

Abstract
Single-cell genomics is emerging as an important tool in cellular biology. We describe for the first time a system to investigate RNA virus quasispecies diversity at the cellular level utilizing hepatitis C virus (HCV) replicons. A high-fidelity nested reverse transcription (RT)-PCR assay was developed, and validation using control transcripts of known copy number indicated a detection limit of 3 copies of viral RNA/reaction. This system was used to determine the cellular diversity of subgenomic JFH-1 HCV replicons constitutively expressed in Huh7 cells. Each cell contained a unique quasispecies that was much less diverse than the quasispecies of the bulk cell population from which the single cells were derived, suggesting the occurrence of independent evolution at the cellular level. An assessment of the replicative fitness of the predominant single-cell quasispecies variants indicated a modest reduction in fitness compared to the wild type. Real-time RT-PCR methods capable of determining single-cell viral loads were developed and indicated an average of 113 copies of replicon RNA per cell, correlating with calculated RNA copy numbers in the bulk cell population. This study introduces a single-cell RNA viral-sequencing method with numerous potential applications to explore host-virus interactions during infection. HCV quasispecies diversity varied greatly between cells in vitro, suggesting different within-cell evolutionary pathways. Such divergent trajectories in vivo could have implications for the evolution and establishment of antiviral-resistant variants and host immune escape mutants.
INTRODUCTION
More than 170 million people worldwide are chronically infected with hepatitis C virus (HCV) and at risk of developing liver disease (1). The virus has a long, relatively asymptomatic period of infection, following which a proportion of patients develop sequelae, including chronic liver disease, cirrhosis, and hepatocellular carcinoma (2). Whereas the incidence of newly acquired HCV infection is decreasing (3), the mortality rate associated with HCV secondary liver disease is growing rapidly (4). There is no vaccine against HCV, and the current standard-of-care therapy (pegylated alpha interferon and ribavirin) is effective in only approximately 40 to 50% of cases (5–7). The recent introduction of two protease inhibitors, telaprevir and boceprevir, can improve the rates of sustained viral response (SVR) to drug therapy by a further 20 to 30% (6, 7), but the development of resistance to these and other HCV-specific direct-acting antivirals requires only a single-nucleotide polymorphism within the viral genome (8).
HCV is an enveloped positive-strand RNA virus that replicates via a negative-strand intermediate. The viral genome, which is 9,600 nucleotides in length and includes flanking, short untranslated regions, encodes a polyprotein that is posttranslationally cleaved to form 10 proteins. The error-prone nature of the RNA-dependent RNA polymerase (RdRP) combined with short generation times and a large population size leads to extensive genetic and antigenic heterogeneity within the viral population. The coexistence of this complex group of closely related genomes, known as a quasispecies, allows rapid adaptation in response to environmental perturbations. Disease progression, viral persistence, and treatment response are all influenced by HCV quasispecies heterogeneity (9). Studies have shown that HCV compartmentalizes within the host, with quasispecies compositions differing between different organs (10, 11). The primary target of HCV is the liver, where it forms distinct foci of infection (12), but it also infects extrahepatic sites, including the brain (10, 11).
While diverse sequences are detected within mixed cell populations in an organ, ultimately, maintenance of the quasispecies pool requires persistence at the single-cell level. In an era of increasing molecular precision, single-cell analysis is an emerging technique for investigating cell heterogeneity (13, 14). This technique, however, has been used in only a limited number of studies investigating viral diversity to date. An early study examined the sequence diversity of HIV DNA amplified from single splenocytes containing 3 or 4 proviruses (15). More recently, similar studies have quantified and explored the genetic relationship of HIV-1 provirus DNA in infected CD4+ T cells (16) and unintegrated HIV-1 DNA molecules in the nuclei of spleen cells (17). The first application of single-cell quantitative-PCR (qPCR) methods to viruses was to determine the DNA copy number and cellular distribution of latent herpes simplex virus 1 and varicella-zoster virus in human trigeminal ganglia (18). Since that time, a few research groups have quantified an assortment of viruses in single cells, including foot and mouth disease virus (19, 20), cauliflower mosaic virus (21, 22), and influenza A virus (23). From a cellular perspective, a recent study has investigated alterations in innate immune defense protein expression in individual murine small intestine cells infected by rotavirus (24). To the best of our knowledge, no studies have previously examined RNA viral quasispecies diversity and evolution at the single-cell level.
Here, we establish single-cell analysis of viral RNA sequence diversity by utilizing a model system for HCV replication. Our findings reveal that compartmentalization of HCV in vitro occurs at the cellular level.
MATERIALS AND METHODS
Isolation of individual Huh7 cells.
Huh7 cells from the cell line 2/1 (25) constitutively expressing the subgenomic replicon (SGR) construct of the JFH-1 genotype (gt) 2a strain (26) were used in this study. This replicon contains a neomycin resistance gene, and addition of neomycin to the culture medium ensures that only cells containing replicons can persist within the culture. A single confluent flask (80 cm2) of cells at passage 15 was trypsinized after 6 days of culture, and an aliquot of cells was diluted to yield approximately 1 cell per 100 μl. The diluted sample was pipetted into a 96-well plate (100 μl/well), and wells containing nonclumping single cells were identified by light microscopy. The contents of 2 wells without any cells were used as negative controls; as expected, no amplification products were observed from these samples. The undiluted cells were retained and constituted the total cell (TC) population.
RNA extraction and nested RT-PCR.
