored for survival. WT, wild type.
We then extended our analysis of the cdc43Δ strain by assessing virulence in a mouse inhalation model of cryptococcal infection. We intranasally inoculated 10 female AJ mice with 5 × 105 cryptococcal cells of the wild-type, cdc43Δ, and cdc43Δ CDC43 strains. There was no statistically significant difference in survival of mice infected with the wild-type or the reconstituted strain, with all mice succumbing to the infection by day 17. In contrast, mice infected with the cdc43Δ mutant displayed significantly longer survival until day 30 (P = <0.0001) (Fig. 2B). These results demonstrate that the Cdc43 protein is required for full virulence in a physiologically relevant model of C. neoformans infection, likely due to its role in growth at elevated temperatures.
Analysis of predicted geranylgeranyltransferase-I substrates.
We hypothesized that the cdc43Δ mutant phenotypes were due to mislocalization of one or more Ggtase-I substrates. Small GTPases constitute an important group of prenylated proteins that require membrane localization to be fully functional (1, 44). In C. neoformans, growth and morphology are maintained at elevated temperatures by the coordinated action of several GTPases. In other eukaryotes, Cdc42 and Rac proteins are classical Ggtase-I substrates, defined by C-terminal CaaX-box motifs ending in leucine. In C. neoformans, Ras1 also terminates in a leucine, designating it as a potential Ggtase-I substrate, even though Ras proteins are generally farnesylated in other eukaryotes (19). These three GTPases are required for mating, morphology, and high-temperature growth in C. neoformans (19, 21, 22, 41). Since Ras, Rac, and Cdc42 proteins in C. neoformans regulate different aspects of morphogenesis, mating, and stress responses, elucidating their roles in the cdc43Δ mutant phenotype could potentially provide insight into which of these proteins require geranylgeranylation for their full activity.
To explore the interaction between protein localization and activity for Ras, Rac, and Cdc42 proteins, we examined the effect of cdc43Δ mutation on their localization. To do this, we utilized fluorescent fusion proteins and chose to initially examine Rac2, Cdc42, and Ras1 as the representative paralogs. In C. neoformans, mCherry-Ras1 is primarily localized to the plasma membrane (19), while GFP-Rac2 localizes to both the plasma membrane and endomembranes (22). Similarly, we observed GFP-Cdc42 localized to the plasma membrane and endomembranes in CDC43 competent cells (Fig. 3). Despite the presence of the CaaL geranylgeranylation motif in Ras1 and Rac2, these proteins maintained wild-type membrane localization in the cdc43Δ mutant background. However, the GFP-Cdc42 protein, which has the same CaaL motif as Rac2, was mislocalized, primarily in the cytoplasm, in the cdc43Δ mutant. Together, these results suggest that Cdc42 requires Cdc43 and geranylgeranylation for full membrane association. Additionally, the lack of mislocalization of the Ras1 and Rac2 proteins in the cdc43Δ mutant strain suggests that the CaaL motif is insufficient for determining prenylation specificity (Fig. 3).
Fig 3.

Localization of Cdc42, Rac2, and Ras1. Wild-type and cdc43Δ strains expressing the indicated GFP fusion proteins were grown in YPD medium at 30°C and imaged by DeltaVision microscopy. The amino acid sequence of the C-terminal -Caax motif encoded by each allele is indicated under each image. Scale bar, 5 μm.
Previously, we demonstrated that C. neoformans Ras1 requires its prenylation site (the CaaX-box cysteine) for membrane localization (19), and mutating this site completely disrupts protein localization and function (19) (Fig. 3). Both Rac2 and Cdc42 contain polybasic regions that potentially aid in membrane localization. To confirm that the Rac2 and Cdc42 proteins also require prenylation for appropriate membrane localization, we mutated the CaaX-box cysteine to an alanine in both proteins. Both the Rac2C195A and Cdc42C190A nonprenylatable mutants localized diffusely throughout the cytoplasm and showed no membrane localization. Therefore, both Rac2 and Cdc42 require their predicted prenylation sites for membrane interaction. These data suggest that, despite containing identical CaaX motifs, the Ras1, Rac2, and Cdc42 proteins are differentially targeted by prenyltransferases in C. neoformans.
Cdc42 function is dependent on prenylation.
In concordance with the altered localization of Cdc42, the cdc43Δ mutant has several phenotypes that mimic disrupted Cdc42 activity, including temperature sensitivity and cytokinesis defects (21). The phenotypic similarities between cdc42Δ and cdc43Δ mutants, combined with the finding that GFP-Cdc42 is unable to fully interact with membranes in the cdc43Δ mutant, suggest that Cdc42 function is dependent on geranylgeranylation. To explore the role of prenylation in Cdc42 function, we examined whether the Cdc42C190A point mutant would be able to complement a cdc42Δ null mutant. We introduced the Cdc42C190A mutant allele into the cdc42Δ strain. In contrast to full phenotypic complementation of the cdc42Δ strain with the wild-type Cdc42 allele, two independent cdc42Δ Cdc42C190A isolates demonstrated no restoration of cdc42Δ mutant phenotypes, including failed growth at 37°C (Fig. 4). We confirmed wild-type levels of expression in these strains using semiquantitative PCR with primers specific to CDC42 mRNA (data not shown). We also sequenced the mutant allele to confirm that there were no additional mutations in the CDC42 coding sequence. These results are consistent with a requirement for geranylgeranylation-dependent membrane localization for full Cdc42 activity.
