

Abstract
Humans have evolved elaborate mechanisms to activate p53 in response to insults that lead to cancer, including the binding and inhibition of Hdm2 by the 60S ribosomal proteins (RPs) RPL5 and RPL11. This same mechanism appears to be activated upon impaired ribosome biogenesis, a risk factor for cancer initiation. As loss of RPL5/RPL11 abrogates ribosome biogenesis and protein synthesis to the same extent as loss of other essential 60S RPs, we reasoned the loss of RPL5 and RPL11 would induce a p53-independent cell cycle checkpoint. Unexpectedly, we found that their depletion in primary human lung fibroblasts failed to induce cell cycle arrest but strongly suppressed cell cycle progression. We show that the effects on cell cycle progression stemmed from reduced ribosome content and translational capacity, which suppressed the accumulation of cyclins at the translational level. Thus, unlike other tumor suppressors, RPL5/RPL11 play an essential role in normal cell proliferation, a function cells have evolved to rely on in lieu of a cell cycle checkpoint.
INTRODUCTION
Living organisms are continuously exposed to environmental insults, many of which result in cellular damage. This has led to the evolution of surveillance mechanisms, which gauge the extent of damage and determine the cell's fate. Many of these responses rely on the activation of the tumor suppressor p53, a master regulator of cell cycle arrest, apoptosis, and senescence (1). Under normal growth conditions, levels of p53 are largely restricted by its rapid degradation, mediated by the E3-ligase, Hdm2, which targets p53 to the proteasome. In turn, levels of p53 rapidly increase upon a cellular insult, principally through direct inhibition of Hdm2. Under such conditions, a number of mechanisms have been implicated in regulating the activity and levels of Hdm2, including phosphorylation, ubiquitination, and the binding of inhibitory cofactors (2). A major insult in normal cells is triggered by oncogenic stress, caused by the overexpression or overactivation of proteins with tumorigenic potential. This leads to the induction of the tumor suppressor ARF, which physically sequesters and inhibits Hdm2, allowing p53 levels to accumulate, restraining the proliferation and survival of tumor cells (3). Recent studies have implicated three additional inhibitory cofactors in addition to ARF that directly bind to and suppress Hdm2-mediated p53 degradation. These include the tumor suppressor NUMB, a negative regulator of Notch 1 (4), and, most recently, two essential 60S ribosomal proteins (RPs), RPL5 and RPL11 (5), which play a central role in mediating p53 stabilization following impaired ribosome biogenesis (6, 7).
RPL5 and RPL11 bind to the central acidic domain of Hdm2 within the highly conserved C4 zinc finger at a site distinct from that bound by ARF (5). The importance of this interaction in tumorigenesis was first suggested by the finding in human osteosarcoma of a C305P mutation in the C4 zinc finger of Hdm2 which disrupted its interaction with RPL5 and RPL11 but not ARF (8). Knock-in mice bearing this mutation were crossed with transgenic mice overexpressing the c-Myc proto-oncogene under the control of the immunoglobulin heavy-chain promoter and enhancer (Eμ-Myc) (5). As c-Myc drives the coordinated biogenesis of nascent ribosomes (9), its overexpression in the Eμ-Myc model is predicted to result in elevated levels of RPL5 and RPL11, inhibition of Mdm2, and induction of p53, which would retard tumor development. Supporting this model, Eμ-Myc mice harboring the Mdm2 C305P knock-in mutation developed more aggressive lymphomas and succumbed more quickly, with a median survival of 9 weeks versus 20 weeks for littermates expressing wild-type Mdm2, despite the absence of any impact on ARF binding to Mdm2 (5). These findings support a role for RPL5/RPL11-dependent inhibition of Hdm2 in protecting the cell from the adverse effects of excessive ribosome biogenesis. Consistent with such tumors being addicted to high levels of nascent ribosome biogenesis, selective inhibition of RNA polymerase I in Eμ-Myc lymphomas led to the induction of p53-dependent apoptosis through the apparent activation of the same RPL5/RPL11-Mdm2-p53 checkpoint (10). Therefore, drugs that disrupt ribosome biogenesis could be exploited to induce selective apoptosis in tumors that are characterized by high rates of ribosome biogenesis.
The studies above underscore the importance of surveillance mechanisms that monitor the status of ribosome biogenesis in order to prevent aberrant cell growth. This same mechanism appears to be implicated under conditions of impaired ribosome biogenesis as either hyper- or hypoactivation of ribosome biogenesis can lead to changes in the pattern of translation, which will ultimately alter the genetic program (11–13). We first described the existence of such a mechanism in livers of adult mice following the conditional deletion of RPS6, an essential component of the 40S ribosomal subunit. The absence of RPS6 and the resulting abrogation of 40S biogenesis blocked the ability of hepatocytes to enter S phase following partial hepatectomy (14). We subsequently showed that this response was mediated by the induction of p53 and that it could be recapitulated in cell culture by the depletion of other essential RPs of the 40S or 60S ribosomal subunit (15). These studies led to the finding that the upregulation of p53 upon impairment of the biogenesis of either subunit was mediated by the binding and inhibition of Hdm2 by RPL5 and RPL11 (7, 15). The inhibitory effects of RPL5 and RPL11 on Hdm2 are mutually dependent on both proteins as depletion of either was sufficient to relieve p53 induction and cell cycle arrest (6, 7). In addition to RPL5 and RPL11, RPS7 and RPL23 have been shown to bind and inhibit Hdm2 under conditions of acute inhibition of rRNA synthesis caused by low doses of actinomycin D (16–19). However, our recent studies suggest that the apparent effects of depletion of either RPS7 or RPL23 on the induction of p53 can be ascribed to a reduction in global translation rather than a decrease in p53 stability (7). Thus, RPL5 and RPL11 appear to be the only RPs required for p53 induction in response to aberrant ribosome biogenesis.
It is generally thought that reduced expression of tumor suppressors allows the cell to evade cell cycle checkpoints and acquire higher proliferative capacity, as observed in cells with decreased ARF or p53 levels (20). Similarly, the disruption of the RPL5/RPL11-Mdm2 checkpoint, due to the Mdm2 C305P mutation, led to accelerated lymphomagenesis in the Eμ-Myc background (5). However, unlike ARF, RPL5 and RPL11 play a dual role in the proliferative response as they are not only negative regulators of Mdm2 but also essential RPs required for the synthesis of 60S ribosomes. Indeed, their depletion impairs global translation to the same extent as that of other essential RPs of the 60S ribosomal subunit (7). In addition, in Drosophila, hypomorphic mutations in RPs, including those in orthologues of RPL5 and RPL11, have been identified as Minute mutations, which result in haploinsufficient mutants which are characterized by delayed development, short thin bristles, and poor viability, phenotypes attributed to reduced ribosome content and translational capacity (21). In humans, hypomorphic mutations in RPL5 and RPL11, as well as other RPs, are causal in the pathology of Diamond Blackfan anemia (DBA) (22), a congenital erythroid aplasia syndrome leading to anemia (23–28). Given that most studies to date have been limited to tumor cells (29), which have lost a number of checkpoints, it is unclear as to whether the reduction in ribosome content and global translation, the loss of a p53 checkpoint, or the activation of an alternative p53-independent cell cycle checkpoint is the underlying mechanism that regulates cell proliferation upon reduction of RPL5 or RPL11 levels.
