

Abstract
Infections with the Gram-negative bacterium Burkholderia pseudomallei (melioidosis) are associated with high mortality, and there is currently no approved vaccine to prevent the development of melioidosis in humans. Infected patients also do not develop protective immunity to reinfection, and some individuals will develop chronic, subclinical infections with B. pseudomallei. At present, our understanding of what constitutes effective protective immunity against B. pseudomallei infection remains incomplete. Therefore, we conducted a study to elucidate immune correlates of vaccine-induced protective immunity against acute B. pseudomallei infection. BALB/c and C57BL/6 mice were immunized subcutaneously with a highly attenuated, Select Agent-excluded purM deletion mutant of B. pseudomallei (strain Bp82) and then subjected to intranasal challenge with virulent B. pseudomallei strain 1026b. Immunization with Bp82 generated significant protection from challenge with B. pseudomallei, and protection was associated with a significant reduction in bacterial burden in lungs, liver, and spleen of immunized mice. Humoral immunity was critically important for vaccine-induced protection, as mice lacking B cells were not protected by immunization and serum from Bp82-vaccinated mice could transfer partial protection to nonvaccinated animals. In contrast, vaccine-induced protective immunity was found to be independent of both CD4 and CD8 T cells. Tracking studies demonstrated uptake of the Bp82 vaccine strain predominately by neutrophils in vaccine-draining lymph nodes and by smaller numbers of dendritic cells (DC) and monocytes. We concluded that protection following cutaneous immunization with a live attenuated Burkholderia vaccine strain was dependent primarily on generation of effective humoral immune responses.
INTRODUCTION
Melioidosis is a serious disease of humans caused by the Gram-negative facultative intracellular bacterium Burkholderia pseudomallei (1–3). The disease is endemic in Southeast Asia, northern Australia, and parts of South and Central America (1, 4–6). B. pseudomallei is being isolated increasingly in other parts of the world as well, likely due to greater awareness and surveillance for the organism (7, 8). Infection with B. pseudomallei can be contracted via several routes, including subcutaneous (s.c.) inoculation, inhalation, and likely ingestion (9, 10). The route of B. pseudomallei infection is correlated with the severity of infection, with inhalational infection generally associated with a more rapid disease course. Bacteremic infection is common with B. pseudomallei, and sepsis in melioidosis patients is associated with high mortality rates (11–13). In patients who develop chronic infection, the disease may manifest as disseminated abscesses in multiple sites, including the spleen, liver, joints, and central nervous system (CNS) (14–18). B. pseudomallei is inherently resistant to multiple classes of antibiotics, most notably aminoglycosides and some beta-lactam drugs (19), due to the expression of efflux pumps and PenA β-lactamase (20–23). A delay in diagnosis is often associated with treatment failures in patients with acute infection (24–27).
Currently, there is no approved vaccine for protection of humans against B. pseudomallei infection. A number of candidate vaccines have been developed and tested in animal models of melioidosis, and the state of melioidosis vaccines has been reviewed recently (28). Briefly, the most effective immunity to date has been achieved by use of live attenuated B. pseudomallei vaccines, including strains lacking ilvL (29, 30); serC (31); aroB (32); purN, purM, BPSS1509, lipB, and pabB (33); and bipD (34), and aroC (35) mutants.
Subunit vaccines have also been developed for immunization against B. pseudomallei, with the most promising protection to date being achieved with purified proteins such as BipD (34) and BipB and BipC (36); with recombinant proteins, including Hcp1, Hcp2, Hcp3, and Hcp6 (37); with lipopolysaccharide (LPS) (38); with purified flagellin (39); with LolC, PotF, and OppA nonmembrane protein (40); with outer membrane vesicles (41); and with recombinant Omp85 protein (42). However, the use of single antigen subunit vaccines for Burkholderia infection is unlikely to generate broad protective immunity against this very genetically diverse and unstable organism (35, 43). Effective immunization with subunit vaccines administered by the subcutaneous route has also not been reported.
While the risk of reversion to virulence is a primary concern with the use of live attenuated bacterial vaccines, the ability to induce rapidly broad protective immunity is a plus for this type of vaccine. Our group previously developed a highly attenuated strain of B. pseudomallei 1026b (strain Bp82), which was recently excluded from Select Agent regulations and which with Institutional Biosafety Committee approval can be used under biosafety level 2 (BSL-2) conditions (44). This ΔpurM strain of B. pseudomallei was extensively tested in several different highly immunocompromised animal strains, and reversion to virulence or persistence of the organism was not found (44). In addition, a purM deletion mutant of the K96243 strain of B. pseudomallei was also highly attenuated and safe in animal studies (though this strain is not yet excluded from Select Agent regulations) (44). The ability of these Select Agent-excluded mutant strains of B. pseudomallei to induce protective immunity from melioidosis has not been previously investigated in animal models. Nor has it been determined whether protection could be achieved by subcutaneous (s.c.) vaccine administration, which is a more practical route of immunization than the intranasal (i.n.) or intraperitoneal (i.p.) routes used in most prior studies of attenuated Burkholderia vaccines.
Therefore, in the present study we intended to determine whether the Bp82 strain of B. pseudomallei was capable of inducing protective immunity following cutaneous immunization. We also sought to elucidate immune mechanisms by which Bp82 immunization could induce protective immunity and to also understand how the Bp82 vaccine antigens were processed by antigen-presenting cells (APC) in lymph nodes (LN). Our findings indicate that the Bp82 vaccine is immunogenic following s.c. immunization and capable of inducing significant protection against acute inhaled B. pseudomallei challenge. Protective immunity was provided primarily by humoral immune responses. Therefore, these new insights into protective immune responses generated by live attenuated vaccines such as Bp82 should help guide the development of newer melioidosis vaccines and clinical evaluation of vaccine efficacy using immune correlates.
MATERIALS AND METHODS
Mice.
Specific-pathogen-free 6- to 8-week-old female BALB/c and C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME). In addition, mutant mouse strains (on the C57BL/6 background), including TCRα/β−/−, uMT−/−, CD4−/−, and CD8−/− strains, were also purchased from Jackson. Mice were used for studies when they were between 8 and 12 weeks of age. Animals were housed in microisolator cages under pathogen-free conditions. All studies were conducted in animal biosafety level 3 (ABSL-3) facilities at the Rocky Mountain Regional Biosafety Laboratory and were approved by the Institutional Animal Care and Use Committee at Colorado State University.
Bacteria and culture conditions.
B. pseudomallei strain 1026b (Bp1026b) is a clinical isolate from a patient with septicemic melioidosis in Thailand (45, 46). Culture stocks were grown overnight in Luria-Bertani broth (LB) (BD Biosciences, San Jose, CA) at 37°C with shaking. Frozen stocks of B. pseudomallei were prepared by adding 15% glycerol (Fisher BioReagent, Pittsburgh, PA) to the overnight culture and dividing the sample into 1-ml aliquots. Aliquots were stored at −80°C, and titers were determined prior to use. The construction and characterization of 1026b ΔpurM strain Bp82 have been previously described (44). A 114-bp fragment was deleted from the purM coding sequence, causing attenuation of the strain for the melioidosis animal infection model (44). Because Bp82 is a purine auxotroph, culture was done in LB medium supplemented with 0.6 mM adenine for optimal bacterial growth. For animal experiments, each strain was thawed just before use, and the bacteria were diluted to obtain the desired numbers of cells using sterile phosphate-buffered saline (PBS) (Sigma-Aldrich, St. Louis, MO).
