

Abstract
Next-generation DNA sequencing can be used to catalog individual organisms within complex, polymicrobial specimens. Here, we utilized deep sequencing of 16S rRNA to implicate Actinomadura madurae as the cause of mycetoma in a diabetic patient when culture and conventional molecular methods were overwhelmed by overgrowth of other organisms.
CASE REPORT
A 50-year-old woman from northern Mexico with a diagnosis of diabetes presented with a 12-year history of a gradually enlarging right foot demonstrating multiple draining lesions. She reported a long course of originally pruritic lesions on the plantar instep of her right foot which over many years developed into multiple bulbous nodules with central pinpoint ulcerations expressing purulent and granular material. Although the patient remained ambulatory throughout the course of her disease, she gradually had to modify her footwear to accommodate the increasing size of her foot, which at the time of presentation was causing her great pain. Approximately 4 years prior to the current presentation, the diagnosis of actinomycotic mycetoma was reportedly established by histology, and she was initiated on antibiotic therapy with penicillin (500 mg orally [p.o.] 4 times daily [QID]) and later transitioned to doxycycline (100 mg p.o. twice per day [BID]). Despite treatment, the lesions continued to steadily grow, and 2 years prior to presentation she received a 6-week course of intravenous penicillin therapy with continuous daily infusions (20 million units intravenously [i.v.] per day [QD]). The patient noted some retreat of the lesions and improvement in her pain and pruritis while on this treatment, and she was transitioned to oral penicillin (500 mg p.o. QID), ciprofloxacin (500 mg p.o. BID), and doxycycline (100 mg p.o. BID) for suppression. No new lesions evolved while she was on this therapy, but approximately 1 year prior to presentation the patient self-discontinued ciprofloxacin and shortly thereafter noted increasing drainage from her foot. Intravenous penicillin (20 million units i.v. QD) was resumed for an 8-week course, but no response in the amount of discharge or reduction in the size of the nodules was achieved and the antibiotic was consequently discontinued. Culture of discharged material reportedly returned with overgrowth of skin flora and provided no evidence of infection with Actinomyces species. The patient repeatedly declined the recommendation of surgical debridement.
The patient was referred to the Infectious Disease clinic at Harborview Medical Center for further evaluation. A punch biopsy specimen of the lesion was submitted to pathology, revealing marked acute and chronic inflammation and an inclusion of abundant filamentous structures consistent with aerobic actinomycetes (Fig. 1). Grocott's methenamine silver (GMS) staining for fungal organisms was negative. Based on these findings, the diagnosis of actinomycotic mycetoma was suggested. A portion of the specimen was concurrently submitted for culture in order to establish the identity of the aerobic actinomycetes-like organism and to evaluate its antibiotic sensitivities; however, the culture quickly became overgrown by Staphylococcus aureus, preventing culture for slower-growing aerobic actinomycetes species. A second biopsy specimen was submitted for culture and was again positive for S. aureus, once more precluding the detection of aerobic actinomycetes through prolonged culture.
Fig 1.
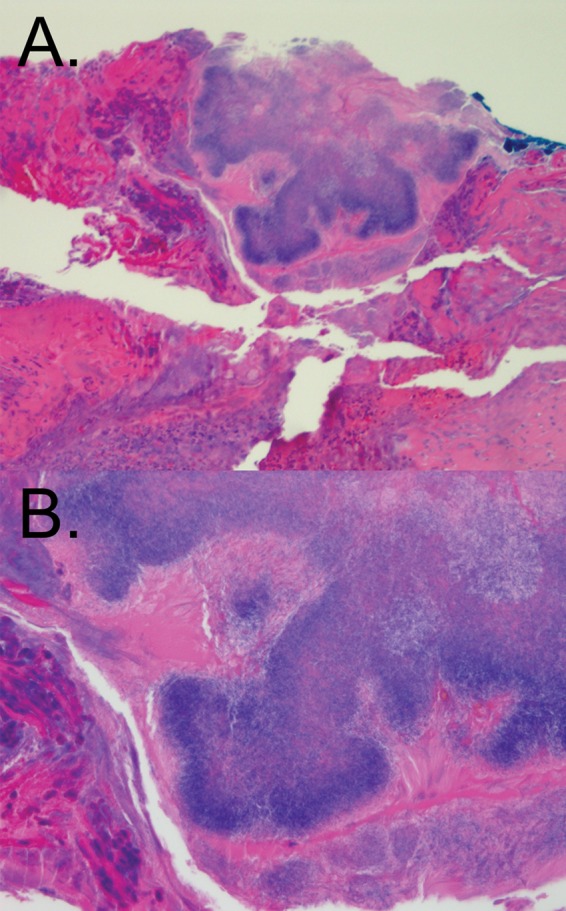
Histological section of actinomycotic mycetoma. (A) Punch biopsy material stained using hematoxylin and eosin. An inclusion of filamentous, basophilic organisms consistent with Actinomyces species is seen. Original magnification, ×100. (B) Same section as shown in panel A; original magnification, ×400.
