

Abstract
Human coronaviruses (CoVs) such as severe acute respiratory syndrome CoV (SARS-CoV) and Middle East respiratory syndrome CoV (MERS-CoV) cause epidemics of severe human respiratory disease. A conserved step of CoV replication is the translation and processing of replicase polyproteins containing 16 nonstructural protein domains (nsp's 1 to 16). The CoV nsp5 protease (3CLpro; Mpro) processes nsp's at 11 cleavage sites and is essential for virus replication. CoV nsp5 has a conserved 3-domain structure and catalytic residues. However, the intra- and intermolecular determinants of nsp5 activity and their conservation across divergent CoVs are unknown, in part due to challenges in cultivating many human and zoonotic CoVs. To test for conservation of nsp5 structure-function determinants, we engineered chimeric betacoronavirus murine hepatitis virus (MHV) genomes encoding nsp5 proteases of human and bat alphacoronaviruses and betacoronaviruses. Exchange of nsp5 proteases from HCoV-HKU1 and HCoV-OC43, which share the same genogroup, genogroup 2a, with MHV, allowed for immediate viral recovery with efficient replication albeit with impaired fitness in direct competition with wild-type MHV. Introduction of MHV nsp5 temperature-sensitive mutations into chimeric HKU1 and OC43 nsp5 proteases resulted in clear differences in viability and temperature-sensitive phenotypes compared with MHV nsp5. These data indicate tight genetic linkage and coevolution between nsp5 protease and the genomic background and identify differences in intramolecular networks regulating nsp5 function. Our results also provide evidence that chimeric viruses within coronavirus genogroups can be used to test nsp5 determinants of function and inhibition in common isogenic backgrounds and cell types.
INTRODUCTION
Coronaviruses (CoVs) are enveloped, positive-strand RNA viruses that infect a wide range of animal hosts. Human CoVs cause illnesses including the common cold and severe acute respiratory syndrome (SARS) as well as the recently identified Middle East respiratory syndrome (MERS) associated with infection of a novel coronavirus (1). Coronaviruses are members of the order Nidovirales, family Coronaviridae, and subfamily Coronavirinae. Among the viruses in Coronavirinae, four main genera have recently been designated (2): alphacoronaviruses, which contain human coronavirus 229E (HCoV-229E) and HCoV-NL63; betacoronaviruses, containing human coronaviruses SARS-CoV, HKU1, MERS-CoV, and OC43; and gammacoronaviruses and deltacoronaviruses, from which no current human coronaviruses have been identified. The betacoronavirus murine hepatitis virus (MHV) is a well-established model for the study of coronavirus replication and pathogenesis. The MHV genome is 32 kb in length and encodes 7 genes (Fig. 1A) (3–5). An essential step of CoV replication is the translation of the ORF1ab replicase polyproteins and the subsequent processing of up to 16 nonstructural proteins (nsp's 1 to 16), including the nsp12 RNA-dependent RNA polymerase (3, 5, 6). CoV nsp5 protease (3CLpro; Mpro) mediates processing at 11 distinct cleavage sites, including its own autoproteolysis, and is indispensable for virus replication (5, 7–13). nsp5 exhibits a conserved three-domain structure containing a chymotrypsin-like fold formed by domains 1 and 2 as well as a third domain of unclear function but which likely is important for the required dimerization of nsp5 (Fig. 1B) (8–10, 12–16).
Fig 1.
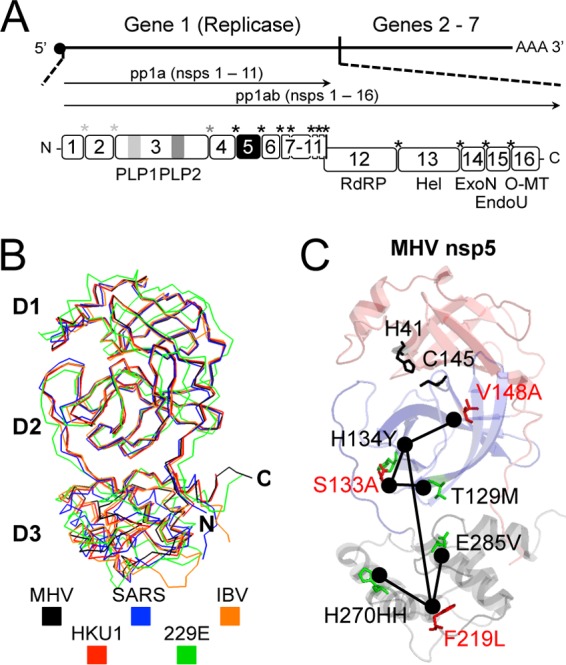
Murine hepatitis virus (MHV) genome and nsp5 protease structure and activity. (A) The MHV genome consists of 7 genes. The replicase gene encodes 16 nonstructural proteins that are processed by viral papain-like protease 1 (PLP1), PLP2, and nsp5 protease. nsp5 is responsible for 11 processing events between nsp's 4 and 16. Cleavage events are color coded and marked by asterisks. RdRP, RNA-dependent RNA polymerase; Hel, helicase; ExoN, exoribonuclease; EndoU, endoribonuclease; O-MT, O-methyltransferase. (B) Alignment of nsp5 crystal structures for HKU1 (PDB accession number 3D23), SARS-CoV (PDB accession number 2H2Z), HCoV-229E (PDB accession number 1P9S), and IBV (PDB accession number 2Q6D) and a model generated by using these structures and Modeller (29) for MHV nsp5. (C) Modeled structure of MHV nsp5 identifying the location of catalytic dyad residues H41 and C145 (black) and previously described temperature-sensitive (red) and second-site suppressor (green) mutations. Lines have been used to denote residues connected by phenotypic reversion.
