

Abstract
Protein exposure to oxidants such as HOCl leads to formation of methionine sulfoxide (MetSO) residues, which can be repaired by methionine sulfoxide reductase (Msr). A Helicobacter pylori msr strain was more sensitive to HOCl-mediated killing than the parent. Because of its abundance in H. pylori and its high methionine content, alkyl hydroperoxide reductase C (AhpC) was hypothesized to be prone to methionine oxidation. AhpC was expressed as a recombinant protein in Escherichia coli. AhpC activity was abolished by HOCl, while all six methionine residues of the enzyme were fully to partially oxidized. Upon incubation with a Msr repair mixture, AhpC activity was restored to nonoxidized levels and the MetSO residues were repaired to methionine, albeit to different degrees. The two most highly oxidized and then Msr-repaired methionine residues in AhpC, Met101 and Met133, were replaced with isoleucine residues by site-directed mutagenesis, either individually or together. E. coli cells expressing variant versions were more sensitive to t-butyl hydroperoxide than cells expressing native protein, and purified AhpC variant proteins had 5% to 39% of the native enzyme activity. Variant proteins were still able to oligomerize like the native version, and circular dichroism (CD) spectra of variant proteins revealed no significant change in AhpC conformation, indicating that the loss of activity in these variants was not related to major structural alterations. Our results suggest that both Met101 and Met133 residues are important for AhpC catalytic activity and that their integrity relies on the presence of a functional Msr.
INTRODUCTION
Helicobacter pylori is a spiral, Gram-negative bacterium that colonizes the gastric epithelium (1) in about 50% of the world's population (2). H. pylori has been shown to be the etiological agent of peptic ulcer disease or chronic atrophic gastritis (3), which can subsequently develop into gastric cancer (4). Colonization of the gastric mucosa by H. pylori induces a strong inflammatory response in the host, who produces harmful reactive nitrogen species (RNS) or oxygen species (ROS) (5); the latter include superoxide anion (O2−), hydrogen peroxide (H2O2), hydroxyl radical (·HO), and hypochlorous acid (HOCl). Increased production of ROS by gastric cells (6) and phagocytes (7) induced by H. pylori has been shown in vitro, and increased levels of ROS in the gastric mucosa have been found in H. pylori-infected patients (8). ROS have the ability to react with and damage bacterial proteins, as well as lipids and DNA. Protein covalent modifications include amino acid modifications or carbonyl group formation, usually leading to partial or total loss of catalytic activity. Among the amino acid modifications, oxidation of the sulfur-containing methionine (Met) into R- and S-diastereoisomers of Met sulfoxide (Met-SO) has been well documented (9–11).
To counter oxidative stress, H. pylori cells have evolved different defense strategies (reviewed in references 12 and 13), including (i) avoidance, (ii) use of antioxidant enzymes such as superoxide dismutase (SOD), catalase (kat), and alkyl hydroperoxide C (AhpC), and (iii) use of repair enzymes such as MutS or methionine sulfoxide reductase (Msr). The latter catalyzes the conversion of Met-SO back to Met (14). While most organisms have two distinct Msr proteins, MsrA and MsrB, shown to reduce the two isomers Met(S)O and Met(R)O of methionine sulfoxide, respectively (15), H. pylori possesses only one Msr protein that consists of MsrA-like and MsrB-like domains (16). H. pylori msr mutants show increased sensitivity to oxidants such as paraquat and H2O2 as well as to nitrosative (GSNO) stress and exhibit attenuated colonization of mice compared to wild-type cells (17). Upon exposure of Msr-deficient mutants to oxidative stress, several methionine-containing proteins were isolated, including GroEL, thioredoxin 1 (Trx1), and catalase (14). Cross-linking interaction studies using purified recombinant proteins revealed that Msr can bind to each of those proteins (14). In addition, oxidation of purified catalase by HOCl generated Met-SO residues within the H2O2-scavenging enzyme and these Met-SO residues were shown to be subsequently repaired upon incubation with an enzyme repair mix that required Msr (10). Recently, the UreG urease accessory protein was also identified in our laboratory as a primary target for Met oxidation (18). Evidence was provided for an Msr-UreG transient interaction as well as for Msr-mediated repair of Met-SO residues within UreG (18). While these findings shed new light on the role of Msr in repairing oxidative stress-damaged proteins, as well as on its interaction with methionine-rich proteins in H. pylori, we sought to find new Msr-repaired targets.