Fig 4.
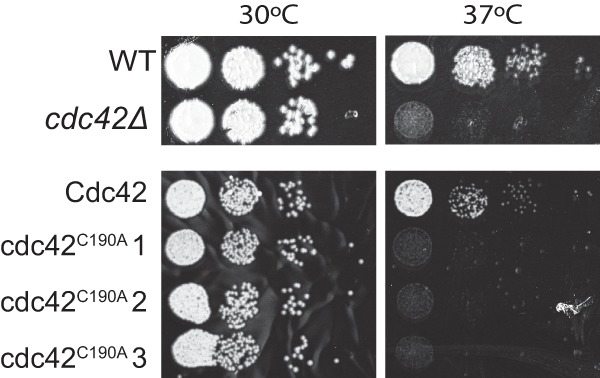
Cdc42 requires its prenylation motif for functionality. The WT, cdc42Δ mutant, cdc42Δ Cdc42 reconstituted strain, and the prenylation defective cdc42Δ Cdc42C190A strains were serially diluted and incubated on YPD medium for 48 h at 30°C or 37°C.
The cdc43Δ mutant does not exhibit cell wall defects.
In S. cerevisiae, Rho1, another member of the Rho-GTPase family, is geranylgeranylated (20). Like Cdc42, Rho1 geranylgeranylation is required for full protein function. In C. neoformans, there are three paralogs of Rho1: Rho1, Rho10, and Rho11. Lam et al. defined the role of each of these paralogs and found that while Rho1 is essential in C. neoformans, each of the paralogs is involved in the cell wall stress response to various degrees. All three Rho proteins contain C-terminal CaaX motifs, with Rho1 and Rho10 containing CaaL sequences. While Rho10 was previously predicted to lack a CaaX motif, analysis of the Rho10 transcript sequence using RNA sequencing data revealed that one splice site was misannotated; the corrected sequence contains a CLIL C-terminal motif (25) (FungiDB.org). To determine whether any of the Rho paralogs requires geranylgeranylation for localization, we analyzed the localization of GFP-Rho fusion proteins in wild-type and cdc43Δ cells. Of the three Rho paralogs, only GFP-Rho10 localized to cellular membranes in wild-type cells (Fig. 5A and data not shown). This membrane localization was slightly disrupted in the cdc43Δ mutant, indicating that the Rho10 protein is likely a Ggtase-I substrate.
Fig 5.
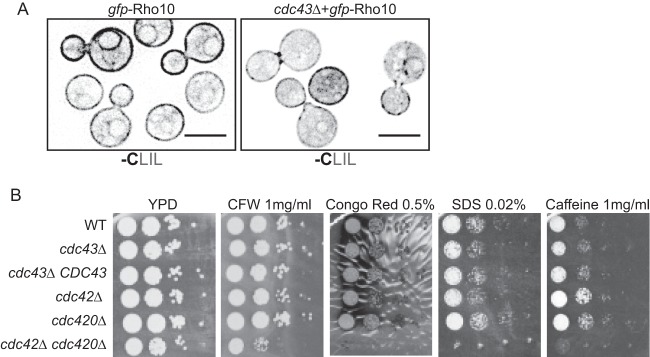
Ggtase-I function is required for proper Rho10 localization but not for cell wall stress tolerance. (A) Wild-type and cdc43Δ cells expressing GFP-Rho10 were cultured in YPD medium at 30°C and imaged using DeltaVision microscopy. The amino acid sequence of the C-terminal -Caax motif encoded by each allele is indicated under each image. Scale bar, 5 μm. Ten-fold serial dilutions of each strain were spotted onto YPD medium containing the indicated cell wall stressor. Plates were incubated at 30°C for sufficient time to visualize colonies. CFW, calcofluor white.
The rho10Δ mutant is sensitive to several cell wall and cell membrane stresses, including caffeine, calcofluor white, and SDS. If the function of Rho10 is significantly disrupted in the cdc43Δ mutant, we hypothesized that the cdc43Δ mutant would be sensitive to cell wall and cell membrane stresses (25). To confirm that any cell wall sensitivities would be due specifically to Rho10 defects, we also examined the growth of the cdc42Δ, cdc42Δ, and cdc42Δ cdc420Δ mutant strains on the same cell wall stressors. We found that the cdc42Δ cdc420Δ double mutant had dramatic growth defects compared to the wild type on all stresses tested, with particular sensitivity to SDS. However, we found that the cdc43Δ mutant grew as well as the wild type on all cell wall and cell membrane stressors (Fig. 5B). These results indicate that Cdc43 and therefore Ggtase-I activity are not required for Rho10 function. Additionally, these results indicate that while Cdc43 is required for Cdc42 and Cdc420 localization and functions in high-temperature growth, it is not required for their full protein function in cell wall integrity.
The cdc43Δ mutant has a mating defect.