RNA was extracted from the TC population (QIAamp Viral RNA Mini Kit; Qiagen) or individual cells (RNeasy Micro Kit; Qiagen). Nested RT-PCR (nRT-PCR) was performed using the SuperScript III One-Step RT-PCR high-fidelity system (Invitrogen) and a KOD hot-start kit (Novagen) sequentially according to the manufacturers' instructions but in a total reaction volume of 20 μl and with an annealing temperature of 55°C. Following completion of the first-round PCR, 1 μl of the reaction mixture was used as a template in the second-round PCR. NS5B JFH-1-specific primers (see Table S1 in the supplemental material) demarcating a 708-bp product were used in this study. This region was selected for analysis because it is the most diverse region within the JFH-1 SGR that does not contain structural genes, including the hypervariable region of the E2 gene. The proofreading enzymes required for high-fidelity amplification generally result in reduced reaction sensitivity; however, their use in this study was essential to discriminate between highly similar sequences. The consensus sequence of the TC population was obtained by direct sequencing of the PCR product.
Determination of quasispecies composition.
NS5B amplicons were ligated into pJET1.2/blunt cloning vectors (CloneJet PCR cloning kit; Fermentas) and cloned into competent cells. Plasmids from randomly selected clones were isolated (QIAprep spin miniprep kit; Qiagen), and the inserts were directly sequenced using the inner antisense primer. To monitor the fidelity of the nRT-PCR procedure, products amplified directly from an aliquot of the JFH-1 transcripts were cloned and sequenced in a similar manner. Potential PCR bias due to low template concentrations was assessed by RT-PCR; cloning of the TC population RNA diluted to 320, 160, 80, 40, and 20 copies/reaction; and determining the quasispecies composition. In total, 525 clones were sequenced: 20 clones derived from each of the 16 single cells (SC), 80 clones from the TC population, 25 clones from the fidelity control, and 20 clones from each of the five dilutions to assess the effect of low template concentrations. The sequences were aligned in SSE version 1.0 (27), and bootstrapped maximum-likelihood trees were generated in MEGA5 (28). Diversity was assessed by the Shannon index and measurements of within-group genetic distances (28). The compositions of the TC and SC replicon populations were examined by median-joining network analysis using Network 4.6.1.0 (29).
Preparation of control transcripts and PCR validation.
Control transcripts were prepared from a plasmid encoding the subgenomic replicon (pSGR) JFH-1 and containing a neomycin resistance gene (pSGR-JFH1-Neo) (26). Stock controls were generated by transforming the plasmids into Escherichia coli competent cells (NEB), overnight growth, and plasmid preparation (PureLink HiPure Plasmid Filter Midiprep Kit; Invitrogen). The isolated plasmid was linearized, blunt ended, purified, and used as a template for in vitro RNA transcription (T7 RiboMAX Express Large Scale RNA Production System; Promega). RNA transcripts were treated with DNase I (Invitrogen) and purified (RNeasy minikit; Qiagen), and the integrity of the resulting RNA was assessed by agarose gel electrophoresis. The RNA concentration was determined using a Nanodrop spectrophotometer and converted to genome copies per μl as described previously (30), and 10-fold dilutions were prepared (31). The sensitivity of the RT-PCR was assessed by performing eight parallel amplifications of suitable transcript dilutions and subsequent probit analysis (31).
Real-time RT-PCR.
Real-time (q)RT-PCR was performed essentially as previously described (32) with pan-HCV primers and a probe targeting a conserved region within the 5′ untranslated region (UTR). The amplification efficiency of the qRT-PCR assay was determined by utilizing 10-fold dilutions of control transcripts (104 to 101 copies/μl RNA) on three separate occasions. Six of the SC viral populations were quantified and compared to the viral loads calculated from dilutions of the TC population.
Variant fitness assays.
Site-directed mutagenesis (Quikchange lightning kit; Agilent) was used to introduce mutations into pSGR-Luc-JFH-1 (26), which was identical to the replicons described above for quasispecies analysis but contained a luciferase gene in place of the neomycin resistance gene. Plasmids were generated and transcribed into RNA as described above for controls, and the resulting replicons were electroporated into Huh7 cells to assess the fitness of three of the major SC variants (V421A, Q436R, and Q441R) compared to the wild type (wt) in transient luciferase assays (26). The luciferase activity in cell lysates was determined after 4, 24, 48, 72, and 96 h using the luciferase assay system (Promega) according to the manufacturer's instructions. qRT-PCR was subsequently performed on RNA extracted from cell lysates sampled at each time point in the luciferase assays as described above. Statistical analysis was performed using analysis of variance (ANOVA) for the 48-h to 96-h period.
RESULTS
Sensitivity, fidelity, and PCR bias of nRT-PCR.
First, the sensitivity of high-fidelity nRT-PCR amplification with primers specific for HCV NS5B sequences from strain JFH-1 was tested using a dilution series of in vitro-synthesized viral transcripts around the assay endpoint (see Table S2 in the supplemental material). The results indicated a 90% detection frequency of 3 copies/reaction (95% confidence interval [CI95], 1 to 13 copies). The system's fidelity was determined by sequencing clones of amplified viral transcripts diluted to 80 copies per reaction; 96% (24/25 clones) were wt. Unlike the clones derived from the cell population in this study, the fidelity control clones were subjected to the potential effect of T7 transcription-induced nucleotide misincorporation events, and therefore, the 96% fidelity rate represents the minimum fidelity of the system. As 20 clones/cell of the SC population were sequenced, there is a low probability that any of these clones contained amplification- or sequencing-induced nucleotide misincorporation errors, whereas the TC population (80 clones sequenced) likely contains errors in <3 clones. Genetic diversity within a quasispecies can be underestimated in populations with a low number of target molecules through the occurrence of resampling during amplification (32). To assess this effect, we sequenced 20 cloned amplicons generated from each of five dilutions of the TC population RNA and assessed the diversity of the populations by within-group p-distances and the Shannon index (Fig. 1). A reduction in diversity by both measures was observed at 20 TC RNA copies per reaction, which was consistent with a “jackpot” effect. This effect was not detected at less dilute RNA concentrations, including copy numbers analogous to those observed in single cells (84 to 160 RNA copies per reaction).
