

Abstract
Protease-activated receptor 2 (PAR2) is implicated in the pathogenesis of chronic inflammatory diseases, including periodontitis; it can be activated by gingipain and produced by Porphyromonas gingivalis and by neutrophil protease 3 (P3). PAR2 activation plays a relevant role in inflammatory processes by inducing the release of important inflammatory mediators associated with periodontal breakdown. The effects of periodontal treatment on PAR2 expression and its association with levels of proinflammatory mediators and activating proteases were investigated in chronic periodontitis patients. Positive staining for PAR2 was observed in gingival crevicular fluid cells and was reflective of tissue destruction. Overexpression of PAR2 was positively associated with inflammatory clinical parameters and with the levels of interleukin-6 (IL-6), IL-8, tumor necrosis factor alpha, matrix metalloprotease 2 (MMP-2), MMP-8, hepatocyte growth factor, and vascular endothelial growth factor. Elevated levels of gingipain and P3 and decreased levels of dentilisin and the protease inhibitors secretory leukocyte protease inhibitor and elafin were also associated with PAR2 overexpression. Healthy periodontal sites from individuals with chronic periodontitis showed diminished expression of PAR2 mRNA and the PAR2 protein (P < 0.05). Furthermore, periodontal treatment resulted in decreased PAR2 expression and correlated with decreased expression of inflammatory mediators and activating proteases. We concluded that periodontal treatment resulted in decreased levels of proteases and that proinflammatory mediators are associated with decreased PAR2 expression, suggesting that PAR2 expression is influenced by the presence of periodontal infection and is not a constitutive characteristic favoring periodontal inflammation.
INTRODUCTION
Proteases are not merely degradative enzymes responsible for hydrolysis of peptide bonds. Recent evidence shows that these molecules allow communication among host cells and between microorganisms and host cells, playing an important role under numerous pathological conditions. Periodontal tissue breakdown can be mediated by some endogenous host enzymes and bacterial proteases found in the periodontal pocket, such as neutrophil serine proteinase 3 (P3), mast cell tryptase, and gingipains from Porphyromonas gingivalis (P. gingivalis). Recently, it was shown that the biological activities of such proteases can be mediated by the activation of protease-activated receptor 2 (PAR2). PAR2 belongs to the family of G-protein-coupled, seven-transmembrane-domain receptors, and its activation occurs through proteolytic cleavage of the N-terminal domain by serine proteinases, resulting in the generation of a new N-terminal “tethered ligand,” which binds to the receptor itself, resulting in its auto-activation (1).
PAR2 is expressed by many cell types found in the periodontal tissues, including immune cells, osteoblasts, oral epithelial cells, and gingival fibroblasts (2–5). Bacterial and host proteases such as gingipains from P. gingivalis, P3, and mast cell tryptase have been reported to activate PAR2, which highlights the significance of the receptor in the pathogenesis of periodontitis.
PAR2 activation-associated enhanced biosynthesis of proinflammatory mediators has been well established (4–10). A previous study by our group demonstrated that PAR2 mediates host cell mechanisms responsible for increased levels of prostaglandin E2, gamma interferon, interleukin-β (IL-1β), and IL-6 and for the resulting increased alveolar bone loss in a periodontitis model of P. gingivalis infection in mice (8). Then, we demonstrated the involvement of PAR2 in human periodontal disease by reporting increased PAR2 expression in chronic periodontitis patients, where higher expression levels of P3 and P. gingivalis were also verified (11). This study also showed that in deeper periodontal pockets, increased PAR2 expression and significantly increased proinflammatory mediators were observed compared to the expression of the receptor in shallower pockets. We also demonstrated that periodontal pockets presenting P. gingivalis show elevated PAR2 expression compared to sites where the bacterium was not observed, thus suggesting that P. gingivalis may disturb the host inflammatory responses not only by regulating PAR2 function but also by enhancing its genetic expression (12). These results clearly suggested that PAR2 overexpression is an essential element in periodontal inflammation severity.
The present study was undertaken in order to answer the question of whether overexpression of the receptor in chronic periodontitis is due to the presence of the disease or to a constitutive characteristic which favors periodontal inflammation. Therefore, the present study aimed to investigate PAR2 expression in healthy periodontal pockets of periodontitis patients and to evaluate whether the impact of nonsurgical periodontal treatment on the levels of endogenous and bacterial PAR2 activators and serine protease inhibitors, as well as proinflammatory mediators associated with periodontal breakdown, is correlated with PAR2 downregulation. An additional aim was to investigate the types of cells which express PAR2 in the gingival crevicular fluid (GCF) of periodontal patients.
MATERIALS AND METHODS
Study design and patient selection.
Subject recruitment was conducted between July 2010 and February 2012 at the periodontal clinic of the University of São Paulo, School of Dentistry. The participants were informed about the nature of the study and signed a consent form previously approved by the Institutional Committee on Research of the School of Dentistry, University of São Paulo (FR337902, protocol 106/2010).
After an initial screening performed in 343 subjects, 31 moderate chronic periodontitis (CP group) (13) and 31 periodontally healthy individuals (control group) who met the inclusion criteria were included in the study. The inclusion criteria required that subjects be of both genders, that they had never smoked (self-reported data), that they be between the ages of 21 and 63 years, and that they be in good overall health. The exclusion criteria included the following: use of an orthodontic appliance; requirement of systemic antibiotic for measures that might cause transitory bacteremia; use of medications such as antibiotics, phenytoin, calcium antagonists, cyclosporine, or anti-inflammatory drugs within the last 6 months before the initial appointment; regular use of hormonal contraceptives or hormone replacement therapy; history of diabetes, hepatitis, or HIV infection or any other disease that compromises the immune functions; pregnancy or lactation; immunosuppressive chemotherapy; and periodontal treatment within the last 6 months before examination.