Mouse vaccination.
Mice were restrained in a tail bleed apparatus allowing access to the hind leg without the need for anesthesia. Mice were vaccinated s.c. in the hind leg with 5 × 106 CFU Bp82, in 100 μl PBS. Mice received a booster immunization, 10 days after the first immunization.
In vitro stimulation assay for T cell immunity.
Mice were euthanized, and single-cell suspensions from spleens were generated via mechanical disruption. Cells were filtered through a 70-μm nylon mesh screen (BD Biosciences, San Jose, CA) and treated with NH4Cl to remove red blood cells. Spleen cells were plated in 24-well plates at a concentration of 2 × 106 cells/ml and restimulated with antigens for 72 h. These antigens included heat-killed Bp82 (2 × 107 cells/ml), Bp82 lysates (5 μg/ml), or recombinant GroEL (12.5 μg/ml) (kindly provided by Katherine Brown, University of Texas Austin). Cell culture supernatants were collected and analyzed for gamma interferon (IFN-γ) and interleukin-17 (IL-17) production via enzyme-linked immunosorbent assay (ELISA), using kits according to the manufacturer's instructions (R&D Systems, Minneapolis, MN).
Determination of antibody titers.
Nunc Maxisorp 96-well plates (Thermo Fisher Scientific, San Jose, CA) were coated with heat-killed Bp82 (5 × 107 bacteria/ml) in carbonate-bicarbonate buffer (pH 9.6) and incubated at 4°C overnight. Plates were washed with PBS and Tween 20 (0.05%) and blocked with PBS-Tween containing 5% nonfat dry milk for 2 h at room temperature. Sera were serially diluted (10-fold dilutions) in blocking buffer and incubated for 90 min at room temperature. All secondary antibodies were conjugated to horseradish peroxidase, including rat anti-mouse IgM and IgG (BD Biosciences, San Jose, CA), goat anti-mouse IgG2a (Abcam, Cambridge, MA), and rat anti-mouse IgG1 (BD Biosciences, San Jose, CA). These antibodies were diluted 1:2,000 in blocking buffer and were incubated on the plate for 1 h at room temperature. 3,3′,5,5′-Tetramethylbenzidine (TMB) (Sigma-Aldrich, St. Louis, MO) substrate was used to develop the plates. The reaction was stopped by the addition of 50 μl of 1 N HCl. The absorbance at 450 nm was determined using a Thermo Multiskan EX spectrophotometer (Thermo Scientific, Rockford, IL).
Serum transfer studies.
To obtain immune serum, naive BALB/c mice were vaccinated with Bp82 (5 × 106 CFU) s.c. and boosted 10 days later. Two weeks after the boost, mice were anesthetized via intraperitoneal (i.p.) injection with 100 mg ketamine/kg of body weight plus 10 mg/kg xylazine diluted in sterile water, and blood was collected via terminal cardiac puncture. Blood was allowed to clot for 30 min at 4°C and then centrifuged to prepare serum, which was collected and stored at −80°C prior to use. Nonimmune serum was also collected from unvaccinated mice in a similar manner. Anti-Burkholderia IgG titers were quantified via ELISA prior to transfer as previously described (47, 48). Recipient mice received 250 μl of immune serum or naive (control) serum given i.p. 1 day prior to challenge.
B. pseudomallei i.n. challenge model.
For intranasal (i.n.) challenge, mice were anesthetized as described above. Bacteria were thawed just prior to use and diluted in sterile PBS (pH 7.4) for inoculation. Mice were challenged with approximately 5 50% lethal doses (LD50) of B. pseudomallei (5 × 103 CFU for BALB/c mice or 1.2 × 104 CFU for C57BL/6 mice), in a total volume of 20 μl administered in sequential droplets on alternating nares. Actual infectious doses delivered to the mice were determined by plating the inoculum.
Determination of bacterial burden in organs.
Lungs, liver, and spleen were harvested at 72 h postinfection in the acute challenge model and at 30 and 60 days postinfection for the chronic challenge model and placed in 4 ml of sterile PBS. Organs were homogenized using a Seward stomacher (Seward, Bohemia, NY). Homogenates (300 μl) were removed, placed in a 96-well plate, and serially diluted in sterile PBS, using 10-fold dilutions. Bacterial counts were determined by plating serial 10-fold dilutions on LB agar and placing them at 37°C, and CFU were enumerated after 48 h of incubation.
Tracking Bp82 uptake in vaccine-draining lymph nodes.
To facilitate tracking the fate of Bp82 in vaccine-draining lymph nodes following immunization, Bp82 was engineered to express green fluorescent protein (GFP) (Bp82-gfp). Briefly, strain Bp82 (44) was labeled with an enhanced green fluorescent protein (eGFP) whose expression is driven by the constitutive Burkholderia thailandensis ribosomal S12 gene promoter PS12 (49) in single copy from the pUC18T-mini-Tn7T-PS12-eGFP plasmid using previously described methods (49). The stable chromosomal insertion of the site-specific mini-Tn7 kanamycin (Kan) resistance element was performed by triparental conjugation of Bp82 with RHO3/pTNS3 and RHO3/pUC18T-mini-Tn7T-PS12-eGFP, and transformants were selected on LB medium containing 1,000 μg/ml kanamycin and 0.6 mM adenine. Isolates were screened by PCR to identify single insertions in the glmS2-associated attTn7 site. An unmarked strain was obtained by Flp-mediated excision of the Kan resistance marker using pFLPe2. Bp82-gfp was injected into the left footpad (1.2 × 108 CFU/mouse) in 50 μl PBS, and popliteal lymph nodes were harvested 10 h after injection and processed as previously described (50). Briefly, lymph nodes were harvested and placed in 5 ml of Hanks' balanced salt solution (HBSS) and digested in collagenase D (Roche, San Francisco, CA) for 30 min at 37°C. Following digestion, the tissue was triturated using a glass pipette and filtered through a 70-μm nylon mesh screen (BD Biosciences, San Jose, CA).
Flow cytometry.