Fig 1.
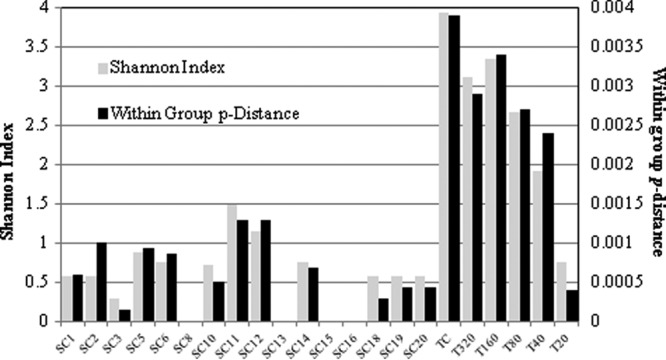
Measures of population diversity for SC and TC populations. The Shannon index is shown on the primary y axis, and the within-group p-distance is shown on the secondary y axis. The diversity of the SC and TC populations is shown, including that of the TC population diluted to 20, 40, 80, 160, and 320 copies per reaction (T20, T40, T80, T160, and T320, respectively).
Quantification of SC and TC populations.
The viral-RNA abundance within individual cells was determined and, in concert with the sensitivity of the nRT-PCR obtained from the assay validation, was used to determine the appropriate number of clones to sequence. The real-time (q)PCR method (33) was first validated using the transcript controls; the mean calculated amplification efficiency of the control dilution series was 101% (range, 91 to 116%), and the correlation coefficient was >0.99 (data not shown). Using this validated assay, the RNA copy number of a proportion of the SCs was determined (Fig. 2), and the results revealed an average viral-RNA copy number per cell of 113 (range, 84 to 160). As the viral load in the individual cells was lower than expected, the TC population was quantified using a range of dilutions (Fig. 2). The average copy number of viral RNA calculated per cell was 111 (range, 79 to 133); therefore, there was no difference in the HCV RNA levels between the TC and SC populations. In correlation with these results, a previous study (34) ascertained that there were approximately 100 copies of JFH-1 replicons per Huh7 cell in cultures maintained for more than 7 days. As the 90% detection frequency of the nRT-PCR was 3 copies/cell and the CI95-calculated minimum copy number for five SC populations was 72 to 118, it was decided to sequence 20 clones/cell. This allowed adequate exploration of diversity within cells containing >60 viral copies. For one of the cells quantified, SC11, large standard errors were obtained due to inconsistent results between replicate assays, and as a result, a CI95 minimum copy number of 25 was obtained. The possibility that this population was subjected to PCR bias cannot be excluded, and this might explain why only this cell population contained codominant sequences (V421A and the wt). Alternatively, prior to the evaluation that this particular aliquot contained a single cell, the viral contents of another cell may have been released due to cell death and unknowingly included in the analysis. Other explanations include active adaptation to an environmental change occurring at the time of sampling or a cellular environment requiring both the wt and the V421A variant for maximum fitness.
Fig 2.
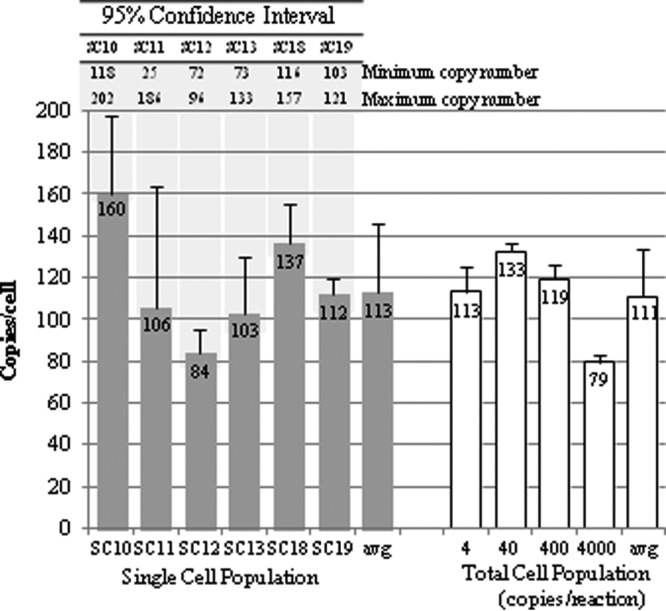
Replicon copy numbers per cell for the SC and TC populations, as determined by qRT-PCR. The number of copies/cell for the TC population was calculated from the dilutions shown on the x axis. The error bars show the standard deviations of three replicates. The copy numbers calculated from 95% confidence intervals are shown above the graph, with shading to indicate the relevant SC populations.
Quasispecies diversity.
We hypothesized that HCV evolves independently within individual cells. To test this, the existence of quasispecies in 16 Huh7 cells harboring the JFH-1 subgenomic replicon was determined, and the diversity was compared to that of the TC population from which they were derived. For the 16 isolated cells, there was considerable intercell diversity in viral sequences (Fig. 3A). Four single cells (SC8, SC13, SC15, and SC16) were composed entirely of wt sequences. The wt predominated in SC1, SC2, and SC18 (90%), SC3 (95%), and SC12 (65%), but these cells also contained other variants as minor components of the viral population. SC2 contained two variants with a double amino acid insertion (PR) between positions 255 and 256, and one of these variants also contained a single amino acid deletion (L) at position 256. The variant V421A was the prevailing sequence for SC5 (80%), SC6 (85%), and SC10 (80%), whereas SC11 consisted of 45% V421A, 35% wt, and 20% with a synonymous mutation at position 376 (syn376). Other dominant variants were Q436R (SC14; 85%), T287A and syn272 (SC20; 90%), and Q441R (SC19; 90%); the last cell was the only one that did not contain any wt clones.
