

Abstract
Some bacterial aconitases are bifunctional proteins that function in the citric acid cycle and act as posttranscriptional regulators in response to iron levels and oxidative stress. We explore the role of aconitase (AcnB) in Helicobacter pylori as a posttranscriptional regulator of the cell wall-modifying enzyme peptidoglycan deacetylase, PgdA. Under oxidative stress, PgdA is highly expressed and confers resistance to lysozyme in wild-type cells. PgdA protein expression as well as transcript abundance is significantly decreased in an acnB mutant. In the wild type, pgdA mRNA half-life was 13 min, whereas the half-life for the acnB strain was 7 min. Based on electrophoretic mobility shift assays and RNA footprinting, the H. pylori apo-AcnB binds to the 3′-untranslated region of the pgdA RNA transcript. Some of the protected bases (from footprinting) were localized in proposed stem-loop structures. AcnB-pgdA transcript binding was abolished by the addition of iron. The acnB strain is more susceptible to lysozyme-mediated killing and was attenuated in its ability to colonize mice. The results support a model whereby apo-AcnB directly interacts with the pgdA transcript to enhance stability and increase deacetylase enzyme expression, which impacts in vivo survival.
INTRODUCTION
Helicobacter pylori is a Gram-negative, microaerophilic bacterium that infects over 50% of the world's population and is the etiological agent for gastritis, peptic ulcer disease, and most gastric cancers (1). During colonization of the human gastric mucosa, H. pylori induces a strong inflammatory response resulting in the production of large amounts of reactive oxygen species (ROS). H. pylori can thrive in the gastric mucosa by employing a battery of diverse antioxidant enzymes that detoxify oxidants and repair essential biomolecules (2, 3). Additionally, H. pylori utilizes other mechanisms to persist in the host, including peptidoglycan (PG) modification (4).
H. pylori PG consists of alternating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) residues connected by β-1,4 bonds and cross-linked via short peptide bridges (5, 6). The β-1,4 bonds are susceptible to hydrolysis by the muramidase lysozyme, which results in decreased cell wall integrity and cell lysis. Lysozyme, an important component of the host innate immune system, is abundant in the mucosal surface and is present in the granules of professional phagocytes (7, 8). Interestingly, bacteria have evolved mechanisms to modify their PG around the lysozyme cleavage site to prevent hydrolysis (9). H. pylori is equipped with a peptidoglycan deacetylase (PgdA) that confers both pure PG and whole bacterial resistance to lysozyme degradation (10). PgdA expression is significantly increased in H. pylori cells when they are exposed to oxidative stress and when in contact with macrophages (10, 11). Furthermore, pgdA mutants have an attenuated ability to colonize the mouse stomach and permit a stronger host immune (cytokine) response (11). This previous work led us to question how PgdA expression is regulated, especially considering that H. pylori lacks many of the oxidative stress response regulators found in most other Gram-negative bacteria. We hypothesized that aconitase plays an important role during oxidative stress by serving as a posttranscriptional regulator for peptidoglycan deacetylase (10).
Aconitases are [4Fe-4S] proteins that catalyze the reversible isomerization of citrate to isocitrate in the citric acid cycle. In eukaryotes, there are two types of aconitase proteins, mitochondrial aconitase (m-Acn) and cytosolic aconitase (c-Acn), also referred to as iron regulatory protein 1 (IRP1). IRP1 is bifunctional, having enzymatic activity when the [4Fe-4S] cluster is intact and acting as a posttranscriptional regulator when the cluster is disassembled (12). Iron deprivation and oxidative stress are known to cause disassembly of the cluster, resulting in the apo-form of IRP1, which undergoes domain rearrangements that allow it to bind to iron-responsive elements (IREs) (12, 13). IREs are approximately 30-nt-long sequences that form stem-loop structures in the untranslated regions (UTRs) of mRNA transcripts. The eukaryotic consensus IRE contains a C bulge in the stem and the sequence CAGUGN in the loop (14). If the IRE is located in the 5′ UTR, binding of IRP1 will inhibit translation; if the IRE is located in the 3′ UTR, binding of IRP1 will increase transcript stability and result in enhanced translation (12, 14).
Several bifunctional bacterial aconitases have been studied thus far, including Escherichia coli AcnA and AcnB, which have been found to enhance and decrease superoxide dismutase (SodA) expression, respectively (15). In Salmonella enterica serovar Typhimurium LT2, AcnB was shown to indirectly regulate the flagellum protein, FliC (16). Bacillus subtilis aconitase (CitB) binds to the 3′ UTR of gerE, a transcriptional activator and repressor involved in sporulation (17). Furthermore, Mycobacterium tuberculosis aconitase (Acn) binds to the thioredoxin (trxC) transcript and to the ideR transcript, an iron-dependent activator and repressor (18). H. pylori possesses one copy of aconitase, acnB, whose gene product is known to have aconitase activity in the citric acid cycle (19), but it has not yet been studied as a posttranscriptional regulator. Here, we present evidence that apo-AcnB acts as a posttranscriptional regulator for PgdA, which is an important PG modification enzyme conferring lysozyme resistance, and that it contributes to survival in the mouse host.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
H. pylori X47 and 43504 wild-type and acnB strains were grown on Brucella agar (Difco) supplemented with 10% defibrinated sheep blood (BA plates) and chloramphenicol (50 μg/ml) at 37°C under constant microaerophilic conditions (2% O2). For mRNA half-life determinations, wild-type and mutant strains were grown in brain heart infusion (BHI) broth with 0.4% β-cyclodextrin. Sealed bottles initially contained 5% CO2, 10% H2, 75% N2, and 10% O2 and were incubated at 37°C with shaking. The H. pylori wild-type and acnB strain growth patterns were similar. E. coli BL21 RIL cells were grown at 37°C aerobically on Luria-Bertani agar or broth (shaking) supplemented with ampicillin (100 μg/ml) and chloramphenicol (50 μg/ml).