Cells from spleen or lymph nodes were suspended in fluorescence-activated cell sorting (FACS) buffer (PBS with 2% fetal bovine serum [FBS] [Atlas Biologicals, Fort Collins, CO] and 0.05% sodium azide [Fisher Scientific, Philadelphia, PA]) and stained as previously described (51). To block nonspecific binding, cells were incubated at room temperature for 15 min in FACS block consisting of normal mouse serum (Jackson ImmunoResearch, West Grove, PA) and human IgG (Jackson ImmunoResearch, West Grove, PA), along with unlabeled anti-mouse CD16/32 (clone 93) (eBioscience, San Diego, CA) prior to staining. After blocking, cells were stained with the following antibodies: anti-Ly6-G (phycoerythrin [PE], clone HK1.4), anti-Ly6-C (PE Cy7, clone RB6-8C5), anti-CD11c (biotin, clone N418), anti-CD11b (allophycocyanin Alexa 750, clone M1/70), anti-CD169 (Alexa Fluor 647, clone MOMA-1). The antibodies were purchased from eBioscience (San Diego, CA), BD Pharmingen (San Diego, CA), or AbD Serotec (Raleigh, NC). Cells were immunostained at room temperature for 30 min with antibodies diluted in FACS buffer. After the primary antibody incubation, cells were washed in FACS buffer and incubated with streptavidin-peridinin chlorophyll protein (PerCP) for 20 min at room temperature. Cell acquisition was done using a Gallios flow cytometer (Beckman Coulter, Brea, CA). Data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Immunohistochemistry.
Lungs and spleen were collected 48 h postinfection and fixed in 4% paraformaldehyde for 48 h. Tissues were paraffin embedded, cut into 5- to 7-μm sections, deparaffinized using EZ-DeWax solution (Biogenex Lab, San Ramon, CA), and transferred into PBS. Sections were blocked with blocking eraser (Biocare Medical, Concord, CA) for 5 min at room temperature and then incubated with appropriately diluted rabbit polyclonal anti-B. pseudomallei antibody (provided by D. Waag from USAMRIID), and the slides were incubated overnight at room temperature. Slides were washed three times with PBS followed by incubation with anti-rabbit Cy3-conjugated secondary antibody (Millipore, Billerica, MA) for 1 h at room temperature. Slides were washed and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (0.5 μg/ml) for 2 min and coverslipped with Prolong Gold mounting medium (Invitrogen, Carlsbad, CA). Sections were examined using a Zeiss 510 confocal microscope and analyzed using Volocity software.
Statistical analyses.
Statistical analyses were performed using GraphPad Prism5 software (La Jolla, CA). A nonparametric t test was used to analyze statistical differences between 2 groups. Comparisons between multiple groups were done using one-way analysis of variance (ANOVA), followed by Tukey's multiple means comparison test. Survival differences were compared using Kaplan-Meier survival curves, followed by log rank test. Statistical significance was defined as P < 0.05.
RESULTS
Immunization with Bp82 protects against lethal B. pseudomallei challenge.
To determine whether s.c. immunization with Bp82 was capable of generating protective immunity against lethal inhaled challenge with B. pseudomallei, BALB/c and C57BL/6 mice were immunized twice with Bp82 (5 × 106 CFU per immunization) and then subjected to lethal i.n. challenge with B. pseudomallei strain 1026b. The immunizing dose of Bp82 selected for these studies was comparable to doses used in previous studies of live attenuated Burkholderia vaccines. Following challenge with B. pseudomallei, mice were monitored closely and euthanized when they met predetermined endpoints for signs of illness. We observed that unvaccinated mice (both C57BL/6 and BALB/c) reached morbidity endpoints within 3 days of challenge and were humanely euthanized. In contrast, there was 100% survival of vaccinated C57BL/6 mice (Fig. 1A) and 80% survival of vaccinated BALB/c mice, for at least 30 days after challenge (Fig. 1B) (P < 0.05 compared to unvaccinated mice). At 60 days postchallenge, Bp82 vaccination conferred 100% protection on C57BL/6 mice (Fig. 1A), whereas 60% of BALB/c mice were still alive and apparently healthy (Fig. 1B).
Fig 1.
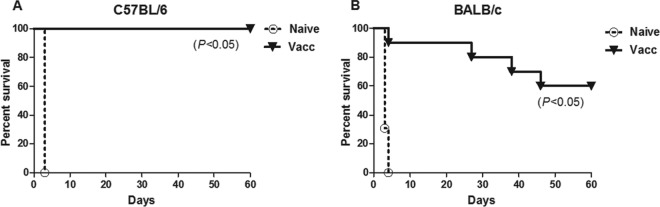
Subcutaneous immunization with Bp82 protects mice from an acutely lethal B. pseudomallei infection. C57BL/6 and BALB/c mice (n = 5) were immunized twice 10 days apart with 5 × 106 CFU of Bp82 suspended in PBS. All animals were then challenged intranasally with ∼5 LD50 (1.2 × 104 [C57BL/6] or 5 × 103 [BALB/c] CFU/mouse) of B. pseudomallei strain 1026b. Animals were euthanized upon reaching a predetermined euthanasia endpoint. Kaplan-Meier survival curves were generated for 60 days of survival for C57BL/6 mice (A) and BALB/c mice (B). Statistical differences in survival were determined by a log rank test. Data are pooled from two combined experiments.
Vaccination with Bp82 reduces bacterial burden.
The evidence that Bp82 is not detectable in mouse organs after 48 h of inoculation was previously considered by the study in reference 44, which challenged mice by the i.n. route with 6 × 103 CFU of Bp82 or B. pseudomallei 1026b. After 48 h of infection, the authors found that Bp82 was below the limit of detection in the lung, liver, and spleen. Therefore, studies were done next to assess the effects of Bp82 vaccination on the B. pseudomallei 1026b bacterial burden in key organs shortly after challenge. We observed that the bacterial burden in the lung and the spleen 72 h after B. pseudomallei challenge was significantly reduced in Bp82-vaccinated animals compared to unvaccinated animals (Fig. 2). In addition, bacteria were not detected in the livers of Bp82-vaccinated animals, compared to the high bacterial burden in the livers of unvaccinated mice (Fig. 2). At 30 and 60 days after challenge, Bp82-vaccinated mice still had detectable B. pseudomallei in the spleen and small amounts in the lung and liver (data not shown).
Fig 2.
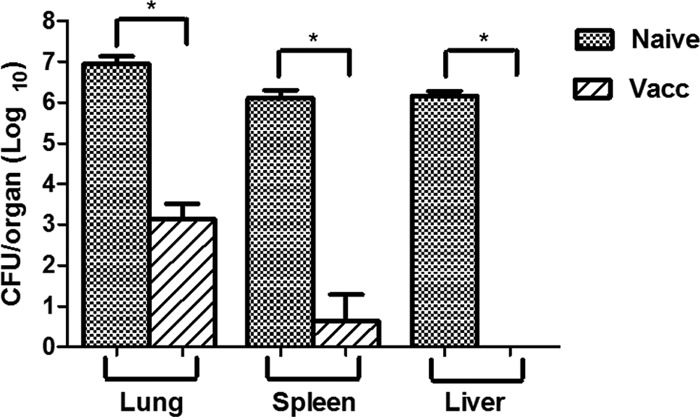
Immunization reduces bacterial burden in vaccinated mice at 72 h after challenge. Bacterial burden in lung, spleen, and liver tissues was determined in naive and vaccinated mice (n = 5) 72 h after intranasal challenge with B. pseudomallei strain 1026b. Statistical comparisons between vaccinated and naive groups were done using a nonparametric t test. *, P < 0.05. Similar results were obtained in one additional experiment.
Induction of antibody responses following Bp82 immunization.