Numerous studies have identified interacting residues within nsp5 and between nsp5 and other replicase nsp's that are essential for nsp5 regulation and function. Studies evaluating SARS-CoV nsp5 dimerization have identified three separate mutations in nsp5 that are >9 Å from known dimerization determinants and which disrupt or abolish nsp5 dimerization in vitro (17–19). Other studies have demonstrated that mutations in nsp3 and nsp10 alter or reduce nsp5-mediated polyprotein processing (20, 21). Mutagenesis of the cleavage site between nsp15 and nsp16 of infectious bronchitis virus (IBV) resulted in the emergence of a second-site mutation near the catalytic site in nsp5 (22). We previously described three separate temperature-sensitive (ts) mutations (S133A, V148A, and F219L) located in domains 2 and 3 of MHV nsp5, which are independent of known catalytic or dimerization residues but which nevertheless result in clear impairment of nsp5-mediated polyprotein processing and virus replication at nonpermissive temperatures (Fig. 1C) (23, 24). Replication at nonpermissive temperatures resulted in the emergence of second-site suppressor mutations, which arose largely in combinations and were distant from their cognate ts residues. One of these second-site mutations, H134Y, was independently selected in all three ts viruses. Collectively, these data support the hypothesis that nsp5 protease activity is extensively regulated by intra- and intermolecular interactions. However, it remains unclear whether intramolecular residue networks or the context of nsp5 in the replicase polyprotein is conserved between closely related coronaviruses.
In this study, we engineered chimeric MHV genomes encoding nsp5 from other alphacoronaviruses and betacoronaviruses to test for conservation of structure-function determinants and intramolecular residue networks. We demonstrate that exchange of nsp5 proteases from HKU1 and OC43, both of which are human betacoronaviruses that share a genogroup (genogroup 2a) with MHV, permits recovery of viruses in MHV with efficient replication. However, both chimeric MHVs were unable to compete with wild-type MHV (WT-MHV) in direct coinfection fitness experiments. Exchange of nsp5 proteases from other genogroups (genogroups 2b and 2c) did not permit recovery in chimeric MHV. To evaluate the conservation of residue determinants of nsp5 function in HKU1 and OC43, we introduced the MHV ts mutations S133A, V148A, and F219L. We show that these mutations result in clear phenotypic differences in the heterologous nsp5. Together, these results demonstrate selection for divergence of nsp5 determinants in conserved structure and function and suggest significant coevolution of nsp5 with other determinants in the genome. The results emphasize the importance of platform approaches for testing of cross-sensitivity of any identified nsp5 inhibitors. Our chimeric substitution of nsp5 proteases constitutes such a platform for evaluating structure-function conservation within a genogroup, providing a system for testing nsp5 inhibitors against human or zoonotic nsp5 proteases in an isogenic cloned background and CoVs for which cultivation is not possible.
MATERIALS AND METHODS
Viruses and cells.
Recombinant WT-MHV strain A59 (GenBank accession number AY910861) was used for all WT-MHV studies and was modified in the generation of recombinant chimeras containing HKU1 (H5-MHV) or OC43 (O5-MHV) nsp5 sequences. Naturally permissive murine delayed brain tumor (DBT) cells and baby hamster kidney 21 cells expressing the MHV receptor (BHK-MHVR) were used for all experiments (25). Dulbecco's modified Eagle medium (DMEM) (Gibco) supplemented with 10% heat-inactivated fetal calf serum (FCS) with and without G418 to maintain selection for MHVR expression in BHK cells was used for all experiments described.
Cloning and recovery of chimeric and mutant viruses.
Viruses were assembled and recovered by using the MHV infectious clone protocol described previously (25). The nsp5-coding sequences for human coronaviruses HKU1 (GenBank accession number NC_006577), OC43 (accession number NC_005147), SARS-CoV (accession number AY278741), 229E (accession number NC_002645), and NL63 (accession number NC_005831) and bat coronavirus HKU4 (accession number NC_009019) were each synthesized in the cloned MHV cDNA genome fragments (BioBasic), and sequences were confirmed prior to attempted virus recovery (26–28). Using the assembly protocol described here, the genomic cDNA fragments were ligated, transcribed, and electroporated into BHK-MHVR cells, which were then added to a subconfluent flask of DBT cells at 37°C (25).
RNA extraction and genomic sequencing.
Confluent monolayers of DBT cells in T25 (25-cm2) flasks were infected at a multiplicity of infection (MOI) of 5 PFU/cell and grown until 30 to 50% of the cells were involved in syncytia. RNA isolation, reverse transcription, and cDNA amplicon synthesis were performed as previously described (24). The complete genomes of H5-MHV and O5-MHV were verified by sequencing. Sequencing of all other viruses involved reading the nsp5-coding region (nucleotides 10160 to 11799).
Analysis of virus replication.
Virus infections were carried out in 6-well plates containing confluent monolayers of DBT cells. Cells were infected at MOIs of 0.01 and 1 PFU/cell at 32°C or 37°C. Samples of supernatants were acquired in triplicate for titer determinations at the time points indicated, and the dishes were supplemented with prewarmed media to ensure a constant volume. Viral titers were determined by plaque assays in duplicate, as previously described (24). Error bars reflect the standard deviations from the means for samples of multiple replicates.
Assay of virus fitness.
Confluent monolayers of DBT cells were coinfected with combinations of WT-MHV, H5-MHV, or O5-MHV in triplicate at ratios of 1:1 or 1:10 to a total MOI of 0.01 PFU/cell in T25 flasks. When infected cells were observed to have 30 to 50% involvement by virus-induced syncytia, the supernatant was removed and stored at −80°C, and the monolayer was treated with TRIzol reagent for total RNA acquisition. To initiate subsequent passages, the medium supernatant was thawed at 4°C, and 5 μl was added to each of three T25 flasks of confluent cells. RNA was isolated and reverse transcribed as previously described (24). Amplicons using oligonucleotides flanking the nsp5-coding region were generated and treated with HKU1 (HincII) and OC43 (BsiWI) sequence-specific restriction enzymes, which resulted in a single digest. The bands were resolved on a 0.8% agarose gel containing ethidium bromide, imaged, and quantified by densitometry.