Given the importance of RPL5 and RPL11 in tumor suppression, we set out to examine the effect of their depletion on global translation, the induction of p53, and cell cycle progression in primary human lung fibroblasts. We demonstrated that depletion of RPL5 or RPL11, unlike depletion of another essential 60S RP, did not induce p53 but repressed cell proliferation, suggesting that an alternative cell cycle checkpoint may regulate cell cycle progression following reduction in their expression. However, RPL5- or RPL11-depleted cells did not accumulate in any specific phase of the cell cycle. Instead, as shown by bromodeoxyuridine (BrdU) pulse-chase experiments, they progressed at a much lower rate through each phase of the cell cycle. This effect was associated with inhibition of global translation such that mRNAs encoding key cyclins, including those of cyclin E1, cyclin A2, and cyclin B1, were present on polysomes of a smaller mean size in RPL5- or RPL11-depleted cells than in control cells. Consistent with this finding, codepletion of p53 and RPL7a, an essential 60S RP, blocked the induction of the p53 cell cycle checkpoint but did not completely rescue cell growth as the effects of RPL7a depletion on global translation persisted. Our findings are consistent with a recent report highlighting the availability of ribosomes as the rate-limiting step in translation initiation (30). Thus, mammalian cells appear to have evolved a general RPL5/RPL11-dependent cell cycle checkpoint in response to impaired or hyperactivated ribosome biogenesis, whereas in the case of lesions in RPL5 or RPL11 they rely on the their essential role in ribosome biogenesis, rather than a cell cycle checkpoint, to limit proliferation.
MATERIALS AND METHODS
Cell culture, siRNA transfection, and synchronization.
MRC5 (Medical Research Council 5), A549, and U2-OS cells were cultured in Dulbecco modified essential medium (DMEM) containing 4,500 mg/ml l-glucose, 4 mM l-glutamine, and 110 mg/liter sodium pyruvate supplemented with 10% fetal bovine serum. Transfection of small interfering RNA (siRNA) was performed as previously described using the calcium phosphate precipitation method (31). For each siRNA, 20 pmol and 60 pmol were transfected in 6- and 10-cm dishes, respectively. The Allstar Negative Control (Qiagen) was used as nonsilencing (NS) control siRNA. The target sequences of the siRNAs against human RPL11, RPL5, RPL7a, and p53 have been previously described (7, 15). For synchronization experiments, MRC5 fibroblasts were first transfected with the siRNAs indicated in Fig. 5. At 6 h posttransfection, the cells were washed twice with Dulbecco's phosphate-buffered saline (DPBS) and incubated in fresh serum-free DMEM for 56 h. Cells were stimulated with DMEM supplemented with 10% fetal bovine serum (FBS) and processed at the indicated time points.
Fig 5.

Delay of entry into S phase and of accumulation of key cyclins and associated kinase activity in RPL11-depleted cells. MRC5 cells transfected with the indicated siRNAs were synchronized in G0/G1 by serum starvation and harvested at the indicated time points following serum stimulation. Parallel samples were processed for cell cycle analysis by flow cytometry (A and B), Western blot analysis of cyclin E and cyclin A protein expression (C), and measurement of cyclin E-associated kinase activity by in vitro kinase assay (D). IP, immunoprecipitation; p-histone H1, phospho-histone H1; siNS, NS siRNA; siL11, siRNA targeting RPL11.
Flow cytometry and BrdU pulse-chase.
Flow cytometry analysis of cell cycle distribution by staining with propidium iodide/RNase solution (Phoenix Flow Systems) and quantification with Modfit (Verity Software House, Inc.) have been previously described (15). Annexin V and 7-aminoactinomycin D (7-AAD) staining (BioLegend) were performed according to the manufacturer's instructions. To perform BrdU pulse-chase experiments, asynchronously growing MRC5 cells transfected with the indicated siRNAs for 48 h were pulse-labeled for 30 min with 10 μM BrdU (BD Pharmingen), washed twice with PBS, and incubated in BrdU-free complete medium for the time points indicated in Fig. 4. Cells were collected by trypsinization, centrifuged, and fixed in ice-cold 70% ethanol. Staining with a fluorescein isothiocyanate-conjugated anti-BrdU antibody (Becton, Dickinson) was performed according to the manufacturer's instructions with the following modification: DNA was denatured by a 30-min incubation at room temperature with 2N HCl with 0.3 mg/ml pepsin. Propidium iodide/RNase solution was added to the cell suspension before the analysis. A Coulter Epics XL (Beckman Coulter) with a 488-nm argon ion laser was used for acquisition of flow cytometry data, as previously described (15).
Fig 4.
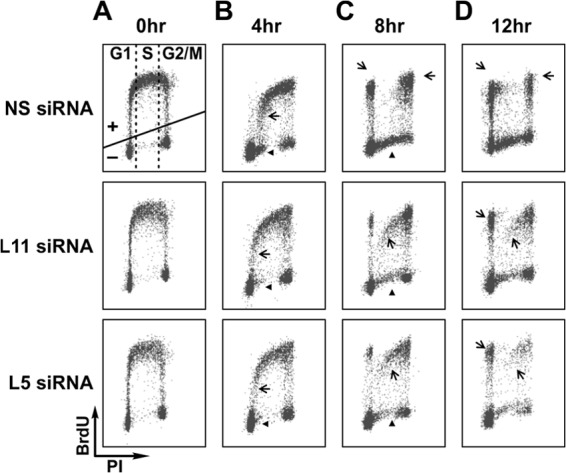
Cells depleted of RPL11 or RPL5 progress through the cell cycle more slowly than control cells. Flow cytometry analysis of BrdU incorporation in MRC5 cells transfected with the indicated siRNAs. Cells were collected at 0, 4, 8, and 12 h following BrdU removal, stained with propidium iodide (PI) and fluorescein-conjugated anti-BrdU antibody, and subjected to flow cytometry analysis to evaluate rates of cell cycle progression. Arrows indicate BrdU-positive cells and arrowheads BrdU-negative cells. (See Results for details.)
Protein extraction and Western blot analysis.
The analysis of protein expression in asynchronous MRC5 cells by Western blotting was performed as previously described (15). For the analysis of protein expression in synchronized MRC5 fibroblasts, cells were washed with ice-cold PBS and lysed in 50 mM Tris-HCl (pH 8), 250 mM NaCl, and 0.1% Nonidet P-40 (NP-40), supplemented with 1 mM dithiothreitol (DTT), protease inhibitor cocktail (Roche), and phosphatase inhibitor cocktails (Sigma). Mouse anti-human cyclin E1 antibody (HE12, catalog number 4129; Cell Signaling Technology) and rabbit anti-human cyclin A antibody (H432; Santa Cruz Biotechnology) were used at dilutions of 1:1,000.
In vitro kinase assay.
A total of 150 μg of protein lysate was incubated for 2 h on ice with rabbit anti-human cyclin E antibody (C-19; Santa Cruz Biotechnology), followed by a 30-min incubation with protein A-Sepharose 6M beads (GE Healthcare) at 4°C. The immunoprecipitated cyclin E complex was washed three times with extraction buffer and once with 1× kinase buffer (20 mM Tris-HCl, pH 7.5, 7.5 mM MgCl2, 1 mM DTT). The in vitro kinase assay reaction was performed for 30 min at 37°C in 15 μl of 1× kinase buffer containing 5 μg of recombinant histone H1 (Sigma) as the substrate, 300 μM cold ATP (Sigma), and 10 μCi of [γ-32P]ATP (PerkinElmer). The reactions were terminated by the addition of Laemmli SDS protein sample buffer, denatured at 99°C for 3 min, and separated on 4 to 20% SDS-PAGE (Bio-Rad) gels before being transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Immobilon-P) and exposed to a phosphor screen. The screen was developed with a Storm 840 phosphorimager.
qRT-PCR analysis of mRNA expression.
Quantitative reverse transcription-PCR (qRT-PCR) analysis of mRNA expression was largely performed as previously described (15). Briefly, total cellular RNA was extracted and purified using an RNeasy kit from Qiagen (catalog number 74104). A description of RNA purification from polysome fractions is found in the paragraph “Polysome profiles.” Following reverse transcription (Superscript III First Strand Synthesis SuperMix, catalog number 11752-050; Invitrogen), cDNAs were used in qRT-PCRs with Fast SYBR green Master Mix (catalog number 4385612; Applied Biosystems) and analyzed on a 7500 Fast Realtime PCR system from Applied Biosystems. Standard curves were generated for each gene using PCR-amplified fragments from each target. Reaction cycle parameters and primers for RPL11 and RPL7a (15), RPL5 and β-actin (7), and 18S rRNA (79) have been previously described. Other primers used include the following: cyclin E1 forward primer, 5′-TGCAGAGCTGTTGGATCTCTGTGT-3′; cyclin E1 reverse primer, 5′-ACCATGGCAAATGGAACCATCCAC-3′; cyclin A2 forward primer, 5′-GCTGGAGCTGCCTTTCATTTAGCA-3′; cyclin A2 reverse primer, 5′-TTGACTGTTGTGCATGCTGTGGTG-3′; cyclin B1 forward primer, 5′-AGGAAGAGCAAGCAGTCAGACCAA-3′; cyclin B1 reverse primer, 5′-GCAGCATCTTCTTGGGCACACAAT-3′; luciferase forward primer, 5′-ATCAGGCAAGGATATGGGCTCACT-3′; luciferase reverse primer, 5′-TCCAGATCCACAACCTTCGCTTCA-3′.