Mutant construction.
The 2.6-kb acnB gene (hp0779) has been determined to be the first gene of an operon containing 3 additional genes: hp0780, hp0781, and hp0782 (20). Ninety-eight percent of the gene was deleted from H. pylori strains X47 and 43504 using overlap extension PCR and was replaced with the chloramphenicol (cat) cassette, which was inserted in the same direction of transcription as that of the native gene. The cat cassette has its own promoter, lacks a transcription terminator, and is routinely used in our laboratory without any polar effect (21, 22). Primers acnB1 (5′-CCCCGCATCAATACGCC-3′) and acnB2 (5′-ATCCACTTTTCAATCTATATCCTAAAAAATCTTTCATCAT-3′) were used to amplify a 327-bp DNA sequence that contained the beginning of hp0778, the intergenic region between hp0778 and hp0779, the beginning of hp0779, and a portion of the cat cassette. Primers acnB3 (5′-CCCAGTTTGTCGCACTGATAAGGAGAATTTCAGGCTCTAG-3′) and acnB4 (5′-CTAGCGCCAATTATAGATATAAGG-3′) were used to amplify a 388-bp sequence containing part of the cat cassette, the end of hp0779, the intergenic region between hp0779 and hp0780, and hp0780. Primers acnB1 and acnB4 were used in the final PCR step to fuse together the product of acnB1/acnB2, the cat cassette, and the product of acnB3/acnB4. This final step yielded a 1.5-kb PCR product that was then cloned into the pGEM-T vector (Promega) to generate pGEMacnB::cat. This plasmid was introduced into the wild-type strain via natural transformation and homologous recombination. acnB mutants were selected on BA plates with chloramphenicol (50 μg/ml) at 2% O2. Insertion of the cat cassette and absence of the acnB gene were confirmed by PCR and sequencing using primers acnB1 and acnB4 and primers specific for the cat cassette (5′-GATATAGATTGAAAAGTGGAT-3′ and 5′-TTATCAGTGCGACAAACTGGG-3′). Also, reverse transcription-PCR (RT-PCR) was performed on the downstream gene, hp0781, to rule out a possible polar effect due to the cat cassette. Since hp0780 is only 273 bp in length, we instead designed primers for hp0781 (1.3 kb). Total RNA was extracted (Aurum total RNA mini kit; Bio-Rad), treated with DNase (Turbo DNA-free kit; Ambion), and used as a template for cDNA synthesis (iScript cDNA synthesis kit; Bio-Rad). The generated cDNA was then used as a template for PCR, and a product of the expected size was obtained for both the wild-type and acnB strains (data not shown).
Western blotting.
H. pylori wild-type and acnB strains were each separately grown under 2% O2 for 36 h and 12% O2 for 72 h. The time for cells grown under 12% O2 was extended, because cell growth is slower at this higher oxygen level. Cell extracts were subjected to SDS-PAGE, and the proteins were electroblotted onto a nitrocellulose membrane. The membrane was then incubated with anti-PgdA (1:500) (Antagene, Inc.) followed by exposure to goat anti-rabbit IgG alkaline phosphatase-conjugated secondary antibody (1:1,000) (Bio-Rad). Anti-UreA (1:10,000) (Santa Cruz Biotechnology) was used as an internal loading control. ImageJ (http://rsbweb.nih.gov/ij/) was used to analyze dried membranes. Student's t test was used for statistical comparisons.
Real-time quantitative PCR.
The Aurum total RNA mini kit (Bio-Rad) was used to extract total RNA from H. pylori wild-type and acnB cells grown under 2 and 12% O2. The Turbo DNA-free kit (Ambion) was used as an additional measure to degrade any remaining DNA. cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad) according to the kit instructions. The iQ SYBR green supermix (Bio-Rad) kit was used for real-time PCR according to the manufacturer's protocol. Primers specific for pgdA (5′-GGATTCGCCTGATGATATTTCG-3′ and 5′-CCTGCATCCACGATCATTTTC-3′) were used along with primers specific for gyrA (5′-GCTAGGATCGTGGGTGATGT-3′ and 5′-TGGCTTCAGTGTAACGCATC-3′), the internal control. Relative transcript abundance was calculated using the 2−ΔCT formula (23). For mRNA half-life determinations, wild-type and acnB strains were grown in BHI for 24 h (early exponential phase) and subjected to 21% O2 for 2 h. Rifampin (500 μg/ml) was added, and after 70 s (t = 0) time points were established (2, 5, 10, 15, 20, and 30 min). For each time point, RNA was extracted as described above and cDNA was synthesized using primers specific for pgdA. The quantitative PCR (qPCR) data were used to calculate the pgdA mRNA half-life as described previously (24). Prism (GraphPad, San Diego, CA) was used with the equation Y = (Y0)e−kt, where Y is the percent pgdA mRNA remaining at time t, to find first-order decay constants (k) by nonlinear regression analysis. Half-lives were calculated using t1/2 = ln2/k (24).
Overexpression and purification of AcnB.