Fig 3.

Median-joining networks analyzed using Network 4.6.1.0 (18) for partial NS5B products of JFH-1 replicon TC (A) and SC (B) populations. The circle size is proportional to the number of clones analyzed. The depiction of the TC population network represents only one potential version of the connectivity within the system; several interpretations of the positioning of the variants containing the mutation H254R can be posited, and similarly for mutants expressing the synonymous substitution at position 272. (C) Positions of mutations within the 236-aa genome fragment of NS5B analyzed in this study occurring in the TC and SC populations. Nonsynonymous mutations are shown in gray, synonymous substitutions in black, and positions with both types of substitution in black and gray.
The predominant sequence of the TC population was the wt (32.5%) (Fig. 3B). This low frequency, however, did not alter the consensus sequence of the TC population (data not shown). Individually, the V421A mutation constituted 10% of the TC population, but the variant appeared to form a subpopulation that, with the additional mutations, comprised 16.2% of the complete TC population. There were two other major variants in the TC population: T287A and syn272 (8.8%) and Q436R (6.2%). All individual mutations were present at a frequency of ≤10% within the population, explaining why the consensus sequence is wt despite the low frequency of the wt within the quasispecies.
Overall, there were 48 sequence types from 400 clones, comprising 21 sequence types from 320 SC population clones (6.5%) and 32 sequence types from 80 TC population clones (40%). The complete list of variants is shown in Table S3 in the supplemental material. The positioning of the mutations along the NS5B fragment in the TC and SC populations followed similar distribution patterns (Fig. 3C), with a higher proportion of mutations observed at the 3′ end of the fragment, in the thumb region of the polymerase. From the 236-amino-acid (aa) region analyzed, there were 61 mutations in 53 positions; in the TC population, this constituted 24 nonsynonymous (NS) mutations and 16 synonymous mutations from 38 positions (Fig. 3C), whereas there were 19 NS and 9 synonymous mutations in the SC population at 26 different positions (Fig. 3C). Synonymous substitutions occurred alone in only 13 clones analyzed (3.2%) but in 51 sequences (12.8%) in conjunction with NS substitutions. The wt was the most frequent type in both the SC populations (58.1%) and the TC population (32.5%). Variants present in greater than 5% of the SC populations were V421A (18.1%), T287A and syn272 (5.9%), Q441R (5.6%), and Q436R (5.3%), and for the TC population, V421A (10%), T287A and syn272 (8.8%), and Q436R (6.2%). Discrepancies between the percentages of sequence types in the SC populations and the TC population would be expected to decrease with increased SC sampling.
The variant T287A was always associated with the synonymous nucleotide substitution (A to G) at position 272 (27/27 sequences), either singly (24 sequences) or with other substitutions (one sequence with Y346C, one with M343T, and one with V421A, F430S, syn448). The syn272 mutation was also associated with other variants (T232S plus syn350, A252T, A252T plus H254R, D332G, and Q436R) and was observed as a single mutation in two clones of the TC population. Synonymous substitutions allow the exploration of genotypic space without amino acid changes, and this aspect of the syn272 substitution was investigated. Allowing one nucleotide change in either the first or second codon position of the syn272 mutation (CAG) and the wt (CAA) results in the same amino acids (P, R, L, K, E, and a stop codon). Using the same criteria with the codons CAC/T at position 272, which were not identified in the SC or TC quasispecies, leads to the amino acids P, R, L, N, D, and Y, changing 50% of the potential amino acid availability compared to the wt.
The diversity of the SC and TC viral populations was assessed by within-group p-distances and the Shannon index (Fig. 1). The viral diversity was substantially less in individual cells than in the TC population by both measures. A maximum of four unique sequences was found in any individual cell, whereas the TC population contained 32 different sequences.
Replicative fitness of variants.
Prior to the production of the variants used in this assay, the genetic background of the variants was assessed by determining the consensus sequence of the entire NS5B region of the TC population. Previously reported dominant NS5B mutations (25) sited outside the genome region analyzed in this study were absent (data not shown). To further assess replication fitness, three dominant SC variants (V421A, Q436R, and Q441R) were compared to the wt replicon by transient-replication assays (Fig. 4). Mutations were inserted into the subgenomic replicon containing a luciferase reporter using site-directed mutagenesis, and replication was assessed by luciferase production and qRT-PCR. Consensus sequencing of each variant at all time points showed that the mutations were maintained throughout the assay and that no other mutations within the NS5B fragment emerged (data not shown). RNA synthesis peaked at 48 h (Fig. 4). The wt subgenomic replicon displayed the greatest fitness, followed by variants Q436R, Q441R, and V421A in descending order of fitness in both the luciferase and qPCR assays; however, no statistical differences were detected for the luciferase assay. Statistical analysis of the data generated from the qPCR assay at the 48-, 72-, and 96-h time points showed that although no difference was detected between the wt and the Q436R variant, the wt was fitter than the V421A variant (P < 0.005) and the Q441R variant (P = 0.079) at the 10% level.
Fig 4.