Formalin-fixed paraffin-embedded (FFPE) material from the biopsy specimen was concurrently submitted for molecular characterization of the pathogen to the University of Washington Molecular Diagnosis Microbiology Section. No fungal DNA was detected by 28S ribosomal DNA (rDNA) and internal transcribed spacer (ITS) sequencing. 16S rRNA sequencing (1) yielded good quality sequence in the reverse direction only, producing 326 bp of unidirectional sequence data with 100% identity and 100% coverage with S. aureus (GenBank accession no. CP005288.1).
In an effort to more fully characterize the organisms present in this specimen, we performed deep sequencing of 16S rRNA PCR amplicon from the sample using an Ion Torrent Personal Genome Machine (PGM) sequencer (Life Technologies). DNA extraction from FFPE material was performed as reported previously (2). DNA was amplified from the specimen in two separate reactions, in which sequencing adapters were incorporated on opposite ends of the amplicon, enabling bidirectional sequencing across the full length of 16S rRNA variable regions 1 and 2 (3). Primer pairs (“reverse orientation” forward primer 5′-CCATCTCATCCCTGCGTGTCTCCGACTCAGTTCCGATAACCTGCTGCCICCCGTAGGAG-3′ and “reverse orientation” reverse primer 5′-CCTCTCTATGGGCAGTCGGTGATAGAGTTTGATCITGGCTCAG-3′; “forward orientation” forward primer 5′-CCATCTCATCCCTGCGTGTCTCCGACTCAGTCTGGCAACGGCAGAGTTTGATCITGGCTCAG-3′ and “forward orientation” reverse primer 5′-CCTCTCTATGGGCAGTCGGTGATCTGCTGCCICCCGTAGGAG-3′ [I = deoxyinosine]) were obtained from IDT and included 10-bp “bar codes” unique to each library, permitting multiplexing on the same sequencing run. PCR conditions for construction of sequencing libraries were as described elsewhere (3) except that an annealing temperature of 60°C was used and 28 cycles were performed. PCR products were purified using 0.7 vol of AMPure beads (Agencourt), eluted in low TE buffer (10 mM Tris [pH 8], 0.1 mM EDTA), and quantified by the use of a Qubit double-stranded DNA (dsDNA) HS kit (Life Technologies). A template procedure for Ion Torrent sequencing was performed using a OneTouch 2 system (Life Technologies), and sequencing was performed on an Ion PGM sequencer (Life Technologies) using a 400-bp sequencing kit and a 318 v2 chip according to the manufacturer's instructions. Base calling was performed using TorrentServer software, version 3.6.2. Sequence data from each of the two PCRs were combined after reverse complementation of those sequences from the “reverse” orientation, after which data processing, denoising, and classification of sequence reads against the Ribosomal Database Project database (RDP, Release 10, Update 3) (4) were performed as described elsewhere (3). Taxonomic assignment was performed for full-length (∼350-bp) consensus sequences that were greater than 99% identical to a reference sequence; those not matching any reference sequence below that threshold, including chimeric sequences, were left unassigned. Taxonomic classifications represented by less than 1% of the total sequence reads were not considered in this analysis. Cumulatively, those low-abundance classifications accounted for 2,592 reads (7.15% of the total reads passing quality filters).
Table 1 details the classification of deep-sequencing reads from the specimen submitted for analysis. The tallies and percent abundance of reads corresponding to each organism are indicated. For some sequences, multiple organisms met the identity threshold for species-level classification; in such cases, species names separated by a slash and genus names separated by a semicolon list the possible taxonomic assignments. Consistent with culture results, reads classified as S. aureus were highly prevalent. A number of other organisms were also detected, reflecting the polymicrobial nature of such specimens (5). Nevertheless, deep sequencing recovered 748 reads (2.07% of the total) that were 99.3% to 100% identical to a sequence representing Actinomadura madurae (GenBank accession no. X97889.1). This pathogen is one of the classical agents of actinomycotic mycetoma and is consistent with the organism visualized histologically.
Table 1.