Polysome profiles.
Preparations of cellular extracts for polysome profiles, sucrose gradient centrifugation, and profile recording have been previously described (15). Heparin was omitted from lysates used to analyze cyclin E1, cyclin A2, and cyclin B1 mRNAs by qRT-PCR due to its inhibitory effect on the PCR. Sucrose gradient fractions were collected by upward displacement, and 50 pg of synthetic luciferase mRNA (catalog number L4561; Promega) and 10 μg of glycogen (catalog number AM9510; Life Technology) were added to each fraction to control for extraction and PCR efficiency and to improve RNA recovery, respectively. Extraction and precipitation of RNA from sucrose fractions have been previously described (15). Precipitated RNA was washed twice with ice-cold 70% ethanol, dried, and resuspended in RNase-free H2O. Equal volumes of RNA from each fraction were subjected to cDNA synthesis and qRT-PCR analysis. Levels of cyclin E1, cyclin A2, and cyclin B1 mRNAs in each fraction were normalized to luciferase mRNA and plotted as the percentage of total mRNAs from all 12 fractions.
Labeling of cells with [35S]methionine.
Cells were labeled for 1 h with 20 μCi of [35S]methionine (PerkinElmer). Extraction and precipitation of proteins and measurement of incorporated radioactivity were performed as previously described (15). For each sample, the number of counts per minute (cpm) was normalized to that of total protein.
Proliferation assay by cell number.
At the time points posttransfection indicated in Fig. 2A, MRC5 cells were trypsinized, collected by centrifugation, and resuspended in known volumes of PBS. The total number of cells in each sample was determined by hemacytometer counting, and the ratio of the final cell number to the initial number of plated cells was determined. The population doubling time was calculated using the algorithm of V. Roth (http://www.doubling-time.com/compute.php).
Fig 2.

RPL11 or RPL5 depletion represses cell proliferation without inducing cell cycle arrest. (A) Proliferation assay of MRC5 cells transfected with the indicated siRNAs and harvested and counted at the indicated time points following transfection. The calculated population doubling times were 34 h, 43 h, and 103 h, respectively, for RPL11-, RPL5-, and RPL7a-depleted cells compared to 20 h for the NS siRNA-treated control cells. (B) Representative cell cycle distribution of the MRC5 cells used in the proliferation assay in panel A at the 24-h time point. Each channel represents the signal intensity of DNA content after amplification. (C) Quantification of the cell cycle distribution of MRC5 cells counted in the proliferation assay in panel A. (D) Representative annexin V/7-AAD staining images of MRC5 cells transfected with indicated siRNAs for 96 h. (E) Quantification of the percentage of annexin V and 7-AAD staining of the samples from panel D. Each bar in panels A, C, and E represents the means ± standard errors of the means from three independent experiments. Statistical significance was determined using a two-tailed t test. ns, not significant.
Statistics.
Statistical significance analysis of the functional studies in Fig. 2C and 3B was carried out using a two-tailed Student's t test in Prism 5.
Fig 3.

p53 and RPL7a codepletion resembles RPL5 or RPL11 depletion. (A) Representative cell cycle distribution of the MRC5 cells used in the proliferation assay in panel C at the 72-h time point. (B) Quantification of the cell cycle distribution of MRC5 cells at the 72-h time point used in the proliferation assay in panel C. Each bar in panel B represents the means ± standard errors of the means from three independent experiments. Statistical significance was determined using a two-tailed t test. (C) Proliferation assay of MRC5 cells transfected with the indicated siRNAs and harvested and counted at the indicated time points following transfection. The calculated population doubling time for cells codepleted of p53 and RPL7a was 28 h compared to 86 h for RPL7a siRNA-transfected cells and 22 and 20 h for NS siRNA- and p53 siRNA-transfected cells, respectively.
RESULTS
Depletion of RPL5 or RPL11 does not induce p53.
Consistent with the role of RPL5 and RPL11 as positive regulators of p53, depletion of either protein in many cell types does not lead to induction of p53 (16, 32, 33). We confirmed these findings in human A549 or U2-OS cells, which express wild-type p53 (Fig. 1A and B) (10, 34). However, given the essential role of RPL5 and RPL11 in 60S ribosome biogenesis, we reasoned that a checkpoint which monitors the effect of their loss may be absent in tumor cell types used in earlier studies. Indeed, in A549 and U2-OS cells, the tumor suppressor ARF is either deleted or silenced (35, 36). Therefore, to address this issue we utilized MRC5 (Medical Research Council 5) primary human lung fibroblasts (37). Compared to MRC5 cells treated with a control nonsilencing (NS) siRNA, treatment with siRNAs against either RPL5 or RPL11 did not induce p53 over basal levels and had no measurable effect on its downstream target, p21 (Fig. 1C). In contrast, an siRNA targeting RPL7a, an essential RP of the 60S ribosomal subunit, caused a significant increase in both p53 and p21 levels (Fig. 1C). These differences in the p53 response were not attributed to the extent of depletion of each transcript as the mRNAs of all three 60S RPs were depleted by more than 85% compared to control cell levels (Fig. 1D).
Fig 1.

Depletion of RPL11 or RPL5 differs from RPL7a depletion in the induction of p53, despite similar effects on nascent ribosome biogenesis and global translation. (A to C) Western blot analysis showing expression levels of p53, p21, and β-actin proteins in (A) A549 cells, (B) U2-OS cells, and (C) MRC5 cells transfected with the indicated siRNAs for 48 h. (D) qRT-PCR analysis showing levels of RPL11 (L11), RPL5 (L5), and RPL7a (L7a) mRNAs in MRC5 cells transfected with the indicated siRNAs for 48 h. Each bar represents the ratio of the indicated mRNA to that of β-actin mRNA, normalized to the ratio obtained for the NS siRNA-treated sample. (E) Polysome profile analysis of extracts of MRC5 cells transfected with the indicated siRNAs. (F) Measurement of [35S]methionine incorporation in MRC5 cells transfected with the indicated siRNAs. The data in panels D and F represent the means ± standard errors of the means from a minimum of two independent experiments.
To determine whether the distinct p53 responses resulted from differential effects on the ribosome content and translational machinery, we analyzed polysome profiles from MRC5 cells treated with an siRNA targeting RPL5, RPL11, or RPL7a. In each case, compared to cells treated with an NS siRNA, there was an equivalent decrease of native 60S ribosomal subunits and a concomitant increase in native 40S ribosomal subunits, accompanied by a small but apparent reduction in the mean polysome size (Fig. 1E). In addition, the reduction in the amount of 60S ribosomal subunits relative to the amount of 40S ribosomal subunits led to the increased binding to mRNAs of 43S preinitiation complexes, detected as halfmer polyribosomes, apparent as pronounced shoulders on the right sides of the 80S monosome and polysomal peaks (38) (Fig. 1E). Because of the impact of the increased formation of halfmers and the inherent difficulty of assigning the baseline to the polysome profile, we turned to 35S-labeled methionine incorporation to better quantitate the difference in translational rates. Consistent with the apparent reduced mean polysome size, translation rates in MRC5 cells depleted of RPL5, RPL11, and RPL7a, as measured by 35S-labeled methionine incorporation into nascent proteins, were decreased by the same extent, ∼30%, compared to control cells (Fig. 1F). Therefore, although RPL5, RPL11, and RPL7a depletion in primary human fibroblasts had similar inhibitory effects on ribosome biogenesis and translation, results differed in that the loss of RPL7a induced p53, whereas loss of either RPL5 or RPL11 did not.