AhpC is a key enzyme to combat oxidative stress: it detoxifies harmful lipid hydroperoxides. In the current study, we hypothesized that this enzyme would be a target for methionine oxidation because (i) other proteins known to combat oxidative stress, such as catalase and Trx1, have been shown to be primary targets for Met-So formation (14), (ii) AhpC is one of the most abundant proteins in H. pylori, and (iii) its Met content (3% of the total amino-acid content) is higher than average. To get a better assessment of the oxidative stress-induced protein damage and to determine which Met residues would be preferentially oxidized and repaired, we exposed recombinant AhpC to HOCl and measured the level of oxidation of each individual methionine residue before and after repair by Msr. Finally, the role of the two most oxidized methionine residues of AhpC was investigated by replacing these two residues with isoleucine residues and studying the effect of both substitutions on the in vitro and in vivo AhpC activity.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Escherichia coli TOP10 (Invitrogen) was used for all cloning experiments, and E. coli BL21(DE3)RIL (Novagen) was used to overexpress recombinant H. pylori AhpC (HpAhpC) proteins. H. pylori wild-type strain 26695 (16) was used as a source of DNA for PCR. H. pylori SS1 (19) and H. pylori SS1 Δmsr (17) were used for all other experiments. Plasmid pET21b (Invitrogen) was used to clone and express native and variant AhpC proteins. All plasmids and PCR products used were sequenced at the Georgia Genomics Facility of the University of Georgia, Athens, GA, and compared to DNA sequences from strain 26695 (16) to ensure that no error had been introduced following PCR amplification.
Growth conditions.
E. coli cells were grown aerobically in Luria-Bertani (LB) medium or plates at 37°C. Ampicillin (100 μg/ml), chloramphenicol (30 μg/ml), and isopropyl-β-d-thiogalactopyranoside (IPTG) (0.1 to 0.2 mM) were added as needed. H. pylori was routinely grown on Brucella agar plates supplemented with 10% defibrinated sheep blood (BA) at 37°C under microaerophilic conditions with 1% to 5% O2, 5% CO2, and 90% to 94% N2, as indicated.
Sensitivity of nongrowing cells to HOCl.
The H. pylori wild type (SS1) and the SS1Δmsr mutant were grown for 36 h on BA plates under 1% O2 conditions, harvested, and resuspended in phosphate-buffered saline (PBS) to an optical density at 600 nm (OD600) of 1. Cells (0.9 ml) were mixed with 0.1 ml freshly prepared HOCl (available as NaClO [Sigma]; final concentrations ranging from 31.25 to 500 μM) and incubated for 30 min at room temperature, with gentle shaking (40 rpm). HOCl-exposed cells were then serially diluted, and 10 μl of each dilution (n = 4) was plated on BA plates. CFU were counted after 3 to 5 days. Data are shown as the averages and standard deviations of the results of quadruplicate assays from one experiment. Another experiment (also done in quadruplicate) confirmed that the mutants were significantly more sensitive to HOCl than the parental cells.
Cloning, expression, and purification of AhpC.
Recombinant native and variant AhpC proteins were expressed as hexahistidine-tagged proteins in E. coli BL21(DE3)RIL. Briefly, primers AhpC1 (5′-CCGGATCCATATGTTAGTTACAAAACTTGCC-3′) and AhpC2 (5′-CACGGTCTCGAGAAGCTTAATGGAATTTTCTTTG-3′) were used to amplify a 600-bp-long DNA sequence containing the whole hp1563 (ahpC) open reading frame (ORF) (without its stop codon), as well as to incorporate a 5′ NdeI restriction site and a 3′ XhoI restriction site, respectively. The PCR product was digested with NdeI and XhoI, gel purified, and cloned into similarly digested pET21b plasmid, generating pET-AhpC. Plasmid pET-AhpC.M101I was generated by overlapping PCR using genomic DNA and primers AhpC1and AhpC2, along with primers AhpC3 (5′-CCTATCGTGGCTGATATTACCA-3′) and AhpC4 (5′-CCACGATAGGGAAAGTTACTTG-3′) (new codons are shown in bold). Plasmid pET-AhpC.M133I was generated by overlapping PCR using genomic DNA and primers AhpC1 and AhpC2, along with primers AhpC5 (5′-AACATCAAAGTAAGGCATGCGG-3′) and AhpC6 (5′-CTTTGATGTTTTTGTCAATCAAAAAAC-3′). Plasmid pET-AhpC.M101I.M133I (double substitution) was generated using pET-AhpC.M101I as the template and primers AhpC1, AhpC2, AhpC5, and AhpC6. BL21(DE3)RIL cells transformed with the appropriate pET-AhpC plasmid were grown at 37°C in 500 ml of LB supplemented with ampicillin and chloramphenicol until an OD600 of 0.5 was reached. Gene expression was induced by the addition of 0.2 mM IPTG followed by incubation at 22°C for 3 to 5 h. Cells were harvested by centrifugation (10,000 × g, 15 min, 4°C), and His-tagged proteins were purified following the manufacturer's instructions (Qiagen, Valencia, CA) and finally dialyzed three times against 1 liter of 50 mM NaH2PO4 buffer (pH 7.5). Purified AhpC wild-type or mutant proteins were purified to a level close to homogeneity (as estimated by SDS-PAGE).
In vitro HOCl oxidation of purified AhpC.
The in vitro oxidation of AhpC was carried out as previously described by Mahawar et al. (10). Briefly, 10 μM purified AhpC was incubated with a 60-fold-molar-excess ratio of HOCl in oxidation buffer (50 mM sodium phosphate [pH 7.5], 50 mM KCl, 10 mM MgCl2) for 15 min at room temperature in the dark. Residual oxidants were quenched by excess free methionine (5 mM) for 10 min on ice and finally dialyzed twice against 1 liter of 50 mM sodium phosphate buffer (pH 7.5) to remove HOCl and free methionine.
