

Abstract
Nonnative viral glycoproteins, including Friend murine leukemia virus envelope (F-MLV Env) are actively recruited to HIV-1 assembly sites by an unknown mechanism. Because interactions with the lipid microenvironment at budding sites could contribute to recruitment, we examined the contribution of the hydrophobicity of the F-MLV Env membrane-spanning domain (MSD) to its incorporation into HIV-1 particles. A series of F-MLV Env mutants that added or deleted one, two, or three leucines in the MSD were constructed. All six mutants retained the ability to be incorporated into HIV-1 particles, but the −1L, −2L, −3L, +1L, and +2L mutants were not capable of producing infectious particles. Surprisingly, the +3L Env glycoprotein was able to produce infectious particles and was constitutively fusogenic. However, when the cytoplasmic tail domains (CTDs) in the Env constructs were deleted, all six of the MSD mutants were able to produce infectious particles. Further mutational analyses revealed that the first 10 amino acids of the CTD is a critical regulator of infectivity. A similar phenotype was observed in HIV-1 Env upon addition of leucines in the MSD, with +1 and +2 leucine mutations greatly reducing Env activity, but +3 leucine mutations behaving similar to the wild type. Unlike F-MLV Env (+1L and +2L), HIV-1 Env (+1L and +2L) infectivity was not restored by deletion of the CTD. We hypothesize that the CTD forms a coiled-coil that disrupts the protein's functionality if it is not in phase with the trimer interface of the ectodomain.
INTRODUCTION
Enveloped viruses assemble by the confluence of structural proteins, genetic material, and surface glycoproteins. Glycoprotein-deficient viruses have the potential to be complemented by diverse, nonnative viral glycoproteins in a process termed pseudotyping (reviewed in references 1 to 4). The molecular mechanisms underlying the recruitment of foreign glycoproteins to budding viral particles are not understood. However, such pseudotyped viruses can be used as a gene delivery tool to direct viruses to a specific cell type (5–10).
Retroviral Env glycoproteins are produced as precursors that trimerize in the endoplasmic reticulum (ER). Subsequently, the precursor protein is cleaved by furin or a furin-like protease into its constituent subdomains: the surface subunit (SU) containing the receptor binding domain and the transmembrane subunit (TM) containing the fusion-promoting domain. The mature Env is thus a trimer of heterodimers. SU and TM are held together by an intersubunit disulfide bond in the case of alpha-, gamma-, and deltaretroviruses (11–16) and noncovalently associated in the case of lentiviruses and betaretroviruses (17, 18). Upon receptor binding by SU, several conformational changes take place in Env, promoting coreceptor binding and/or accession of the host cell membrane by the fusion peptide in TM (19). For gammaretroviruses, fusogenicity is controlled by the cytoplasmic tail domain (CTD) of Env and isomerization of the disulfide bond between SU and TM (20–22). The C termini of these glycoproteins contain a short peptide (R-peptide) that is cleaved by the viral protease during or shortly after viral assembly. The presence of the R-peptide at the C terminus of Env prevents its fusogenic activity (20, 21, 23–27). Cleavage of the R-peptide allows for receptor-dependent fusion of the Env protein to the target cell (25, 28). Recent cryo-electron microscopy (cryo-EM) imaging data suggest that the CTD of murine leukemia virus (MLV) Env holds the MLV Env ectodomain in a tight conformation, but upon cleavage of the R-peptide, the TM legs are splayed, and this allows fusogenic activation of Env (29).
Glycoprotein acquisition by retroviral particles is not well understood, and the physical factors that contribute to this recruitment have not been defined, but multiple viral glycoproteins have been reported to efficiently pseudotype retroviral particles (reviewed in references 1, 2, and 4). Not every glycoprotein-virus pair results in infectious particles, but the incorporation of such a variety of surface proteins would indicate the use of a common mechanism by viruses to obtain their surface glycoprotein. Some reports indicate that a direct interaction between the N-terminal matrix (MA) domain of Gag and the CTD of Env may promote packaging of Env into viral particles (30–33). In the case of HIV-1, several studies have shown the MA domain of HIV-1 Gag to be required for HIV-1 Env incorporation into budding viral particles. Further, compensatory mutations in the MA domain of HIV-1 Gag overcome mutations in HIV-1 Env that cause an incorporation defect (32). However, infectious HIV-1 viral particles can still be produced when the entire CTD of HIV-1 Env is deleted, and in some cases such CTD truncations are naturally selected for when lentiviruses are passaged in tissue culture (34–39). Furthermore, we have demonstrated previously that F-MLV Env with its CTD deleted is efficiently recruited to HIV-1 assembly sites (40). Thus, direct interactions between MA and the glycoprotein CTD are not the sole factors dictating glycoprotein incorporation into viral particles. To identify the elements in Env that contribute to recruitment to viral assembly, we chose to examine the contribution of the hydrophobicity of the MSD in F-MLV Env to glycoprotein incorporation. We hypothesized that the hydrophobicity of the MSD of Env would contribute to the translocation of Env to viral particles. We chose to focus on a stretch of 10 continuous hydrophobic amino acids in the F-MLV Env MSD (see Fig. 1). Contrary to our hypothesis, we found no connection between MSD hydrophobicity and glycoprotein incorporation. Independent of whether glycoproteins were incorporated into viral assembly particles, we found that Env functionality depended on coordination between the ectodomain and CTD of Env.