Serum from vaccinated mice was analyzed 10 days after the last immunization for induction of antibodies against Bp82 intact organisms. In the serum of vaccinated animals, significantly increased titers (P < 0.05) of antibodies against intact Bp82 were detected, compared to unvaccinated control animals (Fig. 3). Vaccinated animals mounted strong IgG responses (Fig. 3A), as well as IgM responses (Fig. 3B), against heat-killed Bp82. In addition, there was significant induction of IgG responses of the IgG1 and IgG2a isotypes (Fig. 3C and D, P < 0.05). Immunized animals also had significantly increased titers against antigens present in Bp82 lysates (data not shown). Immunization with Bp82 also induced antibody responses against certain immunogenic proteins of B. pseudomallei, including GroEL (data not shown), but not other antigens such as BimA (data not shown).
Fig 3.

Humoral immune responses in Bp82-vaccinated mice. Serum was prepared from BALB/c and C57BL/6 mice (n = 5) vaccinated twice s.c. with Bp82, and titers of antibodies to heat-killed Bp82 were determined by endpoint dilution ELISA. Titers of total IgG (A), IgM (B), IgG1 (C), and IgG2a (D) were expressed as the reciprocal of the endpoint dilution. Significant differences (*, P < 0.05; **, P < 0.005; ***, P < 0.0001) were determined by a nonparametric Kruskal-Wallis test followed by Dunnett's multiple comparison test. Data represent pooled data from two independent experiments.
Induction of cellular immune responses following Bp82 immunization.
The ability of the Bp82 vaccine to induce specific T cell responses was evaluated next. Spleens were harvested from immunized and nonimmunized mice 2 weeks after s.c. Bp82 vaccination. Single-cell suspensions of spleen cells were placed in triplicate wells of 96-well plates as noted in Materials and Methods and incubated with heat-killed Bp82, lysed Bp82, or recombinant GroEL protein. Incubation of spleen cells from immunized mice with heat-killed Bp82 generated production of significant increases in IFN-γ and IL-17 production (Fig. 4A and B). However, incubation with Bp82 did not induce production of IL-10 from spleen cells (data not shown). Lysates of Bp82 or recombinant GroEL, however, did not induce significant cytokine production. Thus, immunization with the Bp82 vaccine induced cellular immune responses that appeared to be directly primarily toward surface determinants on Bp82.
Fig 4.

Cellular immune responses to Bp82 immunization. BALB/c mice (n = 5) were immunized twice s.c. with Bp82. Fourteen days after the second immunization, spleen cells from naive or vaccinated animals were restimulated in vitro with heat-killed Bp82 (2 × 107 CFU/ml), lysate from Bp82 (5 μg/ml), recombinant GroEL (12.5 μg/ml), or medium alone for 3 days. Supernatants were assayed by ELISA for IFN-γ (A) and IL-17 (B) production. *, P < 0.05 using a Mann-Whitney U test. Data represent pooled data from two independent experiments.
Role of humoral immunity in vaccine-induced protection against B. pseudomallei.
In previous studies where immune mechanisms of protection have been examined, humoral immunity was found to be a primary mediator of vaccine-induced immune protection against acute B. pseudomallei challenge (52–55). Therefore, the role of antibody-mediated protection was investigated in the Bp82 vaccine model. First, serum transfer experiments were conducted, by transferring serum from Bp82-vaccinated animals to naive animals, which were then subjected to B. pseudomallei challenge. Immune serum transfer generated partial protection to B. pseudomallei challenge, with 38% of immune serum recipient mice protected from lethal challenge, compared to 0% of mice receiving nonimmune serum (Fig. 5A). In addition, B-cell-deficient mice (uMT−/−) were also vaccinated with Bp82 and challenged. In this study, only 50% of the uMT−/− mice were protected from challenge, compared to all of the vaccinated wild-type (WT) animals (Fig. 5B). These results indicated therefore that humoral immunity played an important, but incomplete, role in Bp82 vaccine-induced protection from B. pseudomallei infection.
Fig 5.
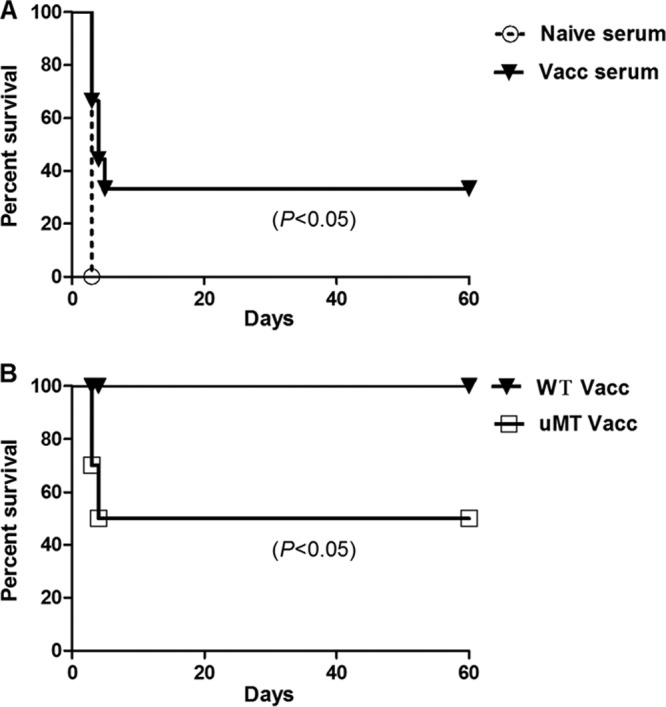
Role of antibody in Bp82 vaccine protection. (A) Serum from immunized and naive animals was transferred to naive BALB/c recipient animals (n = 5 per group), which were then subjected to lethal i.n. challenge with B. pseudomallei 24 h later. Survival percentages were significantly increased in the animals that received immune serum compared to the animals that received nonimmune serum. (B) uMT−/− and WT C57BL/6 mice (n = 5) were immunized twice s.c. with Bp82 and then challenged intranasally with B. pseudomallei 1026b, and survival times were determined. Survival was significantly reduced in uMT−/− mice compared to WT animals. Data represent pooled data from two independent experiments.
Role of T cells in Bp82 vaccine-induced protection.
Studies were done next to elucidate the role of T cells in vaccine-induced immunity to Burkholderia infection. Mice lacking CD4 T cells (CD4−/−) or CD8 T cells (CD8−/−) on the C57BL/6 background were vaccinated s.c. with the Bp82 vaccine and subjected to B. pseudomallei challenge. Survival times were compared to those of WT animals (Fig. 6). At 30 days after challenge, there was no statistical difference in survival times between CD8−/− or CD4−/− vaccinated animals and vaccinated WT mice (Fig. 6A and B). All animals from CD4−/− and TCRα/β−/− groups survived to at least 60 days after challenge and did not manifest signs of infection, whereas unvaccinated C57BL/6 mice were euthanized due to progressive infection 3 days after challenge. Mice lacking all conventional T cells (TCRα/β−/−) were also immunized with the Bp82 vaccine and subjected to challenge. These animals were also significantly protected from infection (Fig. 6C). These results indicated that protection from inhaled B. pseudomallei challenge induced by the Bp82 vaccine was largely independent of T cells and mediated almost entirely by B cells and antibody production. Since CD4−/− mice are unable to produce IgG efficiently, these results also suggest that it is likely that T-cell-independent antibodies such as IgM antibodies may have played a major role in vaccine-induced protection.
