

Abstract
Listeria monocytogenes is a Gram-positive human intracellular pathogen that infects diverse mammalian cells. Upon invasion, L. monocytogenes secretes multiple virulence factors that target host cellular processes and promote infection. It has been presumed, but was not empirically established, that the Sec translocation system is the primary mediator of this secretion. Here, we validate an important role for SecDF, a component of the Sec system, in the secretion of several critical L. monocytogenes virulence factors. A ΔsecDF mutant is demonstrated to exhibit impaired membrane translocation of listeriolysin O (LLO), PlcA, PlcB, and ActA, factors that mediate L. monocytogenes phagosomal escape and spread from cell to cell. This impaired translocation was monitored by accumulation of the factors on the bacterial membrane and by reduced activity upon secretion. This defect in secretion is shown to be associated with a severe intracellular growth defect of the ΔsecDF mutant in macrophages and a less virulent phenotype in mice, despite normal growth in laboratory medium. We further show that SecDF is upregulated when the bacteria reside in macrophage phagosomes and that it is necessary for efficient phagosomal escape. Taken together, these data support the premise that SecDF plays a role as a chaperone that facilitates the translocation of L. monocytogenes virulence factors during infection.
INTRODUCTION
A prerequisite for a pathogen to succeed is an ability to influence the host's cellular processes. In bacteria, this ability is largely mediated by secreting multiple virulence factors that target host cellular components during the course of infection. Whereas Gram-negative pathogenic bacteria are armed with several specialized secretion systems that serve this purpose (types I to VI), Gram-positive bacteria lack these systems and presumably use mainly the canonical Sec translocation system (1). Listeria monocytogenes is a Gram-positive intracellular bacterial pathogen that invades and grows within diverse mammalian cells (2). It is the causative agent of the food-borne disease listeriosis in humans, which can result in meningitis, abortion, or septicemia. During infection, L. monocytogenes invades host cells by inducing its own internalization into a vacuole (or via phagocytosis into a phagosome), from which it rapidly escapes by producing and secreting the pore-forming hemolysin toxin listeriolysin O (LLO; encoded by the hly gene) (3–6). LLO, together with two additional secreted phospholipases (PLC), PlcA, a phosphoinositol-PLC (PI-PLC), and PlcB, a phosphatidylcholine-PLC (PC-PLC), perforate the phagosomal membrane and enable the bacteria to reach the host cell's cytosol (7, 8). In the cytosol, L. monocytogenes replicates and secretes the surface protein, ActA, that recruits the host cell's actin polymerization machinery to propel the bacteria around the cell and to facilitate spread from cell to cell without causing lysis (9, 10). While these virulence factors are essential for L. monocytogenes pathogenesis, currently not much is known about the mechanisms that mediate their secretion during infection.
Most L. monocytogenes secretory proteins are assumed to be secreted via the Sec translocation system based on signal peptide predictions and proteomic studies (11–15). The Sec system is found in both Gram-positive and Gram-negative bacteria and serves as the predominant protein translocation system for integral membrane and secretory proteins. As such, most of the components of the Sec system are highly conserved (also beyond the prokaryotic kingdom) and are essential for growth (16). The Sec system has been studied primarily in Escherichia coli and was shown to comprise a protein-conducting channel (the translocon) and several accessory proteins. The translocon is composed of three integral membrane proteins, SecYEG, which together conduct the translocation of proteins across and into the cytoplasmic membrane (17). Secretory proteins (preproteins) are targeted to the membrane posttranslationally by the SecB chaperone, which stabilizes them in an unfolded conformation. Once localized to the membrane, SecB directs the preproteins to the translocon motor protein, SecA, which threads the unfolded preproteins through the translocon channel using ATP hydrolysis. Across the membrane, at the translocon exit site, the SecD-SecF/YajC complex pulls out the preproteins, completing their translocation (18, 19). Another component of the Sec system is the insertase YidC, which is thought to facilitate mainly the insertion of inner membrane proteins (20). Although the Sec components are generally conserved among bacteria, there are several differences between the Sec systems of Gram-negative and Gram-positive bacteria. Typically, Gram-positive bacteria lack the SecB chaperone, some species encode additional copies of sec genes, such as secA, secY, and yidC (secA2, secY2, and yidC2), and the SecD-SecF complex is fused into a single protein encoded by a single gene (21, 22). In addition, some Gram-positive species encode a chaperone termed PrsA, which is located extracellularly and facilitates the folding of secretory proteins upon their secretion (23). Specifically, L. monocytogenes encodes an additional secA gene (secA2, but not secY2), two yidC genes (yidC1 and yidC2), two prsA genes (prsA1 and prsA2), and a fused secDF gene. SecA2 was shown to facilitate the secretion of several autolytic enzymes, cell wall proteins, and a superoxide dismutase, termed MnSOD, all of which contribute to L. monocytogenes in vivo infection (13, 24, 25). PrsA2 was shown to promote the folding and activity of LLO and PC-PLC upon secretion (26–29); however, the role of SecDF in L. monocytogenes has not been characterized.
L. monocytogenes SecDF is predicted to contain 12 transmembrane segments and two large extracellular loops, which is topologically similar to the E. coli SecDF complex (22). The initial observation that E. coli SecD and SecF are the only Sec components harboring periplasmic domains led to the suggestion that they are involved in later stages of protein export (30). Further studies have implicated SecD and SecF in playing a role in facilitating efficient translocation of preproteins. However, they are not completely necessary for protein export in vivo and in vitro (31–33). This ambiguity concerning the function of SecDF in the protein secretion process was only recently resolved with the publication of the Thermus thermophilus SecDF crystal structure (19). This study demonstrated that SecDF functions as a membrane-integrated chaperone that binds the emerging preprotein and tugs it out of the translocon in a highly precise manner, using proton-motive force as an energy source (19, 34). Nevertheless, the essentiality of SecDF in protein secretion remains mysterious. For example, in E. coli the secDF mutant is characterized by a severe growth defect in rich laboratory medium at 30 and 37°C, yet it grows normally in glucose minimal defined medium at 37°C (31). In Bacillus subtilis and Staphylococcus aureus, secDF deletion mutants exhibit a cold-sensitive growth phenotype (at 15°C) but grow normally at 37°C (22, 35). Notably, the cold sensitivity of the B. subtilis secDF mutant is exacerbated when the bacteria overexpress a secretory protein, yet this phenotype disappears if the bacteria are grown at 37°C (22). These observations highlight the flexibility in the requirement for SecDF during protein export with different bacteria and even under different growth conditions. In this study, we show that L. monocytogenes SecDF is required for in vivo bacterial infection, yet it is dispensable for growth in laboratory-rich medium (at both 15 and 37°C). We further demonstrate that SecDF plays a role in membrane translocation of several critical virulence factors; thus, it contributes to the L. monocytogenes infection process.
MATERIALS AND METHODS
Strains, cells, growth conditions, and reagents.
L. monocytogenes strains used in this study are shown in Table 2. E. coli XL-1 Blue strain was used for vector propagation (Stratagen), and E. coli strain SM-10 (53) was used as a donor when delivering plasmids to L. monocytogenes by conjugation. E. coli strains were grown in Luria-Bertani (LB; BD) media at 37°C, and L. monocytogenes strains were grown in brain heart infusion (BHI; BD) rich media at 37°C. For induction of the virulence genes, bacteria were grown in LB morpholinepropanesulfonic acid (MOPS)-glucose 1-phosphate (LB-Glu-1P) medium (44). Anti-LLO antibody was purchased from Abcam, Inc. (ab43018). Anti-PlcA antibody was a gift from Helen Marquis (Cornell), and anti-PlcB antibody was a gift from Howard Goldfine (University of Pennsylvania) and Daniel Portnoy (Berkeley). HisProbe-horseradish peroxidase (HRP) conjugated for ActA-6His detection was purchased from Thermo Scientific (15165). Anti-GroEL antibody was a gift from Abdussalam Azem (Tel-Aviv University). Peroxidase-conjugated goat anti-rabbit IgG and goat anti-mouse IgG were purchased from Jackson ImmunoResearch Laboratories, Inc. (catalog no. 111-035-003 and 115-035-003, respectively). For infection experiments, L. monocytogenes bacteria were grown overnight in BHI at 30°C without agitation. Bone marrow-derived (BMD) macrophages were isolated from C57BL/6 (6- to 8-week-old) female mice (Harlan Laboratories, Ltd., Israel) and cultured as described previously (3). The ISRE-L929 cell line was a gift from Bruce Beutler (University of Texas Southwestern Medical Center) (38).