Isolation and expansion of suppressor mutants.
A plaque assay was performed by infecting DBT cells in duplicate in 6-well plates with serial dilutions and a 1-h adsorption period at 32°C. The overlay contained a 1:1 mixture of 2% agar and 2× DMEM. The plates were incubated for 48 h until plaques were easily visible. Ten plaques were selected for each virus sample and resuspended in gel saline. Isolated plaques for each virus were used to infect separate T25 flasks for expansion at 40°C. Flasks were removed from nonpermissive temperatures when 70 to 95% of cells were involved in syncytia, and RNA was then isolated as described above.
Sequence analyses and modeling of the MHV nsp5 structures.
A multiple-sequence alignment and a phylogenetic tree of coronavirus nsp5 sequences were generated by using ClustalX and a bootstrap alignment from 1,000 trials. The X-ray crystal structure of HCoV-HKU1 nsp5 (PDB accession number 3D23) was used for structure comparisons and to generate structural models of MHV and OC43 (13). Structural models were generated by using Modeller and MacPyMol (DeLano Scientific) (29). Other coronavirus nsp5 X-ray structures used for alignment and comparisons were SARS-CoV (PDB accession number 2H2Z), HCoV-229E (PDB accession number 1P9S), and IBV (PDB accession number 2Q6D) (9, 10, 12).
RESULTS
MHV replication is supported by nsp5 from HCoV-OC43 and -HKU1.
We engineered chimeric genomes of MHV-A59 by replacing MHV nsp5 with the nsp5 coding sequence of the betacoronaviruses HCoV-HKU1 (genogroup group 2a), HCoV-OC43 (group 2a), SARS-CoV (group 2b), and bat CoV-HKU4 (group 2c) as well as the alphacoronaviruses HCoV-229E and HCoV-NL63. Chimeric MHV-A59 viruses encoding nsp5 from HKU1 (H5-MHV) and OC43 (O5-MHV) were readily recovered at 37°C and exhibited cytopathic effects and recovery similar to those of wild-type MHV on murine delayed brain tumor (DBT) cells (Fig. 2A). No other chimeric viruses were recovered despite multiple attempts at recovery. The complete genome sequences of chimeric H5- and O5-MHV identified an intact heterologous nsp5 sequence and no additional mutations. To compare the replication of the chimeric H5- and O5-MHV viruses to that of wild-type MHV (WT-MHV), replicate plates of murine DBT cells were infected at a low multiplicity of infection (MOI) (0.01 PFU/cell) or a high MOI (1 PFU/cell). In low-MOI infection, WT-MHV and the chimeric H5- and O5-MHV exhibited similar replication and attained peak titers at the same time postinfection (p.i.) (Fig. 2B). During single-cycle infection (MOI = 1 PFU/cell), both H5- and O5-MHV attained peak titers similar to those of WT-MHV; however, O5-MHV and H5-MHV displayed approximately 10-fold and 30-fold reductions in titers, respectively, compared to WT-MHV at 8 h p.i. (Fig. 2C). Overall, the results demonstrated that chimeric viruses containing nsp5 from OC43 and HKU1 mediated all steps required for replication but suggested possible subtle differences in the timing and efficiency of function in a heterologous background.
Fig 2.
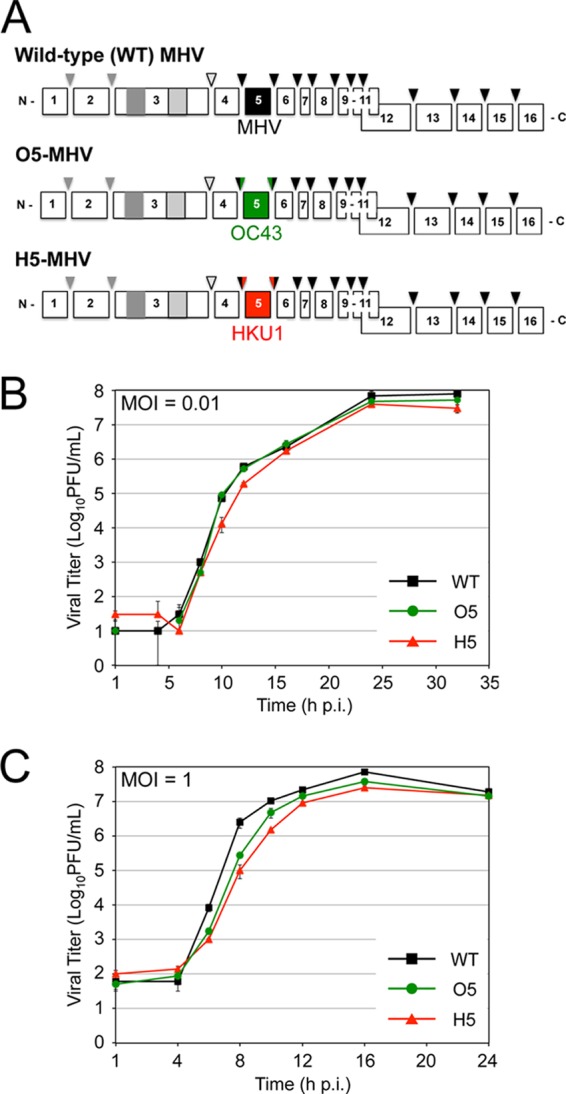
Recovery and virus replication kinetics of H5- and O5-MHV. (A) Virus constructs containing nsp5 protease substitutions of OC43 (green) or HKU1 (red). Proteases and cleavage sites are color coded. (B and C) DBT cells were infected with WT-MHV, H5-MHV, or O5-MHV at 37°C at an MOI of either 0.01 PFU/cell (B) or 1 PFU/cell (C). Samples were acquired in triplicate (n = 3), and titers were determined by plaque assays in duplicate per sample. Error bars reflect standard deviations from the means based on samples from multiple biological replicates for each time point.