To determine whether the Ggtase-I mutant has a mating defect similar to that of the cdc42Δ mutant, we analyzed the hyphae and spore formation in cdc43Δ mutant crosses. The unilateral mating between a cdc43Δ MATα strain and an isogenic wild-type strain of the opposite mating type (MATa) produced filaments and spores indistinguishable from a wild-type cross (data not shown). In contrast, the cdc43Δ MATα/cdc43Δ MATa bilateral mutant cross resulted in delayed mating hypha production (Fig. 6A). Instead of being evenly distributed around the mating reaction, as was observed for the wild-type mating, the cdc43Δ MATα/cdc43Δ MATa hyphae are sporadically distributed, with noticeable gaps between each grouping of hyphae. While there is a notable decrease in mating hypha production at early time points in a cdc43Δ mutant bilateral cross, these mating mixtures eventually produced wild-type levels of mating structures and viable spores (Fig. 6B). Additionally, each hyphal segment maintained two nuclei, as observed in wild-type crosses (Fig. 6C).
Fig 6.
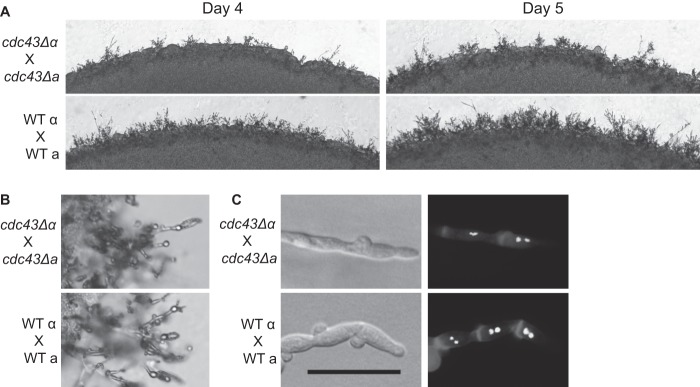
Role of Ggtase-I in mating. (A) The indicated strains were coincubated in the dark on MS medium. The same location on each plate was imaged for 4 and 5 days. (B) Basidia and spore structures were imaged from matings shown in panel A. (C) Mating plugs were cut, permeabilized, and stained with calcofluor white to visualize the cell wall and with SYTOX green to visualize nuclei. Scale bar, 20 μm.
The cdc43Δ mutant is more susceptible to farnesyltransferase inhibitors.
In other eukaryotes, the Ftase enzyme can partially compensate for a cdc43Δ mutation by farnesylating Ggtase-I substrates (20). Therefore, we hypothesized that Ftase inhibition may be more detrimental to cell survival in the absence of a partially redundant prenyltransferase. To test this possibility, we performed antifungal drug susceptibility testing in wild-type and cdc43Δ strains using tipifarnib and manumycin A, two Ftase inhibitors (45). Standard disk diffusion assays revealed enhanced antifungal effects for both of these compounds in the cdc43Δ mutant compared to the isogenic wild-type strain (Fig. 7). Additionally, assessment of the MIC for these strains in liquid medium revealed that the cdc43Δ strain was 4-fold more sensitive to tipifarnib and 2-fold more sensitive to manumycin A than the wild type. Therefore, some degree of cross-prenylation may be protective for basic growth parameters in C. neoformans, even in the absence of significant cell stress.
Fig 7.
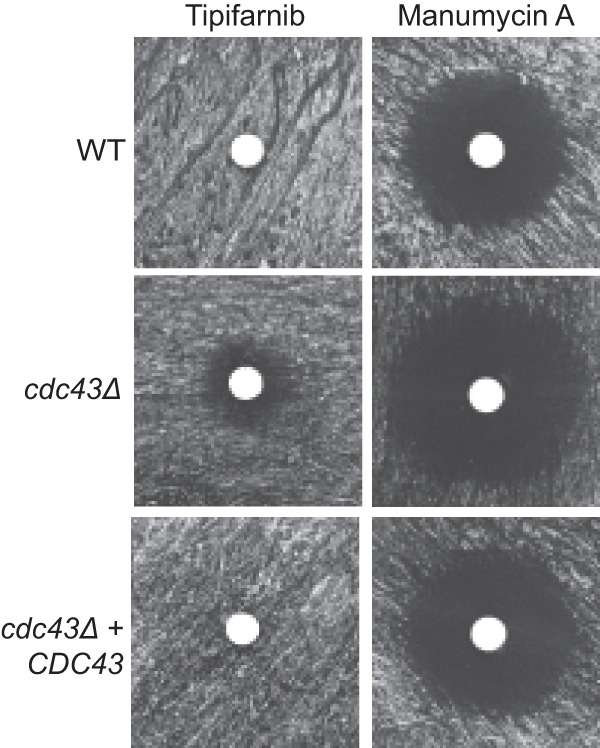
cdc43Δ mutant is hypersensitive to Ftase inhibitors. Wild-type and cdc43Δ cells were incubated at 37°C on yeast nitrogen base plates with sterile cotton disks containing 10 μl of 20 μM tipifarnib or 20 μM manumycin A.