Depletion of RPL5 or RPL11 reduces the rate of proliferation without inducing a cell cycle checkpoint.
Since RPL5 or RPL11 depletion had a pronounced inhibitory effect on 60S ribosome biogenesis and global translation (Fig. 1E and F), we reasoned that under such conditions a p53-independent cell cycle checkpoint may be activated. To address this, we performed a proliferation assay by counting MRC5 cells transfected with NS, RPL11, RPL5, or RPL7a siRNA at 24, 48, and 72 h after transfection (Fig. 2A). Although the rate of proliferation of RPL5- and RPL11-depleted cells was severely affected at 48 h and 72 h posttransfection compared to that of control cells, the effect of RPL7a depletion was more pronounced (Fig. 2A), despite equivalent effects on global translation (Fig. 1E and F). The calculated population doubling time was 34 h, 43 h, and 103 h, respectively, for RPL11-, RPL5-, and RPL7a-depleted cells, compared to 20 h for the NS siRNA-treated control cells. Therefore, if a distinct cell cycle checkpoint was engaged by depletion of RPL5 or RPL11, it was not as strong as that induced by p53. In order to gain more insight into this possibility, we compared by flow cytometry the cell cycle profile of RPL5- or RPL11-depleted cells to that of RPL7a-depleted cells. Unexpectedly, the cell cycle distributions of RPL5- and RPL11-depleted cells were largely indistinguishable from the distribution of NS siRNA-treated cells (Fig. 2B and C). In contrast, RPL7a-depleted cells showed a sharp decrease in the S-phase population, concomitant with an accumulation of cells in G0/G1 and G2/M phases, as early as 24 h posttransfection (Fig. 2B and C). The effects of RP depletion on proliferation were not due to an increase in cell death as greater than 90% of cells depleted of RPL5, RPL11, or RPL7a were negative for annexin V and 7-AAD staining as late as 96 h posttransfection (Fig. 2D and E). Thus, unlike loss of RPL7a, loss of RPL5 or RPL11 decreases cell proliferation without inducing an apparent cell cycle checkpoint or causing cell death.
The depletion of RPL7a impaired ribosome biogenesis and inhibited global translational rates to a similar extent as RPL5 or RPL11 depletion (Fig. 1E and F); however, the inhibitory effects on cell proliferation were more pronounced (Fig. 2A). We reasoned that removing the p53-dependent cell cycle block in RPL7a-depleted cells would rescue their ability to progress through the cell cycle but at a proliferative rate equivalent to that observed for RPL5- or RPL11-depleted cells. Consistent with this hypothesis, codepletion of RPL7a and p53 rescued the G1 and G2/M block (Fig. 3A and B) and partially rescued the inhibition of cell proliferation induced by depletion of RPL7a (Fig. 3C) to levels similar to those observed for RPL5- and RPL11-depleted cells (Fig. 2A). The calculated population doubling time for cells codepleted of p53 and RPL7a was 28 h compared to 86 h for RPL7a siRNA-transfected cells and 22 and 20 h for NS siRNA- and p53 siRNA-transfected cells, respectively. The data demonstrate that in the absence of p53, depletion of an essential RP leads to a defect in cell proliferation, with no obvious activation of a p53-independent cell cycle checkpoint.
RPL5- and RPL11-depleted cells progress through the cell cycle at a lower rate.
The apparent normal cell cycle distribution, despite a decrease in population doubling times, suggests that the progression of RPL5- and RPL11-depleted MRC5 cells through each phase of the cell cycle may be prolonged. To measure the rate of progression through the cell cycle, cells transfected with NS, RPL5, or RPL11 siRNA were first pulse-labeled for 30 min with BrdU and then chased for increasing times with BrdU-free medium. At the beginning of the chase, all cells in S phase were labeled with BrdU, whereas the majority of the cells in G1 and G2/M phases were negative for BrdU staining (Fig. 4A). After a chase of 4 h, BrdU-positive NS control cells were enriched in late S phase, whereas the majority of BrdU-positive RPL5- or RPL11-depleted cells were primarily in early S phase (Fig. 4B, arrows). This delay was also apparent when the BrdU-negative population of cells in S phase was analyzed: in the control sample, BrdU-negative cells appeared after 4 h in early S phase as a cluster protruding from the G1 population (Fig. 4B, arrowheads). This phenomenon was significantly reduced in RPL5- or RPL11-depleted cells (Fig. 4B, arrowheads). After 8 h of chase, almost all BrdU-positive cells in the control sample had exited S phase and entered G2/M (Fig. 4C, arrows), whereas some appeared in G1, indicating that they had undergone a round of cell division (Fig. 4C, arrows). In contrast, at the same time point a significant number of the BrdU-positive RPL5- or RPL11-depleted cells were still trailing in S phase (Fig. 4C, arrows). After 12 h of chase, RPL5- or RPL11-depleted BrdU-positive cells had reached G2/M but were strongly delayed compared to the BrdU-positive control cells, which had accumulated in the next G1 phase, with some entering a second round of S phase (Fig. 4D, arrows). These data demonstrate that depletion of RPL5 or RPL11 reduces the rate of cell cycle progression without activation of a cell cycle checkpoint.
If cells depleted of either RPL5 or RPL11 progress through the cell cycle at a lower rate (Fig. 4), then one would predict that if arrested at any phase of the cell cycle and then released, they would progress to the next phase of the cell cycle with delayed kinetics. To examine this possibility, we transfected cells with NS or RPL11 siRNA, arrested them in G0/G1 by serum deprivation, and then analyzed the rate at which they reentered the cell cycle following serum stimulation. In both samples, nearly 90% of cells accumulated in G0/G1 in response to serum deprivation (Fig. 5A). Within 16 h of serum stimulation, ∼50% of NS control cells had entered S phase, whereas virtually all of the RPL11-depleted cells remained in G0/G1. At 20 h post-serum stimulation, the majority of control cells had progressed toward late S phase and entered G2/M phase, whereas only ∼26% of RPL11-depleted cells had entered early S phase (Fig. 5A). The analyses at time points up to 32 h post-serum stimulation highlight the major difference in rates of cell cycle progression between RPL11-depleted and control cells (Fig. 5B). These data, in conjunction with the BrdU pulse-chase experiment, argue that the consequence of depletion of RPL5 or RPL11 is to hamper, rather than halt, cell cycle progression.
RPL11 depletion leads to delayed cyclin E1 and cyclin A2 accumulation.
If one of the rate-limiting events in the progression of eukaryotic cells from G1 to S phase is the accumulation of cyclin E1 (39, 40), then we would expect this response to be suppressed in RPL11-depleted cells. Indeed, an analysis of the expression of cyclin E1 at sequential time points after serum stimulation revealed a significant delay in the accumulation of cyclin E1 in RPL11-depleted cells compared to that in NS siRNA-treated control cells (Fig. 5C). Consistent with this finding, in vitro kinase assays of cyclin E1 immunoprecipitates showed a shift in the peak of cyclin E1-associated kinase activity from 12 to 16 h post-serum stimulation in control cells to 16 to 24 h post-serum treatment in RPL11-depleted cells (Fig. 5D). The retarded time of activation closely correlated with the delay in the accumulation of cyclin E1 protein levels (Fig. 5C) and entry into S phase (Fig. 5B). Consistent with delayed progression through S phase (Fig. 5B), accumulation of cyclin A2 protein was also retarded in RPL11-depleted cells compared to controls cell levels (Fig. 5C). These results are compatible with the accumulation of cyclin E1 protein dictating the timing of S-phase entry (39, 40) and that of cyclin A2 dictating S-phase progression (41).
Inhibition of global translation in RPL11-depleted cells is responsible for delayed synthesis of cyclins.