MHV temperature-sensitive mutations differ in their phenotypes in chimeric H5-MHV and O5-MHV.
To evaluate the differences and conservation of intramolecular regulation between MHV, HKU1, and OC43 nsp5 proteases, we substituted S133A, V148A, and F219L mutations into the background of the chimeric H5-MHV and O5-MHV genomes. We have previously shown that in MHV, the three temperature-sensitive (ts) mutations impair virus replication and nsp5-mediated polyprotein activity at a nonpermissive temperature of 40°C (23, 24) (Fig. 1C). HKU1 and OC43 nsp5 proteases have identical amino acid residues at the equivalent ts allele positions of MHV and share an isogenic MHV background in the context of the chimeric H5- and O5-MHVs. Three different chimeric viruses containing the MHV temperature-sensitive mutations were successfully recovered at 32°C and sequence confirmed without additional mutations in nsp5: H5-V148A (H5-MHV with a V-to-A mutation at position 148), O5-V148A, and O5-S133A (Fig. 3A). In contrast, the H5-F219L, O5-F219L, and H5-S133A mutants could not be recovered after multiple independent attempts at 32°C, indicating a different response to the conditional mutations in nsp5 between all three viruses.
Fig 3.

Evaluation of temperature sensitivity and replication kinetics of H5- and O5-MHV containing MHV ts alleles S133A and V148A. (A) Virus titers for WT-MHV, H5-MHV, O5-MHV, and H5- and O5-MHV containing either S133A or V148A mutations were determined by plaque assays on DBT cells at either 32°C or 40°C. The EOP was determined as the ratio of the average titers for 40°C over 32°C. (B and C) DBT cells were infected at an MOI of 1 PFU/cell. Samples were acquired from triplicate infections at various time points, and the temperature either remained at 32°C (B) or was shifted to 40°C (C) at 6 h p.i. Titers were determined by plaque assays in duplicate per sample, and the standard deviations from the means are shown for all time points from multiple replicates.
To assess temperature sensitivity of the recovered viruses, infections were performed at 32°C and 40°C, and the efficiency of plating (EOP) (ratio of titers at 40°C to 32°C) was determined. The V148A mutation did not confer a temperature-sensitive phenotype in either the HKU1 or OC43 chimeric virus background at 40°C. In contrast, the O5-S133A mutant was ts, while the H5-S133A mutant could not be recovered. Replication kinetics were determined during infection of DBT cells at an MOI of 1 PFU/cell at 32°C (Fig. 3B) or with a temperature shift at 6 h p.i. to 40°C (Fig. 3C). The chimeric and mutant viruses all replicated similarly to WT-MHV at 32°C. After a shift to 40°C at 6 h p.i., only the O5-S133A mutant exhibited impaired growth, manifesting as no detectable replication from 6 to 10 h p.i., followed by low-level exponential replication with impaired peak titers. Previous studies reported possible intermolecular regulation of nsp5 by nsp3 and nsp10 (20, 21). We therefore sequenced across the nsp3- and nsp10-coding regions. No second-site mutations were detected in the nsp3- and nsp10-coding regions of the H5-V148A, O5-V148A, and O5-S133A populations at peak titers. The delayed recovery of replication in the O5-S133A mutant suggested the emergence of phenotypic revertants, so the O5-S133A virus was passaged at 40°C, and plaque isolates were selected and sequenced for nsp5 mutations, resulting in the identification of two putative suppressor mutations, A116V and N8Y (Fig. 4A). These residues differ from the H134Y suppressor mutant viruses that arose in MHV; however, Y134 is already present in the O5-S133A mutant and did not suppress the ts phenotype. While we did not directly test these residues for the capacity to suppress the S133A phenotype, they were associated with recovery in virus replication and were the only mutations identified individually with retention of the S133A mutation.
Fig 4.

Selection for second-site suppressor mutations of the O5-S133A virus. (A) Location of the S133A ts allele (red) and its identified second-site suppressor mutations (green) and the V148A and Y134H mutations (red), which result in a ts phenotype in combination on a model of OC43 nsp5 generated by using the crystal structure of HKU1 nsp5 (PDB accession number 3D23) and Modeler. (B) Primary sequence alignment of MHV-A59, HCoV-HKU1, and HCoV-OC43. Domain partitions are denoted by dotted lines and are labeled. The catalytic dyad residues are labeled with asterisks. The S133A, V148A, and F219L mutations are highlighted and labeled in red. Second-site mutations identified for individual viruses are shown in green. (C) Virus titers for WT-MHV, the MHV-V148A ts mutant, and O5-MHV containing V148A and Y134H mutations were determined by plaque assays on DBT cells at either 32°C or 40°C. The EOP was determined at the ratio of the average titers for 40°C over 32°C.
Nsp5 protease residue Y134 regulates nsp5 function in MHV and OC43.
We demonstrated previously that a single H134Y suppressor mutation in MHV was capable of partially or fully suppressing independently the phenotype of all three ts mutations (S133A, V148A, and F219L) (23, 24). To test whether the Y134 native residue in nsp5 of OC43 and HKU1 was necessary for the non-ts phenotype of the O5-V148A and H5-V148A viruses, we introduced a Y134H mutation into each virus containing the V148A mutation to recapitulate the MHV wild-type H134 residue (Fig. 4B). The O5-V148A/Y134H virus demonstrated a clear transition to a ts phenotype (Fig. 4C), as evidenced by a substantial decrease in the EOP. In contrast, the H5-V148A/Y134H mutant could not be recovered at either 32°C or 37°C. These data demonstrate that the Y134 residue identified as a second-site suppressor mutation of all three MHV ts mutants also confers resistance to temperature sensitivity in the background of O5-V148A. Surprisingly, introduction of the Y134H mutation in the background of H5-V148A resulted in a lethal phenotype. Collectively, these data demonstrate a critical role of residue 134 in nsp5 function.
H5- and O5-MHV exhibit reduced fitness relative to WT-MHV.