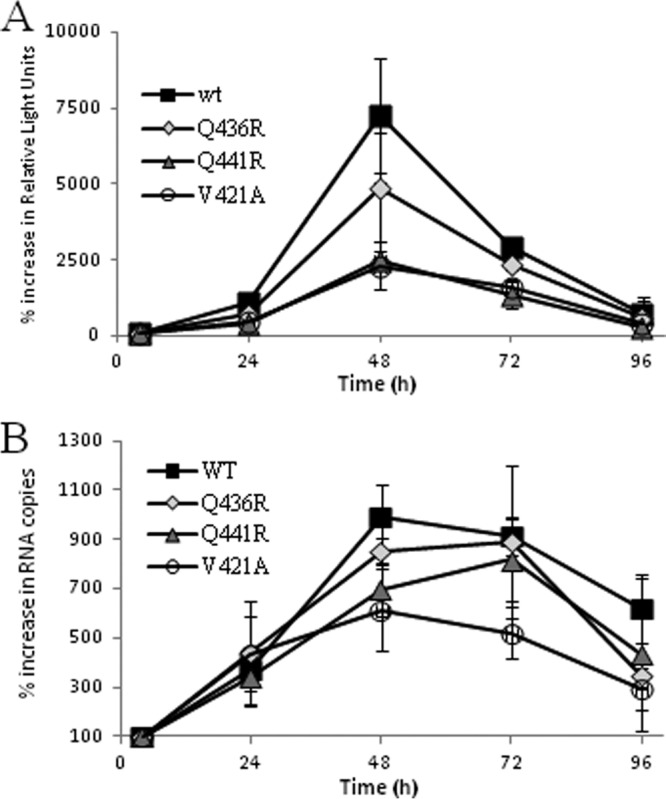
Replication of the wt and variants Q441R, Q436R, and V421A as measured by transient luciferase assay (A) or qRT-PCR (B). The y axes show the percent change in relative light units (A) and the percent increase in RNA copy number (B). Both data sets are normalized to 100 at the 4-h time point values. The actual copy numbers ranged from 2.8 × 105 to 2.7 × 106 copies/μl of RNA (wt), 2.6 × 105 to 2.2 × 106 copies/μl of RNA (Q436R), 1.6 × 105 to 1.3 × 106 copies/μl of RNA (Q441R), and 3.3 × 105 to 2.0 × 106 copies/μl of RNA (V421A). The error bars indicate standard deviations of the means.
DISCUSSION
We report the establishment of a system for high-fidelity amplification of viral RNA genomes and use this method to analyze quasispecies diversity in individual cells. Consistent with our hypothesis, HCV quasispecies compositions differed extensively between cells, indicating cellular compartmentalization of replicon HCV RNA with distinct sequences. It is likely that cells effectively form discrete compartments in which viruses may evolve independently. As Huh7 cells are known to be highly heterogeneous (25), the cellular quasispecies composition probably reflects individual cell environments.
The wt was the most prevalent sequence identified in both the TC and SC populations. In addition, 3 major variants (>5% frequency within the quasispecies) were determined from the SC and TC populations (V421A, Q436R, and T287A plus syn272); the major SC variant of one cell, Q441R, was only a minor variant in the TC population (2.5%). Of all the major variants identified in the current study, only V421A has been previously detected. This variant was noted in HCV gt 2a clinical samples (35), other unpublished gt 2a sequences, and non-gt 2a sequences, including the reference gt 1a strain H77. The HCV gt 2a J6 replicon (36) also contains V421A, but as this variant occurs in the original patient-derived sample HC-J6 (37) from which the J6 replicon was developed, it is unlikely to be a cell culture adaptation. Minor variants found in the current study have been reported in the literature, including Y452H, associated with resistance to a nonnucleoside inhibitor (38).
This study identified insertions in two sequences from the quasispecies of a single cell (SC2). Insertions in the NS5B region of HCV have not previously been reported in the literature, although insertions in the 5′ UTR (39), NS5A (30), and E2 (40) genome regions are known to occur at low frequency, with the last insert being correlated with HCV-associated cryoglobulinemia (41). The possible role played by the NS5B insertions observed in this study has yet to be determined.
The prevalence of a sequence within a quasispecies is known to reflect its relative fitness (9). Accordingly, in the current study, the wt was the most frequently encountered sequence and displayed the greatest replicative fitness among the quasispecies. Despite being the most frequently encountered variant within both the SC and TC populations, V421A displayed the least replicative fitness of the variants analyzed. Epistatic mutations occurring outside the amplified region may have compensated for the apparently lower replication efficiency of this variant. Alternatively, the predominant variants may be specifically adapted to minority cell types within the heterogeneous Huh7 population, and achieving maximum replicative fitness for each of these variants would require clonal selection of the specific cell type. Although perhaps requiring a longer time to allow clonal selection, it would be expected that the variant copy number would eventually match that of the wt. This does not occur, perhaps because the growth rate of Huh7 cells in culture appears to decrease after approximately 48 to 72 h (data not shown). It is possible that there is selective pressure for reduced replication efficiency, as strong replication of JFH-1 is associated with cytostatic effects. It should be noted that, as the viral loads for the SC populations were determined from a continuous-culture system and replicative fitness was assessed using a transient-replication assay, no direct correlations can be made between the SC viral load and the fitness of the dominant variant within each SC population. It might be preferable to assess variant fitness in an assay designed to support persistent replication, such as a colony formation assay; however, as there is very little difference between the replicative capacities of the wild type and variants in the transient system, it is unlikely that such insignificant differences in replicative capacity would be observable in a colony formation assay.