DISCUSSION
In this study, we identified and characterized the role of the putative geranylgeranyltransferase-I enzyme in the pathogenic fungus C. neoformans. Our lab has previously explored the importance of prenylation for C. neoformans survival during infection. Pharmacological inhibition of the Ftase enzyme inhibited mating and also resulted in increased susceptibility to the calcineurin inhibitor FK506 (18). In a subsequent study, the C. neoformans Ras1 protein was shown to require its predicted prenylated cysteine to be functional (19). Appropriate Ras1 membrane localization is absolutely required for adaptation to the elevated temperatures of the host; therefore, Ras1 prenylation is likely essential for C. neoformans virulence.
Previous work on the C. neoformans Ram1 Ftase protein revealed that this enzyme is required for Ras1 localization and function. While Ras1 is a conserved Ftase substrate in most eukaryotes, the C. neoformans Ras1 protein contains a CaaX-box motif normally found on Ggtase-I substrates. Classically, the variable (X) amino acid of the CaaX box determines whether the protein will be an Ftase or a Ggtase-I substrate (4). While this paradigm may represent an oversimplification of prenyltransferase specificity, it is generally accepted that proteins containing CaaL (leucine) motifs are primarily Ggtase-I substrates. Interestingly, C. neoformans Ras1 terminates in a leucine residue, despite being farnesylated by Ftase in vitro and losing membrane localization after Ftase inhibition. Due to the importance of Ftase in C. neoformans infections, we hypothesized that the Ftase may be the major CaaX prenyltransferase in C. neoformans.
To more fully explore the role of prenyltransferases in C. neoformans biology, we identified the gene encoding the predicted Ggtase-I β subunit and characterized its role in cellular proliferation and virulence. We generated several cdc43Δ mutants, demonstrating that in C. neoformans, Ggtase-I activity is not essential. However, this mutant displayed growth defects and morphological abnormalities at elevated temperatures. These results indicate that while C. neoformans Ggtase-I activity may not be completely required for cellular proliferation, Ggtase-I substrates likely regulate many important stress-related processes in the fungal cell.
These phenotypes are in contrast to studies in S. cerevisiae, where the Ftase is dispensable but the Ggtase-I enzyme is essential (46). However, the relative importance of these prenyltransferases appears to vary from species to species, as the C. albicans CDC43 gene is not essential and as the cdc43Δ mutant displays only mild morphological defects (39).
C. neoformans Ggtase-I substrates.
The phenotypes that result from disrupting prenylation are likely to be consequences of altered function in their substrate proteins. While we have no biochemical evidence that the C. neoformans protein is acting as a component of the Ggtase-I complex, our data strongly indicate that this highly conserved protein is required for Ggtase-I function. The commonly studied Ggtase-I substrates belong to the Rho GTPase family, which is comprised of Rho, Cdc42, and Rac proteins. In C. neoformans, these proteins regulate cellular polarity, morphogenesis, and cell wall integrity. C. neoformans encodes two Cdc42 proteins, three Rho proteins, and two Rac proteins. Each of these GTPases contains C-terminal CaaX motifs, and all but Rho11 and Rac1 have CaaL domains and are therefore predicted to be typical Ggtase-I substrates. Therefore, to test the hypothesis that each of these proteins requires geranylgeranylation for appropriate localization, we examined the localization of each via GFP fusion constructs in the background of a cdc43Δ mutant strain that lacks Ggtase-I function. We additionally examined an mCherry-Ras1 fusion protein as the C. neoformans Ras1 also contains a CaaL motif. Although most of these Rho family proteins were predicted Ggtase-I substrates based on their CaaX motifs and have been shown to be geranylgeranylated in other eukaryotes (20, 39, 47), only Cdc42, Cdc420, and Rho10 required the Cdc43 Ggtase-I β subunit for membrane localization in C. neoformans.
We also demonstrated that Cdc42 absolutely requires its prenylation site for both membrane localization and function. It is therefore likely that the cdc43Δ mutant phenotypes result from decreased Cdc42 protein function. In fact, both the cdc42Δ mutant's temperature sensitivity and cytokinesis defects are shared phenotypes of cdc42Δ and cdc42Δ cdc420Δ mutants (21). However, the cdc42Δ mutations cause much more severe temperature sensitivity and virulence defects than the cdc43Δ mutant. Furthermore, the cdc43Δ mutant displays appropriately structured clamp cells during mating, which are abnormal in cdc42Δ cdc420Δ mutants. Finally, the cdc43Δ mutant was not sensitive to cell wall or membrane stress while the cdc42Δ cdc420Δ mutant was highly sensitive to each of these stresses. Taken together, these results indicate that Cdc42 and Cdc420 activity is only partially disrupted in the cdc43Δ mutant. A close examination of GFP-Cdc42 localization in the cdc43Δ mutant shows that membrane localization is not completely abolished in the cdc43Δ mutant, providing some explanation for the intermediate cdc43Δ phenotypes. Potentially, the intact farnesyltransferase may compensate in the absence of Ggtase-I activity. Additionally, it is possible that some of the Cdc42/Cdc420 functions are independent of membrane localization and are thus not affected when this localization is disrupted.