Fig 6.

Role of T cells in vaccine-induced protection against B. pseudomallei. CD8−/− (A), CD4−/− (B), and TCRα/β−/− (C) and WT C57BL/6 mice were immunized twice with Bp82. Naive and immunized animals were then challenged intranasally with 1.2 × 104 CFU of B. pseudomallei 1026b, and survival percentages were determined. Survival percentages for CD8−/−, CD4−/−, and TCRα/β−/− mice compared to WT mice were not significantly different. Survival differences were determined using Kaplan-Meier survival curves and log rank analysis. Data are representative of two independent experiments.
Delivery of Bp82 vaccine to draining LN.
Finally, studies were conducted to assess the uptake and trafficking of the Bp82 vaccine to vaccine-draining lymph nodes (LN). We recently demonstrated that following immunization with conventional adjuvanted vaccines, a significant and rapid influx of inflammatory monocytes into vaccine-draining lymph nodes occurs (56). Therefore, studies were conducted to determine whether the Bp82 vaccine elicited a similar leukocyte response in LN and to also identify potential antigen-presenting cells (APC) responsible for trafficking of Bp82 from the cutaneous inoculation site to the LN. Therefore, a gfp-expressing construct of Bp82 was used to track the early movement of Bp82 vaccine bacteria from the skin site to the nearest draining LN. For these studies, the vaccine was administered s.c. in the footpad and the popliteal LN response was monitored using flow cytometry.
We found that following s.c. administration of the Bp82 vaccine, there was a marked influx of CD11b+ Ly6G+ Ly6C− neutrophils into the nearest draining LN, as well as a much smaller influx of CD11b+ Ly6G− Ly6C+ monocytes (Fig. 7A and B). We also found that approximately 80% of all Bp82-gfp bacteria found in the LN after vaccination were contained within neutrophils (Fig. 7C). In addition, bacteria in the LN were also found within Langerhans dendritic cells (DC) (10%), CD169+ subcapsular macrophages (3%), and inflammatory monocytes (3%), with very few non-cell-associated bacteria being detected.
Fig 7.
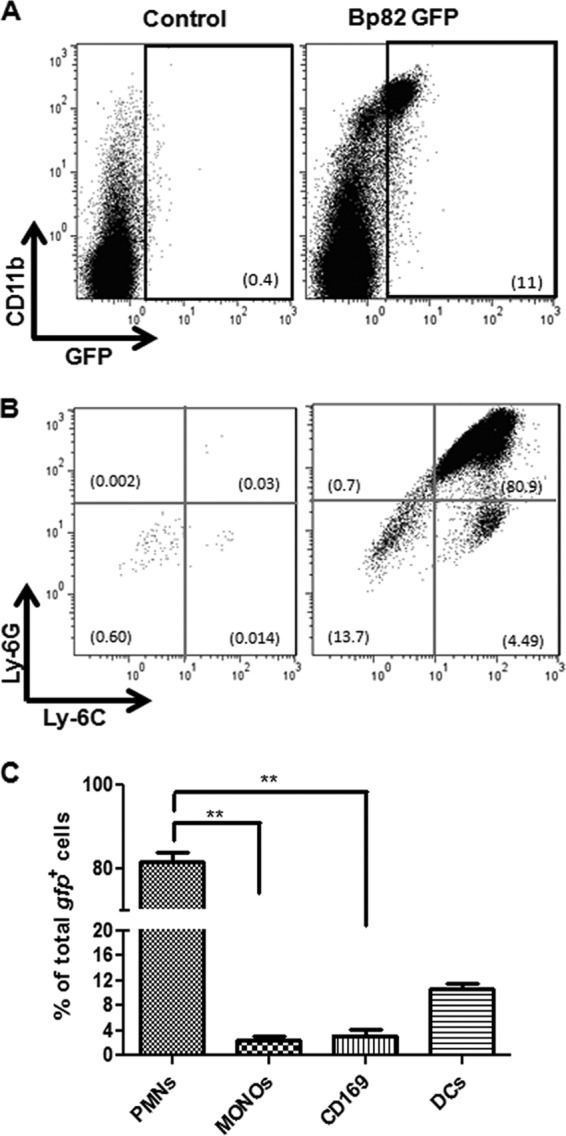
Lymph node cellular response to Bp82 vaccination. BALB/c mice (n = 5 per group) were injected in one rear limb footpad with 1.2 × 108 CFU of Bp82-gfp. Ten hours after inoculation, cells from the ipsilateral popliteal LN were collected and immunostained. Cells from the contralateral popliteal LN served as the control. (A) The appearance of GFP+ CD11b+ cells in the draining LN was determined by flow cytometry at 10 h after injection. (B) The GFP+ cells were further subdivided into polymorphonuclear leukocytes (PMN) (CD11b+ Ly6G+ Ly6C−) and monocytes (CD11b+ Ly6G− Ly6C+). (C) The distribution of Bp82-gfp bacteria in relevant LN APC populations, including neutrophils (PMN), dendritic cells (DC), CD169+ subcapsular macrophages, and monocytes, was calculated. Data are representative of two independent experiments. Statistical differences between Bp82-injected and uninjected contralateral LN were determined by Student's two-tailed t test. **, P < 0.05. For comparisons between 4 groups (C), ANOVA was used, followed by the Tukey post hoc test.
To compare the relative efficiencies of uptake of Bp82-gfp by antigen-presenting cells in the LN, the percentages of each cell population containing Bp82 were calculated. We calculated that 22.12% of all neutrophils in the LN, 1.87% of all DC in the LN, 16.17% of subcapsular macrophages in the LN, and 11.62% of all inflammatory monocytes in the LN contained Bp82-gfp. Thus, neutrophils appeared to be the most efficient antigen-presenting cells at taking up and transporting Bp82 bacteria to the draining LN, and the majority of Bp82 bacteria in the LN were found within neutrophils.
DISCUSSION
A number of different live attenuated and subunit vaccines have been evaluated for their ability to generate protective immunity against B. pseudomallei (29, 30, 32–36, 38–42, 55). However, there is still relatively little known regarding the most effective methods of generating protection against melioidosis or the immune mechanisms of protection. In the present study, we demonstrated long-term (i.e., >60 days) protection against acute B. pseudomallei challenge following cutaneous immunization with a live attenuated strain of B. pseudomallei. Importantly, our study utilized a more conventional route of immunization (i.e., cutaneous immunization) than did previous studies, which have typically relied on intranasal or intraperitoneal routes of immunization. In addition, we found that the Bp82 vaccine did not require an adjuvant for activity (data not shown). Another important consideration is the fact that the Bp82 vaccine strain used in this study has been extensively evaluated for attenuation and safety. Thus, issues of reversion to virulence are potentially less of a concern with the purM deletion mutant of B. pseudomallei.