The replication assays and results from introduction of MHV ts residues into the backgrounds of H5- and O5-MHV indicate that subtle differences in structure or sequence between the proteases can have a significant impact on nsp5 activity and regulation. To directly test the differences between WT-MHV and the chimeric O5- and H5-MHV, we performed competitive fitness assays in which DBT cells were coinfected with WT-, H5-, and O5-MHV at different ratios and at a total MOI of 0.01 PFU/cell. When cells were ∼30% involved in syncytia, total cellular RNA was harvested and used to generate cDNA amplicons containing the nsp5-coding region, as previously described (30). The amplicons were then digested with restriction endonucleases that recognize unique sites in the nsp5 sequence of HKU1 or OC43, followed by electrophoresis and densitometric analysis (Fig. 5A and B). Following coinfection with MHV and either O5-MHV or H5-MHV at an initial ratio of 1:1, WT-MHV represented >75% of supernatant infectious virus after passage 1 (P1), and by P3, almost no chimeric virus was detected (Fig. 5C). Even when H5-MHV or O5-MHV was inoculated with a 10:1 advantage over WT-MHV, the amount of WT-MHV was equivalent to or exceeded the amount of the chimeric mutants by P2, and WT-MHV was dominant in both cases by P3.
Fig 5.

Fitness analysis of H5- and O5-MHV. (A) Confluent monolayers of DBT cells were infected with WT-MHV, H5-MHV, or O5-MHV, and total RNA was isolated. cDNA amplicons containing the nsp5-coding region were generated and digested with HKU1 or OC43 nsp5-specific restriction enzymes and resolved by electrophoresis. (B) WT-MHV, H5-MHV, or either 1:1 or 1:10 mixtures of purified RNA were reverse transcribed, digested with an HKU1 nsp5-specific restriction enzyme, and resolved by gel electrophoresis. (C) Coinfections of either WT- and H5-MHV (red) or WT- and O5-MHV (green) at ratios of 1:1 or 1:10 were carried out at 37°C and at a total MOI of 0.01. The viruses were passaged three times (P1 to P3), each in triplicate (n = 3). Digested amplicons for one of each of the infections as well as the quantification of the ratio of WT-MHV to either H5- or O5-MHV and the ratio of H5-MHV to O5-MHV, as determined by the averages of three replicates at each designated passage, are shown. Error bars represent the standard deviations from the means for each of the relative frequencies shown.
We next directly compared H5-MHV and O5-MHV to determine if there were differences in competitive fitness in the chimeric viruses (Fig. 5C). At a 1:1 ratio for coinfection, the relative amounts of H5:O5 detected were approximately 1:1, with no clear fitness advantage through P3. Even when O5-MHV was given a 10:1 advantage, H5-MHV was still present at P3 at amounts similar to those of the initial coinfection, indicating no significant fitness advantage for O5-MHV over H5-MHV. These results demonstrated that while the replication defects of chimeric H5-MHV and O5-MHV were mild in single infection, introduction of even a closely related nsp5 resulted in a profound loss of competitive fitness compared to WT-MHV. These data demonstrate that nsp5 of closely related coronaviruses can mediate all required activities for replication in culture in the heterologous MHV background but that the mismatch between protease and viral background has profound consequences for the fitness of the recombinant virus.
DISCUSSION
Coronavirus nsp5 proteases play a critical role in the proteolytic processing of the replicase polyproteins and are regulated by both intramolecular residue networks and associations with other nsp's. In this study, we tested the impact of specific conserved residues and genetic backgrounds on the activity of nsp5 using chimeric MHV as a common platform. Our results show successful recovery of chimeric MHV with nsp5 proteases only from very closely related CoVs of the same genogroup (betacoronavirus group 2a). Furthermore, we demonstrate that three previously characterized MHV ts mutations result in clearly different phenotypes in the chimeric virus backgrounds of H5- and O5-MHV. Finally, we show that substitution of analogous nsp5 proteases of the same genogroup, although replication competent, results in fitness impairment during direct competition with wild-type MHV.
These data suggest that differences between proteases and their interactions with their respective virus backgrounds may have provided a pressure for intramolecular reorganization between even closely related coronavirus species. The structures of nsp5 proteases from several divergent CoVs have been solved, providing important insights into the structure-function relationships of nsp5, including the relationships of the N and C termini, and predicted determinants of dimerization (8–10, 12, 13, 15, 17, 31–34). In addition, biochemical studies of purified nsp5 have been used to test the specificity for different cleavage sites in small peptides (7, 11, 33, 35–38). However, the role of nsp5 in regulating replicase polyprotein processing during virus infection remains much less well understood. Specifically, following translation of ORF1a/b, nsp5 must properly fold within the context of the replicase polyprotein and an nsp4-10 intermediate, orchestrate its autoproteolytic processing, dimerize, and recognize and process up to 9 additional cleavage sites (5, 9, 14, 34, 39, 40). Thus, while the general specificity of nsp5 is known for specific cleavage site peptides in vitro, the accessibility, activity, and hierarchy of events during infection are not known. Our study suggests a high degree of both intra- and intermolecular coevolution of nsp5 even among closely related CoVs and potential barriers to genetic exchange across more divergent CoVs. Furthermore, the chimeric-exchange approach used in this study provides a framework for comparing nsp5 proteases between CoVs and identifying intra- and intermolecular determinants of nsp5 function.
Intramolecular regulation of nsp5 activity.