The C. neoformans Rho1, Rho10, and Rho11 proteins function in cell wall maintenance, stress response, and high-temperature growth. Rho1 is the only Rho protein in C. neoformans that appears to be essential. Mutating these proteins dramatically reduces C. neoformans adaptations to cell wall stresses (25). Even though Rho10 membrane localization was mildly disrupted in the cdc43Δ mutant, the cdc43Δ mutant displayed no increased sensitivity to calcofluor white, Congo red, SDS, or caffeine. Therefore, Rho10 activity appears to be sufficient to support cell wall integrity in spite of incomplete membrane localization.
In contrast to commonly accepted models, GFP-Rho1 and GFP-Rho11 proteins were not localized primarily to the plasma membrane in wild-type cells. Membrane localization of Rho proteins has been established in S. cerevisiae, Schizosaccharomyces pombe, and C. albicans (20, 38, 39). There are several possibilities as to why these proteins are not obviously localized to the plasma membrane. First, perhaps only a small percentage of the cellular Rho1 and Rho11 protein population is membrane localized, making it difficult to visualize membrane association. Additionally, we used a histone H3 promoter to drive constitutive expression of the GFP constructs. It is possible that overexpression of Rho1 or Rho11 is detrimental to C. neoformans cells, and we may have inadvertently selected for transformants in which there is decreased membrane localization in order to decrease their activity. Supporting this model, Lam et al. found that Rho1 is detrimental to C. neoformans cells when it is constitutively active (25). However, there is no increase in membrane staining in strains with lower levels of GFP-Rho1 or GFP-Rho11 expression. Therefore, it is also possible that the proteins are sequestered into the cytoplasm via inhibitory proteins, such as guanosine nucleotide dissociation inhibitors (GDIs). These GDIs decrease the effective activity of small GTPases by binding to their prenyl groups and removing these proteins from membranes (48).
In addition to their predicted prenylation sites, most prenylated proteins contain at least one additional membrane-targeting domain. The C. neoformans Ras1 protein is also palmitoylated, and each of the C. neoformans Rho family GTPases contains a polybasic domain upstream from the CaaX-box motif. These domains are also important for membrane localization, but they are rarely sufficient for membrane interaction (49, 50). Therefore, even though both Cdc42 and Rac2 contain polybasic regions, both lose all membrane localization when their prenylation sites are disrupted (Fig. 3), similar to Ras1 (19). Therefore, membrane localization signals distant from the CaaX motif are insufficient to direct membrane localization of these proteins in the absence of prenylation.
The results of this study demonstrate that the C. neoformans Ggtase-I plays a conserved role in Cdc42 and Rho10 protein localization but that it is not likely to be the major CaaX prenyltransferase in this species. The only clear Ggtase-I substrates were the Cdc42 proteins and Rho10 even though there are several other important GTPases that are predicated to be Ggtase-I substrates. We hypothesize that many of the classical Ggtase-I substrates, such as the Rac proteins, are instead Ftase substrates in C. neoformans. Furthermore, the CaaL motif is clearly not a sufficiently conserved Ggtase-I recognition sequence in C. neoformans, which implies that there may be an unknown mechanism for prenyltransferase substrate recognition. These studies and our previous work suggest that the Ftase prenyltransferase has assumed many of the canonical Ggtase-I substrates in this microbial pathogen. Supporting this hypothesis, biochemical analysis of the C. neoformans Ftase revealed a uniquely high affinity for CaaL domains in vitro (45). This is a clear deviation from a similar study on human prenyltransferase substrate specificity, which demonstrated that the hydrophobic X amino acids, primarily leucine, specify Ggtase-I substrates (4).
Although the Rho proteins analyzed here are some of the most studied Ggtase-I substrates, there are other potential substrates that may contribute to the cdc43Δ mutant phenotypes. The chitin synthase regulator (Csr1 to Csr3) proteins, which are involved in chitin and chitosan synthesis, all contain CaaX motifs and likely require membrane localization for their function (51).
C. neoformans Ggtase-I role in mating.
In addition to affecting high-temperature growth and morphology in C. neoformans, many of the prenylation substrates are required for efficient mating (21, 41). In C. neoformans, the Cdc42 and Rac proteins act downstream of Ras1 to regulate septin localization and the establishment of polarized growth, both of which are central processes in the complex mating reaction (21–23). In addition, both a and α pheromones contain CaaX-box domains, suggesting that, as in S. cerevisiae, prenylation is required for pheromone function in C. neoformans (52). We found that the cdc43Δ mutant had a very subtle defect during mating filament formation. This phenotype is also similar to the phenotypes of cdc42Δ and cdc420Δ mutants; therefore, we hypothesized that this cdc43Δ phenotype was primarily due to decreased Cdc42 protein function. However, the cdc43Δ mating filaments had normal clamp cell morphology and dikaryotic nuclear transport, which are normally disrupted when the Cdc42 proteins are mutated. Therefore, as suggested by our studies in fungal cell growth and morphogenesis, Cdc42 protein activity in mating is reduced but not abolished in the absence of the Cdc43 Ggtase-I protein.
Disrupting C. neoformans Ggtase-I increases Ftase inhibitor efficacy.