Table 2.
List of strains and mutants used in this study
| Name | LMRG gene/L. monocytogenes EGDe gene identifier | Description and/or source |
|---|---|---|
| WT | L. monocytogenes 10403S, parental strain used in this study; Daniel A. Portnoy, laboratory stock | |
| Δhly | LMRG_02076/lmo0202 | Deletion of hly gene encoding LLO DP-L2161; 6 |
| ΔplcAB | LMRG_02627/lmo0205 | Deletion of plcA and plcB genes |
| LMRG_02623/lmo0201 | DP-L1936; 8 | |
| ΔsecDF | LMRG_01443/lmo1527 | Deletion of secDF gene, AH-L101; this study |
| ΔsecDF + pLIV2-secDF | ΔsecDF mutant harboring the integrative plasmid pLIV2 encoding the secDF gene under inducible promoter, AH-L102; this study | |
| ΔyidC1 | LMRG_00831/lmo1379 | Deletion of yidC1 gene, AH-L103; this study |
| ΔsecA2 | LMRG_00265/lmo0583 | Deletion of secA2 gene, DP-L4342; 24 |
| WT actA-6his-tag | LMRG_02626/lmo0204 | L. monocytogenes 10403S, encoding 6 histidines at the 3′ end of actA gene (chromosomally), AH-L104; this study |
| WT + pPL2-mCherry | L. monocytogenes 10403S harboring the integrative plasmid pPL2 carrying mCherry gene under a constitutive promoter; this study | |
| ΔsecDF + pPL2-mCherry | ΔsecDF mutant harboring the integrative plasmid pPL2 carrying mCherry gene under a constitutive promoter; this study |
Bacterial genetic screen.
Bacterial genetic screening was performed as described before (36). Briefly, a total of 5,000 Mariner-Tn mutants were grown in BHI medium in 96-well plates overnight at 30°C. BMD macrophages from C57BL/6 mice were plated in 96-well plates; 4 × 104 macrophages per well were infected with 2 × 106 bacterial cells. Thirty minutes postinfection, macrophages were washed and gentamicin was added (50 μg/ml) to kill extracellular bacteria. At 6 h postinfection (h p.i.), macrophage culture supernatants were taken and frozen at −80°C. The amount of beta interferon (IFN-β) in the culture supernatants was detected using the type I interferon reporter cell line (ISRE-L929) (38). Reporter cells were grown in 96-well plates and incubated with a 10× dilution of the macrophage culture supernatants for 4 h. Cells were lysed using Glo lysis buffer (Promega), and luciferase activity was detected using the Beetlejuice D-luciferine kit (PJK) and measured with a luminescence plate reader (Biotek Synergy HT).
L. monocytogenes intracellular growth in macrophages.
L. monocytogenes intracellular growth curves were determined as described in reference 54. Briefly, 2 × 106 cells were seeded on a 60-mm petri dish with glass coverslips. About 8 × 106 L. monocytogenes bacteria were used to infect the BMD macrophages. At 30 min postinfection, the macrophage monolayers were washed and fresh medium was added. At 1 h p.i., gentamicin was added to a final concentration of 5 μg/ml to limit the growth of extracellular bacteria. At each time point, three coverslips were taken and transferred into 2 ml of sterile water to release the intracellular bacteria, and then serial dilutions were plated on BHI agar plates. CFU were counted after 24 h of incubation at 37°C. Error bars represent the standard errors from the triplicate samples. All experiments were repeated at least three times. To analyze the bacterial gene expression of intracellular L. monocytogenes, 25 × 106 BMD macrophages were seeded on 150-mm petri dishes and were infected with 1 × 108 bacteria. At 2.5 and 6 h p.i., the macrophages were lysed in 20 ml of ice-cold water, and then the released bacteria were collected on 0.45-μm hemagglutinin (HA) filters (HAWP04700; Millipore). Bacterial RNA was purified as described below for gene expression analysis.
Construction of L. monocytogenes gene deletion mutants and His-tagged ActA.
Upstream and downstream regions of the selected genes were amplified using Phusion DNA polymerase and cloned into the pKSV7oriT vector (55) (see Table S1 in the supplemental material for primers used in this study). Cloned plasmids were sequenced and then conjugated to L. monocytogenes using E. coli SM-10. L. monocytogenes conjugants were grown at 30°C with chloramphenicol (Cm) and then transferred to 41°C and grown for another 2 days on BHI plates with Cm (20 μg/ml) to stabilize the plasmid in the bacterial chromosome. To cure the integrated plasmids and generate a clean in-frame deletion, bacteria were passed several times in fresh BHI medium without Cm at 30°C. Bacteria were then seeded on BHI plates, and deletion mutants were identified by screening for Cm-sensitive bacteria. Finally, the deletion genotype was validated using PCR. Since the yidC1 complete gene deletion vector was found to be unstable in E. coli XL-1 Blue, the ΔyidC1 mutant was generated by removing the ribosome binding site at the 5′ untranslated region (UTR) and the first 378 coding nucleotides of the yidC1 gene (LMRG_00831). To construct a chromosomal ActA-6His translational fusion, a DNA sequence encoding 6 histidines was fused in-frame at the end of the actA gene (LMRG_02626).
Gene expression analysis.
The transcription levels of bacterial genes and macrophage genes were analyzed using quantitative real time-PCR analysis (RT-qPCR). RNA was purified from bacteria grown exponentially in BHI and from intracellularly grown bacteria at 2.5 and 6 h p.i. using standard phenol-chloroform extraction methods. RNA of intracellularly grown bacteria was amplified using the MessageAmp II (Ambion) bacterial amplification kit according to the manufacturer's instructions. When analyzing the expression of macrophage genes, RNA of infected macrophages was extracted using TRIzol reagent according to standard protocols. In all cases, 1 μg of RNA was reverse transcribed to cDNA using a high-capacity reverse transcription kit (Applied Biosystems). RT-qPCR was performed on 10 ng of cDNA using SYBR green with the StepOnePlus RT-PCR system (Applied Biosystems) (see Table S1 in the supplemental material for RT-qPCR primers). The relative expression of bacterial genes was determined by comparing their transcription levels to that of the bacterial 16S rRNA as a reference. The transcription levels of macrophage cytokine genes were normalized using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a reference gene. Statistical analysis was performed using StepOneV2.1 software. All experiments were performed on at least three independent occasions (n = 3). Error bars represent a 95% confidence interval, i.e., the value is expected to fall within the bar range in 95% of repeat experiments. When the error bars of two samples do not overlap, the significance of the difference (P value) is ≪0.01.
Immunofluorescence microscopy and phagosomal escape assays.