Coronavirus nsp5 proteases display a high degree of tertiary structure conservation and a conserved core of residues around the active site. As we have shown for MHV, there are a number of conserved residues, independent of known catalytic and dimerization regions, which span the protease structure and may be functionally conserved between divergent proteases (24). The structural and functional roles of these residues and other nonconserved residues proximal to them remain largely unknown. We show in this study that the temperature-sensitive residues reported for MHV nsp5 domains 2 and 3 are also important for replication and temperature sensitivity in HKU1 and OC43 (Table 1). However, these results also demonstrate that the genetic divergence of nsp5, even in closely related viruses with similar structures, is associated with subtle refining of the intramolecular communication networks and their role in regulating protease activity. When we modeled the ts mutations in the solved structures of nsp5 proteases of HCoV-HKU1, HCoV-229E, and SARS-CoV, there were no predictions of significant proximal or more distal propagated structural perturbations that might account for the demonstrated changes. This result was similar to the results of our original studies (23, 24). These data suggest that the functional impact of these mutations or their pathways of intramolecular communication may be too subtle to be identified by modeling or may be limited to elevated temperatures. The emergence of unique putative second-site mutations during phenotypic reversion of the O5-S133A virus indicates that the ts and second-site suppressor communication pathways described for MHV differ in the context of chimeric OC43. However, these data do not exclude the possibility that other regulatory networks may be shared between closely related virus species. This is supported by the common role of the Tyr134 residue between MHV and OC43. This residue was also selected for a H134Y substitution during mouse adaptation of the more distantly related SARS-CoV (41). Potential explanations of Y134 function may include stabilization of the active site, disruption of dimerization, or modification of protease specificity. The selection of changes at Y134 in three different and divergent virus backgrounds and conditions emphasizes the importance of this residue. Biochemical characterization of purified mutant proteases, which is under way, will be essential to precisely define the functional role of the tested mutations in thermal stability, protease activity, and cleavage site specificity.
Table 1.
Summary of temperature-sensitive and second-site suppressor mutations in MHV, HKU1, and OC43 nsp5 proteases
| Mutation | Virus phenotype and suppressor mutation(s) detecteda |
||
|---|---|---|---|
| MHV | HKU1 | OC43 | |
| S133A | ts (EOP = 3 × 10−5) | Not recovered | ts (EOP = 6 × 10−3) |
| Sup, T129M, H134Y | Sup, N8Y, A116V | ||
| V148A | ts (EOP = 1 × 10−4) | Not ts (EOP = 3 × 100) | Not ts (EOP = 2 × 100) |
| Sup, S133N, H134Y | With +Y134H, not recovered | +Y134H, ts (EOP = 3 × 10−4) | |
| F219L | ts (EOP = 3 × 10−5) | Not recovered | Not recovered |
| Sup, H134Y, E285V, H270HH | |||
Sup, suppressor mutation(s).
Intermolecular regulation and potential for coronavirus nsp5 exchange.
Previous studies noted that the tertiary structure of nsp5 and cleavage site recognition sequences are largely conserved across all known coronavirus strains (8, 9, 11–13, 42). However, we demonstrate that chimeric substitution of more diverged nsp5 proteases prevents virus recovery. Furthermore, substitution of nsp5 from even the same genogroup (genogroup 2a) is associated with clear fitness costs during direct competition. Amino acid alignments of nsp5 proteases of betacoronaviruses MHV, HKU1, and OC43 exhibit 80 to 84% sequence identity to each other and occupy the same genogroup, genogroup 2a. In contrast, nsp5 proteases of alphacoronaviruses 229E and NL63 and more distantly related betacoronaviruses SARS-CoV and bat HKU4 show no greater than 53% identity (Fig. 6). Bat CoV-HKU4 nsp5 was selected for exchange in this study since it exhibits the highest identity (53%) among coronaviruses outside betacoronavirus group 1 (genogroup 2a) (containing MHV, HKU1, and OC43) and shows high sequence homology (81% identity) to the recently identified human coronavirus MERS-CoV (1). The data presented in this study suggest that the function and regulation of nsp5 protease activity has diverged significantly and that conservation of intermolecular residue interactions may be most closely associated with members of the same genogroup rather than the overall genus.
Fig 6.
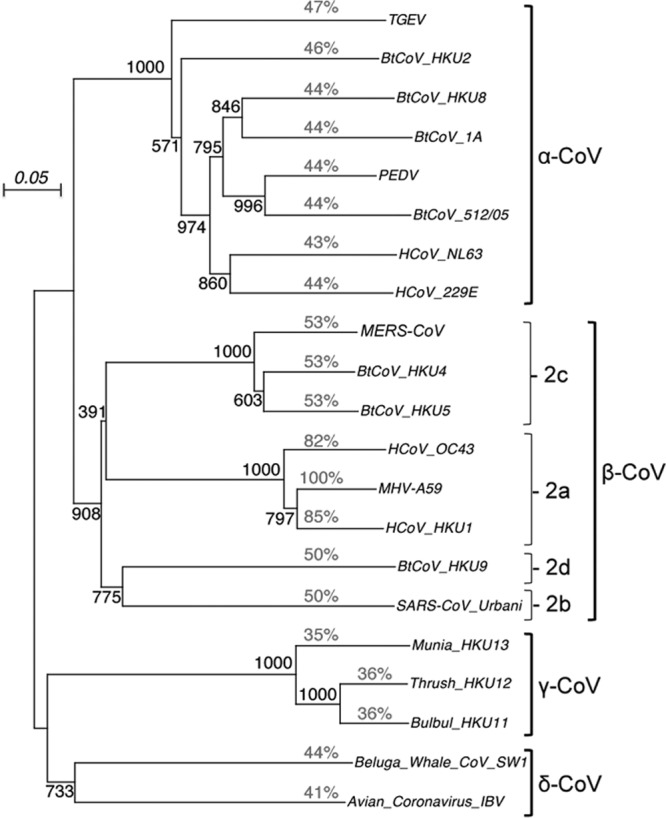
Phylogeny of coronavirus nsp5. The phylogenetic tree was generated by using a bootstrap alignment of CoV nsp5 sequences. The phylogenetic genera and the betacoronavirus subgroups (genogroups) are indicated. The percent amino acid identity relative to MHV nsp5 is shown in lightface type. TGEV, transmissible gastroenteritis virus; PEDV, porcine epidemic diarrhea virus; BtCoV, bat coronavirus.