While some classical Ggtase-I substrates appeared to be fully functional in the Ggtase-I-deficient cdc43Δ mutant, this mutation still resulted in decreased C. neoformans virulence, both in macrophages and in a mouse inhalation model of C. neoformans infection. This decreased virulence is likely due to the thermosensitivity of the cdc43Δ mutant.
While Cdc43 is required for full virulence, all mice infected with the cdc43Δ mutant eventually succumbed to the infection. Therefore, C. neoformans Ggtase-I alone would not be an ideal antifungal drug target. However, previous investigations have shown that farnesyltransferase inhibitors dramatically impair C. neoformans growth. In addition, the results presented in this paper indicate that there may be an unexpected degree of overlap between Ggtase-I and Ftase substrate specificity. Therefore, we studied the concept of cross-prenylation using Ftase inhibitors in strains with disrupted Ggtase-I function. The cdc43Δ mutant was significantly more sensitive to both Ftase inhibitors tested. These results indicate that while Ggtase-I inhibitors are not likely to be effective antifungal agents on their own, they could be combined with Ftase inhibitors to increase their efficacy.
In conclusion, these studies demonstrate that the C. neoformans Ggtase-I performs a conserved role in the prenylation and localization of the Cdc42 proteins and Rho10. This activity is important for allowing growth of this pathogenic fungus at host physiological temperatures. In fact, disruption of Ggtase-I function results in impaired virulence in an animal model of C. neoformans infection. However, these studies also suggest that the CaaX motif is not sufficient to determine the specificity of farnesylation versus geranylgeranylation; some degree of compensatory farnesylation appears to occur with proteins classically predicted to be Ggtase-I substrates. Moreover, between the two types of prenyltransferases, the Ftase is likely more directly required for virulence-associated function. Single or dual prenylation inhibition strategies are therefore promising tools to explore the pathogenesis and drug inhibition of this important pathogen.
ACKNOWLEDGMENTS
We thank Sam Johnson at the Duke University Light Microscopy Core Facility for his assistance with the Delta Vision microscopy. We also thank Christie Hay for strain creation. Finally, we thank Teresa O'Meara for advice and critical reading of the manuscript.
This work was supported by PHS grant AI050128.
Footnotes
Published ahead of print 6 September 2013
REFERENCES
- 1. Nguyen UT, Goody RS, Alexandrov K. 2010. Understanding and exploiting protein prenyltransferases. Chembiochem 11:1194–1201 [DOI] [PubMed] [Google Scholar]
- 2. Basso AD, Kirschmeier P, Bishop WR. 2006. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J. Lipid Res. 47:15–31 [DOI] [PubMed] [Google Scholar]
- 3. Casey PJ, Seabra MC. 1996. Protein prenyltransferases. J. Biol. Chem. 271:5289–5292 [DOI] [PubMed] [Google Scholar]
- 4. Reid TS, Terry KL, Casey PJ, Beese LS. 2004. Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. J. Mol. Biol. 343:417–433 [DOI] [PubMed] [Google Scholar]
- 5. Moores SL, Schaber MD, Mosser SD, Rands E, O'Hara MB, Garsky VM, Marshall MS, Pompliano DL, Gibbs JB. 1991. Sequence dependence of protein isoprenylation. J. Biol. Chem. 266:14603–14610 [PubMed] [Google Scholar]
- 6. Finegold AA, Johnson DI, Farnsworth CC, Gelb MH, Judd SR, Glomset JA, Tamanoi F. 1991. Protein geranylgeranyltransferase of Saccharomyces cerevisiae is specific for Cys-Xaa-Xaa-Leu motif proteins and requires the CDC43 gene product but not the DPR1 gene product. Proc. Natl. Acad. Sci. U. S. A. 88:4448–4452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boyartchuk VL, Ashby MN, Rine J. 1997. Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science 275:1796–1800 [DOI] [PubMed] [Google Scholar]
- 8. Ashby MN, King DS, Rine J. 1992. Endoproteolytic processing of a farnesylated peptide in vitro. Proc. Natl. Acad. Sci. U. S. A. 89:4613–4617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hrycyna CA, Clarke S. 1992. Maturation of isoprenylated proteins in Saccharomyces cerevisiae. Multiple activities catalyze the cleavage of the three carboxyl-terminal amino acids from farnesylated substrates in vitro. J. Biol. Chem. 267:10457–10464 [PubMed] [Google Scholar]
- 10. Hrycyna CA, Sapperstein SK, Clarke S, Michaelis S. 1991. The Saccharomyces cerevisiae Ste14 gene encodes a methyltransferase that mediates C-terminal methylation of a-factor and Ras proteins. EMBO J. 10:1699–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ashby MN, Errada PR, Boyartchuk VL, Rine J. 1993. Isolation and DNA sequence of the STE14 gene encoding farnesyl cysteine: carboxyl methyltransferase. Yeast 9:907–913 [DOI] [PubMed] [Google Scholar]