The H. pylori acnB gene was PCR amplified using wild-type DNA as the template with primers acnBHisF (5′-CGCACCCATATGATGAAAGATTTTTTAGAAG-3′) and acnBHisR (5′-GAAGACCTCGAGGAGCCTGAAATTCTCCATTAAG-3′). The PCR product was cloned into the pET-21b vector (Novagen) and overexpressed as a hexahistidine-tagged protein in E. coli BL21 RIL. Cells were grown in LB broth to an A600 of 0.5 and induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside for 4 h. After harvesting by centrifugation (5,000 × g, 10 min, 4°C) and resuspending and washing the pellet with 50 mM NaH2PO4, 300 mM NaCl, pH 8 (buffer A supplemented with 10 mM imidazole), cells were resuspended in the same buffer and lysed by three passages though a French pressure cell (22). Following centrifugation of the extract (16,000 × g, 30 min), the supernatant was collected and applied to a nickel-nitrilotriacetic acid column (Qiagen). The column was washed with buffer A supplemented with 20 mM imidazole, and the protein was eluted with buffer A supplemented with 250 mM imidazole. Purified protein was analyzed by SDS-PAGE and estimated to be approximately 95% pure. Apo-AcnB was prepared by incubating the purified native protein with dipyridyl (0.5 mM) for 30 min and then dialyzing overnight against 20 mM Tris-Cl, pH 8, 100 mM NaCl, 3% glycerol (18). As expected, the UV-visible absorption spectrum of native AcnB showed absorbance in the 300- to 600-nm range indicative of Fe-S clusters, while absorbance in this range was significantly lower for the dipyridyl-treated protein (data not shown). Protein concentration was determined using the bicinchoninic acid assay kit (Thermo Scientific).
Electrophoretic mobility shift assays.
The entire pgdA mRNA 3′ UTR (45 nt) was synthesized (Integrated DNA Technologies) and radiolabeled at the 5′ end with T4 polynucleotide kinase (Invitrogen) according to the manufacturer's instructions. A 2.5-μl sample of [γ-32P]ATP (10 μCi/μl; 3,000 Ci/mmol) was incubated with 5 pmol of pgdA RNA, 1× forward reaction buffer, T4 polynucleotide kinase, and water for 10 min at 37°C. Labeled RNA was purified by phenol extraction and ethanol precipitation. Size and purity were checked on a denaturing urea polyacrylamide gel. Purified pgdA RNA (50 nM) was incubated with either increasing apo-AcnB (0, 600, 1,200, and 3,000 nM) or bovine serum albumin (BSA) (3,000 nM) in binding buffer (10 mM Tris pH 8.3, 20 mM KCl, and 10% glycerol) with 130-fold nonspecific yeast tRNA for 15 min at room temperature. Apo-AcnB (3,000 nM) was incubated with either 1 mM ammonium iron (II) sulfate plus 10 mM dithiothreitol or 0.5 mM dipyridyl. Unlabeled specific pgdA competitor (50× and 100× molar excess) was added to apo-AcnB (3,000 nM) in some reactions prior to the addition of radiolabeled pgdA RNA and allowed to incubate for 5 min to maximize effectiveness (recommended by Thermo Scientific). As a positive control for the binding reactions, the 5′ UTR human ferritin sequence (5′-GTGAGAGAATTCGGGAGAGGATTTCCTGCTTCAACAGTGCTTGGACGGAACTTTGTCTTGAAGCTTGGAGAG-3′) was synthesized (Integrated DNA Technologies) and cloned into the pGEM-3Z vector (Promega). HindII was used to generate the linearized plasmid, which was then used as a template for in vitro transcription. T7 RNA polymerase (Promega) was used to synthesize radiolabeled RNA using [α-32P]UTP (10 μCi/μl; 800 Ci/mmol) according to the manufacturer's instructions. RNA was purified by phenol extraction and ethanol precipitation. As a negative control, the pGEM-3Z vector alone was linearized and subjected to in vitro transcription. The positive- and negative-control reactions were performed using 3,000 nM apo-AcnB. Reaction products were resolved on a 6% nondenaturing polyacrylamide gel. The gel was incubated overnight in a phosphor screen cassette (Molecular Dynamics), and the screen was scanned using the Typhoon Imager (GE Healthcare).
RNA footprinting.
5′-End-labeled pgdA probe was prepared as described for electrophoretic mobility shift assays. pgdA probe (50 nM) was incubated for 15 min at room temperature with (1 μM) and without apo-AcnB, 1× RNA structure buffer (Ambion), 1 μg of yeast tRNA, and either RNase A (1 ng) or RNase V1 (0.001 U). Digestion products were purified by phenol extraction and ethanol precipitation. Samples were resolved on a 20% denaturing polyacrylamide 7 M urea gel in 1× Tris-borate-EDTA (TBE). The gel was vacuum dried, incubated overnight in a phosphor screen cassette (Molecular Dynamics), and scanned using a Typhoon imager (GE Healthcare).
Lysozyme sensitivity assays.
Wild-type and acnB strains were grown to late exponential phase under microaerophilic conditions, harvested, resuspended in phosphate-buffered saline (PBS) to an optical density at 600 nm (OD600) of 1, and then subjected to incubation for 8 h with lysozyme (50 mg/ml). After incubation, cells were serially diluted and plated, and colonies were counted after 4 days. Significant differences between the wild type and mutant were determined using Student's t test.
Mouse colonization.
All procedural work was approved by the Institutional Animal Care and Use Committee at the University of Georgia, Athens, GA. H. pylori wild-type and acnB strains were grown on BA plates under microaerophilic conditions for 48 h. Cells were suspended in PBS to an OD600 of 1.7, and 3 × 108 cells were administered to each C57BL/6J mouse. Three weeks after inoculation, the mice were sacrificed after withholding food and water for 1.5 to 2 h and the stomachs were removed, weighed, and homogenized. Stomach homogenate dilutions were made in PBS and plated onto BA plates supplemented with amphotericin B (10 μg/ml), bacitracin (100 μg/ml), and vancomycin (10 μg/ml). After 5 days of incubation under microaerophilic conditions, cells were enumerated and the data expressed as log(CFU/g) of stomach. The Wilcoxon signed-rank test was used to determine significant differences in colonization between the wild-type and mutant strains.
