

Abstract
Global proteomic analysis was performed with Shigella flexneri strain 2457T in association with three distinct growth environments: S. flexneri growing in broth (in vitro), S. flexneri growing within epithelial cell cytoplasm (intracellular), and S. flexneri that were cultured with, but did not invade, Henle cells (extracellular). Compared to in vitro and extracellular bacteria, intracellular bacteria had increased levels of proteins required for invasion and cell-to-cell spread, including Ipa, Mxi, and Ics proteins. Changes in metabolic pathways in response to the intracellular environment also were evident. There was an increase in glycogen biosynthesis enzymes, altered expression of sugar transporters, and a reduced amount of the carbon storage regulator CsrA. Mixed acid fermentation enzymes were highly expressed intracellularly, while tricarboxylic acid (TCA) cycle oxidoreductive enzymes and most electron transport chain proteins, except CydAB, were markedly decreased. This suggested that fermentation and the CydAB system primarily sustain energy generation intracellularly. Elevated levels of PntAB, which is responsible for NADPH regeneration, suggested a shortage of reducing factors for ATP synthesis. These metabolic changes likely reflect changes in available carbon sources, oxygen levels, and iron availability. Intracellular bacteria showed strong evidence of iron starvation. Iron acquisition systems (Iut, Sit, FhuA, and Feo) and the iron starvation, stress-associated Fe-S cluster assembly (Suf) protein were markedly increased in abundance. Mutational analysis confirmed that the mixed-acid fermentation pathway was required for wild-type intracellular growth and spread of S. flexneri. Thus, iron stress and changes in carbon metabolism may be key factors in the S. flexneri transition from the extra- to the intracellular milieu.
INTRODUCTION
Shigella species are invasive enteric pathogens that are a significant cause of morbidity and mortality worldwide, with the estimated number of cases exceeding 100 million per year (1). Despite intensive efforts to produce a vaccine, no effective vaccine is currently available, and treatment of the disease is complicated by increased antibiotic resistance. Significant progress has been made in elucidating the mechanisms of pathogenesis of Shigella, particularly Shigella flexneri (2–9). S. flexneri crosses the colonic epithelium via M cells and invades the epithelial cells from the basolateral side (10, 11). Invasion requires a type three secretion system (TTSS), encoded by the mxi and spa genes, to secrete effectors, such as IpaB and IpaC, that induce rearrangement of the host cell cytoskeleton and uptake of the bacteria into vacuoles by epithelial cells (12–15). Once inside the host cells, the bacterium lyses the vacuole membrane (16), replicates in the cytoplasm, and ultimately moves to adjacent cells via mobilization of host cell actin by the bacterial protein IcsA, which is localized to one pole of the bacterial cell (17–19).
The genes required for S. flexneri invasion of host cells are carried on a large plasmid (20, 21). The expression of these genes is regulated in response to environmental cues (22), including changes in temperature, osmolarity, pH, and oxygen or iron levels (23–28). A number of transcription factors critical to the regulation of Shigella virulence genes have been identified. The transcriptional activator VirB and its upstream activator, VirF, are essential for expression of the Shigella invasion genes and these, along with Hfq, Fis, Hns, ArcA, Fnr, and the small RNA RyhB, are responsible for controlling the timing and extent of expression of the virulence genes (24–27, 29–33). In addition, posttranscriptional regulation of Shigella virulence factors has been observed (30, 34).
An understanding of the molecular and physiological basis of invasion of human cells by Shigella spp. has been aided by the use of cell culture assays. An invasion assay to visualize S. flexneri within cultured cells (35, 36) or a plaque assay in which the bacteria invade and spread to adjacent cells, creating visible holes in the monolayer (35, 37), allows rapid assessment of Shigella invasion and cell-to-cell spread. Comparisons of specific mutants with wild-type S. flexneri in these assays have confirmed that ipa genes are required for an early step in invasion (20) and that icsA is needed for actin polymerization and spread (38).
While we are in the process of developing an understanding of the mechanism by which Shigella invades and spreads within the epithelium, we still do not have complete knowledge of the physiology, metabolism, and expression of antigens by the bacterium when growing within the intracellular environment of the host. The metabolic requirements of intracellular bacteria have been inferred from the reduced plaque formation exhibited by bacteria with mutations in specific pathways. Mutations affecting carbon metabolism pathways (csrA and glycolysis) (39), synthesis of aromatic amino acids (aroA, aroC, or aroD) (40, 41), nucleotides (purE, guaAB) (40, 42), and diaminopimelic acid (dapB) (40), and defects in iron transport (sit) (43) all reduce the ability of the bacterium to grow within epithelial cells, suggesting that the bacterium relies on these pathways during some stage of the intracellular lifestyle.
Cultured cells also have been used to probe Shigella gene expression when the bacteria are growing intracellularly (44–46). These studies showed increased expression of genes for iron transport systems, a glucose-phosphate transporter, and amino acid and vitamin biosynthesis. However, these techniques did not detect expression of virulence genes by intracellular bacteria, although it is known that some of these gene products are required for cell-to-cell spread (47). Thus, there are still significant gaps in our knowledge of Shigella in the intracellular environment. Our goal in this study was to characterize the proteome of S. flexneri during intracellular growth in order to determine the antigens the bacterial cells express and the metabolic pathways they use and also to compare the intracellular proteome to the proteome in the extracellular environment and under in vitro growth conditions to determine changes in protein expression. We used both differential two-dimensional (2D) gel display and shotgun proteomics for observation and identification of proteins with altered abundances. Mutational analysis was then used to determine whether selected pathways were required for growth and spread to other cells in culture.
MATERIALS AND METHODS
Bacterial strain and growth conditions.
S. flexneri 2457T stock was maintained frozen in tryptic soy broth (TSB) with 20% glycerol. The frozen culture was streaked on TSB with 1.5% agar and 0.01% Congo red to identify virulent (Congo red-positive) colonies. Red colonies were inoculated into LB medium (10 g tryptone, 5 g yeast extract, and 10 g of NaCl per liter) plus 0.1% deoxycholate (Sigma-Aldrich, St. Louis, MO) to increase invasion, as described previously (48), and grown at 37°C with shaking to mid-exponential phase for infection of cultured cells. A sample of the broth culture, termed in vitro grown (the in vitro sample), was processed for proteome analysis as described below.
Cell culture for isolation of intracellular bacteria.
Henle cells (intestinal 407 cells; American Type Culture Collection, Manassas, VA) were maintained in minimal essential medium (MEM; Invitrogen, Grand Island, NY) supplemented with 10% Bacto tryptone phosphate broth (Difco, Detroit, MI), 10% fetal bovine serum (Invitrogen, Grand Island, NY), 2 mM glutamine (Sigma-Aldrich, St. Louis, MO), and nonessential amino acids (Invitrogen, Grand Island, NY) in a 95% air–5% CO2 humidified atmosphere at 37°C. Prior to infection, Henle cells were subcultured in RoboFlasks (3059; Corning Life Sciences, Tewksbury, MA) at low density and allowed to reach 70 to 95% confluence. The medium was changed prior to infection, and approximately 5 × 109 exponential-phase bacteria were added per RoboFlask at a multiplicity of infection of approximately 200. The flasks were centrifuged at 1,000 × g for 10 min and incubated at 37°C in 5% CO2 for 30 min to allow adherence and invasion. The medium containing extracellular bacteria was removed and centrifuged to harvest the extracellular bacteria (the extracellular sample). After washing the monolayer 4 times to remove the remaining adherent bacteria, the cell culture medium was replaced with fresh medium containing 40 μg/ml gentamicin and incubated for a further 2.5 h to allow growth of the intracellular bacteria. At the time of harvest, infected Henle cells contained 10 to 50 bacteria/cell, as determined by plating a sample of the infected monolayer.
To determine whether the extracellular bacteria retained the ability to invade, subconfluent monolayers of Henle cells (6-well plates) were infected with wild-type S. flexneri 2457T. After 30 min, bacteria remaining in the cell culture medium of each well (the extracellular sample) were gently resuspended, and the contents of each well were transferred to a second uninfected subconfluent monolayer and incubated for 30 min. After infection, each set of monolayers was washed with phosphate-buffered saline (PBS) and incubated for 1 h with medium containing gentamicin. Monolayers were then washed, stained, and scored for invaded cells. A minimum of 300 Henle cells was counted for each well, and cells were scored as infected if the cell contained 3 or more bacteria. Samples of the inoculum and of the extracellular bacteria were diluted and plated on Congo red agar to screen for white colonies, which indicated loss of the virulence plasmid.
Protein sample preparation.
The infected Henle cell monolayers were rinsed 2 times with 10 ml warm PBS-D (26.5 mM KCl, 137 mM NaCl, 1.5 mM KH2PO4, 8 mM K2HPO4; pH 7.5) and lysed with 5 ml per flask of 5% (wt/vol) saponin in PBS-D for 5 min. Lysates were collected on ice, and flasks were rinsed with an additional 5 ml of PBS-D per flask, which was added to the lysate. Care was taken to maintain the samples at 4°C throughout harvest of the bacteria. Lysates were centrifuged for 7 min at 5,800 × g and resuspended in approximately 1.5 ml of saline. Lysates were layered on top of 12 ml of 65% isotonic Percoll (GE Healthcare, Pittsburgh, PA) in an ultracentrifuge tube (Beckman 342413). Tubes were sealed and centrifuged for 15 min at 30,000 × g, 4°C. The bacteria formed a band near the bottom of the tube. The fraction containing the bacteria was collected, added to 25 ml saline, and centrifuged for 7 min at 5,800 × g to remove the separation medium. Pellets were resuspended in 1.5 ml saline, and a sample was removed to determine the number of bacteria, which ranged from 4.1 × 108 to 1.2 ×1010 cells per lysate sample. The remaining bacteria were then pelleted in a 2-ml microcentrifuge tube (intracellular sample). For shotgun proteomic experiments, aliquots of the bacterial samples were suspended in TTE lysis buffer [25 mM Tris-O-acetate (pH 7.8), 0.05% Triton X-100, 5 mM Na-EDTA, 1 mM benzamidine, and 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride]. Samples for 2D gel electrophoresis were suspended in GR lysis buffer {8 M urea, 2 M thiourea, 4% (wt/vol) 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate, 18 mM dithiothreitol, and 0.5% (vol/vol) Bio-Lyte pH 3 to 10 carrier ampholytes}. The samples were frozen at −80°C until further use.
Sample preparation for shotgun proteomics.