Repair of AhpC Met-SO residues by Msr.
Equimolar amounts of HOCl-oxidized AhpC and Msr were incubated for 2 h at 37°C in the presence of 100 μM dithiothreitol (DTT) and a thioredoxin system containing 5 μM purified H. pylori thioredoxin 1 (Trx1), 0.1 μM purified H. pylori thioredoxin reductase (TrxR), and 400 μM NADPH in 50 mM sodium phosphate buffer (pH 7.4). These components plus Msr are referred to here as the repair mixture. Purification of Msr, Trx1, and TrxR was previously described (14).
Peroxide reductase activity of recombinant AhpC.
AhpC activity was measured in a coupled assay by monitoring thioredoxin reductase-dependent oxidation of NADPH at 340 nm, as previously described (20, 21). Briefly, each assay was carried out with 1 μM AhpC (previously unoxidized or HOCl oxidized or HOCl oxidized and Msr repaired) in a final volume of 100 μl containing 50 mM potassium phosphate buffer (pH 7.0), 100 mM ammonium sulfate, and 0.5 mM EDTA in the presence of 0.5 μM Trx1, 0.5 μM TrxR, and 150 μM NADPH; 1 mM H2O2 was added to start the reaction. To account for non-AhpC-dependent NADPH oxidation, our control included all the above except AhpC. To rule out any Msr-dependent NADPH oxidation, a Msr-only control included Trx1, TrxR, and Msr. The decrease of NADPH absorbance (at room temperature) was followed for 2 min using an ELX800 BioTek microplate reader (Winooski, VT). One unit of specific activity is defined as 1 μmol of NADPH oxidized per min per mg of AhpC protein. Data shown are means and standard deviations of the results from 4 to 15 replicate assays.
Detection and quantification of oxidized methionine residues by LC-MS/MS.
For detection and quantification of oxidized Met residues within AhpC, the protein was digested with sequencing-grade GluC, following the recommendations of the manufacturer (Promega, Madison, WI). GluC digests of (i) unoxidized AhpC, (ii) unoxidized AhpC with Msr, (iii) oxidized AhpC, or (iv) oxidized and Msr-repaired AhpC were separated by reversed-phase liquid chromatography (LC) using trapping cartridges coupled to a 50-mm-long, 200-μm-inside-diameter (ID) Halo C18 column (Advanced Materials Technology, Wilmington, DE), and the eluent was analyzed by electrospray ionization-tandem mass spectrometry (ESI-MS/MS) using a linear trap quadrupole-Fourier transform (LTQ-FT) mass spectrometer (Thermo Scientific, West Palm Beach, FL) as described before (10, 18).
Disk inhibition assays.
Disk inhibition assays were done following the method of Lundström and Bölin (22). Briefly, E. coli BL21(DE3)RIL strains with either pET21b vector (control) or one of the pET-AhpC plasmids were grown to an OD600 of 2, and 0.2 ml of cells was mixed with 3.5 ml of top agarose (0.8%) before being spread on top of LB plates supplemented with ampicillin and chloramphenicol and with 0.1 mM IPTG when indicated. Sterile paper disks (7.5-mm diameter) were placed in the center of each plate, and 10 μl of 5% t-butyl hydroperoxide was added to the disk. Cells were allowed to grow for 24 h, and the diameter of the inhibition zone was recorded. Data are shown as means and standard deviations of the results from measurements done on 3 to 5 plates.
CD analysis.
Circular dichroism (CD) spectra of AhpC were obtained on an Aviv 400 spectropolarimeter (Lakewood, NJ) at the Robert Wood Johnson Medical School CD Facility, Piscataway, NJ. Purified recombinant wild-type, Met101Ile, or Met133Ile AhpC proteins dissolved in 10 mM NaH2PO4 buffer (pH 7.5) to a final concentration of ∼0.15 mg/ml were used for the Far-UV CD analysis at from 185 to 260 nm at 20°C in a 0.1-cm-gap cuvette. The molar ellipticity values (θ) were calculated using a molar concentration of 6.4 μM and 206 amino acids.
RESULTS
H. pylori Δmsr mutants are more sensitive to HOCl.
Previous studies in our laboratory showed that nongrowing H. pylori Δmsr mutants were more sensitive to a variety of oxidants (such as O2, H2O2, and GSNO) than the wild type (17). In addition, those mutants were also more sensitive to neutrophil-mediated killing (10). Neutrophils produce HOCl via their robust myeloperoxidase, so the increased sensitivity of the Δmsr mutants was possibly due to an inability to repair HOCl-induced Met-SO residues. In the current study, we aimed at complementing our previous findings by comparing the sensitivities of nongrowing H. pylori wild-type and Δmsr mutant cells to HOCl. Wild-type and Δmsr mutant cells were grown on plates, resuspended in PBS, and mixed with increasing HOCl concentrations before being serially diluted and plated. CFU counts indicated that Δmsr mutants were significantly more sensitive to HOCl than the parental strain (Fig. 1).
Fig 1.