RESULTS
PgdA expression, transcript abundance, and mRNA half-life in the strains upon oxidative stress exposure.
We began this work by investigating the effect of the aconitase deletion on expression of peptidoglycan deacetylase (PgdA). Western blot analyses were performed using anti-PgdA antibody and cell extracts from wild-type and acnB strains grown under both 2% O2 for 36 h and 12% O2 for 72 h. A significant difference in PgdA expression was found for the wild type at 2 versus 12% O2 (Fig. 1), similar to the previous finding that reported PgdA is increased upon subjection of cells to oxidative stress (10). No significant difference in PgdA levels was found in the acnB mutant between 2 and 12% O2. There was a decrease in PgdA expression in the acnB strain under 2% O2 compared to the wild type under the same condition. Interestingly, there was 4-fold less PgdA expression in the acnB strain under 12% O2 compared to the wild type under 12% O2.
Fig 1.
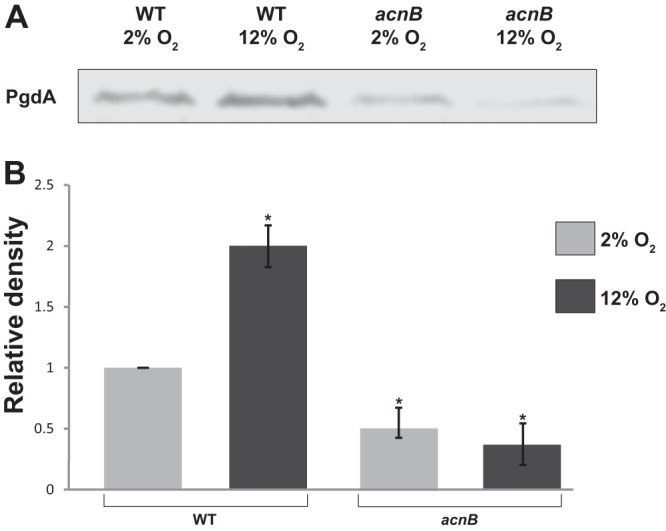
Expression of the PgdA enzyme at two oxygen levels. Western blot (A) and (B) densitometry analysis of PgdA expression from the Western blot are shown. Cell extracts from 43504 wild-type and acnB strains were subjected to SDS-PAGE followed by immunoblotting using specific anti-PgdA antibody. Dried blots were analyzed using ImageJ. Wild-type (2% O2) expression was used as the reference (adjusted to 1.0 U). The experiment included an internal loading control, UreA (not shown), that verified equal amounts of protein were loaded. Data shown are averages and standard deviations (SD) from three independent experiments. Asterisks indicate a significant difference compared to the reference. P < 0.01 as determined by Student's t test.
pgdA transcript abundance was assessed in the acnB strain using quantitative real-time PCR (Fig. 2). Wild-type and acnB strains were subjected to the same growth conditions as those described for the Western blot experiments. There was a 2-fold increase in relative pgdA transcript abundance for the wild type under 12% O2 compared to the 2% O2 condition, which was expected. pgdA transcript levels in the acnB mutant under 12% O2 were 2-fold decreased compared to those at the 2% O2 incubation. A 3-fold decrease in pgdA abundance was found in the acnB strain under 2% O2 versus the wild type under the same condition. Most interestingly, a 9-fold decrease was observed for the pgdA transcript in the acnB strain under 12% O2 versus the wild type under 12% O2.
Fig 2.
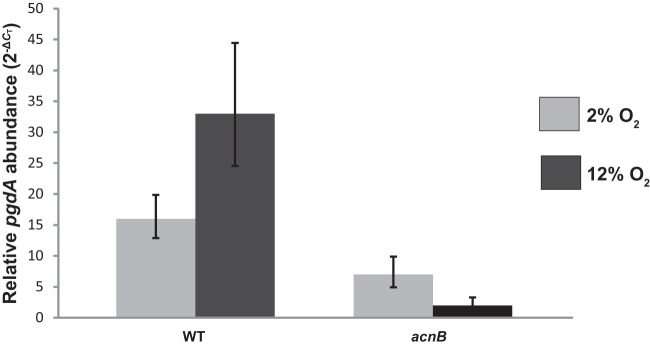
Relative pgdA transcript abundance. Transcript levels were determined by quantitative real-time PCR after growth of each strain under either 2 or 12% O2. The gyrA housekeeping gene was used as an internal control. Results shown are representative data from one experiment performed in triplicate. The entire experiment was repeated with similar results. Error bars represent 2−(ΔCT − SD) − 2−ΔCT and 2−ΔCT − 2−(ΔCT + SD).
To directly determine if AcnB was functioning as a posttranscriptional regulator to stabilize the pgdA message, we used qPCR to calculate the half-life of the pgdA mRNA in the wild-type and acnB strains. Cells were grown under microaerophilic conditions and then exposed to 21% O2 for 2 h. Our results revealed that upon high oxygen exposure, the wild-type pgdA mRNA half-life is 13 min, whereas the acnB mutant pgdA mRNA half-life is 7 min.