Lysates in TTE buffer were thawed, supplemented with chicken lysozyme (150 μg/ml), and agitated at 20°C for 1 h. Protein extraction and size exclusion chromatography (SEC) steps were essentially performed as described previously (49), with the exception that only two SEC protein fractions were collected, one in the mass range above 15 kDa and the other lower than 15 kDa. Based on SDS-PAGE analysis, the lower SEC mass fraction contained most of the lysozyme and was discarded. The higher SEC mass fraction was retained from three biological replicates each of the in vitro, extracellular, and intracellular S. flexneri lysates, and protein concentrations were estimated and averaged based on two methods: the 2D Quant kit (GE Healthcare) method and Coomassie brilliant blue staining of total protein in SDS-PAGE gels. Equivalent biological triplicate samples (in vitro, extracellular, and intracellular) were combined in equal proportions to obtain ∼250 to 300 μg total protein for filter-aided sample preparation (FASP), with the exception of the intracellular sample pool, for which the entire protein amount from two samples (50 and 75 μg) was combined with ∼150 μg of the third replicate; normalization was not possible in the latter case. FASP enzymatic digestions were performed at a protein-to-trypsin ratio of 50:1 (wt/wt). All three FASP digests were fractionated on a nonporous Inertsil C18 reverse-phase (RP) column (4.6 mm by 150 mm; GL Sciences, Rolling Hills Estates, CA) at a flow rate of 0.5 ml/min as follows: 100% buffer A (5 mM NH4HCO2 [pH 9], 2% CH3CN) over 5 min, linear gradient to 100% buffer B (5 mM NH4HCO2 [pH 9], 80% CH3CN) over 45 min; isocratic flow at 100% B for 5 min. The early eluting and late eluting fractions had low peptide contents based on the A214 UV trace and were combined. In total, 36 C18 RP fractions were prepared for lyophilization, dried, and stored at −80°C until use for liquid chromatography-tandem mass spectrometry (LC-MS/MS). Intracellular FASP digests contained lower peptide concentrations in the 36 fractions. Therefore, in vitro and extracellular samples were run in triplicate LC-MS/MS experiments, and the intracellular samples were run in duplicate LC-MS/MS experiments.
Two-dimensional gel electrophoresis and differential gel display.
S. flexneri lysates in GR buffer were thawed, incubated at 20°C for 30 min, vortexed intermittently to complete protein solubilization, and centrifuged at 16,100 × g for 30 min. Protein concentrations in the supernatants were measured using the 2D Quant kit. Pooling of individual S. flexneri lysates was not done. Aliquots of 150 μg protein each were subjected to 2D gel electrophoresis in 12-gel batches by using established experimental methods (50). Large-format slab gels with acrylamide gradients from 8 to 18% total acrylamide monomer concentrations in g/100 ml (25 by 19.5 by 0.15 cm) were fixed, stained with Coomassie brilliant blue G250, destained, and scanned as 6-bit TIFF images. TIFF files were imported into Proteomweaver v.4.0 (Bio-Rad Laboratories), and differential display experiments were conducted as described previously (51). Prematch and pair match-based normalizations were performed for the image analysis experiments to adjust for differential Coomassie brilliant blue staining intensities and so that only the matched spot pairs would be submitted for comparative numerical analysis. The three groups contained 8, 11, and 5 2D gel images for the in vitro, extracellular, and intracellular S. flexneri lysates, respectively, with each group composed of gels derived from three distinct biological replicates. The x-y positions of 25 cytoplasmic protein spots were used as landmarks for mass and pI calibrations of 2D gels. The Mann-Whitney test, a nonparametric two-sample distribution-free t test that determines whether two independent samples of observations come from the same distribution, was used for statistical significance analysis of protein spot differences. Protein spots with P values of <0.05 and abundance ratios of ≥1.5 were generally accepted as differentially abundant in the two-group comparisons. Data were also imported into the Multiple Experiment Viewer (MeV) software (52), with which a multiple testing correction module was used to calculate Benjamin-Hochberg test adjusted P values (53). More than 500 protein spots, including those with P values of <0.05 in binary data comparisons, were processed using in-gel trypsin digestion. Matrix-assisted laser desorption ionization–tandem time of flight analysis and LC-ion trap-MS/MS analysis of 2D gel spots were performed as previously described (54). This resulted in 1,293 successful protein spot analyses, corresponding to 448 unique S. flexneri gene products.
LC-MS/MS analysis.
LC-MS/MS methods published previously were used for shotgun proteomics (36 fractions per sample) (55). An LTQ XL nano-electrospray ionization linear ion trap mass spectrometer (Thermo-Fisher, San Jose, CA) was coupled to an upfront Agilent 1200 solvent delivery system equipped with a BioBasic C18 column (75 μm by 10 cm; New Objective, Woburn, MA) for high-resolution peptide separation. Calibration was performed with 200 nmol human [Glu1]-fibrinopeptide B (molecular weight, 1,570.57); retention times with a CH3CN gradient varied by less than 10%, and peaks represented ion counts with widths at half-heights of <0.25 min, signal/noise ratios of >200, and heights of >107. Following desalting on a C18 trapping cartridge, 53-min binary gradients were run, starting with 97% solvent A (0.1% HCOOH) to 80% solvent B (0.1% HCOOH, 90% acetonitrile). Spectra were acquired in automated MS/MS mode, with the top five parent ions selected for fragmentation in scans of the m/z range from 300 to 1,500 and a dynamic exclusion setting of 70 s. LTQ search parameters (+1 to +3 ions) included mass error tolerances of ±1.4 Da for peptide precursor ions and ±0.5Da for fragment ions (monoisotopic mass values), allowed one missed tryptic cleavage, and were set for Cys carbamidomethyl modification and Met oxidation of peptides as fixed and variable modifications, respectively. The search engine was Mascot v.2.3 (Matrix Science, Boston, MA). Protein sequences were derived from the annotated genome of Shigella flexneri 2a strain 2457T and downloaded from the RefSeq protein sequence data set in NCBI. The MS data sets are also available in the repository PRIDE (accession numbers 18992 to 18999).
Proteome-wide quantification using the APEX tool.
Protein identifications from Mascot v.2.3 searches required at least one unique peptide with an E value of <0.1. Peptide false-discovery rates (FDRs) were determined by searching a randomized S. flexneri protein sequence database in Mascot. The average FDR for eight 2D LC-MS/MS experiments was ∼1%. Following file conversion into the mzXML format, MS data were rescored using the algorithms PeptideProphet and ProteinProphet (56). Data in the prot.xml format were processed using the APEX quantitative proteomics tool v.1.1, which was designed in 2008 (57) and utilizes machine learning-based methods developed in 2007 (58). Using 30 physicochemical peptide properties selectable within the APEX tool set (we used default settings), ARFF files for the estimated MS detectability (Oi) computations (the expected number of unique proteotypic peptides for a given protein) for all protein sequences were generated for a set of 100 abundant bacterial proteins with mass values of >30 kDa to obtain a good representation of tryptic peptides. Observation of tryptic peptides in the training data set was correlated with the 30 selected peptide properties, resulting in the computed Oi values; in this case, 5,397 proteins were annotated in the S. flexneri 2457T sequence database. The following equation was used to calculate APEXi values from 2D LC-MS/MS data (with the concentration in molecules per cell [C], probability scores for protein identification [pi], and spectral counts [ni] as variables): . After setting the protein FDR at 1%, proteins identified at a 99% confidence level were used for spectra counting. The APEX method integrates one normalization step by calculating spectral count-based protein abundances normalized for peptide observability with the mass spectrometer. Based on the estimated number of protein molecules per Escherichia coli cell, with this method a second normalization factor of 2.5 × 106 was integrated, providing the protein abundances as the number of protein molecules per cell (59).
Bioinformatics and statistical methods.
Methods to analyze global S. flexneri proteomic changes included protein and pathway databases such as EcoCyc (ecocyc.org) and Uniprot (www.uniprot.org), as well as bioinformatics tools such as PSORTb (psort.org/psortb/) and GO ontology terms (geneontology.org). Differential protein abundances used in comparing shotgun proteomics data sets from distinct growth conditions were determined using a two-tailed Z-test. A statistical significance threshold value of ≥(±2.2) was accepted.
Construction and testing of mutants.
Mutants were constructed by P1 vir transduction of the deletions from the Keio Collection (60) strains (Table 1) as described previously (61). All mutations were confirmed by PCR amplification and agarose gel analysis. The dppB and adhE mutants were constructed by the method of Datsenko and Wanner (62). Briefly, primers corresponding to those used to create the deletions in the respective genes in the Keio Collection were synthesized, introducing changes as necessary to match the 2457T genomic sequence. Using plasmid pKD13 as the template, PCR products were generated for the respective genes, purified, and electroporated into 2457T cells carrying plasmid pKD46. Resulting mutants were screened by PCR, sequenced, and tested to ensure loss of the pKD46 plasmid.
Table 1.
Strains used in the studya
| Species and strain | Genotype | Source |
|---|---|---|
| E. coli | ||
| JW1806 | manX::kan | 60 |
| JW2154 | fruA::kan | 60 |
| JW1375 | ldhA::kan | 60 |
| JW3580 | lldD::kan | 60 |
| JW0886 | pflB::kan | 60 |
| JW1375 | ldhA::kan | 60 |
| JW5060 | bolA::kan | 60 |
| JW3525 | cspA::kan | 60 |
| S. flexneri | ||
| 2457T | Wild type, serotype 2a | Walter Reed Army Institute of Research |
| SM100 | Serotype 2a Strr | 90 |
| CFS202 | 2457T manX::kan | This study |
| CFS203 | 2457T fruA::kan | This study |
| CFS204 | 2457T lldD::kan | This study |
| CFS212 | 2457T adhE::kan | This study |
| CFS218 | 2457T ldhA::kan | This study |
| CFS217 | 2457T pflB::kan | This study |
| CFS205 | 2457T bolA::kan | This study |
| CFS207 | 2457T cspA::kan | This study |
| CFS209 | 2457T ΔfruA manX::kan | This study |
| CFS224 | 2457T ΔadhE ΔpflB ldhA::kan | This study |
| HGS101 | 2457T dppB::kan | This study |
| SM100ahpC | SM100 ahpC::kan | This study |
Strr, streptomycin resistant.
Double and triple mutants were constructed by first transforming single mutants with the temperature-sensitive plasmid pCP20 (62) in order to remove the Kanr cassette while maintaining the gene deletion. Loss of the Kanr cassette was confirmed by PCR and screening on kanamycin as well as ampicillin plates to ensure loss of pCP20. Resulting Kans mutants were transduced with P1 phage stocks generated from the desired Keio mutant to incorporate the second mutation. The triple mutant adhE pflB ldhA::kan was created by successive removal of the adhE and pflB Kanr cassettes from these genes followed by transduction with the ldhA mutation.
Plaque assay.
The plaque assay was modified from the method of Oaks (63), as described previously (24). Briefly, bacterial strains were grown to mid-log phase at 37°C and diluted to 106 to 104 CFU/ml. Bacterial dilutions (0.1 ml) were added to Henle cells in 6-well plates. After low-speed centrifugation, the infected monolayers were incubated at 37°C in 5% CO2 for 45 min to allow invasion. The monolayers were then washed with PBS and overlaid with fresh medium containing 20 μg gentamicin per ml. The infected monolayers were incubated for 72 h, washed twice with PBS, and stained with Wright-Giemsa stain to visualize the plaques.
RESULTS
S. flexneri 2457T was grown under three different conditions to assess the proteins synthesized by intracellular bacteria compared to that in other environments. Bacteria grown under standard laboratory conditions (in vitro, LB medium with aeration at 37°C to exponential phase) and in cell culture but outside the Henle cells (extracellular) (Fig. 1A) were compared to intracellular bacteria (Fig. 1B) to assess the changes in proteins synthesized in response to invasion of host cells (Fig. 1). To verify that the extracellular bacteria did not represent a population that was unable to invade, samples of extracellular bacteria taken from the cell culture medium of infected monolayers were each added to a second monolayer, and invasion was measured. An average of 30.5% of the Henle cells had 3 or more intracellular bacteria in the primary invasion assay, and the monolayers infected with the transferred, extracellular bacteria had an average of 26.3% infected cells. The bacteria in the original inoculum and the extracellular sample were also plated on Congo red agar to screen for white colonies, indicative of loss of virulence plasmid genes. Less than 1% of the colonies in the inoculum or the extracellular sample were white. These data indicated that the extracellular bacteria did not represent a genetically distinct population that had lost the ability to invade. 2D gel analysis of the bacterial proteins in these samples resulted in identification of 448 unique S. flexneri gene products, 127 of which were altered in abundance (P < 0.05, Kruskal-Wallis statistic) in a three-group comparison (in vitro, extracellular, and intracellular). Details for proteins with different abundances can be found in Data Set S1 in the supplemental material.