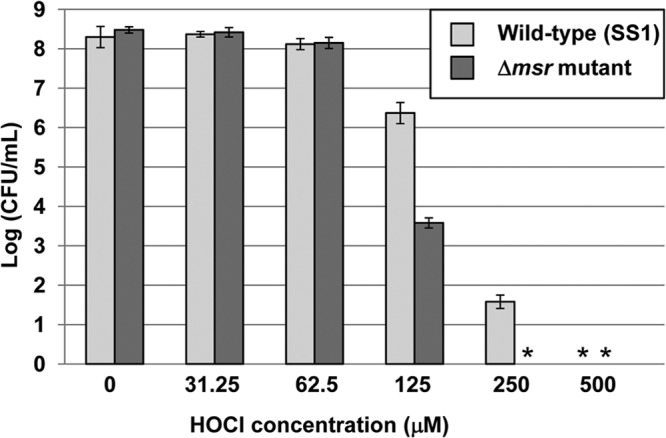
Sensitivity of nongrowing H. pylori wild-type and Δmsr mutant cells to HOCl. H. pylori SS1 (wild-type) and SS1 Δmsr mutant cells were grown on BA plates, harvested, and resuspended in PBS containing the indicated HOCl concentration for 30 min at room temperature. Cells were serially diluted and plated on BA plates, and CFU were counted after 3 to 5 days. Data shown represent the averages and standard deviations of the results of quadruplicate assays from one experiment, and the mutant is significantly more sensitive than the parent at 125 and 250 μM HOCl (P < 0.05, as determined by Student's t test). *, no CFU could be detected (detection limit, 100 CFU/ml).
AhpC inactivation by HOCl can be repaired by Msr.
Among the methionine-rich proteins known to be affected by HOCl treatment in the Δmsr mutant strain are two proteins involved in oxidative stress combat, catalase and Trx1 (14). This finding led us to consider a third oxidative-stress-combating protein, AhpC, as a likely target for methionine oxidation. Indeed, AhpC has a high methionine content (more than 3% of its residues). It is also an abundant protein, making up approximately 3% of the total protein content, according to our own densitometry analysis of resolved peptide bands after gel electrophoresis resolution (data not shown). Therefore, we sought to study further a possible link between oxidative stress and Met oxidation in AhpC. To investigate the functionality of AhpC before and after oxidative repair, we analyzed the peroxide reductase activities of various purified AhpC preparations by following TrxR-dependent NADPH oxidation (Table 1). AhpC was expressed as a hexahistidine-tagged recombinant protein in E. coli and purified to a level close to homogeneity (data not shown). Purified AhpC was divided in three pools: (i) as purified, (ii) oxidized with HOCl, and (iii) HOCl oxidized and subsequently Msr repaired. AhpC-dependent reduction of H2O2 was observed with purified (unoxidized) AhpC; however, treatment of pure AhpC enzyme with excess HOCl abolished the reductase's activity (Table 1). Upon incubation with an enzyme repair mixture requiring Msr, oxidized AhpC fully recovered peroxide reductase activity (at a level similar to that seen with the nonoxidized sample). As a control, the presence of Msr did not affect the peroxidase enzyme activity of the nonoxidized AhpC (Table 1). Also, a Msr repair mixture control had negligible peroxidase activity (approximately 2 nmol NADPH/min/mg of Msr). Taken together, our results strongly suggest that HOCl-oxidized AhpC can be fully repaired by the Msr methionine sulfoxide repair enzyme.
Table 1.
AhpC activity of native and variant AhpC proteinsa
| AhpC protein | AhpC activity (nmoles NADPH oxidized/min/mg protein) |
|---|---|
| Native† | 216 ± 22 |
| Native, oxidized† | 5 ± 5 |
| Native, oxidized, Msr added† | 242 ± 5 |
| Native, Msr added† | 244 ± 22 |
| Native‡ | 144 ± 22 |
| Variant M101I‡ | 23 ± 7 |
| Variant M133I‡ | 57 ± 15 |
| Variants M101I and M133I‡ | 8 ± 7 |
AhpC activity was measured in a coupled assay by monitoring thioredoxin reductase-dependent oxidation of NADPH at 340 nm. “native” refers to the protein as purified from E. coli. † and ‡ indicate 2 independent sets of experiments done with different batches of native AhpC, Trx1, and TrxR. Data represent means and standard deviations from 4 replicates (†) or 10 to 15 replicates (‡).
Identification of oxidized and Msr-repaired Met residues in H. pylori AhpC.
The primary sequence of the AhpC protein (HP1563 in H. pylori strain 26695) contains 198 amino acids, including 6 Met residues (Met1, Met101, Met133, Met152, Met155, and Met180). Each of the three AhpC pools defined above (unoxidized, oxidized, and oxidized and then repaired) and our control (Msr-incubated unoxidized AhpC) were digested with GluC (protease), with peptide identification confirmed by MS/MS (Fig. 2). Oxidation of residues Met152 and Met155, which were located on the same peptide, was determined by LC-MS/MS of the oxidized peptide to determine the fraction of oxidation of that peptide measured by LC-MS to be assigned to each methionine on the peptide. All the other identified Met residues were distributed on different peptides. We identified residue Met101 as the Met residue with the highest oxidation rate as well as the highest repair rate by Msr, followed by Met133 (Fig. 2). Only partial Msr-mediated Met repair was observed for Met155 or Met180.