The study design consisted of two stages. In stage 1 (baseline), periodontal examination and laboratory analyses were performed. A complete periodontal examination was performed by the same certified periodontist (M. Holzhausen), including plaque index (PI) and gingival index (GI) (14), probing pocket depth (PD), clinical attachment level (CAL), and bleeding on probing (BOP) at six sites (mesio-buccal, buccal, disto-buccal, mesio-lingual, lingual, and disto-lingual) per tooth, using a manual periodontal probe (PCPUNC 15; Hu-Friedy, Chicago, IL, USA). BOP was determined by the presence or absence of bleeding assessed 30 s after probing. An intraexaminer calibration was performed by evaluating 10 nonstudy patients who were examined twice for each clinical parameter (kappa value, 0.92).
Based on the periodontal evaluation, the study population was divided into the following groups: (i) control subjects (control group), having <10% sites with BOP, <1% of sites with a PD of ≥5 mm, no sites with a PD of ≥6 mm, <1% of sites with clinical attachment loss of >2 mm, and no evidence of radiographic bone loss (31 individuals); (ii) moderate chronic periodontitis (CP) subjects, having generalized chronic periodontitis with moderate destruction, that is, having more than 30% of the sites presenting PDs from 3 to 6 mm with CAL up to 4 mm and BOP in more than 30% of the sites (31 individuals).
Control and periodontitis groups received oral prophylaxis and oral hygiene instructions. Patients with chronic periodontitis (CP) received nonsurgical periodontal treatment performed at four to six sessions in accordance with the individual characteristics and conditions. The treatment consisted of elimination of iatrogenic factors (restorations and prostheses, if needed), scaling and root planing through manual instruments (Gracey curettes; Hu-Friedy, Chicago, IL, USA) and sonic devices (Minipiezon; EMS, Switzerland), coronal polishing, clinical integration (temporary cavity restoration and hopeless-tooth extraction, if needed), and review of basic procedures. These procedures were conducted by a single experienced periodontist (V. T. Euzebio Alves).
The posttreatment phase lasted for 6 weeks (15). Within this period, patients received weekly professional plaque control (reinforcement of oral hygiene instructions, supragingival scaling, and prophylaxis) until the reassessment.
In stage 2 (6 weeks after the end of stage 1) subjects with chronic periodontitis who received nonsurgical periodontal treatment (treated chronic periodontitis, or TCP, group) were recalled, and all periodontal and laboratorial parameters were reassessed.
GCF sampling.
In the chronic periodontitis group, the deepest site per quadrant (4 mm ≤ PD ≤ 6 mm) was used to collect GCF. In addition, one healthy periodontal site (no attachment loss) from any of the four quadrants was also sampled in this group. After periodontal therapy, GCF was collected from the same sites of these subjects. In the control group, one healthy periodontal site (no attachment loss) per quadrant was sampled.
Supragingival plaque was carefully removed, and periodontal sites were isolated. Periopaper strips (Periopaper Collection Strip; Oraflow, Plainview, NY, USA) were introduced one at a time into the gingival sulcus or periodontal pocket and removed after 30 s. Two strips were used to collect GCF samples from each site and were analyzed by quantitative PCR (qPCR). In addition, another two strips from the same sites were collected on a different day and used for Western blot (WB) analysis. In both qPCR and WB, the four sites were analyzed separately. Pooled samples were used only for Bio-Plex analysis. First, the individual volume of GCF samples was determined by a moisture meter (Periotron 6000; IDE Interstate, Amityville, NY, USA), and then the four strips were combined for the analyses of protease inhibitors and inflammatory biomarkers. GCF samples were discarded for further analysis if they were visibly contaminated with blood. The strips were stored at −80°C. GCF sample collection for flow cytometry analysis was performed using an intracrevicular washing technique (16) at the same sites used for GCF sampling by paper strips.
Gene expression analysis.
PAR2, P3, gingipain, and dentilisin gene expression from crevicular fluid samples was assessed by quantitative PCR (qPCR). Total RNA (tRNA) was obtained by mixing samples of crevicular fluid in TRizol reagent according to the manufacturer's instructions. For tRNA quantification, the pellet was resuspended in 12 μl of 0.01% diethyl pyrocarbonate (DEPC)-treated water; readings were performed using 1 μl of the sample, in duplicate. After quantification, 10 μl of the remaining tRNA was used for first-strand cDNA synthesis using SuperScript II and RNaseOut. Reverse transcriptase samples were submitted to real-time PCR amplification using GoTaq qPCR Master Mix (Promega) and specific oligonucleotides for PAR2, P3, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), gingipain, and dentilisin as well as constitutive bacteria, which were obtained from GenBank (http://www.ncbi.nlm.nih.gov/tools/primer-blast) (Table 1). Real-time PCRs were performed using the Corbett Research system (Corbett Life Sciences, Sydney, Australia). The conditions for PCR were as follows: 95°C for 2 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Expression data were calculated from the cycle threshold (CT) value using the ΔΔCT method for quantification (17). Gene expression of GAPDH mRNA was used for normalizing PAR2 and P3 expression, and expression of the constitutive gene (bacterial 16S rRNA gene) was used for normalizing gingipain and dentilisin expression. Results were expressed in arbitrary units relative to the variation of induction (fold increase) compared to the control group. All oligonucleotides utilized in this protocol were purchased from Invitrogen Co., San Diego, CA.
Table 1.