Fig 1.

Gel display for comparison of extracellular and intracellular bacterial proteomes during coculture with Henle cells. (A) Extracellular S. flexneri cells; (B) intracellular S. flexneri cells. The depicted Coomassie brilliant blue-stained gel images (20 by 25 by 0.15 cm) are representative for their groups (extracellular n = 11; intracellular n = 5). The spots are denoted in the gels with their short protein names, some of which are also listed in Tables 2, 3, 5, and 6. The molecular mass scale is denoted on the ordinate, and the isoelectric point scale (pI) is on the abscissa. Protein spots in red were increased in abundance in the respective gel, and protein spots in black were decreased compared to the other sample. Protein spots in blue were not altered to a statistically significant extent in the differential displays. The figure does not include all of the statistically significant changes (P < 0.05) with ratios of <0.66 or >1.5. More comprehensive data are provided in Data Sets S1 and S2 in the supplemental material.
The 2D gel surveys allowed visualization of differences in amounts of abundant proteins, but they were not sufficiently sensitive for measurements of low-abundance proteins and primarily profiled soluble cytoplasmic and periplasmic S. flexneri proteins. Therefore, a shotgun proteomic strategy was also used. Although shotgun proteomics infers the association of identified peptides with their proteins of origin, it is an established technique that works well for surveys of bacterial proteomes with infrequent observations of degenerate peptides and protein isoforms. A robust protein denaturation and digestion technique (FASP) resulted in identifications of many integral membrane proteins and lipoproteins, and an average of 1,170 S. flexneri proteins were identified per experiment (n = 8). With abundance ratios ≥1.5/≤0.67 and a Z-score of ≥(±2.2), between 297 and 343 proteins were altered in two-group comparisons (see Data Set S2 in the supplemental material). Congruence was generally achieved by comparing protein quantities surveyed with both the 2D gel and LC-MS/MS methods, although large differences in quantitative ratios were observed for a few low-Mr, high-Mr, membrane-attached, and membrane-integrated proteins. Membrane and high-Mr proteins tend to be difficult to quantify reliably in 2D gels due to insufficient solubility and resolution. Proteins with very low native mass values (<15,000 kDa) may be lost in the SEC separation step prior to LC-MS/MS analysis. We identified 190 intracellular bacterial proteins whose abundance was significantly changed compared with that in both the extracellular and in vitro-grown bacteria, supporting the notion of a distinct life stage of S. flexneri inside of epithelial cells. These data were analyzed further to determine patterns of synthesis of known virulence proteins and of proteins for metabolic pathways that might be important to intracellular growth and survival of S. flexneri.
Virulence factors.
Injection of virulence effectors through the TTSS needles is required for invasion of host cells. In a previous study, we noted that there was a reduction in the amount of the invasion proteins (e.g., IpaB and IpaC) immediately after entry into the host cell, with a modest increase in some of the proteins 2 h after invasion (64). At the later time point used in the present study (3 h after invasion), the proteome analysis showed significantly increased levels of these proteins (Table 2). The TTSS components, including MxiC, MxiD, and MxiJ and the effector proteins IpaA, IpaC, IpaD, and VirA, were found at levels as high as or higher than in extracellular bacteria. These proteins are all required for entry into the host cells and are also present in the bacteria used for inoculation of the Henle cells. The relatively higher levels of virulence-related proteins in the intracellular bacteria indicated that the synthesis of these proteins continues or increases as the bacteria replicate intracellularly and begin to move into adjacent cells.
Table 2.
Selected differentially expressed proteins with virulence functions
| Protein name | Protein descriptiona | Avg relative abundanceb |
Trancriptional regulator(s)c | Functional role | ||
|---|---|---|---|---|---|---|
| In vitro | Ext | Int | ||||
| VirA | Cysteine protease-like VirA | 5,913 | 3,041 | 5,528 | VirB+, VirF+, MxiE+ | Interactions with host cells |
| IpaD | Invasin IpaD | 6,607 | 4,982 | 10,432 | VirB+, VirF+ | Interactions with host cells |
| IpaA | Invasin IpaA | 2,405 | 1,076 | 1,322 | VirB+, VirF+ | Interactions with host cells |
| IpaC | Invasin IpaC | 15,197 | 6,605 | 14,728 | VirB+, VirF+ | Interactions with host cells |
| IcsA | OM protein autotransporter IcsA | 426 | 756 | 2,517 | VirF+ | Interactions with host cells |
| IcsB | Virulence protein IcsB | 267 | 284 | 1,076 | VirF+ | Interactions with host cells |
| IcsP | IcsA-specific OM protease | 704 | 158 | 2,349 | VirB+, VirF+ | TTSS effector processing: proteolysis |
| MxiC | Mxi-Spa secretion machinery protein MxiC | 2,519 | 1,520 | 4,427 | VirB+, VirF+ | TTSS substrate specificity factor |
| MxiD | OM protein MxiD | 1,027 | 990 | 2,316 | VirB+, VirF+ | TTSS assembly |
| MxiJ | Lipoprotein MxiJ | 1,186 | 1,223 | 3,713 | VirB+, VirF+ | TTSS assembly |
| SepA | Extracellular serine protease SepA | 690 | 416 | 1,215 | Interactions with host cells: proteolysis | |
| H-NS | DNA-binding protein, nucleoid associated | 21,134 | 30,777 | 13,926 | Negative regulation of virF expression | |
| StpA | DNA-binding protein, nucleoid-associated transcriptional repressor | 2,081 | 3,725 | 69 | Lrp+, H-NS− | Negative regulation of virF expression |
| PldA | Phospholipase A | 572 | 759 | 1,440 | Lipid catabolic process | |
| OmpX | OM protein A | 17,618 | 10,306 | 25,461 | Fnr− | Potential host cell surface adherence |
OM, outer membrane.
Average relative APEXi protein abundance levels for in vitro-grown S. flexneri and for extracellular (Ext) and intracellular (Int) bacteria during coculture with Henle cells. More-detailed analyses are provided in Data Set S2 in the supplemental material.
Known or predicted transcriptional regulator(s) of the indicated gene, based on data for the closely related bacterium E. coli (www.ecocyc.org). +, positive regulation; −, negative regulation.
Virulence proteins required for steps subsequent to invasion also were detected (Table 2). IcsA, which is required for actin-based motility in the host cell (19), and IcsB, which is required for evasion of the host autophagy defense system (65) and for lysis of the double membrane as the bacteria move to adjacent cells (66), were both synthesized at higher levels intracellularly than in either the extracellular or in vitro-grown bacteria. We also noted a decrease in the abundance of the nucleoid structuring proteins H-NS and StpA (67, 68) in the intracellular bacteria (Table 2). Since these are negative regulators of virF, which encodes a positive regulator required for expression of Shigella virulence genes, the reduced levels of H-NS and StpA may account for some of the increase in virulence proteins in the intracellular environment.
Additional proteins that may play a role in S. flexneri virulence were also present at elevated levels during intracellular growth. The serine protease autotransporter SepA, which contributes to tissue invasion (69), and the phospholipase PldA, which is predicted to be in the outer membrane, were markedly increased in abundance intracellularly (Table 2). In 2D gels, peptide mass fingerprints identified only the C-terminal outer membrane-integrated 28-kDa domain of SepA, suggesting that the protease domain was previously released from the S. flexneri cell surface. The outer membrane protein OmpX also increased in abundance intracellularly. There are orthologs of OmpX in Yersinia enterocolitica (Ail and YadA) that play a role in adhesion and in binding to complement factor C4b (70–72), but to our knowledge, a role for OmpX in Shigella virulence has not been reported.
Carbon sources and respiration and fermentation pathways.
Proteins involved in carbohydrate uptake and metabolism were found at high levels in intracellular bacteria (Table 3). To highlight the connectivity of these and other changes in cellular metabolism proteins, which are discussed in the following paragraphs, we have provided a visual representation of the changes in the schematic in Fig. S1 in the supplemental material. Proteins found at high levels in intracellular bacteria include FruA and FruB for fructose import and utilization (also highly abundant in the extracellular milieu) and the mannose importer ManXYZ. To determine whether these pathways for fructose or mannose import are required for intracellular growth, we constructed strains carrying mutations in these genes and tested the mutants for plaque formation. Mutations affecting individual sugar transporters did not prevent the bacteria from replicating intracellularly and forming plaques (Table 4), suggesting that the bacteria are able to use multiple carbon sources inside the host cell. Further, the peptide transporter DppA was strongly induced (Table 3), suggesting that S. flexneri scavenges dipeptides from the cytoplasmic host environment, but as noted with the sugar transporters, loss of dppA alone did not prevent plaque formation (Table 4).
Table 3.