There remain substantial gaps in our knowledge regarding mechanisms of immune protection from melioidosis. The best protective immunity to date has been achieved using live attenuated Burkholderia vaccines, but the mechanisms of protection are not completely understood. There is some evidence that protective immunity is dependent on induction of innate immune responses by live bacterial vaccines (43, 57). Also, there is speculation that live attenuated vaccines generate better immunity due to prolonged antigen persistence in the host (33).
Protection generated by the Bp82 vaccine was found to be mediated almost entirely by antibodies, as revealed by serum transfer studies and by studies in B-cell- and T-cell-deficient mice. These findings are consistent with those of prior studies, where protection from inhaled Burkholderia challenge was found to be mediated largely by humoral immune mechanisms. However, these are the first studies to demonstrate that protection acquired by s.c. immunization is also antibody mediated and that routes of mucosal administration were not required to induce effective levels of immune protection against inhalational challenge.
Interestingly, our studies also revealed a minor role for T cells in vaccine-mediated protection. For example, while CD4−/− and CD8−/− animals did not have a defect in immune protection, mice that lacked both CD4 and CD8 T cells (i.e., TCRα/β−/− mice) did have a small, though not statistically significant, loss of immune protection following Bp82 immunization (Fig. 6C). While the Bp82 vaccine induced both IgG and IgM antibodies, the fact that CD4−/− mice were fully protected following immunization suggests an important potential role for IgM antibodies in mediating vaccine protection, as CD4−/− mice are generally unable to effectively produce antibodies of the IgG subclass (58).
Our findings indicate that administration of a live attenuated vaccine by the s.c. route is effective in generating systemic protection from bacterial challenge by a mucosal route. Though relatively high titers of anti-Burkholderia antibodies were detected in serum of vaccinated animals, it is not clear exactly where bacterial control by vaccine antibodies occurred. It is plausible to suggest that vaccination may protect from lethality mainly by blocking bacterial dissemination from the lungs to other sites (e.g., spleen and liver), rather than by neutralizing bacteria directly in the lungs. The protective antibody immune response in Burkholderia infection has been shown previously to be specific to the lipopolysaccharide (LPS) of B. pseudomallei, which promotes opsonic phagocytic killing (59, 60).
The response of antigen-presenting cells in LN to immunization with the live attenuated Bp82 vaccine was found to be quite different from the response to conventional adjuvanted vaccines, with the primary difference being the much more pronounced neutrophilic response in the case of mice immunized with the Bp82 vaccine. It is unlikely that neutrophils are able to present Bp82 antigens directly to T cells but much more likely that neutrophils containing vaccine bacteria hand the antigens off to professional antigen-presenting cells in the LN, including DC, subcapsular macrophages, or monocytes. Indeed, it been recently observed for a live Mycobacterium bovis BCG vaccine that the vaccine bacteria are taken up primarily by neutrophils but then later handed off to other antigen-presenting cells (e.g., DC) in the LN and peritoneal cavity (61). With the Bp82 vaccine, most of the bacteria were delivered to the LN in neutrophils, but the most avidly phagocytic antigen-presenting cells for Bp82 in the LN were found to be CD169+ macrophages and inflammatory monocytes. Thus, it is likely that initial uptake of live attenuated vaccines such as Bp82 may be mediated by neutrophils, while the bacterial antigens may ultimately be delivered to macrophages and monocytes within the LN for presentation to T cells.
In summary, we have found that s.c. immunization with a highly attenuated strain of B. pseudomallei can generate significant protection against inhalational challenge with virulent B. pseudomallei. These studies suggest, therefore, that it may be possible to develop conventionally delivered vaccines (i.e., vaccines administered by the s.c. or intramuscular [i.m.] route) capable of generating effective humoral immune protection against melioidosis. While subunit vaccines are generally preferred over live attenuated bacterial vaccines, it may be possible as an interim measure for melioidosis protection to use an attenuated vaccine, particularly if vaccine administration is safe and easily accomplished. Our studies in mice indicated that vaccine site reactions did not occur (data not shown), suggesting that the whole-cell Bp82 vaccine did not induce significant local inflammation. Thus, there is reason for optimism that an effective subunit or nonliving bacterial cell vaccine for melioidosis that is safe and easily administered can be developed.
ACKNOWLEDGMENTS
We thank Todd French, University of California Los Angeles, for providing the pUC18T-mini-Tn7T-PS12-eGFP plasmid.
These studies were supported by NIH NIAID grant U54 AI065357-05 to S.W.D. and H.P.S.
Footnotes
Published ahead of print 7 October 2013
REFERENCES
- 1.Cheng AC, Currie BJ. 2005. Melioidosis: epidemiology, pathophysiology, and management. Clin. Microbiol. Rev. 18:383–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wiersinga WJ, van der Poll T, White NJ, Day NP, Peacock SJ. 2006. Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat. Rev. Microbiol. 4:272–282 [DOI] [PubMed] [Google Scholar]
- 3.Wiersinga WJ, Currie BJ, Peacock SJ. 2012. Melioidosis. N. Engl. J. Med. 367:1035–1044 [DOI] [PubMed] [Google Scholar]
- 4.Dance DAB. 2000. Melioidosis as an emerging global problem. Acta Trop. 74:115–119 [DOI] [PubMed] [Google Scholar]
- 5.Dance DA. 1991. Melioidosis: the tip of the iceberg? Clin. Microbiol. Rev. 4:52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Currie BJ, Dance DA, Cheng AC. 2008. The global distribution of Burkholderia pseudomallei and melioidosis: an update. Trans. R. Soc. Trop. Med. Hyg. 102(Suppl 1):S1–S4 [DOI] [PubMed] [Google Scholar]
- 7.Batchelor BI, Paul J, Trakulsomboon S, Mgongo M, Dance DA. 1994. Melioidosis survey in Kenya. Trans. R. Soc. Trop. Med. Hyg. 88:181. [DOI] [PubMed] [Google Scholar]
- 8.Yabuuchi E, Arakawa M. 1993. Burkholderia pseudomallei and melioidosis: be aware in temperate area. Microbiol. Immunol. 37:823–836 [DOI] [PubMed] [Google Scholar]