Deep-sequencing results
| No. of reads | % of reads | Classificationa |
|---|---|---|
| 13,000 | 35.94 | Staphylococcus aureus* |
| 4,402 | 12.17 | Alcaligenes faecalis |
| 3,049 | 8.43 | Acinetobacter bereziniae/A. guillouiae* |
| 1,449 | 4.01 | Comamonas testosteroni*/C. thiooxidans* |
| 1,084 | 3.00 | Acinetobacter lwoffii* |
| 935 | 2.59 | Acinetobacter lwoffii*/A. psychrotolerans |
| 924 | 2.55 | Pseudomonas geniculata/P. hibiscicola; Stenotrophomonas maltophilia |
| 748 | 2.07 | Actinomadura madurae* |
| 747 | 2.07 | Flavobacterium lindanitolerans |
| 738 | 2.04 | Escherichia coli*/E. fergusonii*; Shigella dysenteriae*/S. flexneri* |
| 654 | 1.81 | Campylobacter concisus |
| 476 | 1.32 | Enterobacter cowanii; Escherichia coli*/E. hermannii; Shigella boydii/S. flexneri/S. sonnei |
| 458 | 1.27 | Staphylococcus epidermidis |
| 417 | 1.15 | Ochrobactrum anthropi*/O. cytisi*/O. lupini* |
| 377 | 1.04 | Enterobacter asburiae*/E. cancerogenus*/E. cloacae/E. cowanii; Leclercia adecarboxylata |
| 4,120 | 11.39 | ≤99.0% match to a reference strain |
Asterisks indicate the presence of reads with 100% identity to a reference strain.
Based on this information, a course of sulfamethoxazole-trimethoprim therapy was considered. However, on imaging, the patient's osteomyelitis was found to involve all bones of the foot, including the calcaneus. Given the extent of infection, neither additional antibiotic therapy nor debridement with subsequent reconstruction was felt to be a viable option, and a recommendation was made for below-the-knee amputation. The patient agreed to this procedure, and was discharged on a course of oral sulfamethoxazole-trimethoprim.
Many sites of the human body are colonized by complex communities of microbes in both health and various disease states (6). Chronic infections (7) and diabetic foot ulcers (5, 8, 9), in particular, can contain highly diverse bacterial populations. Polymicrobial specimens may be difficult or even impossible to fully characterize by techniques in common clinical use: culture introduces bias against fastidious or slow-growing organisms (10) and can be practically employed to classify only a limited number of species, while molecular methods such as 16S rRNA gene sequencing (11) may detect only the predominant organism in a sample or may generate a mixed and uninterpretable signal (12). Both of these diagnostic limitations complicated analysis of the patient specimen in this report, a biopsy specimen from a diabetic foot ulcer. Consequently, the likely causative organism of the patient's foot infection could not be identified by existing clinical diagnostic approaches.
In contrast to conventional approaches, deep sequencing (or “next-generation DNA sequencing”) provides independent sequence data from millions of individual DNA molecules, allowing each fragment to be classified independently. Although the application of deep sequencing to microbial communities traces its origins to metagenomics research (13), we recently demonstrated the feasibility of using deep sequencing to interrogate the composition of polymicrobial specimens in a clinical context by sequencing bacterial 16S rRNA amplified directly from patient material (3). In this case report, we have utilized the approach to investigate the causative agent of biopsy-proven actinomycotic mycetoma. Deep sequencing successfully detected the presence of A. madurae, a causative agent of that disorder, with a high-confidence classification, establishing a molecular diagnosis for the patient's disease.
Mycetoma (or “madura foot”) is a clinical syndrome characterized by deformation, cutaneous lesions and sinuses, infection of tissues extending from the cutaneous layer to the underlying fascia, and an indolent course (14, 15). It is considered endemic to Central America, South America, Africa, and India (16). Although the disease can be caused by a number of fungal or bacterial agents that are typically introduced through traumatic inoculation from contaminated soil (15), A. madurae is among the most common agents of mycetoma occurring worldwide (17). Several treatment regimens for actinomycotic mycetoma have been published (18–20), and in general, the condition shows response to a wide range of antibiotics, including aminoglycosides, rifampin, amoxicillin-clavulanic acid, doxycycline, and sulfamethoxazole-trimethoprim, although combination antibiotic therapy is recommended (21). In one small, prospective study, initial treatment with intravenous gentamicin, intravenous penicillin, and oral sulfamethoxazole-trimethoprim followed by oral sulfamethoxazole-trimethoprim and oral amoxicillin maintenance was found effective in empirically treating disease minimally involving the bones, while regimens incorporating intravenous amikacin and oral sulfamethoxazole-trimethoprim for initial treatment and oral sulfamethoxazole-trimethoprim for maintenance therapy were useful in cases of more extensive bony involvement (21). The diagnosis of A. madurae in this case potentially explains the patient's poor response to the initial penicillin therapy, as other reports have noted that this organism has been most effectively treated using therapies that include trimethoprim-sulfamethoxazole or alternative agents such as streptomycin or dapsone (22).