Fig 2.
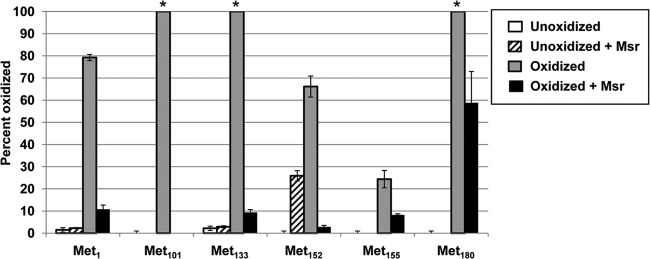
Identification and quantification of HOCl-oxidized and Msr-repaired methionine residues in H. pylori AhpC by LC-MS/MS. AhpC was oxidized with a 60-fold molar excess of HOCl and subsequently repaired (or not) by Msr. Repaired and unrepaired AhpC samples were subjected to in-solution GluC digestion, and Met residues were analyzed and quantified by LC-MS/MS. The values corresponding to the percentages of oxidation (y axis) correspond to the ratios of MetO residues to the total number of times the peptide was found (Met coverage). Error bars represent 1 standard deviation (from three measurements). *, the unoxidized peptide was below our limit of detection.
Met101 and Met133 residues are critical for AhpC activity both in vivo and in vitro.
Since Met101 and Met133 residues were found to be readily oxidized and repaired by Msr (Fig. 2), we hypothesized that both residues might be important for the stability and/or activity of AhpC. Therefore, both Met residues were independently or concomitantly replaced with an isoleucine (I) residue by site-directed mutagenesis. Each construct was cloned into pET21b and introduced into E. coli BL21(DE3)RIL host cells to allow T7-based expression in the presence of the IPTG inducer. First, cells expressing each AhpC variant were characterized for their sensitivity to t-butyl hydroperoxide (t-BH) (Table 2). Although E. coli host cells also have their own AhpC, we hypothesized that expression of active recombinant H. pylori AhpC should confer to them increased resistance to the chemical. While there was no difference between the inhibition zones in the absence of IPTG, upon addition of 0.1 mM IPTG in the medium, E. coli cells expressing native AhpC were indeed more resistant to t-BH (smaller inhibition zone) than E. coli cells with pET21b vector only (Table 2). Interestingly, E. coli cells expressing M101I, M133I, or M101I-M133I AhpC variant proteins were more sensitive to t-BH than cells expressing native AhpC. This phenotype was not due to differences in protein expression, as wild-type and mutant AhpC proteins were expressed to similar levels in the presence of 0.1 mM IPTG, as seen by SDS-PAGE (data not shown). Rather, these results indicate that both Met101 and Met133 residues are important for alkyl hydroperoxide detoxification in vivo.
Table 2.
Diameter of zone of inhibition of E. coli cells expressing H. pylori native or variant AhpC proteinsa
| Plasmid | Diam of zone of inhibition (mm) |
|
|---|---|---|
| No IPTG | 0.1 mM IPTG | |
| pET21b (vector control) | 50 ± 4 | 52 ± 4 |
| pET-AhpC | 48 ± 1 | 41 ± 2 |
| pET-AhpC.M101I | 49 ± 1 | 53 ± 3 |
| pET-AhpC.M133I | 51 ± 1 | 58 ± 5 |
| pET-AhpC.M101I. M133I | 49 ± 1 | 55 ± 2 |
E. coli BL21 RIL cells transformed with the appropriate plasmid were mixed with top agarose and spread on top of LB plates (ampicillin [Amp], chloramphenicol [Cm]) supplemented with 0.1 mM IPTG when indicated. Sterile paper disks (7.5-mm diameter) with t-BH were placed in the center of each plate, and the diameter of the inhibition zone was recorded after 24 h of growth. Results shown represent means and standard deviations of the results of measurements done on 3 to 5 plates, and the inhibition zone for pET-AhpC with IPTG was statistically significantly smaller than for all the other strains (P < 0.05, Student's t test).
The importance of both Met residues was confirmed in vitro, by measuring the AhpC activity of each purified AhpC variant protein. M101I, M133I, and M101I-M133I AhpC proteins were independently purified to a level close to homogeneity on a nickel-nitrilotriacetic acid (Ni-NTA) column (data not shown) and assayed by monitoring the TrxR-dependent oxidation of NADPH at 340 nm, following the exact same protocol as for native AhpC (Table 1). The levels of peroxide reductase activity of M101I, M133I, and M101I-M133I were 16%, 40%, and 6% of wild-type activity, respectively, confirming that the presence of both residues—individually and together—was critical (Table 1). To determine whether the AhpC protein structure was affected by the methionine substitutions, nonreducing SDS-PAGE and CD analyses were conducted. Both the M101I and M133I single variants as well as the double variant were able to form dimers as well as higher-molecular-mass complexes (like native AhpC), as seen by nonreducing SDS-PAGE (Fig. 3). CD analysis (Fig. 4) indicated a slight increase in helicity and decrease in total β-structure for M101I and a slight decrease in helicity and increase in total β-structure for M133I. Overall, these changes are minor and are highly unlikely to indicate a gross destabilization of the structure of these variants. Rather, it appears that these Met residues are important for maintaining catalytic activity.