Fig 1.

Schematic of F-MLV Env. Sequences in the MSD and CTD are shown. The YFP or HA tag is inserted at the indicated position in the WT F-MLV Env. The leucine-rich region that has been modified is shown boxed.
MATERIALS AND METHODS
Plasmids.
The Friend murine leukemia virus envelope (F-MLV Env) and yellow fluorescent protein (YFP)-tagged F-MLV Env expression constructs were a kind gift from Walter Mothes (Yale University). The B-clade consensus, codon-optimized pcDNA 3.1 HIV-1 gp160 expression construct was a kind gift from Beatrice Hahn (University of Pennsylvania) (41). The leucine additions/deletions/mutations and alanine scanning mutations were created by oligonucleotide-mediated mutagenesis. Constructs expressing the truncated form of the F-MLV Env were created by introducing stop codons at the appropriate locations. The hemagglutinin (HA)-tagged F-MLV Env was created by replacement of the DNA fragment encoding YFP with that encoding the HA tag (YPYDVPDYA). HIV-1 Env CTD truncation mutants were created by introducing a stop codon after RVRQGY, a truncation removing 144 amino acids in the HIV-1 Env wild-type (WT) Env.
NL4-3-derived HIV-CMV-GFP was kindly provided by Vineet KewalRamani (National Cancer Institute, Frederick, MD). This proviral vector lacks the genes encoding Vif, Vpr, Vpu, Nef, and Env and has a cytomegalovirus (CMV) immediate early promoter-driven green fluorescent protein (GFP) in the place of Nef.
The lentiviral vector with a CMV-driven, reverse-intron-interrupted Gaussia luciferase (Gluc) was a gift from David Derse. The reporter gene with the intron was amplified and subcloned into the HIV-CMV-GFP vector by PCR in place of the CMV-driven GFP gene between the NotI and XhoI restriction sites. Only infected cells, not transfected cells, can produce an intact luciferase protein, leading to signal specifically from an infection (42, 43).
The gene encoding the tTA (tet-off) protein was cloned into a pQCXIP vector (Clontech) between the NotI and BamHI sites, downstream of the CMV immediate early promoter. Tet-off is a transactivator for the tetracycline response element (TRE) promoter that activates transcription in the absence of tetracycline analogs. The retroviral transfer vector, retro-tight-X-hygro from Clontech, was used to create a TRE-driven Gluc-inducible expression construct (TRE-Gluc). Specifically, the Gluc gene was introduced between the BamHI and NotI sites downstream of the TRE.
Cell culture.
The 293FT cell line was obtained from Invitrogen. The cell line expressing the ecotropic F-MLV Env receptor, 293T mCAT-1, was kindly provided by Walther Mothes. The 293T TVA cell line expressing the receptor for Rous sarcoma virus (RSV) Env was provided by John Young (Scripps Research Institute) (44). TZM-bl cells were obtained from the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. All 293T- and 293FT-based cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM glutamine, 1 mM sodium pyruvate, 10 mM nonessential amino acids, and 0.5 mg/ml G418. In phases of cell culture involving transfection or transduction/infection, G418 was not added to the medium. TZM-bl cells were maintained in DMEM supplemented with 10% fetal bovine serum, 2 mM glutamine and 1% MEM vitamins.
The following procedure was used to create 293T mCAT-1 cells with a stably integrated, tet-off inducible Gluc reporter. 293FT cells were transfected with constructs expressing MLV GagPol, TRE-Gluc (also codes for hygromycin resistance gene) and vesicular stomatitis virus G protein (VSV-G). The supernatant from this transfection was used to transduce 293T mCAT-1 cells. The cells were selected for hygromycin expression. Further, clonal isolates were obtained by following a procedure of cloning by limiting dilution. Six clonal populations were tested, and the best was selected based on level of induction of the reporter and the presence of the murine cationic amino acid transporter 1 (mCAT-1) receptor.
Infectivity assay.