The data above raised the question as to the mechanism responsible for the delay in cyclin E1 accumulation in RPL11-depleted cells. Surprisingly, the induction of cyclin E1 mRNA in RPL11-depleted cells was similar to that observed in NS siRNA-treated cells (Fig. 6A). Earlier studies have shown that the suppression of protein synthesis by cycloheximide delays entry of cells into S phase in a dose-dependent manner (42, 43), leading to the hypothesis that the G1-S transition depends on the continuous translation of proteins with a short half-life (42, 44). Given the short, ∼30-min half-life of cyclin E1 protein (45) and the inhibitory effects of RPL11 depletion on mean polysome size, global rates of protein synthesis (Fig. 1E and F), and accumulation of cyclin E1 protein, we examined the distribution of cyclin E1 mRNA on polysome profiles of NS and RPL11 siRNA-treated MRC5 cells at 12 h post-serum stimulation. In control cells, the majority of cyclin E1 mRNA was present on polysomes with a mean size of 4 to 6 ribosomes, whereas in RPL11-depleted cells, it was distributed on polysomes with a mean size of 3 to 5 ribosomes (Fig. 6B). This phenomenon was not specific to cyclin E1 mRNA as we observed a similar shift in the distribution of RPL5 mRNA (Fig. 6C). Interestingly, RPL5 mRNA was associated with polysomes of a mean size of ∼7 to 8 ribosomes in control cells and of 6 to 7 ribosomes in RPL11-depleted cells, despite its length being 1 kb, which is only one-half of the 2-kb-long cyclin E1 mRNA (Fig. 6B). This observation indicates that cyclin E1 mRNA is approximately three times less efficiently recognized by the translational apparatus than the RPL5 mRNA. Recent studies have argued that the rate-limiting component in protein synthesis is ribosome availability, and transcripts possessing a short 5′ untranslated region (UTR) essentially out-compete longer transcripts with highly structured 5′ UTRs (30). Cyclin E1 mRNA contains a highly structured GC-rich 5′ untranslated region with multiple upstream open reading frames (uORFs), which hampers efficient initiation of translation of the mRNA (46, 47). Therefore, the inherent inefficiency of cyclin E1 mRNA translation, coupled with the short half-life of the encoded protein, would render its accumulation particularly sensitive to small changes in the rate of translation. The shift of cyclin E1 mRNA distribution to smaller polysomes was not specific to cells synchronized at the G1/S transition point nor to RPL11 depletion specifically as we observed a similar small, yet reproducible, shift of cyclin E1 mRNA in asynchronous MRC5 cells depleted for 48 h of either RPL5 or RPL11 (Fig. 6D). Moreover, the percentage of cyclin E1 mRNA that was not associated with polysomes (fractions 1 to 4) was increased in cells depleted of RPL5 or RPL11 (Fig. 6D). We also observed an equivalent phenomenon for mRNAs encoding other key cyclins, including cyclin A2 (Fig. 6E) and cyclin B1 (Fig. 6F), which are longer transcripts with a length of 2.8 kb and 2.2 kb, respectively. Consequently, in RPL11-depleted cells, the inefficient translation of mRNAs encoding cell cycle-promoting proteins is consistent with their progression through the cell cycle at a decreased rate. Thus, general lesions in ribosome biogenesis induce p53 stabilization and cell cycle arrest in an RPL5/RPL11-dependent manner, whereas no such mechanism exists for the same extent of damage in ribosome biogenesis caused by loss of either RPL5 of RPL11 (Fig. 7). Instead, such lesions lead to a reduction in ribosome content and rate of translation, which mediates a slower progression through the cell cycle (Fig. 7).
Fig 6.

RPL11 depletion suppresses cyclins expression through inhibition of global translation. (A) Expression of cyclin E1 mRNA levels in NS siRNA-treated or RPL11 siRNA-treated MRC5 cells from the experiment described in the legend of Fig. 4. For each sample, the arbitrary values represent the ratio of cyclin E1 mRNA levels to that of 18S rRNA levels, normalized to the ratio of the control sample at 0 h. (B) Quantification of cyclin E1 mRNA levels in fractions (Frc) collected from polysome profiles of MRC5 cells transfected with the indicated siRNAs, synchronized in G0/G1, and stimulated with serum for 12 h. (C) Quantification of RPL5 mRNA levels in fractions collected from polysome profiles of MRC5 cells transfected with the indicated siRNAs, synchronized in G0/G1, and stimulated with serum for 12 h. (D) Quantification of cyclin E1 mRNA levels in fractions collected from polysome profiles of asynchronous MRC5 cells transfected with the indicated siRNAs for 48 h. (E) Quantification of cyclin A2 mRNA levels in fractions collected from polysome profiles of asynchronous MRC5 cells transfected with the indicated siRNAs for 48 h. (F) Quantification of cyclin B1 mRNA levels in fractions collected from polysome profiles of asynchronous MRC5 cells transfected with the indicated siRNAs for 48 h. In panels B to F, the traces of the respective polysome profiles have been superimposed on the plots: black, NS siRNA-treated MRC5 cells; gray, RPL11 siRNA-treated MRC5 cells; light gray, RPL5 siRNA-treated MRC5 cells.
Fig 7.

Model of the effects of impaired ribosome biogenesis on cell cycle progression. (A) Ribosome impairment caused by defects other than loss of RPL5 or RPL11 elicit a p53 cell cycle checkpoint in an RPL5/RPL11/5S rRNA-Hdm2-dependent manner. If the p53 response cannot be induced, for reasons such as mutation or deletion, cell cycle progression will resume but at reduced rates due to the defect in global translational capacity that is common among cells which have lost essential RPs or other factors critical for ribosome biogenesis. (B) Deficiency of RPL5 or RPL11 causes a lesion in ribosome biogenesis, reduces global translation, and slows cell cycle progression but fails to induce a p53 cell cycle checkpoint, which depends on RPL5/RPL11/5S rRNA-mediated inhibition of Hdm2. E, empty; F, full.
DISCUSSION
To maintain patterns of translation, the state of the protein synthetic machinery, particularly that of nascent ribosomes, is closely monitored (48). The binding of RPL5 and RPL11 to Hdm2 and the stabilization of p53 have been demonstrated in a number of settings following impairment of ribosome biogenesis (7, 15, 16, 32–34, 49). Consistent with the role of RPL5 and RPL11 as positive regulators of p53, recent studies have also highlighted their importance as novel tumor suppressors (5, 10, 50). In agreement with these reports, RPs have been identified as haploinsufficient tumor suppressors in both Drosophila and zebrafish (51–54). In zebrafish, heterozygous mutations of 17 RPs were shown to lead to the formation of malignant peripheral nerve sheath tumors (MPNSTs) (51). Moreover, RP mutant strains with the highest MPNST incidences are also growth impaired, an observation which led to the hypothesis that a defect in global protein translational rate precedes tumor development and might be predictive of tumorigenesis (52). Likewise, DBA patients have an increased susceptibility to cancer later in life, including acute myelogenous leukemia (AML) and solid tumors (55), highlighting the link between aberrant ribosome biogenesis, impaired global protein translation, and tumorigenesis.
Here, we show that depletion of RPL5 or RPL11 in primary human fibroblasts does not lead to p53 stabilization or cell cycle arrest, despite the fact that such treatments inhibit ribosome biogenesis to an extent similar to depletion of an essential 60S RP, RPL7a. That the loss of either RPL5 or RPL11 does not inhibit Hdm2 and lead to p53 stabilization is consistent with recent findings by our group (7) and those of other investigators (6) showing that in cancer cell lines wild type for p53, RPL5 and RPL11 act in a mutually dependent manner to inhibit Hdm2. Interestingly, despite the lack of p53 induction and cell cycle arrest, MRC5 cells depleted of RPL5 or RPL11 proliferated at a lower rate (Fig. 1, 2, 4, and 5A and B). Moreover, depletion of p53 in RPL7a-deficient cells was sufficient to overcome the cell cycle checkpoint, allowing cells to proliferate at rates equivalent to those of RPL5- or RPL11-depleted cells (Fig. 3). This suggests that delay, but not arrest, of the cell cycle is the underlying response to impaired ribosome biogenesis in the absence of p53 (Fig. 7). Excessive ribosome biogenesis has been speculated to drive proliferation of tumors (12), many of which harbor mutations in the p53 pathway, suggesting that inhibition of this response could be a potential avenue to target cancer cells. Of note, cells of the Drosophila imaginal disc pass through multiple mitotic divisions before giving rise to the adult appendages during morphogenesis (56). Ectopic expression of Drosophila Myc (dMyc), a master regulator of ribosome biogenesis (9), in the cells of the imaginal discs led to their overproliferation at the expense of neighboring cells that expressed dMyc at physiological levels. However, this advantage was suppressed if cells overexpressing dMyc were also heterozygous for dRPL19, indicating that this proliferative advantage was controlled by enhanced rates of ribosome biogenesis and protein synthesis (57). Likewise, the enhanced rates of translation and increase in size of B lymphocytes of transgenic Eμ-Myc mice were restored to normal values when these mice were crossed to a murine minute background, which is hypomorphic for RPL24, causing a delay in the onset of lymphoma. These effects were attributed to diminished ribosome content and a reduced rate of translation (11).