Synonymous substitutions mainly occurred in parallel with NS substitutions, suggesting a compensatory role, although this was not tested directly. This may be the case with the synonymous transitional substitution at position 272 (an A-to-G nucleotide change), which was particularly, although not exclusively, associated with the T287A mutation. Synonymous substitutions are associated with robustness within a quasispecies, allowing exploration of sequence space without phenotypical changes and increasing the potential for rapid adaptation (42). The silent substitution at position 272 did not alter the single-step mutational availability of amino acids compared to the wt. The transitional substitution represented by this mutation is more commonly observed in genomes than the greater structural changes imposed by transversions, perhaps explaining why the codon CAY is not observed in this position but not why the syn272 substitution occurs preferentially to the wt. An additional role of synonymous substitutions is to increase RNA secondary-structure stability, and perhaps the syn272 substitution plays a compensatory role in RNA folding affected by other mutations. As syn272, with its A-to-G nucleotide substitution, was associated predominantly with NS mutations that were also A-to-G substitutions (data not shown), it is unlikely to provide folding compensation within the sequenced region but may be associated with long-range interactions.
HCV replication within infected cells occurs in membranous webs (43) and replication complexes (RCs) (25), specialized structures created through the reorganization of cellular membranes induced by the NS4B protein (44). RCs contain a negative-strand RNA template of the genome that, through the action of the HCV replicase proteins, produces the positive-strand progeny (45). Like other members of the family Flaviviridae, HCV is thought to utilize a stamping mechanism of replication where all progeny are generated from the negative-strand template (46). Speculatively, the dominant sequence within each cell population could represent the faithful copy of the template, whereas the minor variants may be the consequence of random mutagenesis caused by the error-prone RdRP. The existence of these low-level variants is probably transient. In this proposed model, without evolutionary pressure for further adaptation, the predominant sequence in each cell would remain constant but the stochastic nature of the production of the low-level variants would ensure they differed over time. An error-induced variant would become fixed within the population only if it conferred a replicative advantage (Fig. 5). Fixation would first require production of the negative-strand intermediate variant and translation of the replicase proteins containing the mutation from the variant genome, followed by coassembly within an RC. If the variant conferred a replicative advantage, more genome copies of the variant than of the wt would be produced, leading to ever-increasing numbers of variant replicase-template assemblies and eventual dominance of the variant within the cell population. Bottlenecks may also play a role in determining variant fixation through unequal partitioning of RCs during cell division, resulting in daughter cells receiving only a proportion of the RNA.
Fig 5.

Schematic representation of the consequences of the stamping mechanism of replication applied to a randomly generated variant within a population. (A) In the stamping model of replication, the negative-strand RNA template generates all progeny. The error-prone RdRP may introduce a random mutation into the genome, observed as a minor variant within the population. (B and C) The minority variant is translated (B), and a replication complex containing coassembled replicase proteins and an RNA template is formed (C). (D) Replicase proteins derived from an unfit variant result in negligible genome production and will ultimately constitute a minor component of the quasispecies. (E) For a variant that is fit within the cell environment, genome production will be more prolific and further RCs containing this variant will assemble, allowing the variant to become a major component of the cellular population. RdRP errors will produce further mutations or a reversion to wt; the fate of these genomes will depend on their fitness within the cell. RC, replication complex; R, replicase proteins; triangles and circle, genome mutations; dashed lines, negative-strand RNA; solid lines, positive-strand RNA.
Huh7 cells are highly heterogeneous (25), including expression levels of antiviral proteins (47). The results suggest that the between-cell quasispecies variability is likely a consequence of adaptation to these individual cell environments. Replicons lack the structural genes required for productive virus construction, and therefore, this model is a closed system at the cellular level as regards viral spread, suggesting separate intracellular evolutionary pathways. There is some evidence that a similarly closed system operates within the HCV-infected liver. Spatial HCV distribution within the liver is known to be focal (12), and local viral transfer is thought to occur through division of infected cells or cell-to-cell spread (48). Additionally, superinfection exclusion has been shown to prevent viral infection of already-infected cells (49), probably through strain competition at a postentry step (50). It is possible that HCV infection of the liver is a closed system at the focal level, similar to the cellular closed systems in cell culture, and thus, the latter system may provide a good model for examining HCV evolution in the liver. Investigation of cellular or focal evolution occurring in vivo similar to the current study could improve our understanding of the mechanisms involved in viral spread, including variants circumventing host immune defenses and antiviral treatment, and it would be informative to investigate quasispecies partitioning within and between foci in vivo. Foci may consist of infected cells enclosed by a barrier of cells expressing antiviral factors, constraining local cell-to-cell proliferation of virus. Infection of naive cells would thus require extracellular dissemination of productive virus.
We are currently investigating HCV quasispecies cellular compartmentalization in the liver to determine if evolution occurs at the cellular level in a similar manner. The single-cell sequencing system developed here is appropriate for empirical studies for the evolution and spread of antiviral-resistant and host immune escape variants and testing theoretical models of viral evolution. Additionally, studies combining single-cell analysis of viral and host RNAs of infected cells have the potential to become a powerful tool for investigating virus-host interactions.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by grant 63785 from the Medical Research Council.
Footnotes
Published ahead of print 18 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01602-13.