293FT cells were transfected with 500 ng of HIV-CMV-GFP and 500 ng of the wild-type or mutant F-MLV envelope proteins (Envs) using 4 μg of polyethylenimine (PEI) per microgram of DNA (45) in a six-well plate. For transfections with HIV-1 Env and its mutants, 950 ng of HIV-CMV-GFP and 50 ng of the WT or mutant HIV-1 Envs were transfected into 293FT cells in a six-well plate. The medium was changed 6 to 12 h later to remove the residual transfection reagent. Supernatant was collected 24 h after the medium was exchanged and then frozen at −80°C for at least 2 h to lyse any cells in the supernatant. The supernatant was thawed in a 37°C water bath and spun at 1,500 × g for 10 min to pellet any cells or cell debris. Five hundred microliters of the supernatant was added to target cells along with Polybrene to a final concentration of 20 μg/ml. Cells were collected 48 h later, fixed with 4% paraformaldehyde, and analyzed by the Accuri C6 flow cytometer system.
Western blots.
Transfections for Western blots were performed as described above for infectivity assays. HA-tagged F-MLV Env derivatives were used for analysis by Western blotting. Viral samples were pelleted through a 20% sucrose cushion for 2 h at 20,000 relative centrifugal force (RCF) in a refrigerated desktop centrifuge at 4°C. The residual serum was removed by washing the pellet with phosphate-buffered saline (PBS). The pellets were resuspended in 2× SDS-PAGE loading buffer, and the equivalent of 1 ml of viral supernatant was analyzed by 10% discontinuous SDS-PAGE. Cell samples were prepared in 1× SDS-PAGE loading buffer, and 5 to 10% of the total amount of cells was analyzed in parallel with the viral supernatants. Proteins were transferred onto a 0.45-μm polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with 1% nonfat dried milk in PBS containing Tween 20 (PBS-T) and probed with mouse anti-HA antibody diluted 1:1,000 (Sigma) and mouse anti-HIV p24 hybridoma medium diluted 1:500 (obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH; HIV-1 p24 hybridoma [183-H12-5C]) from Bruce Chesebro (46). The blots were then probed with horseradish peroxidase (HRP)-conjugated anti-mouse antibody (Sigma) diluted 1:10,000. Luminata Classico Western HRP substrate from Millipore was used for visualization of the membranes on a chemiluminescence image analyzer, LAS3000 from Fujifilm. The blots were also probed with infrared (IR) dye 700DX anti-mouse IgG and visualized using Odyssey infrared imaging system from LI-COR Biosciences.
Viral capture assay.
293FT cells in a six-well plate were transfected with 900 ng of HIV provirus with the intron-interrupted Gluc reporter, 25 ng of Rous sarcoma virus (RSV) Env ΔCTD and 75 ng of YFP-tagged F-MLV Env or its mutants as described previously. The wells of 96-well enzyme-linked immunosorbent assay (ELISA) plates (Corning) were coated overnight at 4°C with polyclonal anti-GFP antibody from rabbit diluted 1:5,000 in ELISA coating buffer. The antibody was removed and replaced with ELISA blocking buffer for 1 h at room temperature. Viral samples (50 μl) were incubated in the wells after removal of the blocking buffer for 4 h. The wells were then washed with PBS and 10,000 cells of 293T mCAT-1 or 293T TVA cell lines were added to the wells. The experiments were done with or without antibody with both cell lines. Parallel direct transductions of the above-mentioned cell lines were also performed.
Cell-to-cell fusion assay.
293FT cells in a six-well plate were transfected with 500 ng of tet-off expression plasmid and 500 ng of an Env expression construct. The medium was changed 6 to 12 h posttransfection to remove any residual transfection reagent. The transfected cells (100,000 cells) were cocultured with an equal number of 293T mCAT-1 TRE Gluc cells for 48 h. Twenty-microliter portions of the supernatant on the cocultured cells were assayed in duplicate for Gluc with 50 μl of 10 μM coelenterazine in 0.1 M Tris (pH 7.4) and 0.3 M sodium ascorbate. Each experiment was performed along with WT F-MLV Env or filler DNA as negative controls and F-MLV Env lacking the R-peptide (F-MLV ΔR) as a positive control. All data were normalized to F-MLV ΔR Env fusogenicity.
RESULTS
Assembly of Gag at the inner leaflet of the plasma membrane has been proposed to modify the local lipid composition of the bilayer, and this may promote further recruitment of native or nonnative viral glycoproteins (47, 48). This hypothesis would indicate that the MSD of Env plays a critical role in the recruitment process. The MSD of F-MLV Env contains a string of 10 uninterrupted highly hydrophobic amino acids (Fig. 1). While MSDs are always composed of hydrophobic amino acids, this continuous stretch of exclusively hydrophobic amino acids represents an extreme of hydrophobic domains.
Addition or removal of leucines in the MSD of F-MLV Env affects the production of infectious viral particles.