Sequence of primers used for cDNA amplification
| Target | Sequencea | GenBank accession no. | Fragment size (bp) |
|---|---|---|---|
| PAR2 | F, 5′-TGCTAGCAGCCTCTCTCTCC-3′ | NM_053897.2 | 92 |
| R, 5′-TGTGCCATCAACCTTACCAA-3′ | |||
| Proteinase 3 | F, 5′-TCTGCTTCGGAGACTCAGGT-3′ | NM_002777.3 | 111 |
| R, 5′-GCGTGAAGAAGTCAGGGAAA-3′ | |||
| GAPDH | F, 5′-TGGTATCGTGGAAGGACTCATGAC-3′ | NM_002046 | 80 |
| R, 5′-ATGCCAGTGAGCTTCCCGTTCAGC-3′ | |||
| Gingipain | F, 5′-CCTACGTGTACGGACAGAGCTATA-3′ | NC_010729 | 70 |
| R, 5′-AGGATCGCTCAGCGTAGCATT-3′ | |||
| Dentilisin | F, 5′-TCTTACGGAACCGAATTTGC-3′ | AE017226.1 | 82 |
| R, 5′-CGT TACCCA TCGCAATTACC-3′ | |||
| 16S rRNA gene | F, 5′-TCGGTATTGAGGAAGGTTGG-3′ | AB791176.1 | 86 |
| R, 5′-CTGCTGGCACGGAGTTAG-3′ |
F, forward; R, reverse.
Western blot analysis.
Samples of crevicular fluid were homogenized in 60 μl of lysis buffer (50 mM Tris-HCl, pH 7.4, containing 1 mM phenylmethylsulfonyl fluoride [PMSF], 2 mM orthovanadate [Na3VO4], 1 mg/ml leupeptin, 1 mg/ml aprotinin, 1 mg/ml pepstatin, EDTA, and 2 mM Triton X-100 1%). Homogenates were centrifuged at 13,000 × g for 30 min. Twenty micrograms of total proteins was separated by electrophoresis on a 15% polyacrylamide gel and transferred onto a nitrocellulose membrane. Nonspecific binding sites were blocked using a blocking solution (3% bovine albumin serum in Tris-buffered saline solution with 1% Tween) for 1 h at 24°C. Membranes were then incubated overnight at 4°C with anti-PAR2 (1:100; Santa Cruz) diluted in blocking solution and then with horseradish peroxidase (HRP)-conjugated anti-mouse (1:2,000; Santa Cruz) diluted in blocking solution for 1 h at room temperature. The immunoreactive bands were revealed by chemiluminescence using an enhanced chemiluminescence (ECL) kit (Thermo Scientific, USA), visualized by autoradiography, and quantified densitometrically using Image J software (National Institutes of Health). Membranes were then stripped, blocked, and incubated with GAPDH antibody (1:1,000; Santa Cruz) and anti-rabbit (1:5,000; Jackson ImmunoResearch), diluted in blocking solution, for 2 h at room temperature. GAPDH bands were used to normalize PAR2 expression levels. Values were expressed as arbitrary units.
Flow cytometric analysis.
Flow cytometry was performed in order to detect the presence of PAR2 on the GCF cell surface. Samples of GCF, collected by an intracrevicular washing technique (16), were centrifuged at 1,800 rpm at 4°C for 10 min and resuspended in 200 μl of phosphate-buffered saline (PBS; pH 7.2) Gibco-Invitrogen). Ten microliters of samples was used to perform cell counts using a Neubauer chamber. Next, the cells were incubated with 2.5 μl of human TruStain FCX (Fc receptor blocking solution) (BioLegend, San Diego, CA, USA) for 10 min to block nonspecific binding. After cells were washed with PBS, they were incubated for 45 min with 2 μl of specific antibodies for epithelial cells (cytokeratin 19; conjugated to peridinin chlorophyll protein [PerCP]) and PAR2 receptor (PAR-2/SAM-11; conjugated to fluorescein isothiocyanate [FITC]) and 1.5 μl of antibody to leukocytes (CD45; conjugated to phycoerythrin [PE]) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After another washing step, the samples were immediately subjected to flow cytometry analysis.
For each sample, up to 10,000 events were acquired. Analysis by flow cytometry was performed using a FACSCalibur flow cytometer (Becton, Dickinson and Co., USA), and recorded events were analyzed using Cell Quest software (Becton, Dickinson and Co., USA). PAR2 expression in epithelial cells and leukocytes was determined as the percentage of positive cells.
Determination of GCF protease inhibitors and inflammatory biomarkers.
The four strips (one per quadrant) were pooled and eluted in 400 μl of PBS. The samples were vortex mixed three times (30 s each), and the strips were removed before sample centrifugation at 10,000 × g for 10 min at 4°C. The amounts of elafin and secretory leukocyte protease inhibitor (SLPI) within the GCF samples were determined using commercially available enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's instructions. GCF samples were diluted in 100 μl of sterile 0.01 M sodium phosphate buffer, pH 7.4, before being applied to the microplates. The concentrations of the protease inhibitors were calculated by the Softmax data analysis program (Molecular Devices, Menlo Park, CA, USA).
To determine GCF levels of IL-6, IL-8, tumor necrosis factor alpha (TNF-α), hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), matrix metalloprotease 2 (MMP-2), and MMP-8, we used a Bio-Plex cytokine assay kit (Human VersaMAP Multiplex Development System; R&D Systems, Minneapolis, MN). The assay was read on a Bio-Plex suspension array system, and the data were analyzed with Bio-Plex Manager software, version 4.0.
Statistical analysis.
Comparisons between pre- and posttreatment as well as between diseased and healthy sites (within the chronic periodontitis group) were analyzed by a paired t test. The differences between the chronic periodontitis group and control group were analyzed by an unpaired t test. The incidence of BOP among groups was analyzed by a chi-square test. For correlation analysis, a linear correlation test was used. Pearson's correlation coefficient was used to calculate bivariate correlations between the covariates.
The analysis and graphics of this study were carried out using the statistical program GraphPad Prism, version 4.0. A P value of <0.05 was considered statistically significant. Data are expressed as means ± standard deviations (SD).
RESULTS
Patients' characteristics.
Thirty-one patients with generalized moderate chronic periodontitis (CP) were matched for age and gender with each control individual. As shown in Table 2 no significant differences were observed between the CP and control groups with regard to the mean age (P = 0.7601) or with regard to the number of teeth (P = 0.8507).