The higher cell number of RPL11- versus RPL7a-depleted cells in proliferation assays (Fig. 2A) can be attributed to the fact that they progress unimpeded through each phase of the cell cycle, albeit at a lower rate than control cells. That cells adjust the rate of cell cycle progression to translational capacity is consistent with earlier findings based on experiments with low doses of cycloheximide (42, 43, 58, 59), as is the finding that partial inhibition of global translation affects all phases of the cell cycle to similar extents (59). In earlier studies it was hypothesized that the delay in G1/S transition under conditions of suppressed protein translation was due to the rapid turnover of one or more critical proteins which had short half-lives (42, 44). Such a response may be important for the cell to link nutrient availability and environmental cues with the timing and rate of cell cycle progression. Subsequent studies showed that one of the rate-limiting steps in G1/S transition is the accumulation of cyclin E and activation of the associated kinase CDK2 (42, 43, 58, 59), consistent with the finding that overexpression of cyclin E accelerates the rate of G1/S entry and is observed in a number of cancers (60, 61). Importantly, a recent study employing computational simulation modeling argues that the rate-limiting step in protein synthesis is translation initiation, which is dependent on ribosome content (30). Moreover, the probability of translation initiation is different for individual transcripts as mRNAs with short unstructured 5′ UTRs compete more effectively than transcripts with highly structured 5′ UTRs for the initiation complex (30). Not only does cyclin E1 have a short half-life, but the highly structured 5′ UTR and multiple uORFs in its mRNA can also trap translation initiation complexes and render its translation inefficient when global rates of translation are reduced (46, 47). Our findings in RPL11-depleted cells of the delayed expression of cyclin E1 and A2 protein (Fig. 5C) and the association of cyclin E1, A2, and cyclin B1 mRNAs with smaller polysomes (Fig. 6B and D to F) are consistent with a model in which progression through all phases of the cell cycle is hindered by the lesion in ribosome content and translational capacity.
DBA is a congenital disease, with the most severe phenotype affecting survival of erythroid precursors (62). Several reports have pointed to p53 as the underlying mechanism of the anemia (49, 63), with a recent study showing that p53 levels are increased in RPL11-depleted CD34+-derived erythroid cells and in bone marrow cells from a limited number of patients harboring RPL11 mutations (64). However, depletion of RPL11 or RPS19 in a p53−/− murine erythroblast cell line revealed severe defects in erythropoiesis associated with translational repression of mRNAs critical for this process, including Bag1 and Csde1 (65). Moreover, mouse embryos heterozygous for RPS6, which die in utero at embryonic day 5.5 (E5.5), can be rescued in a p53-deficient background but only to E12.5, when they succumb to impaired liver erythropoiesis (66). These findings suggest that a p53-independent mechanism may also contribute to the pathology of DBA. Also, perturbation of cell cycle progression has been implicated in DBA in studies showing that the erythroid expansion phase, prior to terminal differentiation, is associated with the increased expression of cyclin E and synchronous entry of ∼70% of the erythroid progenitors into S phase (67) and that genetic inactivation of specific cyclins in mice results in erythropoietic phenotypes such as anemia (68–70). Thus, the effects on cyclin expression observed here may also operate in the erythroid lineage in response to diminished global translation, contributing to selective cell death and anemia. It seems evident that studies at the molecular level which unravel the mechanisms underlying the impaired erythropoiesis in DBA patients, especially those with mutations in RPL5 and RPL11, will be critical in defining the differential role of RPL5 and RPL11 as tumor suppressors in the Hdm2/Hdm4/p53 pathway.
Among the RPs, only RPL5 and RPL11 appear to have evolved as Hdm2 inhibitors. It may be that this tumor suppressor role was adopted to integrate ribosome biogenesis and cell proliferation during evolution, a response which offered certain advantages under natural selection. Although RPs are highly conserved and although homologues of p53 have been identified in invertebrates, Hdm2 was thought to be restricted to vertebrates as Hdm2 homologues were not identified in Caenorhabditis elegans and Drosophila melanogaster. However, homologues of Hdm2 have been recently identified in an array of other invertebrates, including one of the earliest multicellular organisms, Trichoplax (71–74). Importantly, the zinc finger domain of Hdm2, including C305, critical for RPL5 and RPL11 binding, is highly conserved between this early life form and humans, suggesting that the RPL11/RPL5-Hdm2 dependent interaction may have first appeared over 1 billion years ago (71, 73). Indeed, recent reports demonstrate that multiple tumor suppressors and oncogenes utilize this evolutionarily conserved interaction mechanism (50, 75). These include the proto-oncogene protein interacting with PTEN C terminus 1 (PICT1), which functions by retaining RPL11 in nucleoli away from Hdm2 (50), and the splicing factor SRSF1, which stabilizes the RPL5 and Hdm2 interaction (75). Interestingly, analysis of Hdm2 and Hdm4 homologues in invertebrates further suggests that only one Hdm gene was present in invertebrates and that Hdm2 and Hdm4 were generated through a duplication event at a much later stage but before the emergence of bony vertebrates (74). Thus, it seems surprising that RPL11 and RPL5 are not able to bind Hdm4 (76). However, the situation is much more complex as recent findings from our laboratory (77) show that 5S rRNA is part of the RPL5 and RPL11 complex, which inhibits Hdm2 and activates p53, whereas 5S rRNA is also required for Hdm4-mediated inhibition of p53 (78), underscoring the complexity and the importance of elucidating the interactions between RPL5, RPL11, Hdm2, Hdm4, and p53 in humans.
ACKNOWLEDGMENTS
Flow cytometry was performed at Shriners Hospitals for Children, Cincinnati, OH, supported by a grant from the Shriners of North America (SSF 84070). G.T. is supported by grants from the Spanish Ministry of Science and Innovation (SAF2011-24967), the Instituto de Salud Carlos III (ISIS) (IIS10/00015/P), the CIG European Commission (PCIG10-GA-2011-304160), the Red Temática de Investigación Cooperativa en Cáncer (RTICC-R012/0036/0049), the NIH/NIDDK (1RC1-DK087680), and NCI/NIH (R01CA138647). S.F. is supported by grants from the Ligue Contre le Cancer (RS13/75-36) and the Agence Nationale de la Recherche (ANR-12-BSV2-0006-01).
We are indebted to G. Doerman for preparing the figures.