The percentage of bacteria that escaped from macrophage phagosomes was determined by two immunofluorescence assays as described previously (42, 43). In the first assay, BMD macrophages were infected with wild-type (WT) L. monocytogenes or ΔsecDF mutant bacteria that constitutively express the mCherry fluorescent protein (cloned on the integrative pPL2 plasmid) to visualize all intracellular bacteria. At 3 h p.i. the BMD macrophages were washed three times with KHM buffer (110 mM potassium acetate, 20 mM HEPES, 2 mM MgCl2, pH 7.3), and the plasma membrane was selectively permeabilized by treating the cells with 50 μg/ml digitonin (Sigma) in KHM buffer for 1 min at room temperature. Cells were then washed immediately with KHM buffer, and fluorescein-conjugated anti-L. monocytogenes antibody (Meridian) was delivered to the macrophage cytosol for 10 min at 37°C to label accessible cytosolic bacteria. BMD macrophages were then washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde-PBS, and permeabilized with Triton X-100. Coverslips were mounted with Vectashield mounting media containing 4′,6-diamidino-2-phenylindole (DAPI). Images were taken using a Nikon Eclipse Ti microscope. In the second assay, BMD macrophages were infected with WT L. monocytogenes or ΔsecDF mutant bacteria and then fixed at 3 h p.i. with 4% paraformaldehyde-PBS. The fixed cells were permeabilized with Triton X-100 and incubated with rhodamine phalloidin (Biotium) for actin labeling and with fluorescein-conjugated anti-L. monocytogenes antibody (Meridian) for bacterial labeling. Coverslips were mounted with Vectashield mounting media containing DAPI. Images were taken using a Zeiss LSM 510-META confocal microscope.
Mouse infections.
L. monocytogenes bacteria were grown in BHI medium at 30°C overnight. Bacterial cultures were washed twice in Ringer's lactate solution and counted (∼2 × 109 bacteria per ml). C57BL/6 (6- to 8-week-old) female mice (Harlan Laboratories, Ltd., Israel) were infected via tail vein injections with 4 × 104 bacteria in 200 μl of PBS. Animals were observed daily for any signs of illnesses and were euthanized 72 h postinfection. Spleens and livers were harvested and homogenized in 0.2% saponin, and the numbers of viable bacteria in each organ were determined by plating serial dilutions of homogenates onto BHI agar plates. The experiment involved 6 mice in each group and was repeated twice, yielding similar results.
Analysis of LLO, PlcA, PlcB, and ActA secretion and activity.
L. monocytogenes bacteria were grown in LB-Glu-1P medium at 37°C to an optical density (OD) of 0.4 to 0.7 for 5 to 8 h or overnight (12 h), and supernatants were separated by centrifugation. For Western blot analysis, bacteria from 20 ml culture were lysed by 1 h of incubation with mutanolysin (M9901; Sigma) at 37°C, followed by sonication. Membrane proteins were precipitated by centrifugation at 50,000 rpm for 10 min (4°C) and solubilized by 1% SDS. Cytosolic and total secreted proteins (supernatants) were precipitated by trichloroacetic acid (TCA). Total protein content in each sample was quantified by the Lowry method. Ten μg of total protein was separated on 12.5% SDS-PAGE and probed by a standard immune-blotting technique. The hemolytic activity assay was performed as described previously (43): bacterial supernatants were treated with 5 mM dithiothreitol (DTT), serially diluted in PBS, and incubated with 0.5% sheep blood red cell suspension (NovaMed) following hemolysis at 540 nm. The PI-PLC activity assay was adapted from Geoffroy et al. (46): 1 ml of sodium-cholate (58 mM), CaCl2 (10 mM), and 0.036 g phosphatidyl-inositol (P6636; Sigma) were mixed with 7 ml NaCl (0.15 M). One hundred μl of the assay solution was then mixed with 100 μl of bacterial supernatants and incubated in a plate reader at 37°C for 10 h, following turbidity assessment at 510 nm. PC-PLC activity was assayed using the EnzChek direct phospholipase C assay kit (Molecular Probes) according to the manufacturer's instructions. The fluorescent signal was recorded following overnight incubation. For activity assays on indicative agar plates, 1 μl of overnight bacterial culture was spotted on blood, agar listeria according to Ottaviani and Agosti (ALOA) or egg yolk plates, which were prepared as described previously (45) and incubated for 24 to 48 h at 37°C.
RESULTS
A genetic screen designed to identify genes that contribute to L. monocytogenes intracellular growth.
In a genetic screen designed to identify genes that contribute to L. monocytogenes intracellular growth in macrophages, we identified the secDF gene. Briefly, we used a previously described screening approach that relies on detecting beta interferon (IFN-β) in the supernatants of L. monocytogenes-infected cells (36). IFN-β is a cytokine of the type I interferon innate immune response that is robustly expressed and secreted by mammalian cells upon L. monocytogenes infection. The expression level of IFN-β was shown to correlate with the localization of the bacteria in the cell cytosol and with their ability to further replicate within this niche (37). Based on these observations, in this screen we used IFN-β as a reporter for L. monocytogenes intracellular growth in macrophages. An L. monocytogenes Himar-mariner1 transposon mutant library was used to infect BMD macrophages, and the levels of IFN-β secreted to the culture supernatants were detected using an ISRE-type I interferon reporter cell line, which expresses luciferase in response to type I interferons (28, 36, 38). Approximately 5,000 L. monocytogenes mutants were screened in a 96-well format, revealing 20 mutants to be low inducers of IFN-β compared to wild-type (WT) bacteria. Among the mutants, we identified transposon insertions within genes related to the flagella, the prfA gene, and genes encoding transporters, as well as metabolic and regulatory factors (Table 1). As expected, mutants in the flagella or in the prfA gene, the master regulator of the virulence genes, indeed induce less IFN-β production, since they are impaired in mammalian cell invasion and in intracellular replication, respectively (39, 40). Among the remaining mutants, we identified a mutant in the secDF gene that was particularly impaired in its ability to induce IFN-β; hence, it was chosen for further characterization.
Table 1.
Identification of L. monocytogenes mutants that exhibit low IFN-β response during macrophage infection
| L. monocytogenes 10403S gene | L. monocytogenes EGDe gene identifier | Description | INF-β fold of induction mutant/WT bacteria |
|---|---|---|---|
| LMRG_02622.6 | lmo0200 | PrfA main virulence regulator of L. monocytogenes | 0.159 |
| LMRG_00367.6 | lmo0679 | Flagellar biosynthesis protein FlhB | 0.383 |
| LMRG_00376.6 | lmo0688 | Similar to glycosyl-transferase, group 2 family protein | 0.720 |
| LMRG_00380.6 | lmo0692 | Two-component sensor histidine kinase CheA | 0.486 |
| LMRG_00386.6 | lmo0697 | Similar to flagellar hook protein FlgE | 0.282 |
| LMRG_00395.6 | lmo0706 | Similar to flagellar hook-associated protein 3, FlgL | 0.445 |
| LMRG_0397.6 | lmo0708 | Similar to flagellar biosynthesis protein FliS | 0.561 |
| LMRG_00402.6 | lmo0713 | Flagellar basal-body M-ring protein FliS | 0.594 |
| LMRG_00413.6 | lmo0724 | Similar to B. subtilis YvpB protein | 0.742 |
| LMRG_02270.6 | lmo0847 | Glutamine transporter | 0.493 |
| ∼20bp upstream LMRG_00534.6 | ∼20bp upstream lmo1072 | Highly similar to pyruvate carboxylase | 0.331 |
| LMRG_01443.6 | lmo1527 | Similar to protein-export membrane protein SecDF | 0.354 |
| LMRG_02761.6 | lmo1687 | Hypothetical protein | 0.725 |
| LMRG_02767.6 | lmo1693 | Regulatory protein RecX | 0.830 |
| LMRG_01002.6 | lmo1855 | d-alanyl-d-alanine carboxypeptidase | 0.814 |
| LMRG_01003.6 | lmo1856 | Purine nucleoside phosphorylase | 0.818 |
| LMRG_01264.6 | lmo2110 | Similar to mannose-6 phosphate isomerase | 0.707 |
| LMRG_1717.6 | lmo2531 | ATP synthase F1 alpha subunit | 0.417 |
| LMRG_01981.6 | lmo2715 | ABC transporter CydDC cysteine exporter CydC | 0.840 |
| LMRG_01787.6 and LMRG_01360.6 | lmo2461 and lmo1606 | RNA polymerase sigma-54 factor (sigma-L) and DNA segregation ATPase FtsK/SpoIIIE | 0.506 |
The ΔsecDF mutant is defective in intracellular growth in macrophages yet grows normally in a rich laboratory medium.