In summary, these findings suggested that apo-AcnB directly interacts with the pgdA transcript. The model is that under oxidative stress, the [4Fe-4S] cluster of AcnB is likely to be disassembled and apo-AcnB binds to the pgdA transcript, stabilizing the message and resulting in increased transcript abundance and expression.
Apo-AcnB binds to the pgdA 3′ UTR.
Since it is known that aconitase can function as a posttranscriptional regulator and the results described above suggested aconitase was interacting with the pgdA transcript, we examined the 5′ and 3′ UTRs of the pgdA gene for possible IRE-like sequences. The pgdA 5′ UTR contained only 10 nt (known IREs are approximately 30 nt in length); therefore, the pgdA 3′ UTR was chosen for further investigation. The secondary structure of the 3′ UTR was predicted using the STAR program (25) (see Fig. 4B), revealing two stem-loops that may be involved in aconitase binding. To test if apo-AcnB could bind to the transcript, electrophoretic mobility shift assays were performed. Radiolabeled pgdA probe (50 nM) was incubated with 0, 600, 1,200, and 3,000 nM apo-AcnB protein. As apo-AcnB concentration increased, more pgdA probe was observed to shift (Fig. 3A). The addition of ammonium iron (II) sulfate and dithiothreitol resulted in no shift, while the addition of dipyridyl promoted binding (Fig. 3B). These results agreed with other bacterial studies (18, 26) that demonstrated the RNA-binding ability of aconitase is diminished upon the addition of Fe2+ and restored when iron is not present or the specific iron chelator dipyridyl is added to the reaction. The presence of the [4Fe-4S] cluster in aconitase determines whether it can bind to RNA transcripts (12). Additionally, binding reactions were performed using 50 and 100× molar excesses of specific cold competitor (Fig. 3C). The radiolabeled pgdA probe is effectively outcompeted when unlabeled pgdA 3′ UTR is added at 100× molar excess. When bovine serum albumin, rather than apo-AcnB, was incubated with the radiolabeled pgdA probe, no shift was observed (Fig. 3D). Furthermore, binding reactions were conducted with apo-AcnB (3,000 nM) using a vector-only probe as a negative control, which did not result in a shift, and with the human ferritin IRE as a positive control, which resulted in a shift similar to that of pgdA (data not shown).
Fig 4.
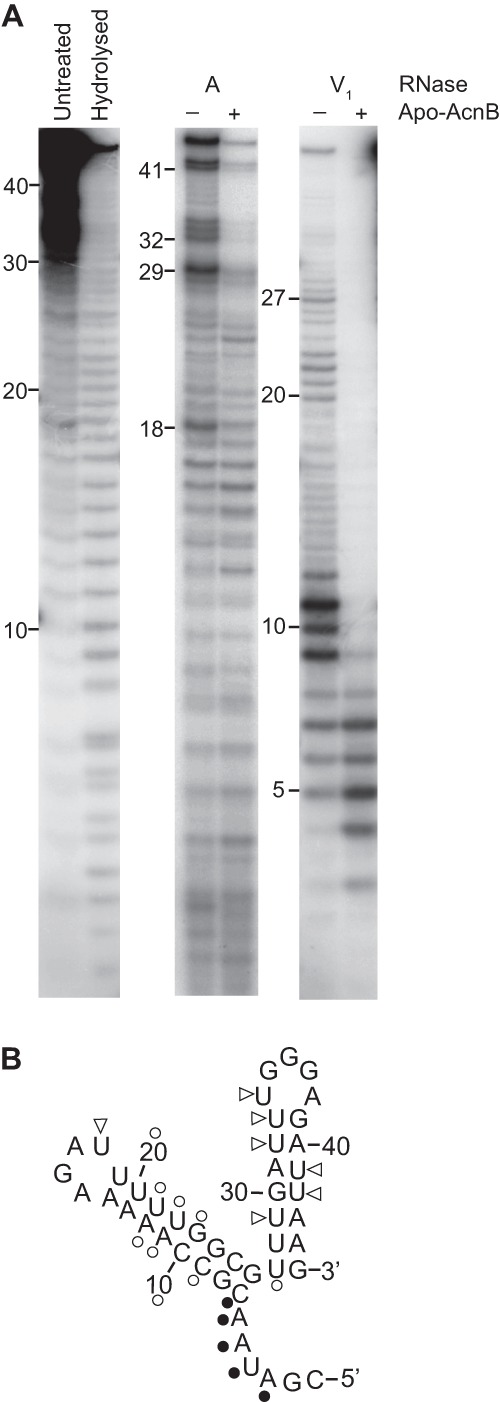
RNA footprinting. (A) 5′-End-labeled 45-nt pgdA RNA was incubated with (+) and without (−) apo-AcnB and subjected to RNase digestion. (B) RNase A (triangles) and RNase V1 (circles) cleavage sites were mapped on the predicted pgdA 3′ UTR structure. Open symbols indicate nucleotides that were more protected from RNase cleavage in the presence of apo-AcnB, whereas filled symbols indicate those nucleotides that were less protected.
Fig 3.