- 12. No JH, de Macedo Dossin F, Zhang Y, Liu YL, Zhu W, Feng X, Yoo JA, Lee E, Wang K, Hui R, Freitas-Junior LH, Oldfield E. 2012. Lipophilic analogs of zoledronate and risedronate inhibit Plasmodium geranylgeranyl diphosphate synthase (GGPPS) and exhibit potent antimalarial activity. Proc. Natl. Acad. Sci. U. S. A. 109:4058–4063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eastman RT, Buckner FS, Yokoyama K, Gelb MH, Van Voorhis WC. 2006. Thematic review series: lipid posttranslational modifications. Fighting parasitic disease by blocking protein farnesylation. J. Lipid Res. 47:233–240 [DOI] [PubMed] [Google Scholar]
- 14. Chakrabarti D, Da Silva T, Barger J, Paquette S, Patel H, Patterson S, Allen CM. 2002. Protein farnesyltransferase and protein prenylation in Plasmodium falciparum. J. Biol. Chem. 277:42066–42073 [DOI] [PubMed] [Google Scholar]
- 15. Song JL, White TC. 2003. RAM2: an essential gene in the prenylation pathway of Candida albicans. Microbiology 149:249–259 [DOI] [PubMed] [Google Scholar]
- 16. Piispanen AE, Bonnefoi O, Carden S, Deveau A, Bassilana M, Hogan DA. 2011. Roles of Ras1 membrane localization during Candida albicans hyphal growth and farnesol response. Eukaryot. Cell 10:1473–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530 [DOI] [PubMed] [Google Scholar]
- 18. Vallim MA, Fernandes L, Alspaugh JA. 2004. The RAM1 gene encoding a protein-farnesyltransferase beta-subunit homologue is essential in Cryptococcus neoformans. Microbiology 150:1925–1935 [DOI] [PubMed] [Google Scholar]
- 19. Nichols CB, Ferreyra J, Ballou ER, Alspaugh JA. 2009. Subcellular localization directs signaling specificity of the Cryptococcus neoformans Ras1 protein. Eukaryot. Cell 8:181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ohya Y, Qadota H, Anraku Y, Pringle JR, Botstein D. 1993. Suppression of yeast geranylgeranyl transferase I defect by alternative prenylation of two target GTPases, Rho1p and Cdc42p. Mol. Biol. Cell 4:1017–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ballou ER, Nichols CB, Miglia KJ, Kozubowski L, Alspaugh JA. 2010. Two CDC42 paralogues modulate Cryptococcus neoformans thermotolerance and morphogenesis under host physiological conditions. Mol. Microbiol. 75:763–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ballou ER, Selvig K, Narloch JL, Nichols CB, Andrew Alspaugh J. 2013. Two Rac paralogs regulate polarized growth in the human fungal pathogen Cryptococcus neoformans. Fungal Genet. Biol. 57:58–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vallim MA, Nichols CB, Fernandes L, Cramer KL, Alspaugh JA. 2005. A Rac homolog functions downstream of Ras1 to control hyphal differentiation and high-temperature growth in the pathogenic fungus Cryptococcus neoformans. Eukaryot. Cell 4:1066–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shen G, Zhou E, Alspaugh JA, Wang P. 2012. Wsp1 is downstream of Cin1 and regulates vesicle transport and actin cytoskeleton as an effector of Cdc42 and Rac1 in Cryptococcus neoformans. Eukaryot. Cell 11:471–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lam WC, Gerik KJ, Lodge JK. 2013. Role of Cryptococcus neoformans Rho1 GTPases in the PKC1 signaling pathway in response to thermal stress. Eukaryot. Cell 12:118–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sherman F. 1991. Getting started with yeast. Methods Enzymol. 194:3–21 [DOI] [PubMed] [Google Scholar]
- 27. Murashige T, Skoog F. 1962. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 15:473–497 [Google Scholar]
- 28. McDade HC, Cox GM. 2001. A new dominant selectable marker for use in Cryptococcus neoformans. Med. Mycol. 39:151–154 [DOI] [PubMed] [Google Scholar]
- 29. Fraser JA, Subaran RL, Nichols CB, Heitman J. 2003. Recapitulation of the sexual cycle of the primary fungal pathogen Cryptococcus neoformans var. gattii: implications for an outbreak on Vancouver Island, Canada. Eukaryot. Cell 2:1036–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Toffaletti DL, Rude TH, Johnston SA, Durack DT, Perfect JR. 1993. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J. Bacteriol. 175:1405–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Meara TR, Norton D, Price MS, Hay C, Clements MF, Nichols CB, Alspaugh JA. 2010. Interaction of Cryptococcus neoformans Rim101 and protein kinase A regulates capsule. PLoS Pathog. 6:e1000776. 10.1371/journal.ppat.1000776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wormley FL, Jr, Perfect JR. 2005. Immunology of infection caused by Cryptococcus neoformans. Methods Mol. Med. 118:193–198 [DOI] [PubMed] [Google Scholar]