Selected differentially expressed proteins with carbon and energy metabolism functions
| Protein name | Protein descriptiona | Avg relative abundanceb |
Transcriptional regulator(s)c | Functional role(s) | ||
|---|---|---|---|---|---|---|
| In vitro | Ext | Int | ||||
| PfkI | 6-Phosphofructokinase 1 | 590 | 3,497 | 1,913 | Cra− | Glycolysis |
| PfkII | 6-Phosphofructokinase 2 | 851 | 552 | 1,334 | Glycolysis | |
| GpmA | Phosphoglyceromutase | 14,384 | 11,311 | 23,438 | Fur− | Glycolysis |
| CsrA | Carbon storage regulator | 31,178 | 33,401 | 6,548 | Regulator of glycolysis, glycogen synthesis and storage, biofilm formation, motility | |
| AdhE | Bifunctional acetaldehyde CoA/alcohol dehydrogenase | 914 | 10,113 | 11,712 | Fnr+, Fis+, NarL−, FruR− | Mixed-acid fermentation: acetate |
| LldD | l-Lactate dehydrogenase | 460 | 909 | 3,497 | ArcA− | Mixed-acid fermentation: l-lactate |
| PflB | Formate acetyltransferase 1 | 9,921 | 15,006 | 15,500 | DksA+, ArgR− | Mixed-acid fermentation: pyruvate |
| FucO | l-1,2-Propanediol oxidoreductase | 1,464 | 1,608 | 2,969 | FruR− | Mixed-acid fermentation: fucose |
| YfiD | Autonomous glycyl radical cofactor | 14,343 | 17,557 | 26,553 | ArcA+, Fis+, Fur− | Mixed-acid fermentation: pyruvate |
| FrdA | Fumarate reductase flavoprotein subunit | 2,912 | 2,582 | 1687 | Fnr+, NarL− | Mixed-acid fermentation: fumarate |
| FocA | Formate transporter | 236 | 1,197 | 0 | ArcA+, Fnr+, Crp+ | Mixed-acid fermentation: formate export; acid stress response |
| SucC | Succinyl CoA synthetase β subunitd | 10,429 | 7,795 | 3,616 | Crp+, Fur+, Fnr− | TCA cycle |
| SucA | α-Ketoglutarate decarboxylase | 5,503 | 3,617 | 1,472 | Crp+, Fur+, Fnr− | TCA cycle |
| IcdA | Isocitrate dehydrogenase | 21,212 | 13,872 | 9,595 | FruR+, ArcA− | TCA cycle |
| SdhA | Succinate dehydrogenase flavoprotein | 8,711 | 4,567 | 1,580 | Crp+, Fur+, Fnr− | TCA cycle |
| SdhB | Succinate dehydrogenase Fe-S subunitd | 5,069 | 2,455 | 637 | Crp+, Fur+, Fnr− | TCA cycle |
| AcnB | Bifunctional aconitate hydratase 2/2-methylisocitrate dehydratased | 15,645 | 11,454 | 3,584 | Crp+, ArcA−, Fis− | TCA cycle |
| GlgB | Glycogen branching enzyme | 1,409 | 457 | 2,131 | PhoP+ | Glycogen biosynthesis and storage |
| GlgC | Glucose-1-phosphate adenylyltransferase | 2,157 | 2,187 | 3,301 | PhoP+ | Glycogen biosynthesis and storage |
| FruK | 1-Phosphofructokinase | 0 | 3,582 | 680 | Cra− | Carbohydrate import and utilization |
| FruA | Fructose-specific PTS IIBC component | 73 | 3,674 | 2,342 | Cra− | Carbohydrate import and utilization |
| FruB | Bifunctional fructose-specific PTS IIA protein | 497 | 8,386 | 4,883 | Cra− | Carbohydrate import and utilization |
| ManZ | Mannose-specific PTS protein IID | 1,718 | 3,277 | 5,693 | Crp+, FruR−, NagC− | Carbohydrate import and utilization |
| ManX | Mannose-specific PTS enzyme IIAB | 7,466 | 10,121 | 18,857 | Crp+, FruR− | Carbohydrate import and utilization |
| GlpD | Glycerol-3-phosphate dehydrogenase | 2,539 | 1,120 | 249 | TrpR− | Phospholipid and glycerol metabolism |
| GlpQ | Periplasmic glycerophosphodiester phosphodiesterase | 5,858 | 3,487 | 2,328 | Fis+, Fnr+, IhfB− | Phospholipid and glycerol metabolism |
| DppA | Dipeptide transport protein | 421 | 385 | 9,775 | IhfB+, Fnr− | Peptide and heme import |
| YdgR | Tripeptide/dipeptide:H+ symporter | 0 | 83 | 1,407 | OmpR+ | Peptide import |
| MglB | Galactose-binding transport protein | 12,567 | 6,840 | 2,926 | Crp+, GalR− | Carbohydrate import and utilization |
| NuoE | NADH dehydrogenase subunit Ed | 2,074 | 2,288 | 117 | Fis+, NarL+, ArcA+ | Major aerobic electron transport chain |
| NuoC | Bifunctional NADH:ubiquinone oxidoreductase subunit C/D | 2,235 | 1,522 | 380 | Fis+, NarL+, ArcA+ | Major aerobic electron transport chain |
| NuoB | NADH dehydrogenase subunit Bd | 994 | 2,033 | 247 | Fis+, NarL+, ArcA+ | Major aerobic electron transport chain |
| VhcB | Predicted cytochrome d ubiquinol oxidase, subunit III | 7,501 | 3,515 | 9,440 | Electron transport in microaerobic milieu | |
| CydB | Cytochrome d terminal oxidase, subunit II | 2,083 | 2,643 | 4,167 | ArcA+, Cra+ | Electron transport, iron limitation |
| FdoG | Formate dehydrogenase O, major subunitd | 2,045 | 1,489 | 232 | Crp+ | Electron transport, oxygen limitation |
| FdoH | Formate dehydrogenase O, Fe-S subunitd | 1,734 | 1,046 | 97 | Crp+ | Electron transport, oxygen limitation |
| NarH | Nitrate reductase A, β subunitd | 2,906 | 2,345 | 139 | Fnr+, NarL+, Fis− | Electron transport, oxygen limitation |
| NarG | Nitrate reductase A, α subunitd | 3,210 | 1,953 | 205 | Fnr+, NarL+, Fis− | Electron transport, oxygen limitation |
| PntA | NAD(P) transhydrogenase α subunit | 902 | 1,133 | 3,055 | Reversible NADP reduction by NADH | |
| PntB | NAD(P) transhydrogenase β subunit | 3,290 | 2,195 | 4,122 | Reversible NADP reduction by NADH | |
| SthA | Pyridine nucleotide transhydrogenase | 778 | 725 | 192 | NADPH reoxidation with NAD+ | |
CoA, coenzyme A; PTS, phosphotransferase system.
Average relative APEXi protein abundance levels for in vitro-grown S. flexneri and for extracellular (Ext) and intracellular (Int) bacteria during coculture with Henle cells. More-detailed analyses are provided in Data Set S2 in the supplemental material.
Known or predicted transcriptional regulator(s) of the indicated gene, based on data for the closely related bacterium E. coli (www.ecocyc.org). +, positive regulation; −, negative regulation.
Fe-S cluster or iron-heme or iron cofactor.
Table 4.
Effects on plaque formation of mutations in genes encoding proteins that are synthesized at higher levels intracellularly or extracellularly
| S. flexneri strain | Mutation(s) | Plaque formation in Henle cell monolayera |
|---|---|---|
| 2457T | None | +++ |
| CFS209 | fruA, manX | +++ |
| HGS101 | dppB | +++ |
| CFS217 | pflB | + |
| CFS218 | ldhA | +++ |
| CFS204 | lldD | +++ |
| CFS212 | adhE | +++ |
| CFS224 | pflB, adhE, ldhA | ± |
| SM100 ahpC | ahpC | +++ |
| CFS205 | bolA | +++ |
| CSF207 | cspA | +++ |
Henle cell monolayers were infected with the indicated mutant, and the size and number of plaques were determined after 3 days. +++, number and size of plaques were the same as for the wild type; +, plaques were much smaller than those for the wild type; ±, small, pinpoint plaques.
Changes in carbon metabolism proteins relative to in vitro-grown bacteria were also noted. The glycolysis enzymes Pfk-1 and Pfk-2 were induced (Table 3), further suggesting that sugars are a source of carbon and energy for intracellular bacteria. pfkA, which encodes Pfk-1, is positively regulated by the RNA-binding protein CsrA (73), which was found at much lower levels intracellularly. Thus, the increase in Pfk-1 is due to regulation by factors other than CsrA. The abundance of glycolysis enzymes is consistent with our previous studies that showed that blocking glycolysis by mutation of pfkA inhibited plaque formation by S. flexneri (39).
There was strong evidence that S. flexneri uses mixed-acid fermentation during intracellular growth. Proteins in the pathways for metabolizing pyruvate, a product of glycolysis, to formate, lactate, and ethanol (e.g., PflB, LdhA, and AdhE) were more abundant in intracellular samples than in in vitro samples (Table 3). The formate exporter FocA was not seen in the intracellular bacteria (Table 3), suggesting that formate was not the primary product of mixed-acid fermentation in the intracellular bacteria. AdhE and PflB were increased in the extracellular sample compared to in vitro-grown cells (Table 3). Therefore, extracellular growth conditions may favor mixed-acid fermentation in the bacteria prior to invasion.
To determine whether these mixed-acid fermentation pathways (Fig. 2A) are required for intracellular growth, the genes for pyruvate formate lyase (pflB), alcohol dehydrogenase E (adhE), and two genes encoding lactate dehydrogenases (ldhA and lldD) were deleted singly or in combination, and the mutants were assessed for plaque formation (Table 4). Individual mutations in ldhA, lldD, or adhE had little effect on the ability of the bacteria to form plaques in Henle cell monolayers. However, mutants defective in pflB singly or in combination with adhE and ldhA produced small plaques, with the triple mutant being more defective than the single mutant. These mutants did not have a general growth defect and grew as well as the wild type in LB medium under aerobic conditions (Fig. 2B). The mutants grew more slowly than the wild type when the cultures were not aerated (Fig. 2C), indicating that mixed-acid fermentation may be more important when the bacteria are in a low-oxygen environment. These data suggest that mixed-acid fermentation, and metabolism of pyruvate in particular, is required for optimal growth of S. flexneri in the host cell cytoplasm. YfiD, which activates PflB, is also elevated in the intracellular bacteria (Table 3), which would increase flux through this pathway (74).
Fig 2.
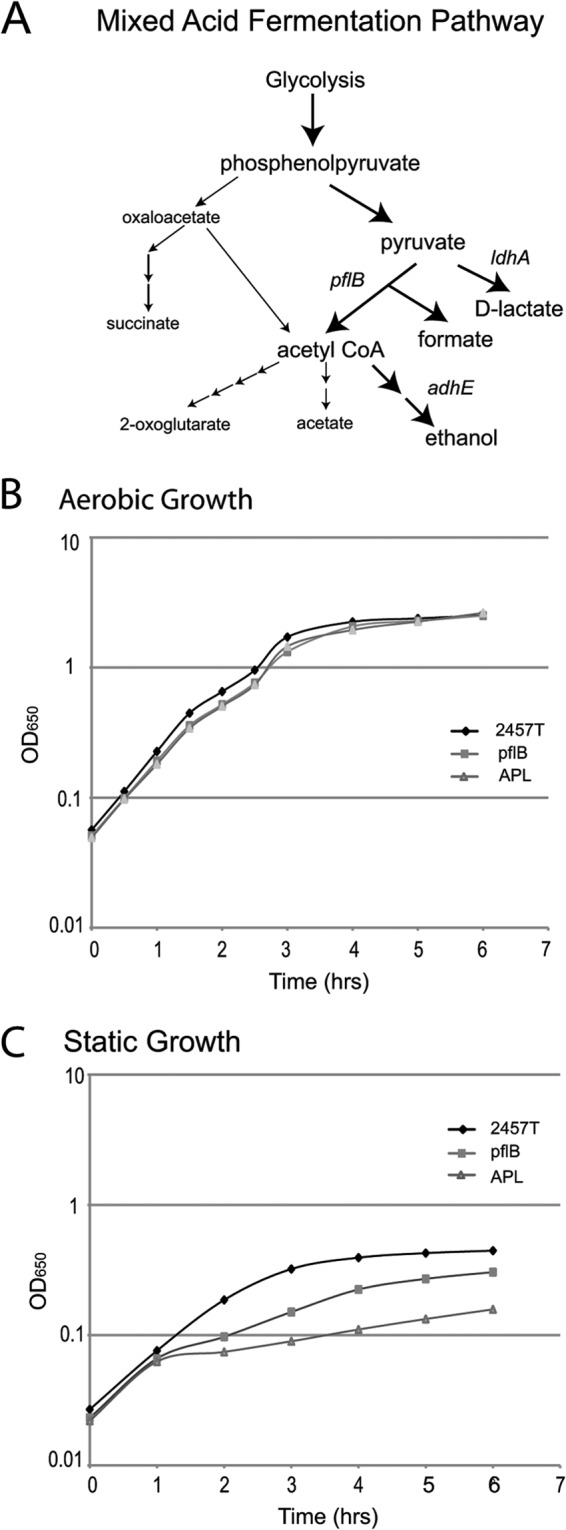
Growth of mixed-acid fermentation pathway mutants in vitro. (A) Simplified schematic of the mixed-acid fermentation pathways. (B and C) Wild-type strain 2457T, the pflB mutant CFS212, and the adhE pflB ldhA triple mutant CFS224 (APL) were grown overnight in LB and diluted 1:50 in fresh LB. Once the cultures reached logarithmic-phase growth, they were diluted to an A650 of 0.05 and grown at 37°C with shaking (B) or statically (C). Growth was monitored by measuring the A650.