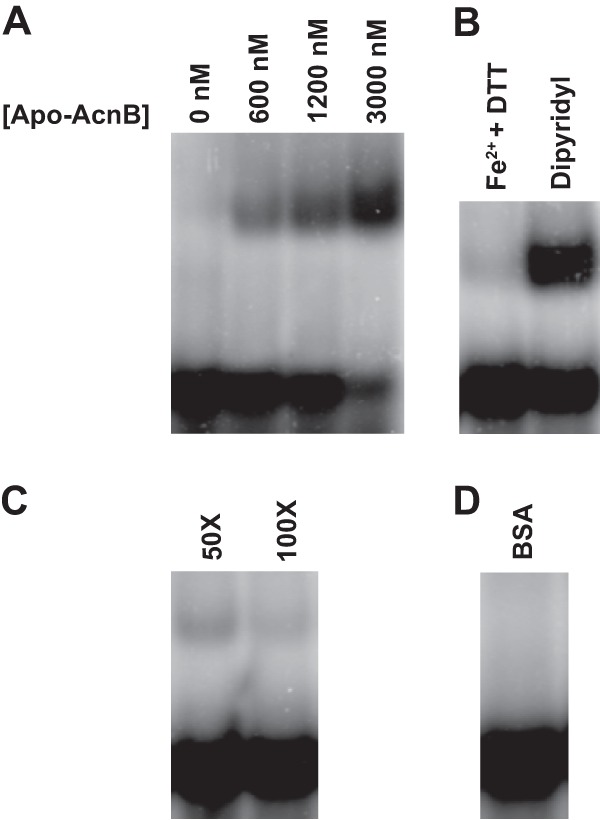
Electrophoretic mobility shift assays. (A) More pgdA transcript (50 nM) bound to apo-AcnB with increasing protein concentration. (B) Addition of iron and reductant abolished binding, whereas the iron chelator dipyridyl promoted binding. DTT, dithiothreitol. (C) Radiolabeled pgdA probe was outcompeted by specific unlabeled competitor RNA at 100× molar excess. (D) Use of nonspecific protein bovine serum albumin (3,000 nM) instead of apo-AcnB did not result in a shift.
RNA footprinting was conducted to elucidate where apo-AcnB (dipyridyl treated; see Materials and Methods) was binding to the pgdA 3′ UTR. 5′ End-labeled pgdA probe was incubated with and without apo-AcnB and subjected to cleavage using two different RNase enzymes (Fig. 4A). RNase A cleaves the 3′ end of single-stranded C and U residues, and RNase V1 cleaves base-paired or stacked nucleotides. RNase A cleavage of nucleotides 18, 29, 32 to 34, and 41 and 42 occurred less frequently in the presence of apo-AcnB. Incubation with RNase V1 revealed that the majority of the pgdA probe is protected when incubated with apo-AcnB, especially nucleotides 9 to 12, 20 to 23, and 27 (Fig. 4B). Nucleotides 3 to 7 were cleaved more frequently by RNase V1 in the presence of apo-AcnB.
Deletion of acnB confers lysozyme sensitivity in H. pylori mutants.
Lysozyme hydrolyzes the β-1,4 bonds connecting GlcNAc and MurNAc residues in bacterial PG, leading to decreased cell wall integrity and subsequent cell lysis. Previously, our laboratory showed that an H. pylori pgdA strain is more sensitive to lysozyme degradation than the wild type (10). Thus far, our findings suggested apo-AcnB regulates PgdA expression. Therefore, we speculated that the acnB strain was more sensitive to lysozyme killing, and this sensitivity was tested over an 8-h period (Fig. 5). The acnB strain is significantly more sensitive to lysozyme killing than the wild type after 4, 6, and 8 h (P < 0.01), as determined by Student's t test. The kill curve for the acnB strain closely resembles that previously published for the pgdA strain (10).
Fig 5.
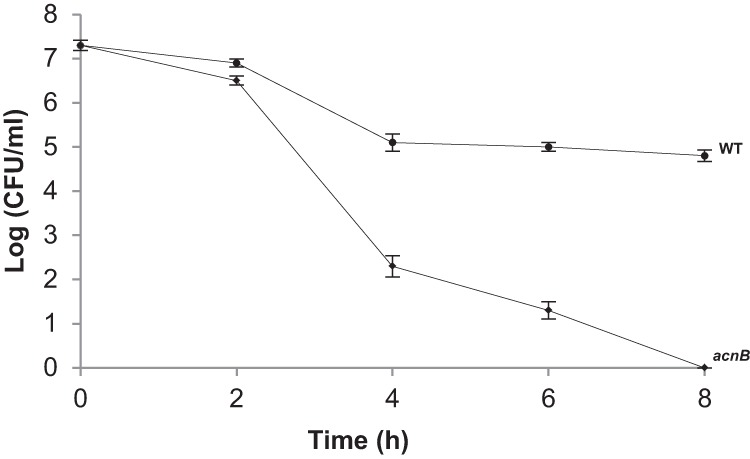
Susceptibility of strains to lysozyme killing. Strains were treated with 50 mg/ml lysozyme, and samples were taken over an 8-h period for comparative viability (CFU determination). Each data point represents averages of combined replicates from two independent experiments (each performed in triplicate). Error bars represent standard deviations. The mutant strain values are significantly lower than the wild-type values at the 4-, 6-, and 8-h time points at P < 0.01 (Student's t test).
The acnB strain has an attenuated ability to colonize the mouse stomach.
We wanted to characterize the physiological role of aconitase in H. pylori by comparing the colonization abilities of the acnB strain to that of the wild type. Each strain was individually inoculated into eight C57BL/6J mice, and after 3 weeks, stomach colonization was examined. The mean colonization for the wild type was 2.1 × 106 CFU/g, whereas the mean for the acnB strain was 4.6 × 105 CFU/g (Fig. 6). Thus, there was a 4.5-fold decrease in mouse colonization by the acnB strain. Using the Wilcoxon signed-rank test, the range of mutant colonization values is significantly smaller than that of the wild type at greater than 95% confidence (P < 0.05). These results demonstrate aconitase contributes to H. pylori survival and colonization in the mouse stomach.
Fig 6.