REFERENCES
- 1.Hanafiah KM, Groeger J, Flaxman AD, Wiersma ST. 2013. Global epidemiology of hepatitis C infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57:1333–1342 [DOI] [PubMed] [Google Scholar]
- 2.Casey LC, Lee WM. 2013. Hepatitis C virus therapy update. Curr. Opin. Gastroenterol. 29:243–249 [DOI] [PubMed] [Google Scholar]
- 3.Centres for Disease Control and Prevention 2009. Viral hepatitis surveillance United States, 2009 http://www.cdc.gov/hepatitis/Statistics/2009Surveillance/PDFs/2009HepSurveillanceRpt.pdf
- 4.Ly KN, Xing J, Klevens M, Jiles RB, Ward JW, Holmberg SD. 2012. The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann. Intern. Med. 156:271–279 [DOI] [PubMed] [Google Scholar]
- 5.Ferenci P, Brunner H, Nachbaur K, Datz C, Gschwantler M, Hofer H, Stauber R, Hackl F, Jessner W, Rosenbeiger M, Munda-Steindl P, Hegenbarth K, Gangl A, Vogel W. 2001. Combination of interferon induction therapy and ribavirin in chronic hepatitis C. Hepatology 34:1006–1011 [DOI] [PubMed] [Google Scholar]
- 6.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416 [DOI] [PubMed] [Google Scholar]
- 7.Poordad F, McCone J, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki J-P. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1195–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trozzi C, Bartholomew L, Ceccacci A, Biasiol G, Pacini L, Altamura S, Narjes F, Muraglia E, Paonessa G, Koch U, De Francesco R, Steinkuhler C, Migliaccio G. 2003. In vitro selection and characterization of hepatitis C virus serine protease variants resistant to an active-site peptide inhibitor. J. Virol. 77:3669–3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Domingo E, Sheldon J, Perales C. 2012. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 76:159–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cabot B, Esteban JI, Martell M, Genesca J, Vargas V, Esteban R, Guardia J, Gomez J. 1997. Structure of replicating hepatitis C virus (HCV) quasispecies in the liver may not be reflected by analysis of circulating HCV virions. J. Virol. 71:1732–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sobesky R, Feray C, Rimlinger F, Derian N, Dos Santos A, Roque-Afonso A-M, Samuel D, Brechot C, Thiers V. 2007. Distinct hepatitis C virus core and F protein quasispecies in tumoral and nontumoral hepatocytes isolated via microdissection. Hepatology 46:1704–1712 [DOI] [PubMed] [Google Scholar]
- 12.Stiffler JD, Nguyen M, Sohn JA, Liu C, Kaplan D, Seeger C. 2009. Focal distribution of hepatitis C virus RNA in infected livers. PLoS One 4:e6661. 10.1371/journal.pone.0006661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang FC, Lao K, Surani MA. 2009. Development and applications of single-cell transcriptome analysis. Nat. Methods 6:377–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M. 2011. Tumour evolution inferred by single-cell sequencing. Nature 472:90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung A, Maier R, Vartanian J-P, Bocharov G, Jung V, Fischer U, Meese E, Wain-Hobson S, Meyerhans A. 2002. Multiply infected spleen cells in HIV patients. Nature 418:144. [DOI] [PubMed] [Google Scholar]
- 16.Josefsson L, King MS, Makitalo B, Brännström J, Shao W, Maldarelli F, Kearney MF, Hu W-S, Chen J, Gaines H, Mellors JW, Alberb J, Coffin JM, Palmer SE. 2011. Majority of CD4+ T cells from peripheral blood of HIV-1-infected individuals contain only one HIV DNA molecule. Proc. Natl. Acad. Sci. U. S. A. 108:11199–11204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suspène R, Meyerhans A. 2012. Quantification of unintegrated HIV-1 DNA at the single cell level in vivo. PLoS One 7:e36246. 10.1371/journal.pone.0036246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K, Lau TY, Morales M, Mont EK, Straus SE. 2005. Laser-capture microdissection: refining estimates of the quantity and distribution of latent herpes simplex virus 1 and varicella-zoster virus DNA in human trigeminal ganglia at the single-cell level. J. Virol. 79:14079–14087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang X, Li Y, Zheng C-Y. 2009. A novel single-cell quantitative real-time RT-PCR method for quantifying foot-and-mouth disease viral RNA. J. Virol. Methods 155:150–156 [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Huang X, Xia B, Zheng C. 2009. Development and validation of a duplex quantitative real-time RT-PCR assay for simultaneous detection and quantitation of foot-and-mouth disease viral positive-stranded RNAs and negative-stranded RNAs. J. Virol. Methods 161:161–167 [DOI] [PubMed] [Google Scholar]
- 21.Yvon M, Monsion B, Martin J-F, Gutiérrez S, Blanc S. 2009. PCR-based amplification and analysis of specific viral sequences from individual plant cells. J. Virol. Methods 159:303–307 [DOI] [PubMed] [Google Scholar]
- 22.Gutiérrez S, Yvon M, Thébaud G, Monsion B, Michalakis Y, Blanc S. 2010. Dynamics of the multiplicity of cellular infection in a plant virus. PLoS Pathog. 6:e1001113. 10.1371/journal.ppat.1001113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson RV, McGill J, Legge KL. 2010. Quantification of the frequency and multiplicity of infection of respiratory- and lymph node-resistant dendritic cells during influenza virus infection. PLoS One 5:e12902. 10.1371/journal.pone.0012902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sen A, Rothenberg ME, Mukherjeee G, Feng N, Kaliskyf T, Nair N, Johnston IM, Clark MF, Greenberg HB. 2012. Innate immune response to homologous rotavirus infection in the small intestinal villous epithelium at single-cell resolution. Proc. Natl. Acad. Sci. U. S. A. 109:20667–20672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato T, Date T, Miyamoto M, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817 [DOI] [PubMed] [Google Scholar]
- 26.Targett-Adams P, McLauchlan J. 2005. Development and characterization of a transient-replication assay for the genotype 2a hepatitis C virus subgenomic replicon. J. Gen. Virol. 86:3075–3080 [DOI] [PubMed] [Google Scholar]