Table 2.
Demographic and clinical parameters of the control group and moderate chronic periodontitis group at baseline and 6 weeks after nonsurgical periodontal treatment
| Parametera | Value for the parameterb |
||
|---|---|---|---|
| Control group (n = 31) | Moderate chronic periodontitis group (n = 31)c |
||
| Baseline | 6 wk posttreatment (n = 31) | ||
| Demographic characteristics | |||
| Age of group (yr [range]) | 43.16 ± 9.60 (24–63) | 44.12 ± 9.08 (21–64) | |
| No. of patients by age | |||
| 20–35 yr | 6 | 6 | |
| 36–50 yr | 18 | 18 | |
| 51–65 yr | 7 | 7 | |
| Gender (no. of patients) | |||
| Male | 17 | 17 | |
| Female | 14 | 14 | |
| No. of teeth (range) | 26.54 ± 1.92 (24–28) | 23.25 ± 3.17 (18–28) | |
| Periodontal characteristics | |||
| PD (mm) | 1.80 ± 0.27 | 2.99 ± 0.65* | 2.35 ± 0.49*† |
| CAL (mm) | 2.31 ± 0.34 | 3.77 ± 0.69* | 3.38 ± 0.74*† |
| BOP (%) | 3.43 ± 3.02 | 63.37 ± 23.3* | 17.64 ± 24.75*† |
| PI | 0.17 ± 0.13 | 1.43 ± 0.45* | 0.31 ± 0.38*† |
| GI | 0.11 ± 0.12 | 1.76 ± 0.42* | 0.54 ± 0.47*† |
PD, probing depth; CAL, clinical attachment level; BOP, bleeding on probing; PI, plaque index; GI, gingival index.
Values for the age of the group, number of teeth, and periodontal characteristics are means ± SD.
*, statistically different compared with the control group (P < 0.05); †, statistically different compared with baseline values (P < 0.0001).
At baseline the mean values of PD, CAL, BOP, PI, and GI were statistically higher (P < 0.0001) in individuals from the CP group than in those from the control group. After periodontal nonsurgical treatment, the individuals showed a significant improvement of all the clinical parameters compared to the baseline values (TCP versus CP, P < 0.0001). However, TCP group mean values for the evaluated clinical parameters were still higher than control values (PD, CAL, and GI, P < 0.0001; BOP, P = 0.0017; PI, P = 0.0407) (Table 2).
Table 3 shows that the clinical parameters (PD and CAL) and GCF volume of the sampled periodontal sites from the CP group were statistically higher (P < 0.05) than those from the control group. Healthy sites at baseline and treated sites (TCP) from the CP group showed significant decreases in PD, CAL, and GCF volume compared with diseased sites at baseline (P < 0.0001).
Table 3.
Clinical parameters and GCF volume of the periodontal sites from control group and moderate chronic periodontitis group at baseline and 6 weeks after nonsurgical periodontal treatment
| Parametera | Value for the parameter (mean ± SD)b |
|||
|---|---|---|---|---|
| Control group | Moderate chronic periodontitis group |
|||
| Baseline | 6 wk posttreatment | Healthy sites | ||
| PD (mm) | 2.08 ± 0.04 | 5.61 ± 0.13* | 3.20 ± 0.13† | 2.65 ± 0.08† |
| CAL (mm) | 2.14 ± 0.05 | 6.53 ± 0.17* | 4.19 ± 0.17*† | 3.18 ± 0.13† |
| GCF vol (μl) | 0.30 ± 0.06 | 0.73 ± 0.05* | 0.41 ± 0.04*† | 0.37 ± 0.05† |
PD, probing depth; CAL, clinical attachment level; GCF vol, gingival crevicular fluid volume.
*, statistically different compared with the control group (P < 0.05); †, statistically different compared with the baseline (P < 0.0001).
PAR2 is downregulated after periodontal treatment.
PAR2 mRNA expression in the gingival crevicular fluid cells in chronic periodontitis patients was significantly higher than in periodontally healthy patients (P = 0.0003) and significantly reduced after nonsurgical periodontal treatment (P < 0.0001) (Fig. 1A).
Fig 1.

(A) Mean PAR2 mRNA expression in the gingival crevicular fluid (GCF) cells of the control group, the periodontitis group before (CP) and after (TCP) nonsurgical periodontal treatment, and healthy sites from the periodontal group. (B) Western blot of PAR2 proteins from control, CP, or TCP group (top panel), quantified by densitometry analysis of the blots (bottom panel). (C) Positive correlation between PAR2 mRNA and PAR2 protein levels. (D) GCF PAR2-expressing epithelial cells and leukocytes from control and periodontitis groups. Data are means ± SD. *, P < 0.05 compared with control values; †, P < 0.05 compared with CP values.
PAR2 protein levels were also elevated in chronic periodontitis patients compared with those of controls (P = 0.0384). Six weeks after periodontal treatment, these levels were significantly reduced (P = 0.0074) (Fig. 1B). Therefore, periodontal treatment not only downregulated the genetic expression of the receptor but also decreased its translated protein levels. Interestingly, there was a very strong positive correlation (r = 0.8935) between PAR2 mRNA expression and PAR2 protein levels (Fig. 1C). Furthermore, healthy periodontal sites from chronic periodontitis individuals showed diminished expression of PAR2 mRNA (P = 0.0092) and PAR2 protein level (P = 0.0413) compared to periodontal sites within the same patient.
There was a strong correlation between PAR2 mRNA and the values for mean PD (r = 0.6308), mean CAL (r = 0.7741), and GCF volume (r = 0.5223).
Moreover, the flow cytometric analysis demonstrated that CP patients had a higher percentage of PAR2-expressing cells than control patients (4.7% ± 0.014% versus 3.3% ± 0.012% for leukocytes and 2.9% ± 0.01% versus 1.5% ± 0.005% for epithelial cells; P < 0.001) (Fig. 1D).