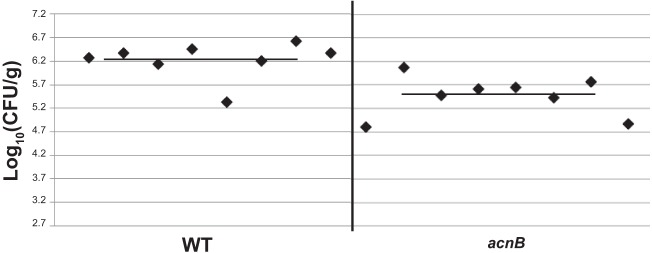
Mouse colonization by strains. Wild-type and acnB strains were separately injected into 8 mice each, and colonization ability (CFU recovered per gram of stomach) was examined after 3 weeks. Each point represents the CFU count from one stomach, and solid horizontal lines represent the means of colonization for each strain. The baseline (log10[CFU/g] = 2.7) is the limit of detection. The experiment was repeated with similar results.
DISCUSSION
During H. pylori colonization, host immune cells mount a strong inflammatory response resulting in the production of large amounts of ROS, including hydrogen peroxide (H2O2) and the superoxide anion (O2−). These can be detrimental to H. pylori, causing damage to DNA, proteins, and lipids. Despite containing a diverse repertoire of antioxidant enzymes (3), the bacterium lacks homologues of the oxidative stress response regulators found in other bacteria, such as OxyR, SoxRS, and the RpoS sigma factor. Thus, it has been hypothesized that posttranscriptional regulation in H. pylori plays a role in the regulation of genes in response to stress (27). We propose a model for the role of aconitase in H. pylori colonization (Fig. 7). Bacterial aconitases are subject to oxidation and Fe/S cluster loss, but the apo-form can still play regulatory roles (16, 18). PgdA has been shown to be upregulated in response to oxidative stress and upon contact with neutrophils (10, 28). Failure of the acnB strain to upregulate PgdA expression under 12% O2 conditions is in contrast to the wild type, and it led us to hypothesize aconitase was playing a role in deacetylase regulation (Fig. 1). Decreased transcript abundance and pgdA mRNA half-life in the acnB strain supported our hypothesis that aconitase is involved in the posttranscriptional regulation of pgdA. Electrophoretic mobility shift assays and RNA footprinting data demonstrated that the regulatory effect of aconitase results from direct interaction between apo-AcnB and the transcript. Furthermore, the acnB strain was more sensitive to lysozyme degradation and was attenuated in its ability to colonize the mouse stomach, so an AcnB role in vivo is proposed (Fig. 7).
Fig 7.
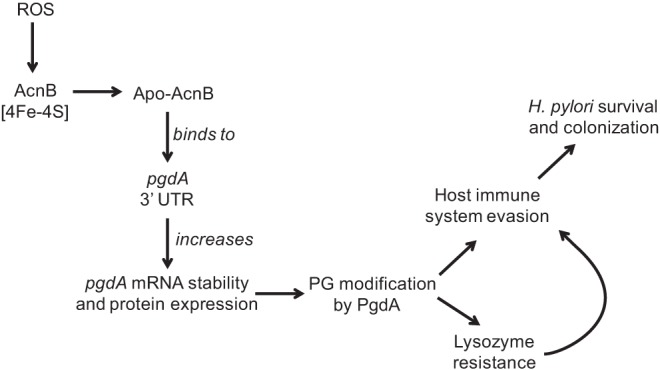
Model of pgdA posttranscriptional regulation and its effects on H. pylori. ROS oxidize the [4Fe-4S] cluster of AcnB, rendering apo-AcnB, which then binds to the pgdA transcript. Increased transcript stability leads to increased expression of the PG-modifying enzyme, PgdA. PG modification is a mechanism H. pylori utilizes to circumvent host immune system detection and resist lysozyme degradation (which further mitigates host immune system recognition), both of which contribute to long-term survival and colonization.
Aconitase-mediated transcript stabilization is a previously described regulatory mechanism. In vertebrates, IRP1 binds to IREs involved in maintaining iron homeostasis (12). The consensus IRE has a C bulge in the stem and the sequence CAGUGN in the loop (12), but the precise structure or sequence recognized by bifunctional bacterial aconitases is less defined. Some aconitase transcript targets contain the eukaryotic consensus sequence, such as thioredoxin (trxC) in M. tuberculosis (18); however, the E. coli acnA, acnB, sodA, and ftsH transcripts as well as the B. subtilis gerE transcript do not have the consensus sequence (15–17, 29). The pgdA 3′ UTR also does not contain this consensus sequence, but gel shift data indicate binding to apo-AcnB (Fig. 3). RNA footprinting revealed which nucleotides of the pgdA 3′ UTR were protected by apo-AcnB (Fig. 4), but it is still unclear which specific nucleotides or sequences are necessary for binding. This appears to be the case for other bacterial aconitase targets as well (16). It is of interest to learn the total repertoire of mRNA targets and the complete role of this tricarboxylic acid (TCA) cycle enzyme.
ACKNOWLEDGMENTS
We thank Bijoy Mohanty for all of his guidance and expertise with the RNA footprinting experiments. We also thank Stéphane Benoit for many helpful discussions and Sue Maier for her help with the mouse colonization experiments.