Fig 3.
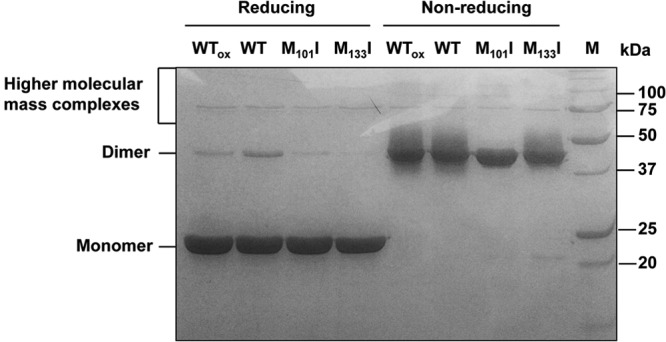
SDS-PAGE of native and variant AhpC proteins. Oxidized native (wild-type) AhpC (WTox), nonoxidized native AhpC (WT), or variant AhpC (M101I, M133I) (2.5 μg per lane) proteins were mixed with reducing buffer and heated at 95°C for 5 min or mixed with nonreducing loading buffer (and not heated) before being separated on an SDS-12.5% polyacrylamide gel. A molecular mass standard (M) is shown on the right, with sizes indicated on the right. Calculated (theoretical) molecular mass of AhpC-His6, 23.3 kDa.
Fig 4.

CD spectra of purified native, M101I variant, or M133I variant AhpC proteins. CD spectra of each purified protein were measured using an Aviv 400 spectropolarimeter in a 0.1-cm-gap cuvette from 185 to 260 nm at 20°C. Mean residue molar ellipticities (θ) were calculated using a molar concentration of 6.4 μM and 206 amino acids.
DISCUSSION
As part of the host defense system against invading microorganisms, neutrophils produce HOCl via the enzyme myeloperoxidase (23) and Met residues are considered primary amino acid targets of HOCl damage via conversion to Met sulfoxide (Met-SO) (24). These Met-So residues can be repaired by Msr. In the current report, we show that nongrowing H. pylori Δmsr mutants are more sensitive to HOCl than the parental strain. This phenotype reflects the inability of Δmsr mutants to repair Met-SO residues in key enzymes, especially enzymes needed to fight against HOCl-generated oxidative stress. Indeed, previous results from our laboratory revealed that catalase and Trx1 were among the proteins most oxidized in Δmsr mutants subjected to oxidative stress (14). Furthermore, H. pylori catalase is inactivated by HOCl treatment but can be repaired upon incubation with purified Msr and MS/MS tandem analysis revealed those Met residues specifically oxidized by HOCl and repaired by Msr (10). Repair of catalase by Msr suggests that a physical interaction occurs between the two proteins: Msr and catalase can indeed interact, as shown by cross-linking (14).
In light of these findings, we sought to investigate whether AhpC (HP1563 in H. pylori strain 26695 [16]), another key enzyme in the fight against oxidative stress, would be oxidized and then subsequently repaired by Msr. Indeed, its high methionine content as well as its overall abundance in H. pylori makes AhpC a prime candidate for methionine oxidation. AhpC is a member of the 2-Cys peroxiredoxin (Prx) family (25) whose catalytic activity involves two conserved cysteine residues. It is commonly accepted that reduced AhpC detoxifies H2O2 or organic peroxides (such as lipid hydroperoxides) by converting them into their corresponding alcohols (26). However, new findings suggest that the role of AhpC might not be solely limited to peroxide detoxification: for instance, AhpC was recently shown to be also required for ferric ion transport and the optimal production of enterobactin in E. coli (27). Interestingly, H. pylori AhpC has been shown to have dual functions: depending on the duration of the oxidative stress, it can switch from a low-molecular-weight (LMW) complex with peroxide reductase activity (short-term oxidative stress) to a high-molecular-weight (HMW) complex with chaperone-like activity (short-term oxidative stress) (28). In addition, HpAhpC differs significantly from other prokaryotes' AhpCs: according to Chuang and coworkers, the sequence of HpAhpC is more homologous to those of mammalian Prxs than to those of other eubacterial AhpC (21). HpAhpC has also been shown to have higher catalytic activity toward H2O2 than other AhpC homologs do (20). Nevertheless, HpAhpC contains two highly conserved cysteine residues (Cys49 and Cys169) which are the hallmarks of the Cys-2 Prx protein family. AhpC is one of the most abundant proteins synthesized by the gastric pathogen. The proteome analysis by Jungblut et al. (29) showed that AhpC (also called TsaA) is the third-most-abundant protein in H. pylori (29); according to our own densitometry analysis using crude extracts as well as known amounts of purified AhpC protein, we estimated that the protein makes up approximately 3% of the total protein content. Its abundance is reflected by the fact that H. pylori mutants with one-third-lower ahpC transcript levels (as controlled by antisense RNA technology) can still colonize mouse stomachs (30), even though H. pylori ahpC mutants fail to colonize, as shown by a different study (31).