PAR2 potential activators and their inhibitors.
Gingipain mRNA expression was significantly lower in control patients than in chronic periodontitis patients (P = 0.0004). After periodontal treatment, both gingipain and dentilisin mRNA expression levels significantly decreased (P = 0.0039 and P = 0.0234, respectively) (Fig. 2A and B).
Fig 2.

(A) Mean expression of gingipain mRNA in the control group and periodontitis group before (CP) and after (TCP) nonsurgical periodontal treatment and at healthy sites from the periodontal group. Levels of dentilisin (B) and P3 (C) mRNAs in the periodontitis group before (CP) and 6 weeks after (TCP) nonsurgical periodontal treatment are shown. Data are means ± SD. *, P < 0.05, compared with control values; †, P < 0.05, compared with CP values.
Gingipain PAR2 mRNA expression was also significantly lower in healthy sites compared to affected periodontal sites in the same subject from the CP group (P = 0.0438). Moreover, periodontal treatment also decreased P3 mRNA expression in patients with moderate chronic periodontitis (P = 0.0108) (Fig. 2C).
The level of SLPI was significantly decreased in the CP group in comparison with control patients (P = 0.0385). After periodontal treatment, levels of SLPI increased; however, this increase was not significant (P > 0.05) (Fig. 3A). On the other hand, elafin levels were not different among groups; in spite of a trend toward greater values for the control group, there were no significant differences (P = 0.1422) (Fig. 3B).
Fig 3.
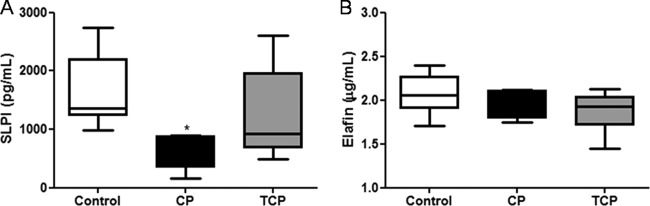
Mean SLPI (A) and elafin (B) GCF levels from the control group and the periodontitis group before (CP) and after (TCP) nonsurgical periodontal treatment are shown. Data are means ± SD (n = 8 per group). *, P < 0.05, compared with control values.
Interestingly, there was a strong correlation between PAR2 mRNA and the expression of gingipain mRNA and P3 mRNA (r = 0.72 and r = 0.49, respectively). In addition, an inverse correlation was observed between PAR2 mRNA and dentilisin mRNA and SLPI levels (r = −0.64 and r = −0.43, respectively).
PAR2 expression is associated with increased levels of inflammatory biomarkers in the GCF.
GCF levels of IL-6 (Fig. 4A), IL-8 (Fig. 4B), TNF-α (Fig. 4C), MMP-1 (Fig. 4D), MMP-2 (Fig. 4E), MMP-8 (Fig. 4F), HGF (Fig. 4G), and VEGF (Fig. 4H) were increased in the gingival crevicular fluid of patients from the CP group compared to levels in the control group (P < 0.05), and they were significantly reduced after periodontal treatment (P < 0.05). Interestingly, a strong correlation was found between PAR2 mRNA and GCF levels of IL-6, IL-8, TNF-α, HGF, and VEGF (r > 0.55).
Fig 4.

GCF levels of IL-6 (A), IL-8 (B), TNF-α (C), MMP-1 (D), MMP-2 (E), MMP-8 (F), HGF (G), and VEGF (H) in patients from the control group and from the periodontitis group before (CP) and after (TCP) nonsurgical periodontal treatment are shown. Data are means ± SD (n = 8 per group). *, P < 0.05, compared with control values; †, P < 0.05, compared with CP values.
DISCUSSION
Protease-activated receptors (PARs) are innate immune receptors that recognize specific bacterial or endogenous serine proteases and initiate defensive immune responses. The receptors from the PAR family have similar structures and mechanisms of activation but can be expressed by different cells and play distinct roles in pathophysiological processes, such as growth, development, inflammation, tissue repair, and pain (18–20). There are four members of this family: PAR1, PAR3, and PAR4, which can be activated by thrombin, and PAR2, which can be activated by serine proteases such as trypsin, neutrophil proteinase 3, tissue factor/factor VIIa/factor Xa, mast cell tryptase, membrane-tethered serine proteinase 1, or gingipains (4, 21).
PAR2 is expressed by epithelial cells, endothelial cells, fibroblasts, osteoblasts, myocytes, neurons, astrocytes, lymphocytes, neutrophils, and mast cells (1, 3, 5, 22–24), where it plays several roles in inflammation (4, 5, 21, 25–29). In fact, PAR2 activation has been associated with several chronic inflammatory conditions (1, 26, 30–32). Furthermore, in vitro and in vivo studies have clearly suggested that PAR2 also plays a role in periodontal inflammation (7, 8, 11, 12). As a novel outcome of the present study, we have clearly demonstrated that epithelial cells and leukocytes present in the gingival crevicular fluid express PAR2 and that the presence of the potential activators, gingipains and P3, and the serine protease inhibitors SLPI and elafin influences its expression.
Overexpression of PAR2 was positively associated with inflammatory clinical parameters and with the levels of IL-6, IL-8, TNF-α, host-derived MMP-2, MMP-8, HGF, and VEGF. Elevated levels of gingipain and P3 and decreased levels of dentilisin and SLPI were also associated with increased PAR2 expression. Healthy sites of periodontitis patients showed decreased PAR2 expression, as did sites of control patients. Furthermore, periodontal treatment resulted in decreased PAR2 expression, correlated with improved clinical parameters, decreased expression of inflammatory mediators and activating proteases, and increased levels of SLPI. We concluded that periodontal treatment resulted in decreased levels of proteases and proinflammatory mediators and is associated with decreased PAR2 expression, suggesting that PAR2 overexpression is due to the presence of periodontal infection and is not a constitutive characteristic favoring periodontal inflammation.