To validate the observed phenotype of the secDF::Tn mutant, we completely deleted the secDF gene in the L. monocytogenes 10403S strain (ΔsecDF) (Table 2). First, the ability of the ΔsecDF mutant to grow in the laboratory rich medium BHI at both 37 and 15°C was examined. Interestingly, we found that unlike the phenotype observed in B. subtilis and S. aureus, the L. monocytogenes ΔsecDF mutant grew similarly to WT bacteria at both temperatures (Fig. 1A and B). Of note, the ΔsecDF mutant did not exhibit a rough colony phenotype or filamentous cells, as has been shown for other Sec-related mutants of L. monocytogenes (24, 41). The induction of IFN-β and two additional proinflammatory cytokines, interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α), next was measured upon infection of macrophages with ΔsecDF mutant versus WT bacteria (at a multiplicity of infection [MOI] of 1) using real-time quantitative PCR (RT-qPCR) analysis at 6 h p.i. As shown in Fig. 1C, the transcription levels of the three cytokines were greatly reduced in the ΔsecDF-infected cells compared to the cells infected with WT bacteria. To test whether the reduced cytokine response is a result of impaired intracellular growth of the ΔsecDF mutant, we monitored its replication in BMD macrophages. Notably, we found that the ΔsecDF mutant is severely defective in intracellular growth compared to WT bacteria and a ΔsecDF-complemented strain, harboring a copy of the secDF gene on an integrative plasmid (pLIV2-secDF) (Fig. 1D). Taken together, these results demonstrated that SecDF is dispensable during extracellular growth under the tested laboratory conditions, but it is required early during L. monocytogenes intracellular growth in macrophages.
Fig 1.

ΔsecDF mutant shows restricted intracellular growth and a decreased cytokine response upon macrophage infection. (A) Growth curves of WT L. monocytogenes and the ΔsecDF mutant grown in BHI laboratory medium at 37°C. Representative growth curves are shown. Error bars represent the standard deviations from triplicate trials and are hidden by the symbols. Growth curves were performed in 3 biological repeats (n = 3). (B) Growth curves of WT L. monocytogenes and ΔsecDF mutant grown in BHI laboratory medium at 15°C. Data shown represent the means from 3 biological repeats (n = 3). Error bars represent the standard deviations. O.D. 600, optical density at 600 nm. (C) RT-qPCR analysis of the IFN-β, IL-6, and TNF-α transcription levels in BMD macrophages infected with WT L. monocytogenes or the ΔsecDF mutant (MOI of 1) at 6 h p.i. Transcription levels are represented as the relative quantity (RQ) relative to the levels in uninfected cells. The data represent 3 biological repeats (n = 3). Error bars represent a 95% confidence interval, i.e., P ≪ 0.01. (D) Intracellular growth curves of the WT L. monocytogenes, ΔsecDF mutant, and ΔsecDF complemented strain harboring pLIV2-secDF plasmid grown in BMD macrophages. Error bars represent the standard deviations from triplicate trials. The data represent 3 independent biological repeats (n = 3).
The secDF gene is upregulated when bacteria reside in macrophage phagosomes and contributes to phagosomal escape.
Since the ΔsecDF mutant did not grow intracellularly in macrophages, we asked whether it is impaired in the ability to escape the macrophage phagosomes or to spread from cell to cell, critical steps in L. monocytogenes infection. To this end, we performed two fluorescence microscopy visualization assays, which are used to assess bacterial phagosomal escape and binding of host actin filaments (42, 43). The first fluorescence assay relies on the selective permeabilization of the plasma membrane, which allows the detection of cytosolic bacteria, and not of phagosomally trapped bacteria, using antibodies. Briefly, BMD macrophages were infected with mCherry-expressing ΔsecDF mutant or WT bacteria (Table 2) and gently treated with digitonin at 3 h p.i. to selectively permeabilize the plasma membrane. Fluorescein-conjugated antilisterial antibody then was added to label accessible cytosolic bacteria. As shown in Fig. 2A, at 3 h p.i. most of the WT bacteria were labeled with mCherry and fluorescein in the macrophage cytosol (green/yellow), whereas more of the ΔsecDF bacteria were labeled solely with mCherry (red), indicating that the ΔsecDF bacteria are impaired in phagosomal escape. Quantifying the percentage of bacteria labeled with mCherry and fluorescein as opposed to mCherry alone confirmed that the proportions of bacteria that escaped the phagosomes were 80 and 50% for WT and ΔsecDF bacteria, respectively (Fig. 2A). The second fluorescence assay relies on the observation that L. monocytogenes recruits host actin filaments in the cytosol, whereas bacteria in phagosomes do not. Briefly, BMD macrophages were infected with ΔsecDF mutant or WT bacteria, fixed at 3 h p.i., and permeabilized with Triton X-100 (permeabilizing both plasma and phagosomal membranes). Cells were stained with rhodamine phalloidin (red) for host actin labeling, with fluorescein-conjugated antilisterial antibody (green) for bacterial visualization and DAPI (blue) for host nuclear staining. As shown in Fig. 2B, at 3 h p.i. most of the WT bacteria were associated with actin filaments in the macrophage cytosol (red/yellow), whereas the majority of the ΔsecDF mutant bacteria were labeled solely with fluorescein (green), suggesting that most of the ΔsecDF mutant bacteria are still located in the phagosomes and/or impaired in actin filament recruitment. Quantifying the percentage of bacteria labeled with rhodamine phalloidin and fluorescein as opposed to fluorescein alone confirmed that 80% of WT bacteria versus 36% of ΔsecDF mutant bacteria were engaged with actin filaments at 3 h p.i. (Fig. 2B). Together, these assays indicated that the ΔsecDF mutant is mostly impaired in phagosomal escape and perhaps in actin binding as well.
Fig 2.

SecDF contributes to phagosomal escape. (A, left) Confocal fluorescence microscope images of digitonin-treated BMD macrophages infected with mCherry expressing WT L. monocytogenes or ΔsecDF bacteria at 3 h p.i. Cytosolic bacteria were labeled with fluorescein-conjugated anti-Listeria antibody (green), and macrophage nuclei were labeled with DAPI (blue). (Right) percentage of bacteria that escaped macrophage phagosomes at 3 h p.i. as determined by a microscopic fluorescence assay based on selective digitonin membrane permeabilization. The results are representative of 10 microscopic images from 2 independent biological repeats for each strain (in each repeat between 300 and 400 bacteria were counted). The P value was calculated using a chi-square test. (B, left) Confocal fluorescence microscope images of BMD macrophages infected with WT L. monocytogenes or ΔsecDF mutant bacteria at 3 h p.i. Bacteria were labeled with fluorescein-conjugated anti-Listeria antibody (green), macrophage nuclei were labeled with DAPI (blue), and macrophage actin filaments were labeled with rhodamine phalloidin (red). (Right) Percentage of bacteria that escaped macrophage phagosomes at 3 h p.i. as determined by a microscopic fluorescence assay based on actin labeling. The results are representative of 10 microscopic images from 3 independent biological repeats for each strain (in each image, between 50 and 100 bacteria were counted). The P value was calculated using a chi-square test. (C) RT-qPCR transcription analysis of different sec-related genes in intracellularly grown (cytosolic) bacteria (WT L. monocytogenes) in macrophages at 6 h p.i. Transcription levels were measured as relative quantity (RQ), i.e., relative to their levels in bacteria grown exponentially in BHI laboratory medium. Data are presented as log2 values of the RQ. The data represent 3 biological repeats (n = 3). Error bars represent a 95% confidence interval. (D) RT-qPCR transcription analysis of different sec-related genes in phagosomally trapped bacteria (Δhly mutant) in macrophages at 2.5 h p.i. Transcription levels were measured as the RQ relative to their levels in bacteria grown exponentially in BHI laboratory medium. Data are presented as log2 values of RQ. The data represent 3 biological repeats (n = 3). Error bars represent a 95% confidence interval, i.e., P ≪ 0.01. (E) Intracellular growth curves of WT L. monocytogenes and the ΔyidC1 and ΔsecA2 mutants in BMD macrophages. Error bars represent the standard deviations from triplicate trials. The data represent 3 independent biological repeats (n = 3).