Footnotes
Published ahead of print 20 September 2013
REFERENCES
- 1.Dunn BE, Cohen H, Blaser MJ. 1997. Helicobacter pylori. Clin. Microbiol. Rev. 10:720–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kusters JG, van Vliet AH, Kuipers EJ. 2006. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19:449–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang G, Alamuri P, Maier RJ. 2006. The diverse antioxidant systems of Helicobacter pylori. Mol. Microbiol. 61:847–860 [DOI] [PubMed] [Google Scholar]
- 4.Wang G, Lo LF, Forsberg LS, Maier RJ. 2012. Helicobacter pylori peptidoglycan modifications confer lysozyme resistance and contribute to survival in the host. mBio 3:e00409–12. 10.1128/mBio.00409-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costa K, Bacher G, Allmaier G, Dominguez-Bello MG, Engstrand L, Falk P, de Pedro MA, Garcia-del Portillo F. 1999. The morphological transition of Helicobacter pylori cells from spiral to coccoid is preceded by a substantial modification of the cell wall. J. Bacteriol. 181:3710–3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mobley HLT, Mendz GL, Hazell SL. 2001. Helicobacter pylori: physiology and genetics. ASM Press, Washington, DC: [PubMed] [Google Scholar]
- 7.Cole AM, Liao HI, Stuchlik O, Tilan J, Pohl J, Ganz T. 2002. Cationic polypeptides are required for antibacterial activity of human airway fluid. J. Immunol. 169:6985–6991 [DOI] [PubMed] [Google Scholar]
- 8.Dziarski R. 2003. Recognition of bacterial peptidoglycan by the innate immune system. Cell. Mol. Life Sci. 60:1793–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis KM, Weiser JN. 2011. Modifications to the peptidoglycan backbone help bacteria to establish infection. Infect. Immun. 79:562–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang G, Olczak A, Forsberg LS, Maier RJ. 2009. Oxidative stress-induced peptidoglycan deacetylase in Helicobacter pylori. J. Biol. Chem. 284:6790–6800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang G, Maier SE, Lo LF, Maier G, Dosi S, Maier RJ. 2010. Peptidoglycan deacetylation in Helicobacter pylori contributes to bacterial survival by mitigating host immune responses. Infect. Immun. 78:4660–4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beinert H, Kennedy MC, Stout CD. 1996. Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev. 96:2335–2374 [DOI] [PubMed] [Google Scholar]
- 13.Dupuy J, Volbeda A, Carpentier P, Darnault C, Moulis JM, Fontecilla-Camps JC. 2006. Crystal structure of human iron regulatory protein 1 as cytosolic aconitase. Structure 14:129–139 [DOI] [PubMed] [Google Scholar]
- 14.Hentze MW, Kuhn LC. 1996. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 93:8175–8182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang Y, Quail MA, Artymiuk PJ, Guest JR, Green J. 2002. Escherichia coli aconitases and oxidative stress: post-transcriptional regulation of sodA expression. Microbiology 148:1027–1037 [DOI] [PubMed] [Google Scholar]
- 16.Tang Y, Guest JR, Artymiuk PJ, Read RC, Green J. 2004. Post-transcriptional regulation of bacterial motility by aconitase proteins. Mol. Microbiol. 51:1817–1826 [DOI] [PubMed] [Google Scholar]
- 17.Serio AW, Pechter KB, Sonenshein AL. 2006. Bacillus subtilis aconitase is required for efficient late-sporulation gene expression. J. Bacteriol. 188:6396–6405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banerjee S, Nandyala AK, Raviprasad P, Ahmed N, Hasnain SE. 2007. Iron-dependent RNA-binding activity of Mycobacterium tuberculosis aconitase. J. Bacteriol. 189:4046–4052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pitson SM, Mendz GL, Srinivasan S, Hazell SL. 1999. The tricarboxylic acid cycle of Helicobacter pylori. Eur. J. Biochem. 260:258–267 [DOI] [PubMed] [Google Scholar]
- 20.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermuller J, Reinhardt R, Stadler PF, Vogel J. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464:250–255 [DOI] [PubMed] [Google Scholar]
- 21.Wang G, Lo LF, Maier RJ. 2011. The RecRO pathway of DNA recombinational repair in Helicobacter pylori and its role in bacterial survival in the host. DNA Repair (Amsterdam) 10:373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benoit SL, Maier RJ. 2011. Mua (HP0868) is a nickel-binding protein that modulates urease activity in Helicobacter pylori. mBio 2(2):e00039-11. 10.1128/mBio.00039-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 24.Ysla RM, Wilson GM, Brewer G. 2008. Chapter 3. Assays of adenylate uridylate-rich element-mediated mRNA decay in cells. Methods Enzymol. 449:47–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abrahams JP, van den Berg M, van Batenburg E, Pleij C. 1990. Prediction of RNA secondary structure, including pseudoknotting, by computer simulation. Nucleic Acids Res. 18:3035–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alen C, Sonenshein AL. 1999. Bacillus subtilis aconitase is an RNA-binding protein. Proc. Natl. Acad. Sci. U. S. A. 96:10412–10417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barnard FM, Loughlin MF, Fainberg HP, Messenger MP, Ussery DW, Williams P, Jenks PJ. 2004. Global regulation of virulence and the stress response by CsrA in the highly adapted human gastric pathogen Helicobacter pylori. Mol. Microbiol. 51:15–32 [DOI] [PubMed] [Google Scholar]
- 28.Fittipaldi N, Sekizaki T, Takamatsu D, de la Cruz Dominguez-Punaro M, Harel J, Bui NK, Vollmer W, Gottschalk M. 2008. Significant contribution of the pgdA gene to the virulence of Streptococcus suis. Mol. Microbiol. 70:1120–1135 [DOI] [PubMed] [Google Scholar]
- 29.Tang Y, Guest JR. 1999. Direct evidence for mRNA binding and post-transcriptional regulation by Escherichia coli aconitases. Microbiology 145:3069–3079 [DOI] [PubMed] [Google Scholar]