In contrast to fermentation pathway enzymes, components of the tricarboxylic acid (TCA) cycle, including SucC, SdhA, and AcnB and electron transport proteins (e.g., NuoB, -C, and -E and FdoG and -H) were decreased in the intracellular bacteria (Table 3). This is consistent with fermentation, rather than respiration, predominating in the intracellular environment. The decreased abundances of the enzymes NarH, NarG, and NapA, components of two different pathways for utilization of nitrogen for respiration under oxygen-deprived conditions, indicated a decrease in nitrate metabolism (intracellular versus extracellular) (Table 3). A number of the genes for fermentation and respiration enzymes are regulated by the oxygen-sensing, two-component transcription regulatory system ArcAB. Levels of ArcB were 10-fold higher in the intracellular bacteria (see Table 6, below), and this increase may help the cell respond to the changes in oxygen levels intracellularly versus in vitro conditions. A decreased abundance of the respiratory enzymes intracellularly also appeared to be linked to iron starvation, as many of their oxidoreductive subunits harbor iron-sulfur clusters (Table 3; see also Fig. S1 in the supplemental material).
Table 6.
Selected differentially expressed proteins with stress response and cell wall functions
| Protein name | Protein description | Avg relative abundancea |
Trancriptional regulator(s)b | Functional role(s) | ||
|---|---|---|---|---|---|---|
| In vitro | Ext | Int | ||||
| HmpA | Nitric oxide dioxygenasec | 1,384 | 908 | 416 | MetR+, Fnr−, Fur− | Detoxification of reactive nitrogen species |
| KatG | Catalase; hydroperoxidase HPIc | 15,847 | 10,449 | 4,759 | Fnr+, Fur+, OxyR+ | Detoxification of reactive oxygen species |
| AhpC | Alkyl hydroperoxide reductase subunit C | 33,936 | 23,158 | 40,364 | OxyR+ | Detoxification of reactive oxygen species |
| AhpF | Alkyl hydroperoxide reductase F52a subunit | 2,349 | 2,559 | 4,244 | OxyR+ | Detoxification of reactive oxygen species |
| SodB | Superoxide dismutasec | 31,190 | 16,455 | 5,986 | Crp−, IhfB− | Detoxification of reactive oxygen species |
| HslV | ATP-dependent protease, subunit of HslUV | 3,275 | 1,362 | 4,781 | Stress response to protein aggregates | |
| CspA | Major cold shock protein CspA | 14,234 | 81,065 | 10,679 | RNA chaperone during cold shock | |
| DksA | RNA polymerase-binding transcription factor | 17,818 | 11,389 | 28,409 | Crp+ | Transcriptional regulator at low pH |
| OsmC | Osmotically inducible peroxidase OsmC | 1,505 | 1,736 | 4,099 | RcsB+, H-NS− | Osmotic stress response |
| OmpR | Osmolarity response regulator OmpR | 2,943 | 2,622 | 715 | IhfB− | Regulator: osmotic stress response |
| BolA | Transcriptional regulator BolA | 2,586 | 18,931 | 715 | H-NS−, OmpR− | Regulator: extracellular changes, carbon starvation, and cell division |
| RcsB | Transcriptional regulator RcsB | 1,192 | 2,301 | 607 | H-NS− | Regulator: extracellular changes, colonic acid capsule, and motility |
| ArcA | ArcA two-component response regulator | 4,733 | 5,088 | 3,678 | Regulator under microaerobic conditions | |
| ArcB | ArcB sensory histidine kinase | 305 | 286 | 3,622 | Regulator under microaerobic conditions | |
| Pal | Peptidoglycan-associated outer membrane lipoprotein Pal | 20,238 | 15,267 | 7,959 | H-NS− | OM barrier function, cell shape stabilizing |
| RfaD | ADP-l-glycero-d-mannoheptose-6-epimerase | 4,315 | 5,447 | 3,665 | LPS: core oligosaccharide biosynthesis | |
| KdsC | 3-Deoxy-d-manno-octulosonate 8-phosphate phosphatase | 2,045 | 2,009 | 1,100 | LPS core oligosaccharide biosynthesis | |
| RfbA | Glucose-1-phosphate thymidylyltransferase | 1,312 | 3,206 | 2,092 | LPS: O-antigen biosynthesis | |
| RfbB | dTDP-glucose 4,6 dehydratase | 1,577 | 5,910 | 2,450 | LPS: O-antigen biosynthesis | |
| RfbD | dTDP-4-dehydrorhamnose reductase | 324 | 1,344 | 498 | LPS: O-antigen biosynthesis | |
| Rph | RNase PH | 300 | 2,247 | 653 | tRNA processing and metabolism | |
| Tgt | tRNA-guanine transglycosylase | 1,321 | 2,141 | 484 | tRNA processing and metabolism | |
| MiaB | Isopentenyl-adenosine A37 tRNA methylthiolasec | 1,431 | 2,040 | 863 | tRNA processing and metabolism | |
| YbiJ | Predicted periplasmic protein YbiJ | 0 | 887 | 7,698 | DNA damage stimulus response | |
| OmpW | Outer membrane protein W | 13,607 | 16,223 | 4,560 | Fnr+ | OM barrier function, colicin receptor |
| OmpX | Outer membrane protein X | 17,618 | 10,306 | 25,461 | Fnr− | Interactions with host cells: adhesion |
| MlaD | Outer membrane lipid asymmetry protein | 1,900 | 718 | 1,966 | UlaR+, IhfB− | OM barrier function, preventing phospholipid accumulation in OM |
| PldA | Outer membrane phospholipase A | 572 | 759 | 1,440 | Extracellular phospholipid metabolism | |
| YbjP | Predicted lipoprotein YbjP | 271 | 142 | 2,480 | Unknown | |
Average relative APEXi protein abundance levels for in vitro-grown S. flexneri and for extracellular (Ext) and intracellular (Int) bacteria during coculture with Henle cells. More-detailed analyses are provided in Data Set S2 in the supplemental material.
Known or predicted transcriptional regulator(s) of the indicated gene, based on data for the closely related bacterium E. coli (www.ecocyc.org). +, positive regulation; −, negative regulation.
Fe-S cluster or iron-heme or iron cofactor.
Iron transport.
S. flexneri has both ferrous and ferric iron transport systems (75). These systems are repressed during growth in iron-rich medium, such as that used for the in vitro bacteria, and detection of iron transporters is indicative of iron starvation. The ferrous iron transport proteins SitA and FeoB were strongly increased in intracellular and extracellular compared to in vitro samples, and SitA was also increased in intracellular compared to extracellular samples (Table 5). This was consistent with our previous genetic and expression data that indicated that sitA is induced intracellularly (46) and that the Sit ferrous iron transport system is important for intracellular growth and virulence of S. flexneri (43). Proteins required for the biosynthesis (IucA, -B, -C, and -D) and transport (IutA) of the S. flexneri siderophore aerobactin (76) and the ferrichrome receptor FhuA were also increased in the intracellular bacteria (Table 5), demonstrating iron starvation in Henle cells. As would be expected under conditions of iron starvation, there was reduced synthesis of the iron storage protein ferritin. The dipeptide permease DppA, described above, also functions as a heme permease in E. coli (77), but it is not known whether its increased abundance is linked to iron starvation or peptide transport.
Table 5.
Selected differentially expressed proteins with roles in iron metabolism or metabolism of other metals
| Protein name | Protein description | Avg relative abundancea |
Transcriptional regulator(s)b | Functional role(s) | ||
|---|---|---|---|---|---|---|
| In vitro | Ext | Int | ||||
| SitA | Fe/Mn transport periplasmic protein | 578 | 5,087 | 15,015 | Fur− | Fe and Mn import |
| FeoB | Ferrous iron transport protein B | 158 | 1,837 | 1,542 | Fnr+, ArcA+, Fur− | Ferrous iron import |
| IucA | Aerobactin biosynthesis | 0 | 45 | 502 | Fur− | Siderophore biosynthesis |
| IucB | Aerobactin biosynthesis | 0 | 648 | 2,556 | Fur− | Siderophore biosynthesis |
| IucC | Aerobactin biosynthesis | 0 | 48 | 308 | Fur− | Siderophore biosynthesis |
| IucD | Aerobactin biosynthesis | 0 | 517 | 2,744 | Fur− | Siderophore biosynthesis |
| IutA | Ferric aerobactin receptor | 34 | 419 | 6,042 | Fur− | Metal ion and siderophore import |
| FhuA | Ferrichrome outer membrane transporter | 0 | 96 | 306 | Fur− | Siderophore import |
| FhuC | Iron-hydroxamate transporter ATP-binding subunit | 0 | 66 | 276 | Fur− | Metal ion and siderophore import |
| ExbB | TonB energy transducing system subunit B | 0 | 1,659 | 508 | Fur− | Energy supply for siderophore and cofactor import |
| ExbD | TonB energy transducing system subunit D | 0 | 567 | 543 | Fur− | Energy supply for siderophore and cofactor import |
| CorA | Mn/Ni/Co transporter | 30 | 521 | 758 | Metal ion import and utilization | |
| TolC | Outer membrane channel protein TolC | 5,125 | 3,665 | 6,710 | PhoP+, SoxS− | Multidrug and siderophore efflux systems |
| IscU | Fe-S assembly scaffold protein IscU | 4,299 | 7,389 | 5,793 | IscR− | Fe-S cluster assembly |
| IscS | Cysteine desulfurase IscS | 848 | 1,918 | 1,393 | Crp+, Fis− | Fe-S cluster assembly |
| SufC | Cysteine desulfurase SufC | 0 | 201 | 1,829 | IhfB+, IscR+, OxyR+, Fur− | Fe-S cluster assembly under iron starvation and oxidative stress |
| SufB | Cysteine desulfurase activator complex subunit SufB | 0 | 76 | 306 | IhfB+, IscR+, OxyR+, Fur− | Fe-S cluster assembly under iron starvation and oxidative stress |
| SufA | Fe-S cluster assembly scaffold protein SufA | 0 | 55 | 661 | IhfB+, IscR+, OxyR+, Fur− | Fe-S cluster assembly under iron starvation and oxidative stress |
| Ftn | Ferritin | 25,840 | 15,482 | 4,031 | Fur+, H-NS− | Cytoplasmic iron storage |
Average relative APEXi protein abundance levels for in vitro-grown S. flexneri and for extracellular (Ext) and intracellular (Int) bacteria during coculture with Henle cells. More-detailed analyses are provided in Data Set S2 in the supplemental material.
Known or predicted transcriptional regulator(s) of the indicated gene, based on data for the closely related bacterium E. coli (www.ecocyc.org). +, positive regulation; −, negative regulation.
Outer membrane proteins.