Footnotes
Published ahead of print 23 September 2013
REFERENCES
- 1.Vousden KH, Lane DP. 2007. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 8:275–283 [DOI] [PubMed] [Google Scholar]
- 2.Wade M, Wang YV, Wahl GM. 2010. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 20:299–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sherr CJ. 1998. Tumor surveillance via the ARF-p53 pathway. Genes Dev. 12:2984–2991 [DOI] [PubMed] [Google Scholar]
- 4.Colaluca IN, Tosoni D, Nuciforo P, Senic-Matuglia F, Galimberti V, Viale G, Pece S, Di Fiore PP. 2008. NUMB controls p53 tumour suppressor activity. Nature 451:76–80 [DOI] [PubMed] [Google Scholar]
- 5.Macias E, Jin A, Deisenroth C, Bhat K, Mao H, Lindstrom MS, Zhang Y. 2010. An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 Interaction. Cancer Cell 18:231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bursac S, Brdovcak MC, Pfannkuchen M, Orsolic I, Golomb L, Zhu Y, Katz C, Daftuar L, Grabusic K, Vukelic I, Filic V, Oren M, Prives C, Volarevic S. 2012. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc. Natl. Acad. Sci. U. S. A. 109:20467–20472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fumagalli S, Ivanenkov VV, Teng T, Thomas G. 2012. Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev. 26:1028–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindstrom MS, Jin A, Deisenroth C, White Wolf G, Zhang Y. 2007. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol. Cell. Biol. 27:1056–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Riggelen J, Yetil A, Felsher DW. 2010. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer. 10:301–309 [DOI] [PubMed] [Google Scholar]
- 10.Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K, Huser N, Proffitt C, Bliesath J, Haddach M, Schwaebe MK, Ryckman DM, Rice WG, Schmitt C, Lowe SW, Johnstone RW, Pearson RB, McArthur GA, Hannan RD. 2012. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 22:51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barna M, Pusic A, Zollo O, Costa M, Kondrashov N, Rego E, Rao PH, Ruggero D. 2008. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456:971–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruggero D, Pandolfi PP. 2003. Does the ribosome translate cancer? Nat. Rev. Cancer. 3:179–192 [DOI] [PubMed] [Google Scholar]
- 13.Topisirovic I, Sonenberg N. 2011. mRNA translation and energy metabolism in cancer: the role of the MAPK and mTORC1 pathways. Cold Spring Harbor Symp. Quant. Biol. 76:355–367 [DOI] [PubMed] [Google Scholar]
- 14.Volarevic S, Stewart MJ, Ledermann B, Zilberman F, Terracciano L, Montini E, Grompe M, Kozma SC, Thomas G. 2000. Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 288:2045–2047 [DOI] [PubMed] [Google Scholar]
- 15.Fumagalli S, Di Cara A, Neb-Gulati A, Natt F, Schwemberger S, Hall J, Babcock GF, Bernardi R, Pandolfi PP, Thomas G. 2009. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat. Cell Biol. 11:501–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dai MS, Lu H. 2004. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 279:44475–44482 [DOI] [PubMed] [Google Scholar]
- 17.Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. 2004. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell. Biol. 24:7654–7668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin A, Itahana K, O'Keefe K, Zhang Y. 2004. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol. Cell. Biol. 24:7669–7680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Y, Poyurovsky MV, Li Y, Biderman L, Stahl J, Jacq X, Prives C. 2009. Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol. Cell 35:316–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sherr CJ, Weber JD. 2000. The ARF/p53 pathway. Curr. Opin. Genet. Dev. 10:94–99 [DOI] [PubMed] [Google Scholar]
- 21.Marygold SJ, Roote J, Reuter G, Lambertsson A, Ashburner M, Millburn GH, Harrison PM, Yu Z, Kenmochi N, Kaufman TC, Leevers SJ, Cook KR. 2007. The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol. 8:R216. 10.1186/gb-2007-8-10-r216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gazda HT, Sheen MR, Vlachos A, Choesmel V, O'Donohue MF, Schneider H, Darras N, Hasman C, Sieff CA, Newburger PE, Ball SE, Niewiadomska E, Matysiak M, Zaucha JM, Glader B, Niemeyer C, Meerpohl JJ, Atsidaftos E, Lipton JM, Gleizes PE, Beggs AH. 2008. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am. J. Hum. Genet. 83:769–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cmejla R, Cmejlova J, Handrkova H, Petrak J, Pospisilova D. 2007. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum. Mutat. 28:1178–1182 [DOI] [PubMed] [Google Scholar]
- 24.Doherty L, Sheen MR, Vlachos A, Choesmel V, O'Donohue MF, Clinton C, Schneider HE, Sieff CA, Newburger PE, Ball SE, Niewiadomska E, Matysiak M, Glader B, Arceci RJ, Farrar JE, Atsidaftos E, Lipton JM, Gleizes PE, Gazda HT. 2010. Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am. J. Hum. Genet. 86:222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, Dianzani I, Ball S, Tchernia G, Klar J, Matsson H, Tentler D, Mohandas N, Carlsson B, Dahl N. 1999. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 21:169–175 [DOI] [PubMed] [Google Scholar]
- 26.Farrar JE, Nater M, Caywood E, McDevitt MA, Kowalski J, Takemoto CM, Talbot CC, Jr, Meltzer P, Esposito D, Beggs AH, Schneider HE, Grabowska A, Ball SE, Niewiadomska E, Sieff CA, Vlachos A, Atsidaftos E, Ellis SR, Lipton JM, Gazda HT, Arceci RJ. 2008. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood 112:1582–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farrar JE, Vlachos A, Atsidaftos E, Carlson-Donohoe H, Markello TC, Arceci RJ, Ellis SR, Lipton JM, Bodine DM. 2011. Ribosomal protein gene deletions in Diamond-Blackfan anemia. Blood 118:6943–6951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gazda HT, Grabowska A, Merida-Long LB, Latawiec E, Schneider HE, Lipton JM, Vlachos A, Atsidaftos E, Ball SE, Orfali KA, Niewiadomska E, Da Costa L, Tchernia G, Niemeyer C, Meerpohl JJ, Stahl J, Schratt G, Glader B, Backer K, Wong C, Nathan DG, Beggs AH, Sieff CA. 2006. Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am. J. Hum. Genet. 79:1110–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Lu H. 2009. Signaling to p53: ribosomal proteins find their way. Cancer Cell 16:369–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah P, Ding Y, Niemczyk M, Kudla G, Plotkin JB. 2013. Rate-limiting steps in yeast protein translation. Cell 153:1589–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen CA, Okayama H. 1988. Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques 6:632–638 [PubMed] [Google Scholar]
- 32.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. 2003. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3:577–587 [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y. 2003. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 23:8902–8912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Donati G, Bertoni S, Brighenti E, Vici M, Trere D, Volarevic S, Montanaro L, Derenzini M. 2011. The balance between rRNA and ribosomal protein synthesis up- and downregulates the tumour suppressor p53 in mammalian cells. Oncogene 30:3274–3288 [DOI] [PubMed] [Google Scholar]
- 35.Lu W, Lin J, Chen J. 2002. Expression of p14ARF overcomes tumor resistance to p53. Cancer Res. 62:1305–1310 [PubMed] [Google Scholar]
- 36.Park YB, Park MJ, Kimura K, Shimizu K, Lee SH, Yokota J. 2002. Alterations in the INK4a/ARF locus and their effects on the growth of human osteosarcoma cell lines. Cancer Genet. Cytogenet. 133:105–111 [DOI] [PubMed] [Google Scholar]
- 37.Jacobs JP, Jones CM, Baille JP. 1970. Characteristics of a human diploid cell designated MRC-5. Nature 227:168–170 [DOI] [PubMed] [Google Scholar]
- 38.Rotenberg MO, Moritz M, Woolford JL., Jr 1988. Depletion of Saccharomyces cerevisiae ribosomal protein L16 causes a decrease in 60S ribosomal subunits and formation of half-mer polyribosomes. Genes Dev. 2:160–172 [DOI] [PubMed] [Google Scholar]
- 39.Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, Elledge SJ, Reed SI. 1994. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 76:1013–1023 [DOI] [PubMed] [Google Scholar]