Other studies have reported that nsp5 is part of a cistron containing nsp's 5 to 16, which share a functional role in the replication of the virus (21, 43). Three different studies have shown a critical regulation of nsp5 by other elements of the replication complex or the polyprotein backbone. Introduction of mutations into nsp's 3 and 10 were associated with direct decreases in nsp5-mediated polyprotein processing, and alteration of the nsp15/16 cleavage site in IBV resulted in the emergence of a second-site mutation in nsp5 at residue 166, which was previously shown to regulate dimerization in SARS-CoV (20–22). These studies suggest that allostery and cleavage site divergence may be critical elements of the replicase gene that regulate nsp5 activity. It is known that nsp5 is capable of functioning in the absence of other elements of the replication complex, as shown through biochemical purification and analysis (14, 33, 39). However, the data presented in this study suggest that nsp5 exhibits critical interactions or potential allostery with other elements of the replication complex. Subsequent analyses must consider the potential role of these interactions when evaluating protease differences and specificity.
Platforms for testing nsp5 inhibitors.
Coronaviruses are known to undergo host species movement, as evidenced by the recent emergence of MERS-CoV (1, 44, 45). However, efforts to study nsp5 inhibitors of zoonotic coronaviruses from bats and other species as well as some HCoVs such as HKU1 and NL63 have been limited by the lack of an ability to readily cultivate viruses in culture or establish reverse genetic systems. Our results show that chimeric substitution of nsp5 proteases into the cloned genome background of a well-studied system such as MHV or SARS-CoV may constitute a robust platform for evaluating structural and functional differences in an isogenic replicating virus. This approach can be used to identify key conserved determinants that may facilitate future development of nsp5 inhibitors. Coronavirus nsp5 proteases are primary inhibitor targets due to their central role in the processing and formation of viral replication. Efforts aimed at designing coronavirus nsp5 inhibitors have focused largely on targeting conserved active-site or substrate binding-site determinants across coronaviruses. However, the more promising inhibitor candidates have been limited to low μM concentrations and have yet to be tested against a replicating virus. The identification of common determinants of function within the closely related MHVs HKU1 and OC43 suggests that other noncatalytic, non-active-site determinants of nsp5 function likely exist, and further efforts should be made to evaluate these regions for potential inhibitor design. However, the promise of creating a broadly effective nsp5 inhibitor may not be feasible with current approaches due to the high level of divergence associated with intra- and intermolecular regulation between coronaviruses. These data suggest that inhibitor design efforts may be more directly suitable to designing and optimizing group-specific inhibitors rather than a single inhibitor that will be effective for all coronaviruses. The chimeric approach described here may constitute a platform for testing inhibitors against the proteases of zoonotic or emergent viruses.
ACKNOWLEDGMENTS
We thank Dia Beachboard, Michelle Becker, Megan Culler, Aimee Eggler, Wayne Hsieh, Lindsay Maxwell, Rose Njoroge, Allison Norlander, Clint Smith, and Gokhan Ünlü for technical aid and assistance in regard to manuscript organization and preparation.
This work was supported by National Institutes of Health grant R01 AI26603 (M.R.D., C.C.S., and A.D.M.) from the National Institute of Allergy and Infectious Diseases, virology training grant T32 AI089554 (C.C.S.) through Vanderbilt University, and the Elizabeth B. Lamb Center for Pediatric Research.
Footnotes
Published ahead of print 11 September 2013
REFERENCES
- 1.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367:1814–1820 [DOI] [PubMed] [Google Scholar]
- 2.Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, Bai R, Teng JL, Tsang CC, Wang M, Zheng BJ, Chan KH, Yuen KY. 2012. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 86:3995–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorbalenya AE, Koonin EV, Donchenko AP, Blinov VM. 1989. Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. 17:4847–4861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee HJ, Shieh CK, Gorbalenya AE, Koonin EV, La Monica N, Tuler J, Bagdzhadzhyan A, Lai MM. 1991. The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and RNA polymerase. Virology 180:567–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7:439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brierley I, Digard P, Inglis SC. 1989. Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell 57:537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ziebuhr J, Snijder EJ, Gorbalenya AE. 2000. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 81:853–879 [DOI] [PubMed] [Google Scholar]
- 8.Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R. 2002. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 21:3213–3224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. 2003. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300:1763–1767 [DOI] [PubMed] [Google Scholar]
- 10.Xue X, Yang H, Shen W, Zhao Q, Li J, Yang K, Chen C, Jin Y, Bartlam M, Rao Z. 2007. Production of authentic SARS-CoV M(pro) with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J. Mol. Biol. 366:965–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bacha U, Barrila J, Gabelli SB, Kiso Y, Mario Amzel L, Freire E. 2008. Development of broad-spectrum halomethyl ketone inhibitors against coronavirus main protease 3CL(pro). Chem. Biol. Drug Des. 72:34–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, Li J, Zhou Z, Ding Y, Zhao Q, Zhang XC, Liao M, Bartlam M, Rao Z. 2008. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 82:2515–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Q, Li S, Xue F, Zou Y, Chen C, Bartlam M, Rao Z. 2008. Structure of the main protease from a global infectious human coronavirus, HCoV-HKU1. J. Virol. 82:8647–8655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu Y, Denison MR. 1997. Determinants of mouse hepatitis virus 3C-like proteinase activity. Virology 230:335–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi J, Song J. 2006. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 273:1035–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi J, Sivaraman J, Song J. 2008. Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J. Virol. 82:4620–4629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrila J, Bacha U, Freire E. 2006. Long-range cooperative interactions modulate dimerization in SARS 3CLpro. Biochemistry 45:14908–14916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barrila J, Gabelli SB, Bacha U, Amzel LM, Freire E. 2010. Mutation of Asn28 disrupts the dimerization and enzymatic activity of SARS 3CL(pro). Biochemistry 49:4308–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng SC, Chang GG, Chou CY. 2010. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys. J. 98:1327–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donaldson EF, Graham RL, Sims AC, Denison MR, Baric RS. 2007. Analysis of murine hepatitis virus strain A59 temperature-sensitive mutant TS-LA6 suggests that nsp10 plays a critical role in polyprotein processing. J. Virol. 81:7086–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stokes HL, Baliji S, Hui CG, Sawicki SG, Baker SC, Siddell SG. 2010. A new cistron in the murine hepatitis virus replicase gene. J. Virol. 84:10148–10158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang S, Shen H, Wang J, Tay FP, Liu DX. 2010. Functional and genetic studies of the substrate specificity of coronavirus infectious bronchitis virus 3C-like proteinase. J. Virol. 84:7325–7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sparks JS, Donaldson EF, Lu X, Baric RS, Denison MR. 2008. A novel mutation in murine hepatitis virus nsp5, the viral 3C-like proteinase, causes temperature-sensitive defects in viral growth and protein processing. J. Virol. 82:5999–6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stobart CC, Lee AS, Lu X, Denison MR. 2012. Temperature-sensitive mutants and revertants in the coronavirus nonstructural protein 5 protease (3CLpro) define residues involved in long-distance communication and regulation of protease activity. J. Virol. 86:4801–4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yount B, Denison MR, Weiss SR, Baric RS. 2002. Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59. J. Virol. 76:11065–11078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394–1399 [DOI] [PubMed] [Google Scholar]
- 27.Vijgen L, Keyaerts E, Moes E, Thoelen I, Wollants E, Lemey P, Vandamme AM, Van Ranst M. 2005. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 79:1595–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BH, Poon RW, Cai JJ, Luk WK, Poon LL, Wong SS, Guan Y, Peiris JS, Yuen KY. 2005. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 79:884–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eswar N, Eramian D, Webb B, Shen MY, Sali A. 2008. Protein structure modeling with MODELLER. Methods Mol. Biol. 426:145–159 [DOI] [PubMed] [Google Scholar]
- 30.Graham RL, Becker MM, Eckerle LD, Bolles M, Denison MR, Baric RS. 2012. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med. 18:1820–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen H, Wei P, Huang C, Tan L, Liu Y, Lai L. 2006. Only one protomer is active in the dimer of SARS 3C-like proteinase. J. Biol. Chem. 281:13894–13898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen S, Hu T, Zhang J, Chen J, Chen K, Ding J, Jiang H, Shen X. 2008. Mutation of Gly-11 on the dimer interface results in the complete crystallographic dimer dissociation of severe acute respiratory syndrome coronavirus 3C-like protease: crystal structure with molecular dynamics simulations. J. Biol. Chem. 283:554–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grum-Tokars V, Ratia K, Begaye A, Baker SC, Mesecar AD. 2008. Evaluating the 3C-like protease activity of SARS-coronavirus: recommendations for standardized assays for drug discovery. Virus Res. 133:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen S, Jonas F, Shen C, Hilgenfeld R. 2010. Liberation of SARS-CoV main protease from the viral polyprotein: N-terminal autocleavage does not depend on the mature dimerization mode. Protein Cell 1:59–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hegyi A, Ziebuhr J. 2002. Conservation of substrate specificities among coronavirus main proteases. J. Gen. Virol. 83:595–599 [DOI] [PubMed] [Google Scholar]
- 36.Fan K, Ma L, Han X, Liang H, Wei P, Liu Y, Lai L. 2005. The substrate specificity of SARS coronavirus 3C-like proteinase. Biochem. Biophys. Res. Commun. 329:934–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goetz DH, Choe Y, Hansell E, Chen YT, McDowell M, Jonsson CB, Roush WR, McKerrow J, Craik CS. 2007. Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry 46:8744–8752 [DOI] [PubMed] [Google Scholar]
- 38.Chuck CP, Chow HF, Wan DC, Wong KB. 2011. Profiling of substrate specificities of 3C-like proteases from group 1, 2a, 2b, and 3 coronaviruses. PLoS One 6:e27228. 10.1371/journal.pone.0027228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu X, Lu Y, Denison MR. 1996. Intracellular and in vitro-translated 27-kDa proteins contain the 3C-like proteinase activity of the coronavirus MHV-A59. Virology 222:375–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okamoto DN, Oliveira LC, Kondo MY, Cezari MH, Szeltner Z, Juhasz T, Juliano MA, Polgar L, Juliano L, Gouvea IE. 2010. Increase of SARS-CoV 3CL peptidase activity due to macromolecular crowding effects in the milieu composition. Biol. Chem. 391:1461–1468 [DOI] [PubMed] [Google Scholar]
- 41.Roberts A, Deming D, Paddock CD, Cheng A, Yount B, Vogel L, Herman BD, Sheahan T, Heise M, Genrich GL, Zaki SR, Baric R, Subbarao K. 2007. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 3:e5. 10.1371/journal.ppat.0030005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woo PC, Lau SK, Huang Y, Yuen KY. 2009. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. (Maywood) 234:1117–1127 [DOI] [PubMed] [Google Scholar]
- 43.Sawicki SG, Sawicki DL, Younker D, Meyer Y, Thiel V, Stokes H, Siddell SG. 2005. Functional and genetic analysis of coronavirus replicase-transcriptase proteins. PLoS Pathog. 1:e39. 10.1371/journal.ppat.0010039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu G, Liu D. 2012. SARS-like virus in the Middle East: a truly bat-related coronavirus causing human diseases. Protein Cell 3:803–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anthony SJ, Ojeda-Flores R, Rico-Chavez O, Navarrete-Macias I, Zambrana-Torrelio CM, Rostal MK, Epstein JH, Tipps T, Liang E, Sanchez-Leon M, Sotomayor-Bonilla J, Aguirre AA, Avila-Flores R, Medellin RA, Goldstein T, Suzan G, Daszak P, Lipkin WI. 2013. Coronaviruses in bats from Mexico. J. Gen. Virol. 94:1028–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]