Treatment of purified AhpC by HOCl led to full inactivation of the enzyme, which could be restored upon incubation with purified Msr, as previously shown for catalase (10) and UreG (17). When the two most oxidized and successfully Msr-repaired Met residues, Met101 and Met133, were independently or concomitantly replaced with isoleucine, the Ahpc enzyme activity of the variant proteins was significantly decreased, and those recombinant AhpC variant enzymes confer lower t-BH resistance to E. coli cells. While these mutations are not in the vicinity of the essential Cys49 and Cys169 cysteine residues, there was a possibility that the overall conformation of AhpC would be disturbed. However, when run on nonreducing SDS-polyacrylamide gels, both single variants as well as the M101I-M133I double variant formed dimers and higher-molecular-mass complexes, similar to native AhpC (Fig. 3), suggesting that the oligomerization capacity is not affected in these variants. In addition, CD analysis confirmed there was only a slight modification of the secondary structure in these variant proteins (Fig. 4). Therefore, one has to conclude that the loss of activity in these variants is not related to major structural alterations. The involvement of these methionine residues in AhpC activity is yet to be understood.
So far, only a few proteins have been shown to be repaired by Msr in bacteria. In H. pylori, the list now includes GroEL, SSR (site-specific recombinase), catalase, UreG, and AhpC (10, 14, 18). In E. coli, Msr was found to protect GroEL and Ffh from oxidative damage (32, 33). As the list expands, so does our knowledge on Msr target specificity; this will eventually lead to a better understanding of Msr-mediated repair mechanisms. Finally, methionine oxidation of a protein is usually considered detrimental to the protein's activity; however, a recent study from Drazic and coworkers actually suggested otherwise: those authors showed that methionine oxidation activates a hypochloride-responsive transcription factor (HypT) in E. coli (34). In this case, Msr repair of the oxidized HypT protein leads to an inactive protein. This finding suggests that Msr can almost be considered a master regulator at the posttranslational level, able to reactivate but also deactivate key enzyme activities in the cell.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (grant number RO1 AI077569).
J.S.S. acknowledges support from the National Institute of General Medical Sciences of the National Institutes of Health through the Research Resource for Integrated Glycotechnology (P41GM103390).
Footnotes
Published ahead of print 4 October 2013
REFERENCES
- 1.Marshall BJ, Warren JR. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet i:1311–1315 [DOI] [PubMed] [Google Scholar]
- 2.Covacci A, Telford JL, Del Giudice G, Parsonnet J, Rappuoli R. 1999. Helicobacter pylori virulence and genetic geography. Science 284:1328–1333 [DOI] [PubMed] [Google Scholar]
- 3.Blaser MJ. 1995. The role of Helicobacter pylori in gastritis and its progression to peptic ulcer disease. Aliment. Pharmacol. Ther. 9(Suppl 1):27–30 [DOI] [PubMed] [Google Scholar]
- 4.Sipponen P, Hyvarinen H, Seppala K, Blaser MJ. 1998. Review article: pathogenesis of the transformation from gastritis to malignancy. Aliment. Pharmacol. Ther. 12(Suppl 1):61–71 [DOI] [PubMed] [Google Scholar]
- 5.Baik SC, Youn HS, Chung MH, Lee WK, Cho MJ, Ko GH, Park CK, Kasai H, Rhee KH. 1996. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res. 56:1279–1282 [PubMed] [Google Scholar]
- 6.Bagchi D, Bhattacharya G, Stohs SJ. 1996. Production of reactive oxygen species by gastric cells in association with Helicobacter pylori. Free Radic. Res. 24:439–450 [DOI] [PubMed] [Google Scholar]
- 7.Ramarao N, Gray-Owen SD, Meyer TF. 2000. Helicobacter pylori induces but survives the extracellular release of oxygen radicals from professional phagocytes using its catalase activity. Mol. Microbiol. 38:103–113 [DOI] [PubMed] [Google Scholar]
- 8.Davies GR, Simmonds NJ, Stevens TR, Sheaff MT, Banatvala N, Laurenson IF, Blake DR, Rampton DS. 1994. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut 35:179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee WL, Gold B, Darby C, Brot N, Jiang X, de Carvalho LP, Wellner D, St John G, Jacobs WR, Jr, Nathan C. 2009. Mycobacterium tuberculosis expresses methionine sulphoxide reductases A and B that protect from killing by nitrite and hypochlorite. Mol. Microbiol. 71:583–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahawar M, Tran V, Sharp JS, Maier RJ. 2011. Synergistic roles of Helicobacter pylori methionine sulfoxide reductase and GroEL in repairing oxidant-damaged catalase. J. Biol. Chem. 286:19159–19169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winter J, Ilbert M, Graf PC, Ozcelik D, Jakob U. 2008. Bleach activates a redox-regulated chaperone by oxidative protein unfolding. Cell 135:691–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stent A, Every AL, Sutton P. 2012. Helicobacter pylori defense against oxidative attack. Am. J. Physiol. Gastrointest. Liver Physiol. 302:G579–G587 [DOI] [PubMed] [Google Scholar]