In addition to the transporters noted above, other outer membrane proteins were differentially expressed in intracellular versus extracellular or in vitro samples (see Table 6, below), suggesting that intracellular bacteria remodel their surface in response to the host environment. PldA, an outer membrane phospholipase (78), was found at higher levels in intracellular bacteria. OmpC, which is required for intracellular growth (79), was also more abundant in intracellular bacteria. Bernardini et al. (79) showed that S. flexneri ompC, unlike E. coli ompC, was not regulated by osmolarity. Thus, the increase in OmpC intracellularly is likely a response to signals other than increased osmolarity intracellularly versus extracellularly. Interestingly, OmpW, which has been shown in pathogenic E. coli strains to play a role in resistance to phagocytosis (80), was less abundant in intracellular bacteria. Reduced amounts of this outer membrane protein may be a consequence of iron starvation, since it has been shown that OmpW is not detected in an E. coli K99 strain grown under iron-limiting conditions (80). OmpW has been reported to be growth phase regulated, and Zhu et al. (81) found OmpW to be one of the most abundant proteins in stationary-phase cells grown in LB, while it is almost undetectable in exponentially growing cells. In contrast, OmpX was decreased in the stationary phase (81). The outer membrane profile observed in the intracellular bacteria, i.e., low OmpW and high OmpX, more closely resembles that of exponentially growing rather than stationary-phase cells.
Stress responses.
Analysis of stress response proteins showed relatively few changes (Table 6), suggesting that the bacteria were not subjected to highly stressful conditions within the cells. OsmC, a protein induced by increased osmolarity, was more highly expressed in intracellular bacteria, consistent with the cytosol having high osmolarity. AhpC and AhpF, the components of the alkyl hydroperoxidase system, were modestly induced, while catalase (KatG) was found at lower levels intracellularly, suggesting that low levels of peroxide, which can be detoxified by the alkylperoxidase, were present in the cell but that the high levels associated with catalase induction were not present. Deletion of ahpC did not significantly affect plaque formation (Table 4), indicating that AhpC is not required for intracellular survival in Henle cells and that peroxides are at subtoxic levels. DksA, which we showed previously to be required for S. flexneri plaque formation (31), was induced intracellularly (Table 6). The superoxide dismutase SodB, which harbors a Fe2+/Fe3+ cofactor, was strongly decreased intracellularly, consistent with the evidence of iron starvation (Table 5). The cold shock protein CspA was highly expressed in the extracellular sample (Table 6), but a mutation of cspA did not affect invasion and plaque formation (Table 4).
Other indicators of the cytoplasm being a low-stress environment are the low levels of BolA and HmpA in the intracellular samples (Table 6). BolA is induced in logarithmically growing cells by sudden carbon starvation, heat shock, osmotic shock, or oxidative stress (82). The reduction in BolA suggests that the bacteria do not encounter these stressors during intracellular growth. To verify this, a bolA mutant was constructed and tested in the plaque assay; the bolA mutant formed wild-type plaques (Table 4). Similarly, the lack of induction of nitric oxide dioxygenase (HmpA) suggested an absence of a stress response to nitric oxide in epithelial cells.
DISCUSSION
The gut is a complex environment for potential pathogens such as S. flexneri. There are numerous niches that are highly diverse with respect to oxygen, nutrients, and competitors. The environment of the lumen is quite distinct from that of the loose mucosal layer, an area rich in carbohydrates that is the site of most commensal multiplication. Pathogens, such as enteropathogenic E. coli, are able to penetrate the tight mucosal layer and exploit the niches adjacent to the cell surface, while others are able to invade the cells and occupy a modified vacuole or replicate free in the cytoplasm. A pathogen such as Shigella would have to deal with more than one of these microenvironments during its residence in the host and might experience changes in cell surface characteristics and metabolism during the course of infection. For Shigella, this means transiting from the lumen of the colon, through the M cells, and encountering macrophages and other host immune cells before gaining entry to the colonic epithelial cells. It then replicates in the cytoplasm, which is a distinctly different environment from that elsewhere in the gut or in laboratory media.
We are now beginning to obtain a systems biology view of the changes S. flexneri induces during growth in the host cell cytoplasm. By analyzing the proteins synthesized by intracellular Shigella, we can determine the metabolic pathways used and also learn more about the host environment in which these bacteria live. By combining these data with previously published transcriptome and gene fusion data and with analyses of mutants defective in specific pathways, we can determine not only which pathways are expressed but also those that are essential during intracellular growth. An overview of the pathways that are significantly increased or decreased in intracellular bacteria is shown in Fig. S1 in the supplemental material.
Our data suggest that the intracellular bacteria scavenge a wide range of carbon sources. Transport systems for a variety of sugars and peptides are expressed, and loss of any one of these systems is apparently compensated by the ability of the other transporters to bring in sufficient carbon sources to meet the cells' needs. Thus, mutation of fruA, manX, or dpp did not have a significant effect on the ability of S. flexneri to grow within cultured cells. S. flexneri appears to metabolize carbon sources primarily through glycolysis and mixed-acid fermentation. We confirmed the importance of mixed-acid fermentation to intracellular growth of S. flexneri by constructing and testing mutants defective in these pathways. Mutants blocked for the metabolism of pyruvate to formate and lactate could not form wild-type plaques, suggesting that they do not grow well in the intracellular environment. The use of fermentation pathways by intracellular S. flexneri was somewhat unexpected, since the energy yield is lower than with respiration. It is possible that in the low-oxygen environment of the host, there are alternative electron acceptors, such as nitrate, available for anaerobic respiration by Shigella. Oxidation of NO to nitrate has been shown to occur in epithelial cells (83) and could provide the alternative electron acceptor. However, the nitrate reductase composed of NarG and NarH is strongly reduced in abundance intracellularly (Table 3).
Additional evidence for the importance of mixed-acid fermentation during infection is the detection of these enzymes in the proteome analysis of Shigella dysenteriae isolated from the large bowel of infected gnotobiotic piglets (84, 85). While similarities were noted in the proteomes of the intracellular bacteria and those isolated from the piglets, the two studies are not entirely comparable, since the bacteria obtained from the piglet intestine likely included extracellular and intracellular bacteria. The dramatic shift toward mixed-acid fermentation in S. dysenteriae in the bowel environment suggested that this adaptation pertains to both intracellular and extracellular bacteria in the host and is similar to our observations in cell culture.
Many of the changes in protein levels seen in the intracellular bacteria were indicative of iron starvation, which was also noted in studies of gene expression by intracellular S. flexneri (44, 46). All of the iron transport systems were more highly expressed in the intracellular bacteria. These systems are all repressed by iron via the iron-binding, transcriptional repressor Fur (75). They are also regulated by oxygen, with Feo being induced anaerobically and both the Sit and aerobactin transport systems induced in the presence of oxygen (24). The fact that all three systems were expressed suggests that the iron starvation was sufficient to overcome any effects of oxygen on the induction of these transporters. The levels of iron transport proteins were higher than we observed previously (86). This may reflect differences under the conditions used for the proteome analysis, with higher numbers of bacteria per cell and a longer infection time allowing iron starvation to become more apparent.
There was also an increase in Suf proteins for Fe-S cluster assembly. It had been shown previously that the suf genes are induced intracellularly and likely respond to low iron levels, since the operon is Fur regulated (87). The Isc Fe-S cluster assembly proteins are not highly increased in abundance, but they play a greater role in intracellular survival than the Suf proteins, since an isc but not a suf mutant was found defective in plaque formation (88).
The reduced levels of TCA cycle enzymes may also be a consequence, in part, of iron starvation. Many of the TCA cycle enzymes contain iron or an Fe-S cluster cofactor. Reduced availability of iron results in extensive changes in transcription in response to loss of Fur-mediated repression, including derepression of the small RNA RyhB (87). RyhB inhibits production of iron-containing proteins, including succinate dehydrogenase and other TCA cycle enzymes, NADH dehydrogenase, and the iron superoxide dismutase SodB, all of which were reduced in the intracellular bacteria.
Adaptation of Shigella to the intracellular environment is a dynamic process. The host cell environment changes in response to the bacterial invasion, and continued intracellular growth of the bacterium alters conditions in the cytoplasm. Changes also were seen in the expression of S. flexneri virulence factors. We had previously observed reduced synthesis of the Ipa proteins immediately after bacterial entry into the cytoplasm compared to levels in extracellular bacteria (64). Schuch et al. (47) demonstrated that the TTSS and effector proteins were needed for escape from the double membrane formed during intercellular spread, indicating that the virulence proteins must be made before the bacterium can spread to adjacent cells. Consistent with this, we noted high levels of Ipa, Mxi, and Ics proteins, among other virulence factors, in intracellular bacteria. In an in vitro proteomic analysis of S. flexneri grown at different temperatures, Zhu et al. (89) observed that IpaA, IpaC, IpaD, IpgC, and SepA were increased at 37°C versus 30°C. Our 2D gel analysis revealed a C-terminal 28-kDa posttranslational truncation product of the protease SepA (Fig. 1), which was also observed by Zhu et al. (89). SepA was quantitatively increased intracellularly.
The extracellular sample had lower levels of some of the virulence proteins, e.g., IpaC and MxiC, than either the intracellular or in vitro bacteria. We considered the possibility that the extracellular sample may be enriched for bacteria in the population that had low virulence gene expression and were unable to invade, while those with higher levels of virulence proteins invaded the Henle cells. However, extracellular bacteria were able to invade epithelial cells at near-wild-type levels when transferred to a fresh monolayer, indicating that these were not bacteria that had lost the ability to invade.
In contrast to our proteome results, transcriptome analyses showed a decrease in expression of virulence genes over an 8-hour time course of intracellular growth (44). A recent study by Morris et al. (45) also showed a decrease in virulence gene expression relative to that in in vitro-grown cells at 3 h postinfection. The reason for the difference between gene expression and protein levels is not clear. We used a time point similar to that examined by Morris et al. (45), but there may have been differences in the in vitro growth conditions or other experimental differences that influenced the relative amounts of virulence gene transcripts and proteins. Differences between the transcriptome and proteome may also indicate that transcript levels do not correlate perfectly with protein levels, and these differences highlight the need for use of multiple approaches to fully characterize the physiology and metabolism of intracellular bacteria.
Other differences between the proteome and transcriptome for intracellular S. flexneri were also noted. The proteome showed an increase in OmpC and OmpX. In contrast, the transcriptome showed decreased expression of ompC and ompX during intracellular growth (44). OmpC is required for cell-to-cell spread and plaque formation by S. flexneri (79), which is consistent with our observation of its abundance in intracellular bacteria.
Based on our observations of protein abundances and analysis of the effects of specific mutations on the ability of S. flexneri to form plaques in Henle cell monolayers, we propose a general model for the adaptation of Shigella to the intracellular environment (see Fig. S1 in the supplemental material). After invasion, Shigella encounters the host cytosol, where iron is sequestered and carbohydrate and peptide sources are altered compared to the external environment. Iron starvation triggers Fur- and RyhB-mediated gene expression changes that lead to increased expression of iron uptake systems and activation of the stress-responsive Fe-S cluster assembly system Suf. Low intracellular iron triggers reduced expression of proteins for iron storage (e.g., Ftn and Bfr) and the primary Fe-S cluster assembly system Isc. Reduced intracellular availability of Fe-S clusters results in transcriptionally and posttranscriptionally controlled changes in many energy metabolism pathways, including the TCA cycle, electron transport chains (each with several enzymes containing Fe-S cofactors), mixed-acid fermentation, and amino acid metabolism. Multiple carbon sources may be used during intracellular residence, as there was increased expression of importers for sugars and peptides. Mixed-acid fermentation pathways and the CydAB electron transport chain are likely to assume major energy generation functions in the intracellular bacteria. The increased abundances of PntA and PntB versus the decreased abundance of SthA suggest that S. flexneri activates regeneration of intracellular NADPH to support essential biosynthetic processes, potentially due to lowered NADH dehydrogenase and TCA cycle activity during intracellular survival.