- 27.Simmonds P. 2012. SSE: a nucleotide and amino acid sequence analysis platform. BMC Res. Notes 5:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bandelt H-J, Forster P, Rohl A. 1999. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16:37–48 [DOI] [PubMed] [Google Scholar]
- 30.McLeish N, Witteveldt J, Clasper L, McIntyre C, McWilliam Leitch EC, Hardie A, Bennett S, Gunson R, Carman WF, Feeney SA, Coyle PV, Vipond B, Muir P, Benschop K, Wolthers K, Waris M, Osterback R, Johannessen I, Templeton K, Harvala H, Simmonds P. 2012. Development and assay of RNA transcripts of Enterovirus species A to D, Rhinovirus species A to C, and human Parechovirus: assessment of assay sensitivity and specificity of real-time screening and typing methods. J. Clin. Microbiol. 50:2910–2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bennett S, Harval H, Witteveldt J, McWilliam Leitch EC, McLeish N, Templeton K, Gunson R, Carman WF, Simmonds P. 2011. Rapid simultaneous detection of Enterovirus and Parechovirus RNAs in clinical samples by one-step real-time reverse transcription-PCR assay. J. Clin. Microbiol. 49:2620–2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu SL, Rodrigo AG, Shankarappa R, Learn GH, Hsu L, Davidov O, Zhao LP, Mullins JI. 1996. HIV quasispecies and resampling. Science 273:415–416 [DOI] [PubMed] [Google Scholar]
- 33.Jones DM, Domingues P, Targett-Adams P, McLauchlan J. 2010. Comparison of U2OS and Huh-7 cells for identifying host factors that affect hepatitis C virus RNA replication. J. Gen. Virol. 91:2238–2248 [DOI] [PubMed] [Google Scholar]
- 34.Miyamoto M, Kato T, Date T, Mizokami M, Wakita T. 2006. Comparison between subgenomic replicons of hepatitis C virus genotypes 2a (JFH-1) and 1b (Con1 NK5.1). Pathophysiology 49:37–43 [DOI] [PubMed] [Google Scholar]
- 35.Okamoto H, Okada S, Sugiyama Y, Kurai K, Iizuka H, Machida A, Miyakawa Y, Mayumi M. 1991. Nucleotide sequence of the genomic RNA of hepatitis C virus isolated from a human carrier: comparison with reported isolates for conserved and divergent regions. J. Gen. Virol. 72:2697–2704 [DOI] [PubMed] [Google Scholar]
- 36.Li YP, Ramirez S, Gottwein JM, Scheel TKH, Mikkelsen L, Purcell RH, Bukh J. 2012. Robust full-length hepatitis C virus genotype 2a and 2b infectious cultures using mutations identified by a systematic approach applicable to patient strains. Proc. Natl. Acad. Sci. U. S. A. 109:E1101–E1110. 10.1073/pnas.1203829109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yanagi M, Purcell RH, Emerson SU, Bukh J. 1997. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc. Natl. Acad. Sci. U. S. A. 94:8738–8743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shih IH, Vliegen I, Peng B, Yang H, Hebner C, Paeshuyse J, Pürstinger G, Fenaux M, Tian Y, Mabery E, Qi X, Bahador G, Paulson M, Lehman LS, Bondy S, Tse W, Reiser H, Lee WA, Schmitz U, Neyts J, Zhong W. 2011. Mechanistic characterization of GS-9190 (Tegobuvir), a novel nonnucleoside inhibitor of hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 55:4196–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Yamada O, Ito T, Akiyama M, Hashimoto Y, Yoshida H, Makino R, Masago A, Uemura H, Araki H. 1999. A single nucleotide insertion in the 5′-untranslated region of hepatitis C virus leads to enhanced cap-independent translation. Virology 261:263–270 [DOI] [PubMed] [Google Scholar]
- 40.Torres-Puente M, Cuevas JM, Jiménez-Hernández N, Bracho MA, García-Robles I, Carnicer F, del Olmo J, Ortega E, Moya A, González-Candelas F. 2007. Contribution of insertions and deletions to the variability of hepatitis C virus populations. J. Gen. Virol. 88:2198–2203 [DOI] [PubMed] [Google Scholar]
- 41.Gerotto M, Dal Pero F, Loffreda S, Bianchi FB, Alberti A, Lenzi M. 2001. A 385 insertion in the hypervariable region 1 of hepatitis C virus E2 envelope protein is found in some patients with mixed cryoglobulinemia type 2. Blood 98:2657–2663 [DOI] [PubMed] [Google Scholar]
- 42.Novella IS, Presloid JB, Beech C, Wilke CO. 2013. Congruent evolution of fitness and genetic robustness in vesicular stomatitis virus. J. Virol. 87:4923–4928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinkert D, Bartenschlager R, Lohmann V. 2005. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 79:13594–13605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kopek BG, Perkins G, Miller DJ, Ellisman MH, Ahlquist P. 2007. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 5:e220. 10.1371/journal.pbio.0050220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lindenbach BD, Tellinghuisen TL. 2009. Hepatitis C virus genome replication, p 61–88 In Cameron CE, Gotte M, Raney KD. (ed), Viral genome replication. Springer Science and Business Media, LLC, New York, NY [Google Scholar]
- 46.Sardanyes J, Santiago EF. 2011. Quasispecies spatial models for RNA viruses with different replication modes and infection strategies. PLoS One 6:e24884. 10.1371/journal.pone.0024884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Q, Denard B, Huang H, Ye J. 2013. Epigenetic silencing of antiviral genes renders clones of Huh-7 cells permissive for hepatitis C virus replication. J. Virol. 87:659–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Webster B, Wissing S, Herker E, Ott M, Greene WC. 2013. Rapid intracellular competition between hepatitis C viral genomes as a result of mitosis. J. Virol. 87:581–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laskus T, Wang LF, Radkowski M, Nowicki M, Wilkinson J, Rakela J. 2001. Exposure of hepatitis C virus-negative recipients to >= 2 infected blood donors. J. Infect. Dis. 183:666–669 [DOI] [PubMed] [Google Scholar]
- 50.Schaller T, Appel N, Koutsoudakis G, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2007. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J. Virol. 81:4591–4603 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.