- 13.Wang G, Alamuri P, Maier RJ. 2006. The diverse antioxidant systems of Helicobacter pylori. Mol. Microbiol. 61:847–860 [DOI] [PubMed] [Google Scholar]
- 14.Alamuri P, Maier RJ. 2006. Methionine sulfoxide reductase in Helicobacter pylori: interaction with methionine-rich proteins and stress-induced expression. J. Bacteriol. 188:5839–5850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kauffmann B, Aubry A, Favier F. 2005. The three-dimensional structures of peptide methionine sulfoxide reductases: current knowledge and open questions. Biochim. Biophys. Acta 1703:249–260 [DOI] [PubMed] [Google Scholar]
- 16.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547 [DOI] [PubMed] [Google Scholar]
- 17.Alamuri P, Maier RJ. 2004. Methionine sulphoxide reductase is an important antioxidant enzyme in the gastric pathogen Helicobacter pylori. Mol. Microbiol. 53:1397–1406 [DOI] [PubMed] [Google Scholar]
- 18.Kuhns LG, Mahawar M, Sharp JS, Benoit S, Maier RJ. 2013. Role of Helicobacter pylori methionine sulfoxide reductase in urease maturation. Biochem. J. 450:141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee A, O'Rourke J, De Ungria MC, Robertson B, Daskalopoulos G, Dixon MF. 1997. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112:1386–1397 [DOI] [PubMed] [Google Scholar]
- 20.Baker LMS, Raudonikiene A, Hoffman PS, Poole LB. 2001. Essential thioredoxin-dependent peroxiredoxin system from Helicobacter pylori: genetic and kinetic characterization. J. Bacteriol. 183:1961–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chuang M-H, Wu M-S, Lo W-L, Lin J-T, Wong C-H, Chiou S-H. 2006. The antioxidant protein alkylhydroperoxide reductase of Helicobacter pylori switches from a peroxide reductase to a molecular chaperone function. Proc. Natl. Acad. Sci. U. S. A. 103:2552–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lundström AM, Bölin I. 2000. A 26 kDa protein of Helicobacter pylori shows alkyl hydroperoxide reductase (AhpC) activity and the mono-cistronic transcription of the gene is affected by pH. Microb. Pathog. 29:257–266 [DOI] [PubMed] [Google Scholar]
- 23.Weiss SJ. 1989. Tissue destruction by neutrophils. N. Engl. J. Med. 320:365–376 [DOI] [PubMed] [Google Scholar]
- 24.Rosen H, Klebanoff SJ, Wang Y, Brot N, Heinecke JW, Fu X. 2009. Methionine oxidation contributes to bacterial killing by the myeloperoxidase system of neutrophils. Proc. Natl. Acad. Sci. U. S. A. 106:18686–18691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wood ZA, Schroder E, Robin Harris J, Poole LB. 2003. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 28:32–40 [DOI] [PubMed] [Google Scholar]
- 26.Poole LB. 2005. Bacterial defenses against oxidants: mechanistic features of cysteine-based peroxidases and their flavoprotein reductases. Arch. Biochem. Biophys. 433:240–254 [DOI] [PubMed] [Google Scholar]
- 27.Ma L, Payne SM. 2012. AhpC is required for optimal production of enterobactin by Escherichia coli. J. Bacteriol. 194:6748–6757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang CH, Chuang MH, Wu YH, Chuang WC, Jhuang PJ, Chiou SH. 2010. Characterization of site-specific mutants of alkylhydroperoxide reductase with dual functionality from Helicobacter pylori. J. Biochem. 147:661–669 [DOI] [PubMed] [Google Scholar]
- 29.Jungblut PR, Bumann D, Haas G, Zimny-Arndt U, Holland P, Lamer S, Siejak F, Aebischer A, Meyer TF. 2000. Comparative proteome analysis of Helicobacter pylori. Mol. Microbiol. 36:710–725 [DOI] [PubMed] [Google Scholar]
- 30.Croxen MA, Ernst PB, Hoffman PS. 2007. Antisense RNA modulation of alkyl hydroperoxide reductase levels in Helicobacter pylori correlates with organic peroxide toxicity but not infectivity. J. Bacteriol. 189:3359–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olczak AA, Seyler RW, Jr, Olson JW, Maier RJ. 2003. Association of Helicobacter pylori antioxidant activities with host colonization proficiency. Infect. Immun. 71:580–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ezraty B, Grimaud R, El Hassouni M, Moinier D, Barras F. 2004. Methionine sulfoxide reductases protect Ffh from oxidative damages in Escherichia coli. EMBO. J. 23:1868–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khor HK, Fisher MT, Schoneich C. 2004. Potential role of methionine sulfoxide in the inactivation of the chaperone GroEL by hypochlorous acid (HOCl) and peroxynitrite (ONOO-). J. Biol. Chem. 279:19486–19493 [DOI] [PubMed] [Google Scholar]
- 34.Drazic A, Miura H, Peschek J, Le Y, Bach NC, Kriehuber T, Winter J. 2013. Methionine oxidation activates a transcription factor in response to oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 110:9493–9498 [DOI] [PMC free article] [PubMed] [Google Scholar]