As we develop an understanding of the proteins and processes used by Shigella growing within host cells, we may be able to predict potential targets for the immune response or pathways for therapeutic intervention. Our data sets are providing a rich resource of information to determine the essentiality of proteins for the intracellular life stage of this pathogen. There is a strong correlation between genes previously shown to be important for intracellular growth of Shigella and increased levels of the encoded proteins in the intracellular environment relative to other growth conditions. This was noted for the virulence proteins, OmpC, and SitA, -B, -C, and –D, among others. Additionally, we have shown that data regarding the intracellular proteome can be used to predict essential intracellular pathways, by constructing and testing mutants defective in pathways highly expressed in the intracellular environment. By using this approach, we confirmed that mixed-acid fermentation is crucial for growth of S. flexneri in the intracellular environment.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the efforts of David Clark and Hamid Alami who performed the protein identification experiments for 2D gel spots. We thank Li Ma for construction and testing of the ahpC mutant and Hannah Giauque for the dppB mutant. We also thank Elizabeth Wyckoff and Alexandra Mey for critical reading of the manuscript.
This study was supported by the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), under contract number N01-AI-30050 and by grant AI16935.
Footnotes
Published ahead of print 7 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00975-13.
REFERENCES
- 1.Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, Adak GK, Levine MM. 1999. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull. World Health Organ. 77:651–666 [PMC free article] [PubMed] [Google Scholar]
- 2.Ashida H, Ogawa M, Kim M, Suzuki S, Sanada T, Punginelli C, Mimuro H, Sasakawa C. 2011. Shigella deploy multiple countermeasures against host innate immune responses. Curr. Opin. Microbiol. 14:16–23 [DOI] [PubMed] [Google Scholar]
- 3.Jennison AV, Verma NK. 2004. Shigella flexneri infection: pathogenesis and vaccine development. FEMS Microbiol. Rev. 28:43–58 [DOI] [PubMed] [Google Scholar]
- 4.Menard R, Dehio C, Sansonetti PJ. 1996. Bacterial entry into epithelial cells: the paradigm of Shigella. Trends Microbiol. 4:220–226 [DOI] [PubMed] [Google Scholar]
- 5.Nhieu GT, Sansonetti PJ. 1999. Mechanism of Shigella entry into epithelial cells. Curr. Opin. Microbiol. 2:51–55 [DOI] [PubMed] [Google Scholar]
- 6.Ogawa M, Handa Y, Ashida H, Suzuki M, Sasakawa C. 2008. The versatility of Shigella effectors. Nat. Rev. Microbiol. 6:11–16 [DOI] [PubMed] [Google Scholar]
- 7.Ogawa M, Sasakawa C. 2006. Intracellular survival of Shigella. Cell. Microbiol. 8:177–184 [DOI] [PubMed] [Google Scholar]
- 8.Sansonetti PJ. 2001. Rupture, invasion and inflammatory destruction of the intestinal barrier by Shigella, making sense of prokaryote-eukaryote cross-talks. FEMS Microbiol. Rev. 25:3–14 [DOI] [PubMed] [Google Scholar]
- 9.Sansonetti PJ, Egile C. 1998. Molecular bases of epithelial cell invasion by Shigella flexneri. Antonie Van Leeuwenhoek 74:191–197 [DOI] [PubMed] [Google Scholar]
- 10.Mounier J, Vasselon T, Hellio R, Lesourd M, Sansonetti PJ. 1992. Shigella flexneri enters human colonic Caco-2 epithelial cells through the basolateral pole. Infect. Immun. 60:237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sansonetti PJ, Phalipon A. 1999. M cells as ports of entry for enteroinvasive pathogens: mechanisms of interaction, consequences for the disease process. Semin. Immunol. 11:193–203 [DOI] [PubMed] [Google Scholar]
- 12.Blocker A, Gounon P, Larquet E, Niebuhr K, Cabiaux V, Parsot C, Sansonetti P. 1999. The tripartite type III secretion of Shigella flexneri inserts IpaB and IpaC into host membranes. J. Cell Biol. 147:683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blocker A, Jouihri N, Larquet E, Gounon P, Ebel F, Parsot C, Sansonetti P, Allaoui A. 2001. Structure and composition of the Shigella flexneri “needle complex,” a part of its type III secretion. Mol. Microbiol. 39:652–663 [DOI] [PubMed] [Google Scholar]
- 14.Menard R, Prevost MC, Gounon P, Sansonetti P, Dehio C. 1996. The secreted Ipa complex of Shigella flexneri promotes entry into mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 93:1254–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watarai M, Tobe T, Yoshikawa M, Sasakawa C. 1995. Contact of Shigella with host cells triggers release of Ipa invasins and is an essential function of invasiveness. EMBO J. 14:2461–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sansonetti PJ, Ryter A, Clerc P, Maurelli AT, Mounier J. 1986. Multiplication of Shigella flexneri within HeLa cells: lysis of the phagocytic vacuole and plasmid-mediated contact hemolysis. Infect. Immun. 51:461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldberg MB, Bârzu O, Parsot C, Sansonetti PJ. 1993. Unipolar localization and ATPase activity of IcsA, a Shigella flexneri protein involved in intracellular movement. J. Bacteriol. 175:2189–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldberg MB, Sansonetti PJ. 1993. Shigella subversion of the cellular cytoskeleton: a strategy for epithelial colonization. Infect. Immun. 61:4941–4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldberg MB, Theriot JA. 1995. Shigella flexneri surface protein IcsA is sufficient to direct actin-based motility. Proc. Natl. Acad. Sci. U. S. A. 92:6572–6576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maurelli AT, Baudry B, d'Hauteville H, Hale TL, Sansonetti PJ. 1985. Cloning of plasmid DNA sequences involved in invasion of HeLa cells by Shigella flexneri. Infect. Immun. 49:164–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sasakawa C, Kamata K, Sakai T, Makino S, Yamada M, Okada N, Yoshikawa M. 1988. Virulence-associated genetic regions comprising 31 kilobases of the 230-kilobase plasmid in Shigella flexneri 2a. J. Bacteriol. 170:2480–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marteyn B, Gazi A, Sansonetti P. 2012. Shigella: a model of virulence regulation in vivo. Gut Microbes 3:104–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernardini ML, Fontaine A, Sansonetti PJ. 1990. The two-component regulatory system ompR-envZ controls the virulence of Shigella flexneri. J. Bacteriol. 172:6274–6281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boulette ML, Payne SM. 2007. Anaerobic regulation of Shigella flexneri virulence: ArcA regulates Fur and iron acquisition genes. J. Bacteriol. 189:6957–6967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hromockyj AE, Tucker SC, Maurelli AT. 1992. Temperature regulation of Shigella virulence: identification of the repressor gene virR, an analogue of hns, and partial complementation by tyrosyl transfer RNA (tRNA1(Tyr)). Mol. Microbiol. 6:2113–2124 [DOI] [PubMed] [Google Scholar]
- 26.Marteyn B, West NP, Browning DF, Cole JA, Shaw JG, Palm F, Mounier J, Prévost M-C, Sansonetti P, Tang CM. 2010. Modulation of Shigella virulence in response to available oxygen in vivo. Nature 465:355–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy ER, Payne SM. 2007. RyhB, an iron-responsive small RNA molecule, regulates Shigella dysenteriae virulence. Infect. Immun. 75:3470–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakayama S, Watanabe H. 1995. Involvement of cpxA, a sensor of a two-component regulatory system, in the pH-dependent regulation of expression of Shigella sonnei virF gene. J. Bacteriol. 177:5062–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Falconi M, Prosseda G, Giangrossi M, Beghetto E, Colonna B. 2001. Involvement of FIS in the H-NS-mediated regulation of virF gene of Shigella and enteroinvasive Escherichia coli. Mol. Microbiol. 42:439–452 [DOI] [PubMed] [Google Scholar]
- 30.Mitobe J, Morita-Ishihara T, Ishihama A, Watanabe H. 2009. Involvement of RNA-binding protein Hfq in the osmotic-response regulation of invE gene expression in Shigella sonnei. BMC Microbiol. 9:110. 10.1186/1471-2180-9-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma AK, Payne SM. 2006. Induction of expression of hfq by DksA is essential for Shigella flexneri virulence. Mol. Microbiol. 62:469–479 [DOI] [PubMed] [Google Scholar]
- 32.Tobe T, Nagai S, Okada N, Adler B, Yoshikawa M, Sasakawa C. 1991. Temperature-regulated expression of invasion genes in Shigella flexneri is controlled through the transcriptional activation of the virB gene on the large plasmid. Mol. Microbiol. 5:887–893 [DOI] [PubMed] [Google Scholar]
- 33.Tobe T, Yoshikawa M, Mizuno T, Sasakawa C. 1993. Transcriptional control of the invasion regulatory gene virB of Shigella flexneri: activation by VirF and repression by H-NS. J. Bacteriol. 175:6142–6149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitobe J, Yanagihara I, Ohnishi K, Yamamoto S, Ohnishi M, Ishihama A, Watanabe H. 2011. RodZ regulates the post-transcriptional processing of the Shigella sonnei type III secretion system. EMBO Rep. 12:911–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hale TL. 1986. Invasion of epithelial cells by shigellae. Ann. Inst. Pasteur Microbiol. 137A:311–314 [DOI] [PubMed] [Google Scholar]
- 36.Hale TL, Formal SB. 1981. Protein synthesis in HeLa or Henle 407 cells infected with Shigella dysenteriae 1, Shigella flexneri 2a, or Salmonella typhimurium W118. Infect. Immun. 32:137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerber DF, Watkins HM. 1961. Growth of shigellae in monolayer tissue cultures. J. Bacteriol. 82:815–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernardini ML, Mounier J, d'Hauteville H, Coquis-Rondon M, Sansonetti PJ. 1989. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc. Natl. Acad. Sci. U. S. A. 86:3867–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gore AL, Payne SM. 2010. CsrA and Cra influence Shigella flexneri pathogenesis. Infect. Immun. 78:4674–4682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cersini A, Salvia AM, Bernardini ML. 1998. Intracellular multiplication and virulence of Shigella flexneri auxotrophic mutants. Infect. Immun. 66:549–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noriega FR, Wang JY, Losonsky G, Maneval DR, Hone DM, Levine MM. 1994. Construction and characterization of attenuated delta aroA delta virG Shigella flexneri 2a strain CVD 1203, a prototype live oral vaccine. Infect. Immun. 62:5168–5172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kotloff KL, Noriega FR, Samandari T, Sztein MB, Losonsky GA, Nataro JP, Picking WD, Barry EM, Levine MM. 2000. Shigella flexneri 2a strain CVD 1207, with specific deletions in virG, sen, set, and guaBA, is highly attenuated in humans. Infect. Immun. 68:1034–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fisher CR, Davies NMLL, Wyckoff EE, Feng Z, Oaks EV, Payne SM. 2009. Genetics and virulence association of the Shigella flexneri sit iron transport system. Infect. Immun. 77:1992–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lucchini S, Liu H, Jin Q, Hinton JC, Yu J. 2005. Transcriptional adaptation of Shigella flexneri during infection of macrophages and epithelial cells: insights into the strategies of a cytosolic bacterial pathogen. Infect. Immun. 73:88–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morris CR, Grassel CL, Redman JC, Sahl JW, Barry EM, Rasko DA. 2013. Characterization of intracellular growth regulator icgR by utilizing transcriptomics to identify mediators of pathogenesis in Shigella flexneri. Infect. Immun. 81:3068–3076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Runyen-Janecky LJ, Payne SM. 2002. Identification of chromosomal Shigella flexneri genes induced by the eukaryotic intracellular environment. Infect. Immun. 70:4379–4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schuch R, Sandlin RC, Maurelli AT. 1999. A system for identifying post-invasion functions of invasion genes: requirements for the Mxi-Spa type III secretion pathway of Shigella flexneri in intercellular dissemination. Mol. Microbiol. 34:675–689 [DOI] [PubMed] [Google Scholar]