Sequence reads originating from A. madurae comprised only a small fraction of the overall reads, with those corresponding to S. aureus being the most prevalent (Table 1). This finding is consistent with the results of both culture and Sanger sequencing for the specimen, which were dominated by S. aureus. The portion of the biopsy material sequenced may have contained little of the aerobic actinomycetes inclusion, which represented a focal area within the larger sample (Fig. 1). Further, it should be noted that read counts in 16S rRNA amplicon deep-sequencing studies are semiquantitative and correlate only roughly with the relative abundances of organisms due to bias introduced through PCR (23, 24), differing DNA extraction efficiencies for particular organisms (25), and organism-specific differences in 16S rRNA operon counts (26). Thus, the relative abundance of A. madurae within this particular polymicrobial sample, and within the patient's lesion, in general, may have been higher than that suggested from the read count alone.
Deep sequencing has previously been used to characterize cultured bacterial isolates (27) and to explore the composition of microbial populations in metagenomic research (28, 29). Here, we have extended the application of next-generation DNA sequencing technologies to perform molecular diagnosis in a clinical context. The case exemplifies several capabilities of deep sequencing as a clinical diagnostic tool, specifically, deconvoluting the identity of individual organisms within polymicrobial samples, classifying organisms directly from patient specimens without the need for culture, and characterizing nonviable or unculturable organisms (in this case, organisms killed by FFPE processing prior to pathology examination). Relatively inexpensive “benchtop” next-generation DNA sequencers are becoming more accessible to clinical laboratories, and we anticipate that molecular diagnosis using deep sequencing will become increasingly common in the future (30, 31).
Nucleotide sequence accession number.
The consensus sequence determined in this work has been submitted to GenBank under accession no. KF680773.
Footnotes
Published ahead of print 9 October 2013
REFERENCES
- 1.Kattar MM, Chavez JF, Limaye AP, Rassoulian-Barrett SL, Yarfitz SL, Carlson LC, Houze Y, Swanzy S, Wood BL, Cookson BT. 2000. Application of 16S rRNA gene sequencing to identify Bordetella hinzii as the causative agent of fatal septicemia. J. Clin. Microbiol. 38:789–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrington AT, Creutzfeldt CJ, Sengupta DJ, Hoogestraat DR, Zunt JR, Cookson BT. 2009. Diagnosis of neurocysticercosis by detection of Taenia solium DNA using a global DNA screening platform. Clin. Infect. Dis. 48:86–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salipante SJ, Sengupta DJ, Rosenthal C, Costa G, Spangler J, Sims EH, Jacobs MA, Miller SI, Hoogestraat DR, Cookson BT, McCoy C, Matsen FA, Shendure J, Lee CC, Harkins TT, Hoffman NG. 2013. Rapid 16S rRNA next-generation sequencing of polymicrobial clinical samples for diagnosis of complex bacterial infections. PLoS One 8:e65226. 10.1371/journal.pone.0065226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Citron DM, Goldstein EJ, Merriam CV, Lipsky BA, Abramson MA. 2007. Bacteriology of moderate-to-severe diabetic foot infections and in vitro activity of antimicrobial agents. J. Clin. Microbiol. 45:2819–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peters BM, Jabra-Rizk MA, O'May GA, Costerton JW, Shirtliff ME. 2012. Polymicrobial interactions: impact on pathogenesis and human disease. Clin. Microbiol. Rev. 25:193–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhoads DD, Wolcott RD, Sun Y, Dowd SE. 2012. Comparison of culture and molecular identification of bacteria in chronic wounds. Int. J. Mol. Sci. 13:2535–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dowd SE, Wolcott RD, Sun Y, McKeehan T, Smith E, Rhoads D. 2008. Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP). PLoS One 3:e3326. 10.1371/journal.pone.0003326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts AD, Simon GL. 2012. Diabetic foot infections: the role of microbiology and antibiotic treatment. Semin. Vasc. Surg. 25:75–81 [DOI] [PubMed] [Google Scholar]