- 9.Wiersinga WJ, van der Poll T. 2009. Burkholderia pseudomallei tropism and the melioidosis road map. J. Infect. Dis. 199:1720–1722 [DOI] [PubMed] [Google Scholar]
- 10.Barnes JL, Ketheesan N. 2005. Route of infection in melioidosis. Emerg. Infect. Dis. 11:638–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White NJ. 2003. Melioidosis. Lancet 361:1715–1722 [DOI] [PubMed] [Google Scholar]
- 12.Tiangpitayakorn C, Songsivilai S, Piyasangthong N, Dharakul T. 1997. Speed of detection of Burkholderia pseudomallei in blood cultures and its correlation with the clinical outcome. Am. J. Trop. Med. Hyg. 57:96–99 [DOI] [PubMed] [Google Scholar]
- 13.Chaowagul W, White NJ, Dance DA, Wattanagoon Y, Naigowit P, Davis TM, Looareesuwan S, Pitakwatchara N. 1989. Melioidosis: a major cause of community-acquired septicemia in northeastern Thailand. J. Infect. Dis. 159:890–899 [DOI] [PubMed] [Google Scholar]
- 14.Rode JW, Webling DD. 1981. Melioidosis in the Northern Territory of Australia. Med. J. Aust. 1:181–184 [DOI] [PubMed] [Google Scholar]
- 15.Thin RN, Brown M, Stewart JB, Garrett CJ. 1970. Melioidosis: a report of ten cases. Q. J. Med. 39:115–127 [PubMed] [Google Scholar]
- 16.Ngauy V, Lemeshev Y, Sadkowski L, Crawford G. 2005. Cutaneous melioidosis in a man who was taken as a prisoner of war by the Japanese during World War II. J. Clin. Microbiol. 43:970–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vatcharapreechasakul T, Suputtamongkol Y, Dance DA, Chaowagul W, White NJ. 1992. Pseudomonas pseudomallei liver abscesses: a clinical, laboratory, and ultrasonographic study. Clin. Infect. Dis. 14:412–417 [DOI] [PubMed] [Google Scholar]
- 18.Wong KT, Puthucheary SD, Vadivelu J. 1995. The histopathology of human melioidosis. Histopathology 26:51–55 [DOI] [PubMed] [Google Scholar]
- 19.Schweizer HP. 2012. Mechanisms of antibiotic resistance in Burkholderia pseudomallei: implications for treatment of melioidosis. Future Microbiol. 7:1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rholl DA, Papp-Wallace KM, Tomaras AP, Vasil ML, Bonomo RA, Schweizer HP. 2011. Molecular investigations of PenA-mediated beta-lactam resistance in Burkholderia pseudomallei. Front. Microbiol. 2:139. 10.3389/fmicb.2011.00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mima T, Schweizer HP. 2010. The BpeAB-OprB efflux pump of Burkholderia pseudomallei 1026b does not play a role in quorum sensing, virulence factor production, or extrusion of aminoglycosides but is a broad-spectrum drug efflux system. Antimicrob. Agents Chemother. 54:3113–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore RA, DeShazer D, Reckseidler S, Weissman A, Woods DE. 1999. Efflux-mediated aminoglycoside and macrolide resistance in Burkholderia pseudomallei. Antimicrob. Agents Chemother. 43:465–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trunck LA, Propst KL, Wuthiekanun V, Tuanyok A, Beckstrom-Sternberg SM, Beckstrom-Sternberg JS, Peacock SJ, Keim P, Dow SW, Schweizer HP. 2009. Molecular basis of rare aminoglycoside susceptibility and pathogenesis of Burkholderia pseudomallei clinical isolates from Thailand. PLoS Negl. Trop. Dis. 3:e519. 10.1371/journal.pntd.0000519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carruthers MM. 1981. Recrudescent melioidosis mimicking lung abscess. Am. Rev. Respir. Dis. 124:756–758 [DOI] [PubMed] [Google Scholar]
- 25.Chan CK, Hyland RH, Leers WD, Hutcheon MA, Chang D. 1984. Pleuropulmonary melioidosis in a Cambodian refugee. Can. Med. Assoc. J. 131:1365–1367 [PMC free article] [PubMed] [Google Scholar]
- 26.Pit S, Chea FK, Jamal F. 1988. Melioidosis with brain abscess. Postgrad. Med. J. 64:140–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson JW, Ashdown LR, Richards MJ, Sutherland AD, Cade JF. 1987. Subacute pulmonary melioidosis in a temperate climate. Med. J. Aust. 147:95–96 [DOI] [PubMed] [Google Scholar]
- 28.Silva EB, Dow SW. 2013. Development of Burkholderia mallei and pseudomallei vaccines. Front. Cell. Infect. Microbiol. 3:10. 10.3389/fcimb.2013.00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atkins T, Prior R, Mack K, Russell P, Nelson M, Prior J, Ellis J, Oyston PC, Dougan G, Titball RW. 2002. Characterisation of an acapsular mutant of Burkholderia pseudomallei identified by signature tagged mutagenesis. J. Med. Microbiol. 51:539–547 [DOI] [PubMed] [Google Scholar]
- 30.Haque A, Chu K, Easton A, Stevens MP, Galyov EE, Atkins T, Titball R, Bancroft GJ. 2006. A live experimental vaccine against Burkholderia pseudomallei elicits CD4+ T cell-mediated immunity, priming T cells specific for 2 type III secretion system proteins. J. Infect. Dis. 194:1241–1248 [DOI] [PubMed] [Google Scholar]
- 31.Rodrigues F, Sarkar-Tyson M, Harding SV, Sim SH, Chua HH, Lin CH, Han X, Karuturi RK, Sung K, Yu K, Chen W, Atkins TP, Titball RW, Tan P. 2006. Global map of growth-regulated gene expression in Burkholderia pseudomallei, the causative agent of melioidosis. J. Bacteriol. 188:8178–8188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cuccui J, Easton A, Chu KK, Bancroft GJ, Oyston PC, Titball RW, Wren BW. 2007. Development of signature-tagged mutagenesis in Burkholderia pseudomallei to identify genes important in survival and pathogenesis. Infect. Immun. 75:1186–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breitbach K, Kohler J, Steinmetz I. 2008. Induction of protective immunity against Burkholderia pseudomallei using attenuated mutants with defects in the intracellular life cycle. Trans. R. Soc. Trop. Med. Hyg. 102(Suppl 1):S89–S94 [DOI] [PubMed] [Google Scholar]
- 34.Stevens MP, Haque A, Atkins T, Hill J, Wood MW, Easton A, Nelson M, Underwood-Fowler C, Titball RW, Bancroft GJ, Galyov EE. 2004. Attenuated virulence and protective efficacy of a Burkholderia pseudomallei bsa type III secretion mutant in murine models of melioidosis. Microbiology 150:2669–2676 [DOI] [PubMed] [Google Scholar]
- 35.Srilunchang T, Proungvitaya T, Wongratanacheewin S, Strugnell R, Homchampa P. 2009. Construction and characterization of an unmarked aroC deletion mutant of Burkholderia pseudomallei strain A2. Southeast Asian J. Trop. Med. Public Health 40:123–130 [PubMed] [Google Scholar]
- 36.Druar C, Yu F, Barnes JL, Okinaka RT, Chantratita N, Beg S, Stratilo CW, Olive AJ, Soltes G, Russell ML, Limmathurotsakul D, Norton RE, Ni SX, Picking WD, Jackson PJ, Stewart DI, Tsvetnitsky V, Picking WL, Cherwonogrodzky JW, Ketheesan N, Peacock SJ, Wiersma EJ. 2008. Evaluating Burkholderia pseudomallei Bip proteins as vaccines and Bip antibodies as detection agents. FEMS Immunol. Med. Microbiol. 52:78–87 [DOI] [PubMed] [Google Scholar]