To gain further insight into the importance of SecDF during infection, we analyzed the transcription levels of the secDF gene and additional genes of the Sec system in both intracellularly grown (WT) and phagosomally trapped (Δhly mutant) bacteria and compared them to levels for WT bacteria grown extracellularly in BHI medium. We observed that the transcription levels of the Sec system genes, e.g., secY, secG, secDF, secA1, secA2, yajC, yidC1, and yidC2, generally were lower during intracellular growth (6 h p.i.) than during exponential growth in BHI medium (Fig. 2C). Exceptional in this regard was the secA2 gene, which showed similar transcription levels under both conditions (Fig. 2C). Notably, this transcription profile was largely altered when the bacteria were located in phagosomes (i.e., in the Δhly mutant). Here, we found that secDF, secA2, and yidC1 in particular were transcribed at a high level inside the phagosomes compared to their levels during extracellular bacterial growth in BHI medium (Fig. 2D). To assess whether SecA2 and YidC1 also contribute to L. monocytogenes intracellular growth like SecDF, ΔsecA2 and ΔyidC1 mutants (Table 2) were grown in BMD macrophages and compared to WT bacteria. Remarkably, we found that although these components were highly induced inside the phagosome they were dispensable, since the ΔsecA2 and ΔyidC1 mutants grew like WT bacteria in the macrophages (Fig. 2E). Taken together, these observations suggested a role for SecDF early within the phagosome compartment, possibly in the secretion of virulence factors that contribute to L. monocytogenes phagosomal escape and intracellular growth.
SecDF contributes to the proper translocation and activity of LLO, PlcA, PlcB, and ActA.
To determine whether SecDF contributes to membrane translocation of the critical virulence factors that mediate phagosomal escape and spread from cell to cell, i.e., LLO, PlcA, PlcB, and ActA, we compared the subcellular distribution of these proteins within ΔsecDF and WT bacteria. Briefly, ΔsecDF mutant and WT bacteria were grown in LB-MOPS-glucose 1P charcoal medium (LB-Glu-1P), which is known to activate the production and secretion of L. monocytogenes virulence factors (44). This activation specifically occurs when the bacteria switch from utilizing glucose to glucose 1-phosphate. At different time points during growth, bacteria were harvested and the bacterial membranes, cytosol, and supernatants fractionated and subjected to semiquantitative Western blot analysis using specific antibodies to LLO, PlcA, PlcB, 6His-tagged ActA, and GroEL, with the latter serving as a control protein. Notably, under these growth conditions the ΔsecDF mutant grew better than WT bacteria in a reproducible manner (Fig. 3A). Initially, the distribution of LLO preproteins was examined after 5.5 and 7 h (Fig. 3A, arrows), at which time the production of virulence factors is known to occur (44). As shown in Fig. 3B, at both time points LLO preproteins were observed to accumulate in the cytosol and on the membrane of the ΔsecDF mutant, unlike WT bacteria, whereas the levels of GroEL remained constant. Notably, despite the observed accumulation of LLO preproteins, the levels of secreted LLO in the supernatants of ΔsecDF mutant and WT bacteria were comparable. Similar results were observed with PlcA, PlcB, and ActA preproteins (Fig. 3C and D). This accumulation of preproteins on the membrane suggests that in the absence of SecDF their translocation is delayed, presumably within the translocons, leading to the observed accumulation in the cytosol. Accumulation of preprotein precursors on the membrane could be expected to stimulate stress responses that suspend growth. However, GroEL levels were comparable in WT and ΔsecDF mutant bacteria, as assessed by Western blot analysis (Fig. 3B to D), indicating the absence of a stress response, since GroEL is a chaperone typically upregulated under protein-related stresses. Currently it remains unclear why the ΔsecDF mutant grows better than WT bacteria under these conditions. The possibility that the bacteria form chains or long filaments that can affect OD measurements was excluded.
Fig 3.

SecDF promotes LLO, PlcA, PlcB, and ActA membrane translocation. (A) Growth curves of WT L. monocytogenes and the ΔsecDF mutant in LB-Glu-1P media. Data are representative of 5 biological experiments (n = 5). (B) Western blot analysis of LLO preproteins' subcellular distribution within the membrane, cytosol, and supernatant fractions of WT L. monocytogenes and the ΔsecDF mutant grown in LB-Glu-1P media using anti-LLO antibodies. GroEL was used as a control protein. The data are representative of three biological repeats (n = 3). (C) Western blot analysis of PlcA and PlcB preproteins' subcellular distribution in the membrane, cytosol, and supernatant fractions of WT L. monocytogenes and ΔsecDF mutant grown in LB-Glu-1P media using specific antibodies. GroEL was used as a control protein. The data are representative of three biological repeats (n = 3). (D) Western blot analysis of 6His-tagged ActA preprotein subcellular distribution in the membrane, cytosol, and supernatant fractions of WT L. monocytogenes and the ΔsecDF mutant grown in LB-Glu-1P media using anti-His tag antibodies. GroEL was used as a control protein. The data are representative of three biological repeats (n = 3).
Since the Western blot analysis demonstrated similar levels of secreted proteins in the supernatants of ΔsecDF mutant and WT bacteria, we examined the possibility that the activity of the secreted proteins is impaired in the ΔsecDF mutant. To this end, assays for LLO, PlcA, and PlcB activity were performed using indicative agar plates. ΔsecDF and WT bacteria were spotted on blood, ALOA, and egg yolk plates, where a zone of hemolysis or opacity is indicative of LLO, PlcA, and PlcB activity, respectively, upon secretion to the agar medium. As controls, the Δhly (LLO−) and ΔplcAB mutants were used. As shown in Fig. 4A, the ΔsecDF mutant exhibited decreased LLO activity, as manifested by a reduced hemolysis zone surrounding the bacterial patch on the blood plate compared to the zone observed with WT bacteria (25% reduction in diameter). Similarly, it also exhibited reduced PlcA and PlcB phospholipase activity (lecithinase activity) on the ALOA and egg yolk plates, as manifested by the smaller white precipitate zones (30% reduction in diameter) (45). These results confirmed that the virulence factors secreted by the ΔsecDF mutant indeed exhibit less activity. To measure directly the activity of LLO, PlcA, and PlcB in the supernatants of ΔsecDF mutant and WT bacteria grown in LB-Glu-1P medium, quantitative activity assays were performed for each of the proteins. Supernatants were collected and subjected to a hemolysis assay (43), a phosphatidyl-inositol assay (46), and a phospholipase C assay (Molecular Probes) that measures LLO, PlcA, and PlcB activity, respectively. Similar to what we observed on the indicative agar plates, the different assays evidenced that LLO, PlcA, and PlcB secreted by the ΔsecDF mutant exhibit lower activity levels (Fig. 4B to D). Given that ΔsecDF mutant bacteria secrete normal levels of LLO, PlcA, and PlcB to the supernatant and yet a substantial portion of the secreted proteins is nonactive (∼30%), we conclude that SecDF plays an important chaperone role, promoting the translocation of the virulence factors in a way that facilitates their secretion in a fully active state.