- 48.Pope LM, Reed KE, Payne SM. 1995. Increased protein secretion and adherence to HeLa cells by Shigella spp. following growth in the presence of bile salts. Infect. Immun. 63:3642–3648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pieper R, Zhang Q, Clark DJ, Huang S-T, Suh M-J, Braisted JC, Payne SH, Fleischmann RD, Peterson SN, Tzipori S. 2011. Characterizing the Escherichia coli O157:H7 proteome including protein associations with higher order assemblies. PLoS One 6(11):e26554. 10.1371/journal.pone.0026554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gatlin CL, Pieper R, Huang S-T, Mongodin E, Gebregeorgis E, Parmar PP, Clark DJ, Alami H, Papazisi L, Fleischmann RD, Gill SR, Peterson SN. 2006. Proteomic profiling of cell envelope-associated proteins from Staphylococcus aureus. Proteomics 6:1530–1549 [DOI] [PubMed] [Google Scholar]
- 51.Pieper R, Huang S-T, Parmar PP, Clark DJ, Alami H, Fleischmann RD, Perry RD, Peterson SN. 2010. Proteomic analysis of iron acquisition, metabolic and regulatory responses of Yersinia pestis to iron starvation. BMC Microbiol. 10:30. 10.1186/1471-2180-10-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. 2006. TM4 microarray software suite. Methods Enzymol. 411:134–193 [DOI] [PubMed] [Google Scholar]
- 53.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Methodol. 57:289–300 [Google Scholar]
- 54.Pieper R, Gatlin-Bunai CL, Mongodin EF, Parmar PP, Huang S-T, Clark DJ, Fleischmann RD, Gill SR, Peterson SN. 2006. Comparative proteomic analysis of Staphylococcus aureus strains with differences in resistance to the cell wall-targeting antibiotic vancomycin. Proteomics 6:4246–4258 [DOI] [PubMed] [Google Scholar]
- 55.Kuntumalla S, Braisted JC, Huang S-T, Parmar PP, Clark DJ, Alami H, Zhang Q, Donohue-Rolfe A, Tzipori S, Fleischmann RD, Peterson SN, Pieper R. 2009. Comparison of two label-free global quantitation methods, APEX and 2D gel electrophoresis, applied to the Shigella dysenteriae proteome. Proteome Sci. 7:22. 10.1186/1477-5956-7-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74:5383–5392 [DOI] [PubMed] [Google Scholar]
- 57.Braisted JC, Kuntumalla S, Vogel C, Marcotte EM, Rodrigues AR, Wang R, Huang S-T, Ferlanti ES, Saeed AI, Fleischmann RD, Peterson SN, Pieper R. 2008. The APEX quantitative proteomics tool: generating protein quantitation estimates from LC-MS/MS proteomics results. BMC Bioinformatics 9:529. 10.1186/1471-2105-9-529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu P, Vogel C, Wang R, Yao X, Marcotte EM. 2007. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 25:117–124 [DOI] [PubMed] [Google Scholar]
- 59.Neidhardt FC, Umbarger HE. 1996. Chemical composition of Escherichia coli, p 13–16 In Neidhardt FC, Curtiss R, III, Ingraham JL, Riley M, Schaechter M, Umbarger HE. (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. American Society for Microbiology, Washington, DC [Google Scholar]
- 60.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio Collection. Mol. Syst. Biol. 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Purdy GE, Fisher CR, Payne SM. 2007. IcsA surface presentation in Shigella flexneri requires the periplasmic chaperones DegP, Skp, and SurA. J. Bacteriol. 189:5566–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oaks EV, Wingfield ME, Formal SB. 1985. Plaque formation by virulent Shigella flexneri. Infect. Immun. 48:124–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Headley VL, Payne SM. 1990. Differential protein expression by Shigella flexneri in intracellular and extracellular environments. Proc. Natl. Acad. Sci. U. S. A. 87:4179–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kayath CA, Hussey S, El hajjami, Nagra NK, Philpott D, Allaoui A. 2010. Escape of intracellular Shigella from autophagy requires binding to cholesterol through the type III effector, IcsB. Microbes Infect. 12:956–966 [DOI] [PubMed] [Google Scholar]
- 66.Allaoui A, Mounier J, Prévost MC, Sansonetti PJ, Parsot C. 1992. icsB: a Shigella flexneri virulence gene necessary for the lysis of protrusions during intercellular spread. Mol. Microbiol. 6:1605–1616 [DOI] [PubMed] [Google Scholar]
- 67.Beloin C, Dorman CJ. 2003. An extended role for the nucleoid structuring protein H-NS in the virulence gene regulatory cascade of Shigella flexneri. Mol. Microbiol. 47:825–838 [DOI] [PubMed] [Google Scholar]
- 68.Beloin C, Deighan P, Doyle M, Dorman CJ. 2003. Shigella flexneri 2a strain 2457T expresses three members of the H-NS-like protein family: characterization of the Sfh protein. Mol. Genet. Genomics 270:66–77 [DOI] [PubMed] [Google Scholar]
- 69.Benjelloun-Touimi Z, Sansonetti PJ, Parsot C. 1995. SepA, the major extracellular protein of Shigella flexneri: autonomous secretion and involvement in tissue invasion. Mol. Microbiol. 17:123–135 [DOI] [PubMed] [Google Scholar]
- 70.Ackermann N, Tiller M, Anding G, Roggenkamp A, Heesemann J. 2008. Contribution of trimeric autotransporter C-terminal domains of oligomeric coiled-coil adhesin (Oca) family members YadA, UspA1, EibA, and Hia to translocation of the YadA passenger domain and virulence of Yersinia enterocolitica. J. Bacteriol. 190:5031–5043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kirjavainen V, Jarva H, Biedzka-Sarek M, Blom AM, Skurnik M, Meri S. 2008. Yersinia enterocolitica serum resistance proteins YadA and Ail bind the complement regulator C4b-binding protein. PLoS Pathog. 4(8):e1000140. 10.1371/journal.ppat.1000140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miller VL, Falkow S. 1988. Evidence for two genetic loci in Yersinia enterocolitica that can promote invasion of epithelial cells. Infect. Immun. 56:1242–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Romeo T. 1998. Global regulation by the small RNA-binding protein CsrA and the non-coding RNA molecule CsrB. Mol. Microbiol. 29:1321–1330 [DOI] [PubMed] [Google Scholar]
- 74.Zhu J, Shalel-Levanon S, Bennett G, San K-Y. 2007. The YfiD protein contributes to the pyruvate formate-lyase flux in an Escherichia coli arcA mutant strain. Biotechnol. Bioeng. 97:138–143 [DOI] [PubMed] [Google Scholar]
- 75.Payne SM, Wyckoff EE, Murphy ER, Oglesby AG, Boulette ML, Davies NM. 2006. Iron and pathogenesis of Shigella: iron acquisition in the intracellular environment. Biometals 19:173–180 [DOI] [PubMed] [Google Scholar]
- 76.Lawlor KM, Payne SM. 1984. Aerobactin genes in Shigella spp. J. Bacteriol. 160:266–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Letoffe S, Delepelaire P, Wandersman C. 2006. The housekeeping dipeptide permease is the Escherichia coli heme transporter and functions with two optional peptide binding proteins. Proc. Natl. Acad. Sci. U. S. A. 103:12891–12896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Homma H, Chiba N, Kobayashi T, Kudo I, Inoue K, Ikeda H, Sekiguchi M, Nojima S. 1984. Characteristics of detergent-resistant phospholipase A overproduced in E. coli cells bearing its cloned structural gene. J. Biochem. 96:1645–1653 [DOI] [PubMed] [Google Scholar]
- 79.Bernardini ML, Sanna MG, Fontaine A, Sansonetti PJ. 1993. OmpC is involved in invasion of epithelial cells by Shigella flexneri. Infect. Immun. 61:3625–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu X-B, Tian L-H, Zou H-J, Wang C-Y, Yu Z-Q, Tang C-H, Zhao F-K, Pan J-Y. 2013. Outer membrane protein OmpW of Escherichia coli is required for resistance to phagocytosis. Res. Microbiol. 164:848–855 [DOI] [PubMed] [Google Scholar]
- 81.Zhu L, Liu X-K, Zhao G, Zhi Y-D, Bu X, Ying T-Y, Feng E-L, Wang J, Zhang X-M, Huang P-T, Wang H-L. 2007. Dynamic proteome changes of Shigella flexneri 2a during transition from exponential growth to stationary phase. Genomics Proteomics Bioinformatics 5:111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Santos JM, Freire P, Vicente M, Arraiano CM. 1999. The stationary-phase morphogene bolA from Escherichia coli is induced by stress during early stages of growth. Mol. Microbiol. 32:789–798 [DOI] [PubMed] [Google Scholar]
- 83.Zhao X-J, Wang L, Shiva S, Tejero J, Wang J, Frizzell S, Gladwin MT. 2013. Mechanisms for cellular NO oxidation and nitrite formation in lung epithelial cells. Free Radic. Biol. Med. 61C:428–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pieper R, Zhang Q, Parmar PP, Huang S-T, Clark DJ, Alami H, Donohue-Rolfe A, Fleischmann RD, Peterson SN, Tzipori S. 2009. The Shigella dysenteriae serotype 1 proteome, profiled in the host intestinal environment, reveals major metabolic modifications and increased expression of invasive proteins. Proteomics 9:5029–5045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kuntumalla S, Zhang Q, Braisted JC, Fleischmann RD, Peterson SN, Donohue-Rolfe A, Tzipori S, Pieper R. 2011. In vivo versus in vitro protein abundance analysis of Shigella dysenteriae type 1 reveals changes in the expression of proteins involved in virulence, stress and energy metabolism. BMC Microbiol. 11:147. 10.1186/1471-2180-11-147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Headley V, Hong M, Galko M, Payne SM. 1997. Expression of aerobactin genes by Shigella flexneri during extracellular and intracellular growth. Infect. Immun. 65:818–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Massé E, Gottesman S. 2002. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 99:4620–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Runyen-Janecky L, Daugherty A, Lloyd B, Wellington C, Eskandarian H, Sagransky M. 2008. Role and regulation of iron-sulfur cluster biosynthesis genes in Shigella flexneri virulence. Infect. Immun. 76:1083–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu L, Zhao G, Stein R, Zheng X, Hu W, Shang N, Bu X, Liu X, Wang J, Feng E, Wang B, Zhang X, Ye Q, Huang P, Zeng M, Wang H. 2010. The proteome of Shigella flexneri 2a 2457T grown at 30 and 37°C. Mol. Cell. Proteomics 9:1209–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Payne SM, Niesel DW, Peixotto SS, Lawlor KM. 1983. Expression of hydroxamate and phenolate siderophores by Shigella flexneri. J. Bacteriol. 155:949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.