Gingipains have been shown to activate PAR2 in immune inflammatory cells and in cells from the oral epithelial barrier, leading to increased production of proinflammatory mediators (4, 8, 10, 33–35) and activation of signaling pathways associated with increased inflammatory responses (36). In addition, neutrophil protease 3 was also shown to activate host cells through PAR2, inducing the release of proinflammatory cytokines (6), which not only have a direct effect on periodontal destruction but can also act indirectly by upregulating MMP expression (37, 38). Therefore, there is compelling evidence in the literature showing that both P. gingivalis, through its gingipains, and neutrophil P3 make use of host cell PAR2 to exacerbate the inflammation seen in chronic periodontal disease. Accordingly, in our present study, chronic periodontitis patients presented increased PAR2 expression associated with increased expression of proteases and increased levels of proinflammatory mediators responsible for periodontal tissue breakdown.
Secretory leukocyte protease inhibitor (SLPI) is expressed by epithelial and immune cells, where it plays a role as an “alarm” proteinase inhibitor mediating anti-inflammatory and antimicrobial effects. In the present study, SLPI levels correlated inversely with the severity of periodontal inflammation. Thus, decreased levels of SLPI were found in chronic periodontitis patients, whereas periodontal treatment led to its upregulation. Since serine protease-derived activities are crucial for the activation of PAR2, in our study, reduced levels of SLPI were associated with increased expression of the proteases gingipain and P3 and increased PAR2 expression. Similar to our data, a results of a previous study also demonstrated that reduced SLPI levels and higher serine protease activities in the gastric mucosa of Helicobacter pylori-infected individuals were correlated with PAR2 overexpression (39). The decreased levels of SLPI at the sites with P. gingivalis infection might be explained by the ability of the arginine-specific gingipains (Rgps) not only to decrease its secretion but also to degrade it (40–42). The reduced concentrations of SLPI may be associated with the loss of the host protective capacity and increased susceptibility to breakdown from chronic infection. These data reinforce the role played by P. gingivalis on PAR2-mediated periodontal inflammation (12).
In addition, in the present study we demonstrated that systemically healthy periodontitis patients have elevated levels of HGF in the crevicular fluid, which is in agreement with other studies from the literature (43–45). We also observed decreased HGF concentration after periodontal treatment. HGF is a cytokine produced by human gingival and ligament fibroblasts upon stimulation with proinflammatory cytokines and bacterial virulence factors, including gingipains of P. gingivalis. Interestingly, it was shown that production of HGF by human gingival fibroblasts upon stimulation with Rgp occurred through PARs, specifically PAR1 and PAR2 (46). Accordingly, in the present study elevated levels of HGF were associated with increased MMP-2 and MMP-8, and VEGF levels in the crevicular fluid of periodontitis patients were correlated with PAR2 overexpression. Furthermore, this increased expression was also associated with elevated levels of gingipain expression and proinflammatory mediators. Then, these results suggest that gingipains may activate PAR2 in gingival crevicular fluid cells, leading to HGF secretion in inflamed periodontal sites.
The oral bacterial organism Treponema denticola (T. denticola) is an anaerobic spirochete particularly associated with severe and refractory periodontal disease. T. denticola produces an outer membrane-associated chymotrypsin-like protease, named dentilisin, which can degrade a variety of humoral proteins, including basement membrane components, serum proteins, and bioactive peptides (47). Also, it has been suggested that dentilisin may disarm PAR2 or inhibit further activation (8). Interestingly, we have made the novel finding of an inverse relationship between PAR2 expression and the expression of dentilisin in the periodontal sites of patients with moderate chronic periodontitis. Thus, it can be suggested that bacterial proteases produced by other periodontal pathogens could also play a role in activation or suppression of PAR2 function or expression. Whether other PAR2-interfering bacterial proteases exist needs to be further investigated in order to explore their effects on PAR2-mediated periodontal inflammation.
In conclusion, we have shown that PAR2 expression in GCF cells is reflective of periodontal tissue destruction and that periodontal treatment results in its downregulation. Our results link the expression of PAR2 with its known activators and with several tissue breakdown mediators. Therefore, our data support the development of antagonists of human PAR2 or inhibitors of PAR2-activating proteases as potential disease-modifying therapeutic agents for chronic periodontitis.
ACKNOWLEDGMENTS
This work was supported by the São Paulo State Research Foundation (FAPESP, São Paulo, SP, Brazil), research grant 2010/16605-0. V.T.E.A. is a recipient of a FAPESP scholarship. H.A.B.d.S. received a scholarship from the Coordination for the Improvement of Upper Education Personnel (CAPES, Brasília, DF, Brazil). B.N.d.F. received a scholarship from the Research and Technology National Council (CNPq, Brasília, DF, Brazil).
We are extremely grateful for the technical assistance of Meire Hiyane from the Department of Immunology and Adriana C. Levada from the Department of Physiology, Institute of Biomedical Sciences, University of São Paulo, São Paulo, SP, Brazil.