Fig 4.

ΔsecDF mutant exhibits low activity of LLO, PlcA, and PlcB. (A) Activity assays of LLO, PlcA, and PlcB in WT L. monocytogenes and the ΔsecDF mutant grown on blood, ALOA, and egg yolk indicative agar plates, respectively. The halo (clear or opaque) zone around the bacterial patch is proportional to the LLO, PlcA, and PlcB activity. The pictures shown are representative of 3 independent experiments (n = 3). (B) LLO activity hemolysis assay based on lysis of sheep red blood cells. LLO activity was measured in the supernatants of WT L. monocytogenes and the ΔsecDF mutant grown overnight in LB-Glu-1P media. The Δhly mutant was used as a control. The data are representative of three biological repeats (n = 3). The error bars represent standard deviations from triplicate trials. (C) PI-PLC (PlcA) activity assay, measuring reaction turbidity (adapted from Geoffroy et al. [46]). PI-PLC activity was measured in supernatants of WT L. monocytogenes and ΔsecDF mutant grown overnight in LB-Glu-1P media. The ΔplcAB mutant was used as a control. The data are representative of three biological repeats (n = 3). Error bars represent the standard deviations from triplicate trials. (D) A measurement of PC-PLC (PlcB) activity using the EnzChek direct phospholipase C assay kit (Molecular Probes). PC-PLC activity was measured in supernatants of WT L. monocytogenes and the ΔsecDF mutant grown overnight in LB-Glu-1P media. The ΔplcAB mutant was used as a control. The data are representative of three biological repeats (n = 3). Error bars represent the standard deviations from triplicate trials.
SecDF contributes to L. monocytogenes virulence in mice.
Finally, we examined the contribution of SecDF to L. monocytogenes virulence in mice. C57BL/6 young female mice were injected intravenously with 4 × 104 of ΔsecDF mutant or WT bacteria, and bacterial counts in the spleens and livers of the infected mice were analyzed at 72 h p.i. (6 mice in each group). As shown in Fig. 5, the ΔsecDF mutant colonized the livers and spleens of the infected mice to a lesser extent, exhibiting an ∼30-fold decrease in CFU recovered from both organs compared to WT bacteria. These results clearly demonstrate that SecDF is necessary to promote L. monocytogenes virulence.
Fig 5.
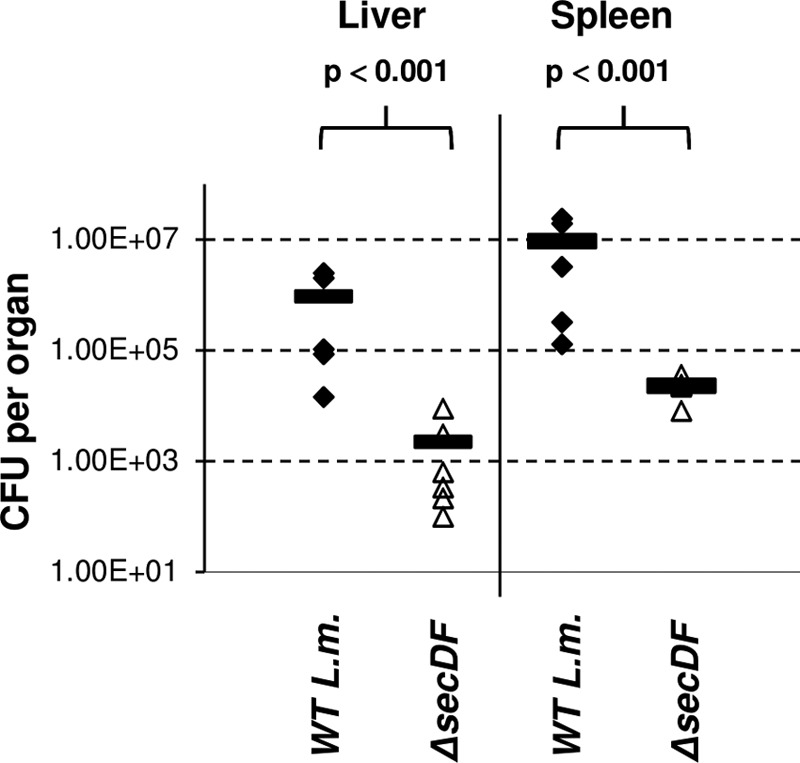
ΔsecDF mutant is less virulent in mice. Intravenous infection of C57BL/6 female mice with WT L. monocytogenes and ΔsecDF mutant bacteria. Bacterial CFU were numerated at 72 h p.i. from livers and spleens taken from 6 infected mice per group. Horizontal bars represent the means. The P value was calculated using a t test.
DISCUSSION
In this study, we demonstrate that the L. monocytogenes SecDF protein is not required for growth, as reported for other Gram-positive strains. Nevertheless, we reveal that it possesses an auxiliary role in facilitating the translocation of several virulence factors, LLO, PlcA, PlcB, and ActA, which are key to L. monocytogenes phagosomal escape and spread in mammalian cells. In the absence of SecDF, precursors of these preproteins accumulate on the bacterial membrane and within the cytosol, in line with impairment in their translocation process. Although reduced secretion levels were not detected, roughly 30% of the secreted proteins were found to be inactive. In line with these findings, the ΔsecDF mutant exhibited a reduced ability to escape macrophage phagosomes and grew less efficiently intracellularly in vitro and in vivo in mice. This study not only highlights SecDF as an important determinant of L. monocytogenes virulence but also provides important insights into the role of SecDF in protein secretion.
Until recently, the role of SecDF in protein export was not well defined. While it was found to be essential for protein secretion in vivo (in E. coli), it was largely dispensable during in vitro Sec-reconstituted translocation experiments (31). Recent crystal structures of SecDF and in vitro structure-function studies have provided novel insights concerning the role of this protein as a membrane chaperone. These studies demonstrated that during translocation the emerging preprotein is captured by the extracellular P1 domain of SecDF, which then rotates and moves 75 Å apart, to further pull out another ∼25 amino acids of the unfolded preprotein (19). This action, presumed to occur repetitively until the full protein is translocated, was hypothesized to enhance the translocation process by preventing a backward movement of the preprotein. In the present study, we provide in vivo observations to support this premise. The accumulation of virulence factor preproteins on the membrane (and in the cytosol) clearly reflects improper translocation and inability to cross the membrane efficiently, perhaps due to sliding backwards through the translocon channel. Such a forward and backward movement, in the absence of SecDF, could be expected to interfere with proper folding of the secreted proteins and explain the observed reduction in the factors' activities. In summary, our results support the premise that SecDF is an accessory chaperone that facilitates the translocation and proper folding of secretory proteins.
In this study, we identified a requirement for SecDF only during L. monocytogenes growth in mammalian cells. This finding is in accordance with prior observations that SecDF is particularly important under conditions of hypersecretion (22). Indeed, during infection L. monocytogenes produces and secretes many secretory and surface-anchored proteins that mediate invasion, phagosomal escape, intracellular replication, and spread from cell to cell (15, 40). Notably, most of these virulence factors are coregulated by the master virulence activator, PrfA, which, in response to various host signals, such as temperature and availability of nutrients, becomes activated and coordinates upregulated expression of the virulence factors (44, 47, 48). It is generally thought that L. monocytogenes initiates the secretion of most virulence factors during phagocytosis, which could explain the upregulation of SecDF within the phagosome. Although in this report we demonstrated the effect of secDF deletion mainly on factors that mediate phagosomal escape, we predict that the secretion of additional virulence factors is also affected, including factors that promote intracellular growth. Our observation that the ΔsecDF mutant does not grow well intracellularly, although 30 to 50% of the bacteria succeed to escape to the macrophage cytosol, largely supports this notion. Taken together, the data link the secretion of L. monocytogenes virulence factors to the Sec system and evidence the importance of this system in L. monocytogenes pathogenesis (24). Of note, the possibility that SecDF assembles with other secretion systems has not been ruled out.