- 33. Casadevall A, Cleare W, Feldmesser M, Glatman-Freedman A, Goldman DL, Kozel TR, Lendvai N, Mukherjee J, Pirofski L-a, Rivera J, Rosas AL, Scharff MD, Valadon P, Westin K, Zhong Z. 1998. Characterization of a murine monoclonal antibody to Cryptococcus neoformans polysaccharide that is a candidate for human therapeutic studies. Antimicrob. Agents Chemother. 42:1437–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mukherjee S, Lee S, Mukherjee J, Scharff MD, Casadevall A. 1994. Monoclonal antibodies to Cryptococcus neoformans capsular polysaccharide modify the course of intravenous infection in mice. Infect. Immun. 62:1079–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cox GM, Mukherjee J, Cole GT, Casadevall A, Perfect JR. 2000. Urease as a virulence factor in experimental cryptococcosis. Infect. Immun. 68:443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adams AE, Johnson DI, Longnecker RM, Sloat BF, Pringle JR. 1990. CDC42 and CDC43, two additional genes involved in budding and the establishment of cell polarity in the yeast Saccharomyces cerevisiae. J. Cell Biol. 111:131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arellano M, Coll PM, Yang W, Duran A, Tamanoi F, Perez P. 1998. Characterization of the geranylgeranyl transferase type I from Schizosaccharomyces pombe. Mol. Microbiol. 29:1357–1367 [DOI] [PubMed] [Google Scholar]
- 38. Diaz M, Sanchez Y, Bennett T, Sun CR, Godoy C, Tamanoi F, Duran A, Perez P. 1993. The Schizosaccharomyces pombe cwg2+ gene codes for the beta subunit of a geranylgeranyltransferase type I required for beta-glucan synthesis. EMBO J. 12:5245–5254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kelly R, Card D, Register E, Mazur P, Kelly T, Tanaka K-I, Onishi J, Williamson JM, Fan H, Satoh T, Kurtz M. 2000. Geranylgeranyltransferase I of Candida albicans: null mutants or enzyme inhibitors produce unexpected phenotypes. J. Bacteriol. 182:704–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nichols CB, Perfect ZH, Alspaugh JA. 2007. A Ras1-Cdc24 signal transduction pathway mediates thermotolerance in the fungal pathogen Cryptococcus neoformans. Mol. Microbiol. 63:1118–1130 [DOI] [PubMed] [Google Scholar]
- 41. Alspaugh JA, Cavallo LM, Perfect JR, Heitman J. 2000. RAS1 regulates filamentation, mating and growth at high temperature of Cryptococcus neoformans. Mol. Microbiol. 36:352–365 [DOI] [PubMed] [Google Scholar]
- 42. Waugh MS, Nichols CB, DeCesare CM, Cox GM, Heitman J, Alspaugh JA. 2002. Ras1 and Ras2 contribute shared and unique roles in physiology and virulence of Cryptococcus neoformans. Microbiology 148:191–201 [DOI] [PubMed] [Google Scholar]
- 43. Johnston SA, May RC. 2013. Cryptococcus interactions with macrophages: evasion and manipulation of the phagosome by a fungal pathogen. Cell Microbiol. 15:403–411 [DOI] [PubMed] [Google Scholar]
- 44. Lane KT, Beese LS. 2006. Thematic review series: lipid posttranslational modifications. Structural biology of protein farnesyltransferase and geranylgeranyltransferase type I. J. Lipid Res. 47:681–699 [DOI] [PubMed] [Google Scholar]
- 45. Hast MA, Nichols CB, Armstrong SM, Kelly SM, Hellinga HW, Alspaugh JA, Beese LS. 2011. Structures of Cryptococcus neoformans protein farnesyltransferase reveal strategies for developing inhibitors that target fungal pathogens. J. Biol. Chem. 286:35149–35162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ohya Y, Goebl M, Goodman LE, Petersen-Bjørn S, Friesen JD, Tamanoi F, Anraku Y. 1991. Yeast CAL1 is a structural and functional homologue to the DPR1 (RAM) gene involved in ras processing. J. Biol. Chem. 266:12356–12360 [PubMed] [Google Scholar]
- 47. Roberts PJ, Mitin N, Keller PJ, Chenette EJ, Madigan JP, Currin RO, Cox AD, Wilson O, Kirschmeier P, Der CJ. 2008. Rho family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 283:25150–25163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Price MS, Nichols CB, Alspaugh JA. 2008. The Cryptococcus neoformans Rho-GDP dissociation inhibitor mediates intracellular survival and virulence. Infect. Immun. 76:5729–5737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hancock JF, Paterson H, Marshall CJ. 1990. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 63:133–139 [DOI] [PubMed] [Google Scholar]
- 50. Hancock JF, Cadwallader K, Paterson H, Marshall CJ. 1991. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 10:4033–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Banks IR, Specht CA, Donlin MJ, Gerik KJ, Levitz SM, Lodge JK. 2005. A chitin synthase and its regulator protein are critical for chitosan production and growth of the fungal pathogen Cryptococcus neoformans. Eukaryot. Cell 4:1902–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schafer WR, Rine J. 1992. Protein prenylation: genes, enzymes, targets, and functions. Annu. Rev. Genet. 26:209–237 [DOI] [PubMed] [Google Scholar]
- 53. Perfect JR, Lang SD, Durack DT. 1980. Chronic cryptococcal meningitis: a new experimental model in rabbits. Am. J. Pathol. 101:177–194 [PMC free article] [PubMed] [Google Scholar]
- 54. Nielsen K, Cox GM, Wang P, Toffaletti DL, Perfect JR, Heitman J. 2003. Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and α isolates. Infect. Immun. 71:4831–4841 [DOI] [PMC free article] [PubMed] [Google Scholar]