- 10.Schlaberg R, Simmon KE, Fisher MA. 2012. A systematic approach for discovering novel, clinically relevant bacteria. Emerg. Infect. Dis. 18:422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarridge JE., III 2004. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 17:840–862, table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drancourt M, Bollet C, Carlioz A, Martelin R, Gayral JP, Raoult D. 2000. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J. Clin. Microbiol. 38:3623–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fournier PE, Raoult D. 2011. Prospects for the future using genomics and proteomics in clinical microbiology. Annu. Rev. Microbiol. 65:169–188 [DOI] [PubMed] [Google Scholar]
- 14.Stelzner A. 1988. Mycetoma, p 80–118 In Rippon J. (ed), Medical mycology: the pathogenic fungi and pathogenic actinomycetes, 3rd ed. W. B. Saunders Co., Philadelphia, PA [Google Scholar]
- 15.Padhi S, Uppin SG, Uppin MS, Umabala P, Challa S, Laxmi V, Prasad VB. 2010. Mycetoma in South India: retrospective analysis of 13 cases and description of two cases caused by unusual pathogens: Neoscytalidium dimidiatum and Aspergillus flavus. Int. J. Dermatol. 49:1289–1296 [DOI] [PubMed] [Google Scholar]
- 16.Lumley J. 1997. Hamilton Bailey's demonstrations of physical signs in clinical surgery, 18th ed. CRC Press, Boca Raton, FL [Google Scholar]
- 17.McNeil MM, Brown JM, Scalise G, Piersimoni C. 1992. Nonmycetomic Actinomadura madurae infection in a patient with AIDS. J. Clin. Microbiol. 30:1008–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welsh O, Sauceda E, Gonzalez J, Ocampo J. 1987. Amikacin alone and in combination with trimethoprim-sulfamethoxazole in the treatment of actinomycotic mycetoma. J. Am. Acad. Dermatol. 17:443–448 [DOI] [PubMed] [Google Scholar]
- 19.Ramam M, Garg T, D'Souza P, Verma KK, Khaitan BK, Singh MK, Banerjee U. 2000. A two-step schedule for the treatment of actinomycotic mycetomas. Acta Derm. Venereol. 80:378–380 [DOI] [PubMed] [Google Scholar]
- 20.Ramam M, Bhat R, Garg T, Sharma VK, Ray R, Singh MK, Banerjee U, Rajendran C. 2007. A modified two-step treatment for actinomycetoma. Indian J. Dermatol. Venereol. Leprol. 73:235–239 [DOI] [PubMed] [Google Scholar]
- 21.Agarwal US, Besarwal RK, Gupta R, Agarwal P. 26 November 2012. Treatment of actinomycetoma foot—our experience with ten patients. J. Eur. Acad. Dermatol. Venereol. [Epub ahead of print.] 10.1111/jdv.12036 [DOI] [PubMed] [Google Scholar]
- 22.Tight RR, Bartlett MS. 1981. Actinomycetoma in the United States. Rev. Infect. Dis. 3:1139–1150 [DOI] [PubMed] [Google Scholar]
- 23.Suzuki MT, Giovannoni SJ. 1996. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 62:625–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Claesson MJ, Wang Q, O'Sullivan O, Greene-Diniz R, Cole JR, Ross RP, O'Toole PW. 2010. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 38:e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willner D, Daly J, Whiley D, Grimwood K, Wainwright CE, Hugenholtz P. 2012. Comparison of DNA extraction methods for microbial community profiling with an application to pediatric bronchoalveolar lavage samples. PLoS One 7:e34605. 10.1371/journal.pone.0034605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klappenbach JA, Saxman PR, Cole JR, Schmidt TM. 2001. rrndb: the Ribosomal RNA Operon Copy Number Database. Nucleic Acids Res. 29:181–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snitkin ES, Zelazny AM, Thomas PJ, Stock F, Henderson DK, Palmore TN, Segre JA. 2012. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci. Transl. Med. 4:148ra116. 10.1126/scitranslmed.3004129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenblum S, Turnbaugh PJ, Borenstein E. 2012. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc. Natl. Acad. Sci. U. S. A. 109:594–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huebinger RM, Liu MM, Dowd SE, Rivera-Chavez FA, Boynton J, Carey C, Hawkins K, Minshall CT, Wolf SE, Minei JP, Barber RC. 2013. Examination with next-generation sequencing technology of the bacterial microbiota in bronchoalveolar lavage samples after traumatic injury. Surg. Infect. (Larchmt) 14:275–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wain J, Mavrogiorgou E. 2013. Next-generation sequencing in clinical microbiology. Expert Rev. Mol. Diagn. 13:225–227 [DOI] [PubMed] [Google Scholar]
- 31.Jünemann S, Prior K, Szczepanowski R, Harks I, Ehmke B, Goesmann A, Stoye J, Harmsen D. 2012. Bacterial community shift in treated periodontitis patients revealed by ion torrent 16S rRNA gene amplicon sequencing. PLoS One 7:e41606. 10.1371/journal.pone.0041606 [DOI] [PMC free article] [PubMed] [Google Scholar]