Footnotes
Published ahead of print 16 September 2013
REFERENCES
- 1.Ossovskaya VS, Bunnett NW. 2004. Protease-activated receptors: contribution to physiology and disease. Physiol. Rev. 84:579–621 [DOI] [PubMed] [Google Scholar]
- 2.Lourbakos A, Chinni C, Thompson P, Potempa J, Travis J, Mackie EJ, Pike RN. 1998. Cleavage and activation of proteinase-activated receptor-2 on human neutrophils by gingipain-R from Porphyromonas gingivalis. FEBS Lett. 435:45–48 [DOI] [PubMed] [Google Scholar]
- 3.Abraham LA, Chinni C, Jenkins AL, Lourbakos A, Ally N, Pike RN, Mackie EJ. 2000. Expression of protease-activated receptor-2 by osteoblasts. Bone 26:7–14 [DOI] [PubMed] [Google Scholar]
- 4.Lourbakos A, Potempa J, Travis J, D'Andrea MR, Andrade-Gordon P, Santulli R, Mackie EJ, Pike RN. 2001. Arginine specific protease from Porphyromonas gingivalis activates protease-activated receptors on human oral epithelial cells and induces interleukin-6 secretion. Infect. Immun. 69:5121–5130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uehara A, Muramoto K, Takada H, Sugawara S. 2003. Neutrophil serine proteinases activate human nonepithelial cells to produce inflammatory cytokines through protease-activated receptor 2. J. Immunol. 170:5690–5696 [DOI] [PubMed] [Google Scholar]
- 6.Uehara A, Sugawara S, Muramoto K, Takada H. 2002. Activation of human oral epithelial cells by neutrophil proteinase 3 through protease-activated receptor-2. J. Immunol. 169:4594–4603 [DOI] [PubMed] [Google Scholar]
- 7.Holzhausen M, Spolidorio LC, Vergnolle N. 2005. Proteinase-activated receptor-2 (PAR2) agonist causes periodontitis in rats. J. Dent. Res. 84:154–159 [DOI] [PubMed] [Google Scholar]
- 8.Holzhausen M, Spolidorio LC, Ellen RP, Jobin MC, Steinhoff M, Andrade-Gordon P, Vergnolle N. 2006. Protease-activated receptor-2 activation: a major role in the pathogenesis of Porphyromonas gingivalis infection. Am. J. Pathol. 168:1189–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uehara A, Naito M, Imamura T, Potempa J, Travis J, Nakayama K, Takada H. 2008. Dual regulation of interleukin-8 production in human oral epithelial cells upon stimulation with gingipains from Porphyromonas gingivalis. J. Med. Microbiol. 57:500–507 [DOI] [PubMed] [Google Scholar]
- 10.Giacaman RA, Asrani AC, Ross KF, Herzberg MC. 2009. Cleavage of protease-activated receptors on an immortalized oral epithelial cell line by Porphyromonas gingivalis gingipains. Microbiology 155:3238–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holzhausen M, Cortelli JR, da Silva VA, Franco GCN, Cortelli SC, Vergnolle N. 2010. Protease-activated receptor-2 (PAR2) in human periodontitis. J. Dental Res. 89:948–953 [DOI] [PubMed] [Google Scholar]
- 12.Fagundes JA, Monoo LD, Euzebio Alves VT, Pannuti CM, Cortelli SC, Cortelli JR, Holzhausen M. 2011. Porphyromonas gingivalis is associated with protease-activated receptor-2 up-regulation in chronic periodontitis. J. Periodontol. 82:1596–1601 [DOI] [PubMed] [Google Scholar]
- 13.Armitage GC. 1999. Development of a classification system for periodontal diseases and conditions. Ann. Periodontol. 4:1–6 [DOI] [PubMed] [Google Scholar]
- 14.Löe H. 1967. The gingival index, the plaque index and the retention index systems. J. Periodontol. 38(Suppl):610–616 [DOI] [PubMed] [Google Scholar]
- 15.Segelnick SL, Weinberg MA. 2006. Reevaluation of initial therapy: when is the appropriate time? J. Periodontol. 77:1598–1601 [DOI] [PubMed] [Google Scholar]
- 16.Salonen JI, Paunio KU. 1991. An intracrevicular washing method for collection of crevicular contents. Scand. J. Dent. Res. 99:406–412 [DOI] [PubMed] [Google Scholar]
- 17.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Déry O, Corvera CU, Steinhoff M, Bunnett NW. 1998. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am. J. Physiol. 274:C1429–C1452 [DOI] [PubMed] [Google Scholar]
- 19.Coughlin SR. 2000. Thrombin signalling and protease-activated receptors. Nature 407:258–264 [DOI] [PubMed] [Google Scholar]
- 20.O'Brien PJ, Molino M, Kahn M, Brass LF. 2001. Protease activated receptors: theme and variations. Oncogene 20:1570–1581 [DOI] [PubMed] [Google Scholar]
- 21.Vergnolle N, Wallace JL, Bunnett NW, Hollenberg MD. 2001. Protease-activated receptors in inflammation, neuronal signaling and pain. Trends Pharmacol. Sci. 22:146–152 [DOI] [PubMed] [Google Scholar]
- 22.Bohm SK, Kong W, Bromme D, Smeekens SP, Anderson DC, Connolly A, Kahn M, Nelken NA, Coughlin SR, Payan DG, Bunnett NW. 1996. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem. J. 314:1009–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. 1994. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. U. S. A. 91:9208–9212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cocks TM, Moffatt JD. 2000. Protease-activated receptors: sentries for inflammation? Trends Pharmacol. Sci. 21:103–108 [DOI] [PubMed] [Google Scholar]
- 25.Cenac N, Andrews CN, Holzhausen M, Chapman K, Cottrell G, Andrade-Gordon P, Steinhoff M, Barbara G, Beck P, Bunnett NW, Sharkey KA, Ferraz JG, Shaffer E, Vergnolle N. 2007. Role for protease activity in visceral pain in irritable bowel syndrome. J. Clin. Invest. 117:636–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coughlin SR, Camerer E. 2003. PARticipation in inflammation. J. Clin. Invest. 111:25–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vergnolle N, Bunnett NW, Sharkey KA, Brussee V, Compton SJ, Grady EF, Cirino G, Gerard N, Basbaum AI, Andrade-Gordon P, Hollenberg MD, Wallace JL. 2001. Proteinase activated receptor-2 and hyperalgesia: a novel pain pathway. Nat. Med. 7:821–826 [DOI] [PubMed] [Google Scholar]