- 40.Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. 1995. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell. Biol. 15:2612–2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yam CH, Fung TK, Poon RY. 2002. Cyclin A in cell cycle control and cancer. Cell. Mol. Life Sci. 59:1317–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brooks RF. 1977. Continuous protein synthesis is required to maintain the probability of entry into S phase. Cell 12:311–317 [DOI] [PubMed] [Google Scholar]
- 43.Brooks RF. 1976. Regulation of fibroblast cell cycle by serum. Nature 260:248–250 [DOI] [PubMed] [Google Scholar]
- 44.Pardee AB. 1989. G1 events and regulation of cell proliferation. Science 246:603–608 [DOI] [PubMed] [Google Scholar]
- 45.Won KA, Reed SI. 1996. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J. 15:4182–4193 [PMC free article] [PubMed] [Google Scholar]
- 46.Lai MC, Chang WC, Shieh SY, Tarn WY. 2010. DDX3 regulates cell growth through translational control of cyclin E1. Mol. Cell. Biol. 30:5444–5453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prober DA, Edgar BA. 2000. Ras1 promotes cellular growth in the Drosophila wing. Cell 100:435–446 [DOI] [PubMed] [Google Scholar]
- 48.Teng T, Thomas G, Mercer CA. 2013. Growth control and ribosomopathies. Curr. Opin. Genet. Dev. 23:63–71 [DOI] [PubMed] [Google Scholar]
- 49.Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, Wilson FH, Currie T, Khanna-Gupta A, Berliner N, Kutok JL, Ebert BL. 2011. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 117:2567–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sasaki M, Kawahara K, Nishio M, Mimori K, Kogo R, Hamada K, Itoh B, Wang J, Komatsu Y, Yang YR, Hikasa H, Horie Y, Yamashita T, Kamijo T, Zhang Y, Zhu Y, Prives C, Nakano T, Mak TW, Sasaki T, Maehama T, Mori M, Suzuki A. 2011. Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat. Med. 17:944–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA, Hopkins N. 2004. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2:E139. 10.1371/journal.pbio.0020139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lai K, Amsterdam A, Farrington S, Bronson RT, Hopkins N, Lees JA. 2009. Many ribosomal protein mutations are associated with growth impairment and tumor predisposition in zebrafish. Dev. Dyn. 238:76–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stewart MJ, Denell R. 1993. Mutations in the Drosophila gene encoding ribosomal protein S6 cause tissue overgrowth. Mol. Cell. Biol. 13:2524–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watson KL, Konrad KD, Woods DF, Bryant PJ. 1992. Drosophila homolog of the human S6 ribosomal protein is required for tumor suppression in the hematopoietic system. Proc. Natl. Acad. Sci. U. S. A. 89:11302–11306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vlachos A, Rosenberg PS, Atsidaftos E, Alter BP, Lipton JM. 2012. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood 119:3815–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taylor J, Adler PN. 2008. Cell rearrangement and cell division during the tissue level morphogenesis of evaginating Drosophila imaginal discs. Dev. Biol. 313:739–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moreno E, Basler K. 2004. dMyc transforms cells into super-competitors. Cell 117:117–129 [DOI] [PubMed] [Google Scholar]
- 58.Okuda A, Kimura G. 1988. Elongation of G1 phase by transient exposure of rat 3Y1 fibroblasts to caffeine during the previous and present generations. J. Cell Sci. 89:379–386 [DOI] [PubMed] [Google Scholar]
- 59.Okuda A, Kimura G. 1988. Non-specific elongation of cell cycle phases by cycloheximide in rat 3Y1 cells, and specific reduction of G1 phase elongation by simian virus 40 large T antigen. J. Cell Sci. 91:295–302 [DOI] [PubMed] [Google Scholar]
- 60.Donnellan R, Chetty R. 1999. Cyclin E in human cancers. FASEB J. 13:773–780 [DOI] [PubMed] [Google Scholar]
- 61.Hwang HC, Clurman BE. 2005. Cyclin E in normal and neoplastic cell cycles. Oncogene 24:2776–2786 [DOI] [PubMed] [Google Scholar]
- 62.Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, Ramenghi U, Meerpohl J, Karlsson S, Liu JM, Leblanc T, Paley C, Kang EM, Leder EJ, Atsidaftos E, Shimamura A, Bessler M, Glader B, Lipton JM. 2008. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br. J. Haematol. 142:859–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McGowan KA, Pang WW, Bhardwaj R, Perez MG, Pluvinage JV, Glader BE, Malek R, Mendrysa SM, Weissman IL, Park CY, Barsh GS. 2011. Reduced ribosomal protein gene dosage and p53 activation in low-risk myelodysplastic syndrome. Blood 118:3622–3633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moniz H, Gastou M, Leblanc T, Hurtaud C, Cretien A, Lecluse Y, Raslova H, Larghero J, Croisille L, Faubladier M, Bluteau O, Lordier L, Tchernia G, Vainchenker W, Mohandas N, Da Costa L. 2012. Primary hematopoietic cells from DBA patients with mutations in RPL11 and RPS19 genes exhibit distinct erythroid phenotype in vitro. Cell Death Dis. 3:e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Horos R, Ijspeert H, Pospisilova D, Sendtner R, Andrieu-Soler C, Taskesen E, Nieradka A, Cmejla R, Sendtner M, Touw IP, von Lindern M. 2012. Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood 119:262–272 [DOI] [PubMed] [Google Scholar]
- 66.Panic L, Tamarut S, Sticker-Jantscheff M, Barkic M, Solter D, Uzelac M, Grabusic K, Volarevic S. 2006. Ribosomal protein S6 gene haploinsufficiency is associated with activation of a p53-dependent checkpoint during gastrulation. Mol. Cell. Biol. 26:8880–8891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dai MS, Mantel CR, Xia ZB, Broxmeyer HE, Lu L. 2000. An expansion phase precedes terminal erythroid differentiation of hematopoietic progenitor cells from cord blood in vitro and is associated with up-regulation of cyclin E and cyclin-dependent kinase 2. Blood 96:3985–3987 [PubMed] [Google Scholar]
- 68.Ciemerych MA, Kenney AM, Sicinska E, Kalaszczynska I, Bronson RT, Rowitch DH, Gardner H, Sicinski P. 2002. Development of mice expressing a single D-type cyclin. Genes Dev. 16:3277–3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kalaszczynska I, Geng Y, Iino T, Mizuno S, Choi Y, Kondratiuk I, Silver DP, Wolgemuth DJ, Akashi K, Sicinski P. 2009. Cyclin A is redundant in fibroblasts but essential in hematopoietic and embryonic stem cells. Cell 138:352–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sankaran VG, Ludwig LS, Sicinska E, Xu J, Bauer DE, Eng JC, Patterson HC, Metcalf RA, Natkunam Y, Orkin SH, Sicinski P, Lander ES, Lodish HF. 2012. Cyclin D3 coordinates the cell cycle during differentiation to regulate erythrocyte size and number. Genes Dev. 26:2075–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lane DP, Cheok CF, Brown C, Madhumalar A, Ghadessy FJ, Verma C. 2010. Mdm2 and p53 are highly conserved from placozoans to man. Cell Cycle 9:540–547 [DOI] [PubMed] [Google Scholar]
- 72.Lane DP, Cheok CF, Brown CJ, Madhumalar A, Ghadessy FJ, Verma C. 2010. The Mdm2 and p53 genes are conserved in the Arachnids. Cell Cycle 9:748–754 [DOI] [PubMed] [Google Scholar]
- 73.Lane DP, Verma C. 2012. Mdm2 in evolution. Genes Cancer 3:320–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Momand J, Villegas A, Belyi VA. 2011. The evolution of MDM2 family genes. Gene 486:23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fregoso OI, Das S, Akerman M, Krainer AR. 2013. Splicing-factor oncoprotein SRSF1 stabilizes p53 via RPL5 and induces cellular senescence. Mol. Cell 50:56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gilkes DM, Chen L, Chen J. 2006. MDMX regulation of p53 response to ribosomal stress. EMBO J. 25:5614–5625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Donati G, Peddigari S, Mercer CA, Thomas G. 2013. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 4:87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li M, Gu W. 2011. A critical role for noncoding 5S rRNA in regulating Mdmx stability. Mol. Cell 43:1023–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carnevalli LS, Masuda K, Frigerio F, Le Bacquer O, Um SH, Gandin V, Topisirovic I, Sonenberg N, Thomas G, Kozma SC. 2010. S6K1 plays a critical role in early adipocyte differentiation. Dev Cell 18:763–774 [DOI] [PMC free article] [PubMed] [Google Scholar]