- 37.Burtnick MN, Brett PJ, Harding SV, Ngugi SA, Ribot WJ, Chantratita N, Scorpio A, Milne TS, Dean RE, Fritz DL, Peacock SJ, Prior JL, Atkins TP, Deshazer D. 2011. The cluster 1 type VI secretion system is a major virulence determinant in Burkholderia pseudomallei. Infect. Immun. 79:1512–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelson M, Prior JL, Lever MS, Jones HE, Atkins TP, Titball RW. 2004. Evaluation of lipopolysaccharide and capsular polysaccharide as subunit vaccines against experimental melioidosis. J. Med. Microbiol. 53:1177–1182 [DOI] [PubMed] [Google Scholar]
- 39.Brett PJ, Mah DC, Woods DE. 1994. Isolation and characterization of Pseudomonas pseudomallei flagellin proteins. Infect. Immun. 62:1914–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harland DN, Chu K, Haque A, Nelson M, Walker NJ, Sarkar-Tyson M, Atkins TP, Moore B, Brown KA, Bancroft G, Titball RW, Atkins HS. 2007. Identification of a LolC homologue in Burkholderia pseudomallei, a novel protective antigen for melioidosis. Infect. Immun. 75:4173–4180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nieves W, Asakrah S, Qazi O, Brown KA, Kurtz J, Aucoin DP, McLachlan JB, Roy CJ, Morici LA. 2011. A naturally derived outer-membrane vesicle vaccine protects against lethal pulmonary Burkholderia pseudomallei infection. Vaccine 29:8381–8389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su YC, Wan KL, Mohamed R, Nathan S. 2010. Immunization with the recombinant Burkholderia pseudomallei outer membrane protein Omp85 induces protective immunity in mice. Vaccine 28:5005–5011 [DOI] [PubMed] [Google Scholar]
- 43.Atkins T, Prior RG, Mack K, Russell P, Nelson M, Oyston PC, Dougan G, Titball RW. 2002. A mutant of Burkholderia pseudomallei, auxotrophic in the branched chain amino acid biosynthetic pathway, is attenuated and protective in a murine model of melioidosis. Infect. Immun. 70:5290–5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Propst KL, Mima T, Choi KH, Dow SW, Schweizer HP. 2010. A Burkholderia pseudomallei deltapurM mutant is avirulent in immunocompetent and immunodeficient animals: candidate strain for exclusion from select-agent lists. Infect. Immun. 78:3136–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeShazer D, Brett PJ, Carlyon R, Woods DE. 1997. Mutagenesis of Burkholderia pseudomallei with Tn5-OT182: isolation of motility mutants and molecular characterization of the flagellin structural gene. J. Bacteriol. 179:2116–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayden HS, Lim R, Brittnacher MJ, Sims EH, Ramage ER, Fong C, Wu Z, Crist E, Chang J, Zhou Y, Radey M, Rohmer L, Haugen E, Gillett W, Wuthiekanun V, Peacock SJ, Kaul R, Miller SI, Manoil C, Jacobs MA. 2012. Evolution of Burkholderia pseudomallei in recurrent melioidosis. PLoS One 7:e36507. 10.1371/journal.pone.0036507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frey A, Di Canzio J, Zurakowski D. 1998. A statistically defined endpoint titer determination method for immunoassays. J. Immunol. Methods 221:35–41 [DOI] [PubMed] [Google Scholar]
- 48.Sutherland MD, Goodyear AW, Troyer RM, Chandler JC, Dow SW, Belisle JT. 2012. Post-exposure immunization against Francisella tularensis membrane proteins augments protective efficacy of gentamicin in a mouse model of pneumonic tularemia. Vaccine 30:4977–4982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi KH, Mima T, Casart Y, Rholl D, Kumar A, Beacham IR, Schweizer HP. 2008. Genetic tools for select-agent-compliant manipulation of Burkholderia pseudomallei. Appl. Environ. Microbiol. 74:1064–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitchell LA, Hansen RJ, Beaupre AJ, Gustafson DL, Dow SW. 2013. Optimized dosing of a CCR2 antagonist for amplification of vaccine immunity. Int. Immunopharmacol. 15:357–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bosio CM, Goodyear AW, Dow SW. 2005. Early interaction of Yersinia pestis with APCs in the lung. J. Immunol. 175:6750–6756 [DOI] [PubMed] [Google Scholar]
- 52.Liu B, Koo GC, Yap EH, Chua KL, Gan YH. 2002. Model of differential susceptibility to mucosal Burkholderia pseudomallei infection. Infect. Immun. 70:504–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bottex C, Gauthier YP, Hagen RM, Finke EJ, Splettstosser WD, Thibault FM, Neubauer H, Vidal DR. 2005. Attempted passive prophylaxis with a monoclonal anti-Burkholderia pseudomallei exopolysaccharide antibody in a murine model of melioidosis. Immunopharmacol. Immunotoxicol. 27:565–583 [DOI] [PubMed] [Google Scholar]
- 54.Bryan LE, Wong S, Woods DE, Dance DA, Chaowagul W. 1994. Passive protection of diabetic rats with antisera specific for the polysaccharide portion of the lipopolysaccharide isolated from Pseudomonas pseudomallei. Can. J. Infect. Dis. 5:170–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones SM, Ellis JF, Russell P, Griffin KF, Oyston PC. 2002. Passive protection against Burkholderia pseudomallei infection in mice by monoclonal antibodies against capsular polysaccharide, lipopolysaccharide or proteins. J. Med. Microbiol. 51:1055–1062 [DOI] [PubMed] [Google Scholar]
- 56.Mitchell LA, Henderson AJ, Dow SW. 2012. Suppression of vaccine immunity by inflammatory monocytes. J. Immunol. 189:5612–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lazar Adler NR, Govan B, Cullinane M, Harper M, Adler B, Boyce JD. 2009. The molecular and cellular basis of pathogenesis in melioidosis: how does Burkholderia pseudomallei cause disease? FEMS Microbiol. Rev. 33:1079–1099 [DOI] [PubMed] [Google Scholar]
- 58.Nash AA, Jayasuriya A, Phelan J, Cobbold SP, Waldmann H, Prospero T. 1987. Different roles for L3T4+ and Lyt 2+ T cell subsets in the control of an acute herpes simplex virus infection of the skin and nervous system. J. Gen. Virol. 68:825–833 [DOI] [PubMed] [Google Scholar]
- 59.Ho M, Schollaardt T, Smith MD, Perry MB, Brett PJ, Chaowagul W, Bryan LE. 1997. Specificity and functional activity of anti-Burkholderia pseudomallei polysaccharide antibodies. Infect. Immun. 65:3648–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang S, Feng SH, Li B, Kim HY, Rodriguez J, Tsai S, Lo SC. 2011. In vitro and in vivo studies of monoclonal antibodies with prominent bactericidal activity against Burkholderia pseudomallei and Burkholderia mallei. Clin. Vaccine Immunol. 18:825–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, Fiette L, Saeland S, Gicquel B, Winter N. 2005. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood 106:1843–1850 [DOI] [PubMed] [Google Scholar]