- 28.Coelho AM, Vergnolle N, Guiard B, Fioramonti J, Bueno L. 2002. Proteinases and proteinase-activated receptor 2: a possible role to promote visceral hyperalgesia in rats. Gastroenterology 122:1035–1047 [DOI] [PubMed] [Google Scholar]
- 29.Schmidlin F, Amadesi S, Dabbagh K, Lewis DE, Knott P, Bunnett NW, Gater PR, Geppetti P, Bertrand C, Stevens ME. 2002. Protease-activated receptor 2 mediates eosinophil infiltration and hyperreactivity in allergic inflammation of the airway. J. Immunol. 169:5315–5321 [DOI] [PubMed] [Google Scholar]
- 30.Ferrell WR, Lockhart JC, Kelso EB, Dunning L, Plevin R, Meek SE, Smith AJ, Hunter GD, McLean JS, McGarry F, Ramage R, Jiang L, Kanke T, Kawagoe J. 2003. Essential role for proteinase-activated receptor-2 in arthritis. J. Clin. Invest. 111:35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindner JR, Kahn ML, Coughlin SR, Sambrano GR, Schauble E, Bernstein D, Foy D, Hafezi-Moghadam A, Ley K. 2000. Delayed onset of inflammation in protease-activated receptor-2-deficient mice. J. Immunol. 165:6504–6510 [DOI] [PubMed] [Google Scholar]
- 32.Uehara A, Imamura T, Potempa J, Travis J, Takada H. 2008. Gingipains from Porphyromonas gingivalis synergistically induce the production of proinflammatory cytokines through protease-activated receptors with Toll-like receptor and NOD1/2 ligands in human monocytic cells. Cell Microbiol. 10:1181–1189 [DOI] [PubMed] [Google Scholar]
- 33.Moraes TJ, Martin R, Plumb JD, Vachon E, Cameron CM, Danesh A, Kelvin DJ, Ruf W, Downey GP. 2008. Role of PAR2 in murine pulmonary pseudomonal infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 294:L368–L377 [DOI] [PubMed] [Google Scholar]
- 34.Lee KE, Kim JW, Jeong KY, Kim KE, Yong TS, Sohn MH. 2007. Regulation of German cockroach extract-induced IL-8 expression in human airway epithelial cells. Clin. Exp. Allergy 37:1364–1373 [DOI] [PubMed] [Google Scholar]
- 35.Yun LW, Decarlo AA, Hunter N. 2007. Blockade of protease-activated receptors on T cells correlates with altered proteolysis of CD27 by gingipains of Porphyromonas gingivalis. Clin. Exp. Immunol. 150:217–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. 2001. Proteinase-activated receptors. Pharmacol. Rev. 53:245–282 [PubMed] [Google Scholar]
- 37.Gemmell E, Marshall RI, Seymour GJ. 1997. Cytokines and prostaglandins in immune homeostasis and tissue destruction in periodontal disease. Periodontol. 2000 14:112–143 [DOI] [PubMed] [Google Scholar]
- 38.Dennison DK, Van Dyke TE. 1997. The acute inflammatory response and the role of phagocytic cells in periodontal health and disease. Periodontol. 2000 14:54–78 [DOI] [PubMed] [Google Scholar]
- 39.Kandulski A, Kuester D, Mönkemüller K, Fry L, Malfertheiner P, Wex T. 2011. Protease-activated receptor-2 (PAR2) in human gastric mucosa as mediator of proinflammatory effects in Helicobacter pylori infection. Helicobacter 16:452–458 [DOI] [PubMed] [Google Scholar]
- 40.Into T, Inomata M, Kanno Y, Matsuyama T, Machigashira M, Izumi Y, Imamura T, Nakashima M, Noguchi T, Matsushita K. 2006. Arginine-specific gingipains from Porphyromonas gingivalis deprive protective functions of secretory leucocyte protease inhibitor in periodontal tissue. Clin. Exp. Immunol. 145:545–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kantyka T, Latendorf T, Wiedow O, Bartels J, Gläser R, Dubin G, Schröder JM, Potempa J, Meyer-Hoffert U. 2009. Elafin is specifically inactivated by RgpB from Porphyromonas gingivalis by distinct proteolytic cleavage. Biol. Chem. 390:1313–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yin L, Swanson B, An J, Hacker BM, Silverman GA, Dale BA, Chung WO. 2010. Differential effects of periopathogens on host protease inhibitors SLPI, elafin, SCCA1, and SCCA2. J. Oral Microbiol. 2010:2. 10.3402/jom.v2i0.5070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagaraja C, Pradeep AR. 2007. Hepatocyte growth factor levels in gingival crevicular fluid in health, disease, and after treatment. J. Periodontol. 78:742–747 [DOI] [PubMed] [Google Scholar]
- 44.Rudrakshi C, Srinivas N, Mehta DS. 2011. A comparative evaluation of hepatocyte growth factor levels in gingival crevicular fluid and saliva and its correlation with clinical parameters in patients with and without chronic periodontitis: a clinico-biochemical study. J. Indian Soc. Periodontol. 15:147–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lönn J, Johansson CS, Nakka S, Palm E, Bengtsson T, Nayeri F, Ravald N. 18 April 2013. High concentration but low biological activity of hepatocyte growth factor in patients with periodontitis. J. Periodontol. 10.1902/jop.2013.130003 [DOI] [PubMed] [Google Scholar]
- 46.Uehara A, Muramoto K, Imamura T, Nakayama K, Potempa J, Travis J, Sugawara S, Takada H. 2005. Arginine-specific gingipains from Porphyromonas gingivalis stimulate production of hepatocyte growth factor (scatter factor) through protease-activated receptors in human gingival fibroblasts in culture. J. Immunol. 175:6076–6084 [DOI] [PubMed] [Google Scholar]
- 47.Chi B, Qi M, Kuramitsu HK. 2003. Role of dentilisin in Treponema denticola epithelial cell layer penetration. Res. Microbiol. 154:637–643 [DOI] [PubMed] [Google Scholar]