Lastly, in this report we identified differential expression of the various Sec system components during L. monocytogenes growth in macrophages. Overall the core translocon (SecYEG) was moderately downregulated, while some accessory proteins were upregulated (e.g., SecDF and SecA2). These observations suggest that the bacteria rearrange translocon composition and/or stoichiometry in order to adapt to the hypersecretion state during infection. In this regard it was shown recently that the translocon stoichiometry varies upon translocation of different precursors, for example, outer membrane versus periplasmic protein (49). In light of these findings and the numerous reports that the SecYEG translocon can be found in various types of oligomers (50–52), it is tempting to speculate that bacteria modify the composition and/or the stoichiometry of the translocon apparatuses as a mechanism to cope with different secretion/translocation requirements. Such a strategy could enable L. monocytogenes to manage the enhanced secretion of virulence and housekeeping proteins during infection. Further study is required to characterize the Sec translocon during L. monocytogenes infection and the mechanisms that control its expression and assembly.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ilya Borovok for critically reviewing the manuscript. We thank Daniel Portnoy, Howard Goldfine, Helen Marquis, and Abdussalam Azem for supplying us with antibodies.
This work was partially supported by the Marie Curie IRG-FP7 and Israel Science Foundation grants to A.A.H., by the Legacy Heritage grant (1640/08) from the Israeli Science Foundation to R.N.-P., and by the Constantiner Institute for T.B.G.
Footnotes
Published ahead of print 20 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00697-13.
REFERENCES
- 1.Desvaux M, Hebraud M. 2006. The protein secretion systems in Listeria: inside out bacterial virulence. FEMS Microbiol. Rev. 30:774–805 [DOI] [PubMed] [Google Scholar]
- 2.Hamon M, Bierne H, Cossart P. 2006. Listeria monocytogenes: a multifaceted model. Nat. Rev. Microbiol. 4:423–434 [DOI] [PubMed] [Google Scholar]
- 3.Portnoy DA, Jacks PS, Hinrichs DJ. 1988. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 167:1459–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kathariou S, Metz P, Hof H, Goebel W. 1987. Tn916-induced mutations in the hemolysin determinant affecting virulence of Listeria monocytogenes. J. Bacteriol. 169:1291–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cossart P, Vicente MF, Mengaud J, Baquero F, Perez-Diaz JC, Berche P. 1989. Listeriolysin O is essential for virulence of Listeria monocytogenes: direct evidence obtained by gene complementation. Infect. Immun. 57:3629–3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones S, Portnoy DA. 1994. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect. Immun. 62:5608–5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marquis HDV, Portnoy DA. 1995. The broad-range phospholipase C and a metalloprotease mediate listeriolysin O-independent escape of Listeria monocytogenes from a primary vacuole in human epithelial cells. Infect. Immun. 63:4531–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith GA, Marquis H, Jones S, Johnston NC, Portnoy DA, Goldfine H. 1995. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect. Immun. 63:4231–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. 1992. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68:521–531 [DOI] [PubMed] [Google Scholar]
- 10.Tilney LG, Portnoy DA. 1989. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J. Cell Biol. 109:1597–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, Berche P, Bloecker H, Brandt P, Chakraborty T, Charbit A, Chetouani F, Couve E, de Daruvar A, Dehoux P, Domann E, Dominguez-Bernal G, Duchaud E, Durant L, Dussurget O, Entian KD, Fsihi H, Garcia-del Portillo F, Garrido P, Gautier L, Goebel W, Gomez-Lopez N, Hain T, Hauf J, Jackson D, Jones LM, Kaerst U, Kreft J, Kuhn M, Kunst F, Kurapkat G, Madueno E, Maitournam A, Vicente JM, Ng E, Nedjari H, Nordsiek G, Novella S, de Pablos B, Perez-Diaz JC, Purcell R, Remmel B, Rose M, Schlueter T, Simoes N, Tierrez A, Vazquez-Boland JA, Voss H, Wehland J, Cossart P. 2001. Comparative genomics of Listeria species. Science 294:849–852 [DOI] [PubMed] [Google Scholar]
- 12.Trost M, Wehmhoner D, Karst U, Dieterich G, Wehland J, Jansch L. 2005. Comparative proteome analysis of secretory proteins from pathogenic and nonpathogenic Listeria species. Proteomics 5:1544–1557 [DOI] [PubMed] [Google Scholar]
- 13.Renier S, Chambon C, Viala D, Chagnot C, Hebraud M, Desvaux M. 2013. Exoproteomic analysis of the SecA2-dependent secretion in Listeria monocytogenes EGD-e. J. Proteomics 80C:183–195 [DOI] [PubMed] [Google Scholar]
- 14.Desvaux M, Dumas E, Chafsey I, Chambon C, Hebraud M. 2010. Comprehensive appraisal of the extracellular proteins from a monoderm bacterium: theoretical and empirical exoproteomes of Listeria monocytogenes EGD-e by secretomics. J. Proteome Res. 9:5076–5092 [DOI] [PubMed] [Google Scholar]
- 15.Bierne H, Cossart P. 2007. Listeria monocytogenes surface proteins: from genome predictions to function. Microbiol. Mol. Biol. Rev. 71:377–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lycklama ANJA, Driessen AJ. 2012. The bacterial Sec-translocase: structure and mechanism. Philos. Trans. R. Soc. London Ser. B Biol. Sci. 367:1016–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veenendaal AK, van der Does C, Driessen AJ. 2004. The protein-conducting channel SecYEG. Biochim. Biophys. Acta 1694:81–95 [DOI] [PubMed] [Google Scholar]
- 18.Economou A, Pogliano JA, Beckwith J, Oliver DB, Wickner W. 1995. SecA membrane cycling at SecYEG is driven by distinct ATP binding and hydrolysis events and is regulated by SecD and SecF. Cell 83:1171–1181 [DOI] [PubMed] [Google Scholar]
- 19.Tsukazaki T, Mori H, Echizen Y, Ishitani R, Fukai S, Tanaka T, Perederina A, Vassylyev DG, Kohno T, Maturana AD, Ito K, Nureki O. 2011. Structure and function of a membrane component SecDF that enhances protein export. Nature 474:235–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dalbey RE, Wang P, Kuhn A. 2011. Assembly of bacterial inner membrane proteins. Annu. Rev. Biochem. 80:161–187 [DOI] [PubMed] [Google Scholar]
- 21.Schneewind O, Missiakas DM. 2012. Protein secretion and surface display in Gram-positive bacteria. Philos. Trans. R. Soc. London Ser. B Biol. Sci. 367:1123–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolhuis A, Broekhuizen CP, Sorokin A, van Roosmalen ML, Venema G, Bron S, Quax WJ, van Dijl JM. 1998. SecDF of Bacillus subtilis, a molecular Siamese twin required for the efficient secretion of proteins. J. Biol. Chem. 273:21217–21224 [DOI] [PubMed] [Google Scholar]
- 23.Hyyrylainen HL, Marciniak BC, Dahncke K, Pietiainen M, Courtin P, Vitikainen M, Seppala R, Otto A, Becher D, Chapot-Chartier MP, Kuipers OP, Kontinen VP. 2010. Penicillin-binding protein folding is dependent on the PrsA peptidyl-prolyl cis-trans isomerase in Bacillus subtilis. Mol. Microbiol. 77:108–127 [DOI] [PubMed] [Google Scholar]
- 24.Lenz LL, Mohammadi S, Geissler A, Portnoy DA. 2003. SecA2-dependent secretion of autolytic enzymes promotes Listeria monocytogenes pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 100:12432–12437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Archambaud C, Nahori MA, Pizarro-Cerda J, Cossart P, Dussurget O. 2006. Control of Listeria superoxide dismutase by phosphorylation. J. Biol. Chem. 281:31812–31822 [DOI] [PubMed] [Google Scholar]
- 26.Alonzo F, III, Freitag NE. 2010. Listeria monocytogenes PrsA2 is required for virulence factor secretion and bacterial viability within the host cell cytosol. Infect. Immun. 78:4944–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alonzo F, III, Whisstock XBJC, Freitag NE. 2011. Functional analysis of the Listeria monocytogenes secretion chaperone PrsA2 and its multiple contributions to bacterial virulence. Mol. Microbiol. 80:1530–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zemansky JKB, Woodward JJ, Leber JH, Marquis H, Portnoy DA. 2009. Development of a mariner-based transposon and identification of Listeria monocytogenes determinants, including the peptidyl-prolyl isomerase PrsA2, that contribute to its hemolytic phenotype. J. Bacteriol. 191:3950–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forster BM, Zemansky J, Portnoy DA, Marquis H. 2011. Posttranslocation chaperone PrsA2 regulates the maturation and secretion of Listeria monocytogenes proprotein virulence factors. J. Bacteriol. 193:5961–5970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pogliano KJ, Beckwith J. 1994. Genetic and molecular characterization of the Escherichia coli secD operon and its products. J. Bacteriol. 176:804–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pogliano JA, Beckwith J. 1994. SecD and SecF facilitate protein export in Escherichia coli. EMBO J. 13:554–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wickner W, Driessen AJ, Hartl FU. 1991. The enzymology of protein translocation across the Escherichia coli plasma membrane. Annu. Rev. Biochem. 60:101–124 [DOI] [PubMed] [Google Scholar]
- 33.Matsuyama S, Fujita Y, Mizushima S. 1993. SecD is involved in the release of translocated secretory proteins from the cytoplasmic membrane of Escherichia coli. EMBO J. 12:265–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsukazaki T, Mori H, Fukai S, Numata T, Perederina A, Adachi H, Matsumura H, Takano K, Murakami S, Inoue T, Mori Y, Sasaki T, Vassylyev DG, Nureki O, Ito K. 2006. Purification, crystallization and preliminary X-ray diffraction of SecDF, a translocon-associated membrane protein, from Thermus thermophilus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62:376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quiblier C, Zinkernagel AS, Schuepbach RA, Berger-Bachi B, Senn MM. 2011. Contribution of SecDF to Staphylococcus aureus resistance and expression of virulence factors. BMC Microbiol. 11:72. 10.1186/1471-2180-11-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crimmins GT, Herskovits AA, Rehder K, Sivick KE, Lauer P, Dubensky TW, Jr, Portnoy DA. 2008. Listeria monocytogenes multidrug resistance transporters activate a cytosolic surveillance pathway of innate immunity. Proc. Natl. Acad. Sci. U. S. A. 105:10191–10196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Riordan M, Yi CH, Gonzales R, Lee KD, Portnoy DA. 2002. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. U. S. A. 99:13861–13866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, Freudenberg M, Beutler B. 2005. CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 6:565–570 [DOI] [PubMed] [Google Scholar]
- 39.Way SS, Thompson LJ, Lopes JE, Hajjar AM, Kollmann TR, Freitag NE, Wilson CB. 2004. Characterization of flagellin expression and its role in Listeria monocytogenes infection and immunity. Cell Microbiol. 6:235–242 [DOI] [PubMed] [Google Scholar]
- 40.Dussurget O, Pizarro-Cerda J, Cossart P. 2004. Molecular determinants of Listeria monocytogenes virulence. Annu. Rev. Microbiol. 58:587–610 [DOI] [PubMed] [Google Scholar]
- 41.Machata S, Hain T, Rohde M, Chakraborty T. 2005. Simultaneous deficiency of both MurA and p60 proteins generates a rough phenotype in Listeria monocytogenes. J. Bacteriol. 187:8385–8394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chong A, Wehrly TD, Nair V, Fischer ER, Barker JR, Klose KE, Celli J. 2008. The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect. Immun. 76:5488–5499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Glomski IJ, Gedde MM, Tsang AW, Swanson JA, Portnoy DA. 2002. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J. Cell Biol. 156:1029–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ripio MT, Brehm K, Lara M, Suarez M, Vazquez-Boland JA. 1997. Glucose-1-phosphate utilization by Listeria monocytogenes is PrfA dependent and coordinately expressed with virulence factors. J. Bacteriol. 179:7174–7180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yeung PS, Zagorski N, Marquis H. 2005. The metalloprotease of Listeria monocytogenes controls cell wall translocation of the broad-range phospholipase C. J. Bacteriol. 187:2601–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geoffroy C, Raveneau J, Beretti JL, Lecroisey A, Vazquez-Boland JA, Alouf JE, Berche P. 1991. Purification and characterization of an extracellular 29-kilodalton phospholipase C from Listeria monocytogenes. Infect. Immun. 59:2382–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johansson J, Mandin P, Renzoni A, Chiaruttini C, Springer M, Cossart P. 2002. An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell 110:551–561 [DOI] [PubMed] [Google Scholar]
- 48.Lobel L, Sigal N, Borovok I, Ruppin E, Herskovits AA. 2012. Integrative genomic analysis identifies isoleucine and CodY as regulators of Listeria monocytogenes virulence. PLoS Genet. 8:e1002887. 10.1371/journal.pgen.1002887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mao C, Cheadle CE, Hardy SJ, Lilly AA, Suo Y, Sanganna Gari RR, King GM, Randall LL. 2013. Stoichiometry of SecYEG in the active translocase of Escherichia coli varies with precursor species. Proc. Natl. Acad. Sci. U. S. A. 110:11815–11820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mori H, Tsukazaki T, Masui R, Kuramitsu S, Yokoyama S, Johnson AE, Kimura Y, Akiyama Y, Ito K. 2003. Fluorescence resonance energy transfer analysis of protein translocase. SecYE from Thermus thermophilus HB8 forms a constitutive oligomer in membranes. J. Biol. Chem. 278:14257–14264 [DOI] [PubMed] [Google Scholar]
- 51.Scheuring J, Braun N, Nothdurft L, Stumpf M, Veenendaal AK, Kol S, van der Does C, Driessen AJ, Weinkauf S. 2005. The oligomeric distribution of SecYEG is altered by SecA and translocation ligands. J. Mol. Biol. 354:258–271 [DOI] [PubMed] [Google Scholar]
- 52.Breyton C, Haase W, Rapoport TA, Kuhlbrandt W, Collinson I. 2002. Three-dimensional structure of the bacterial protein-translocation complex SecYEG. Nature 418:662–665 [DOI] [PubMed] [Google Scholar]
- 53.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vitro genetic engineering: transposon mutagenesis in Gram negative bacteria. Biotechnology 1:784–791 [Google Scholar]
- 54.Herskovits AA, Auerbuch V, Portnoy DA. 2007. Bacterial ligands generated in a phagosome are targets of the cytosolic innate immune system. PLoS Pathog. 3:e51. 10.1371/journal.ppat.0030051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lauer P, Hanson B, Lemmens EE, Liu W, Luckett WS, Leong ML, Allen HE, Skoble J, Bahjat KS, Freitag NE, Brockstedt DG, Dubensky TW., Jr 2008. Constitutive activation of the PrfA regulon enhances the potency of vaccines based on live-attenuated and killed but metabolically active Listeria monocytogenes strains. Infect. Immun. 76:3742–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.