We hypothesized that the hydrophobic region in the F-MLV Env would contribute to its recruitment to viral particles. To test this hypothesis, the MSD was mutated to contain 1, 2, or 3 fewer or additional leucines (termed −1L, −2L, −3L, +1L, +2L, or +3L, respectively; WT Env is termed 0L) within the hydrophobic block of amino acids, highlighted in Fig. 1. Leucine, being a hydrophobic amino acid, would be expected to change the net hydrophobicity of the MSD in addition to changing the MSD length. Initially, infectivity of an Env-defective HIV-1 provirus (HIV-CMV-GFP) cotransfected with these Envs was tested. We expected many of these Env clones, particularly those with large changes, to be nonfunctional. Surprisingly, functionality among these mutants did not appear to be correlated with hydrophobicity of the MSD. Only WT Env (0L) and +3L Env were able to generate infectious particles with HIV-1 cores; all others were essentially nonfunctional in infectivity assays (Fig. 2A). In addition to this approach, we also altered the hydrophobicity of the F-MLV Env by replacement of three leucines in the hydrophobic core of the MSD with three isoleucines (same hydrophobicity) or three alanines (decreased hydrophobicity). In such cases, where the length of the MSD was not affected and hydrophobicity was altered, the infectivity was equivalent to WT F-MLV Env and again suggested that the hydrophobicity of the Env MSD did not dictate Env functionality (data not shown).
Fig 2.
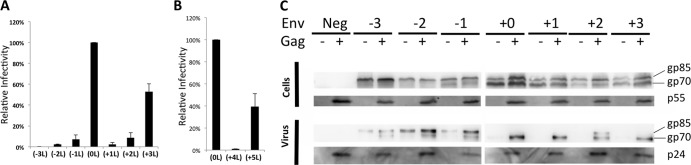
Infectivity of mutant F-MLV Envs. (A) Infectivity relative to WT F-MLV Env glycoproteins of the various mutants. Data shown here are averages of at least three experiments. Error bars indicate the standard deviations (SD) in the experiments. (B) Relative infectivity of +4L and +5L F-MLV Env mutants is shown as an average of three experiments, and the error bars indicate SD in the experiments. (C) Western blot of cell lysate and viral supernatant of cells expressing mutant Envs. A representative image shows antibody probing against HA (Env) and CA (immature p55 and mature p24). gp85 represents the Env precursor, and gp70 represents the SU from Env. Transfection of a Gag-encoding construct (+) or its absence (−) has been indicated.
MSDs of transmembrane proteins usually assume a helical conformation, and secondary structure prediction algorithms predict the F-MLV Env MSD to be helical (49, 50). Since the addition of three leucines would introduce an additional helical turn, this could explain why this was the only mutant with significant activity. To explore this further, two additional mutants with four (+4L) or five (+5L) leucines added to the MSD were assayed for infectivity (Fig. 2B). While the +4L mutant was noninfectious, the +5L mutation partially restored infectivity to the Env glycoprotein, suggesting that the phasing of the MSD, rather than the net hydrophobicity, contributes to Env functionality in this data set. For this reason, the remainder of the study focused on mutants −3L to +3L.
Leucine additions or deletions do not affect viral incorporation of Env.
To determine whether the changes in the transmembrane domain affect the incorporation of Env into viral particles, Western blotting was carried out on cell lysate and viral supernatant from cells expressing each mutant Env along with HIV-CMV-GFP. The HA epitope tag (YPYDVPDYA) was introduced into the mutant Envs in the variable region of the proline-rich region, a region in F-MLV Env where insertions of peptides are tolerated without any effect on Env function (51–53). F-MLV Env is produced as a gp85 precursor and processed by a cellular protease into gp70 (SU) and p15E (TM) (54). In the case of −1L, −2L, −3L, and +2L mutants, there was a noticeable reduction in the amount of processed Env. However, all seven of the Env proteins were incorporated into viral particles. Unexpectedly, transfected cells expressing −1L, −2L, and −3L mutants released some processed and unprocessed Env in a pelletable form even in the absence of the structural proteins. However, the addition of HIV-1 GagPol to the transfection increased the amount of both processed and unprocessed Env released into the medium (Fig. 2C). We suspect that the glycoproteins released in the absence of GagPol are in pelletable exosomal vesicles, but other explanations are possible.
To rule out any ambiguities in the incorporation of Env into viral particles, we performed an alternative infectivity-based assay to quantify Env incorporation into viral particles. For this assay, outlined in Fig. 3A, particles are produced in the presence of one of the mutant F-MLV Env proteins as well as RSV Env ΔCTD, which we demonstrated previously to be randomly incorporated into HIV-1 particles (55). These particles are bound to the wells of a tissue culture plate with an antibody against F-MLV Env, and unbound virus is washed away. Bound virus is then overlaid with 293T TVA cells, which are susceptible to fusion with RSV Env, but not F-MLV Env. Thus, infectivity of the 293T TVA cells reflects incorporation of F-MLV Env into particles.
Fig 3.
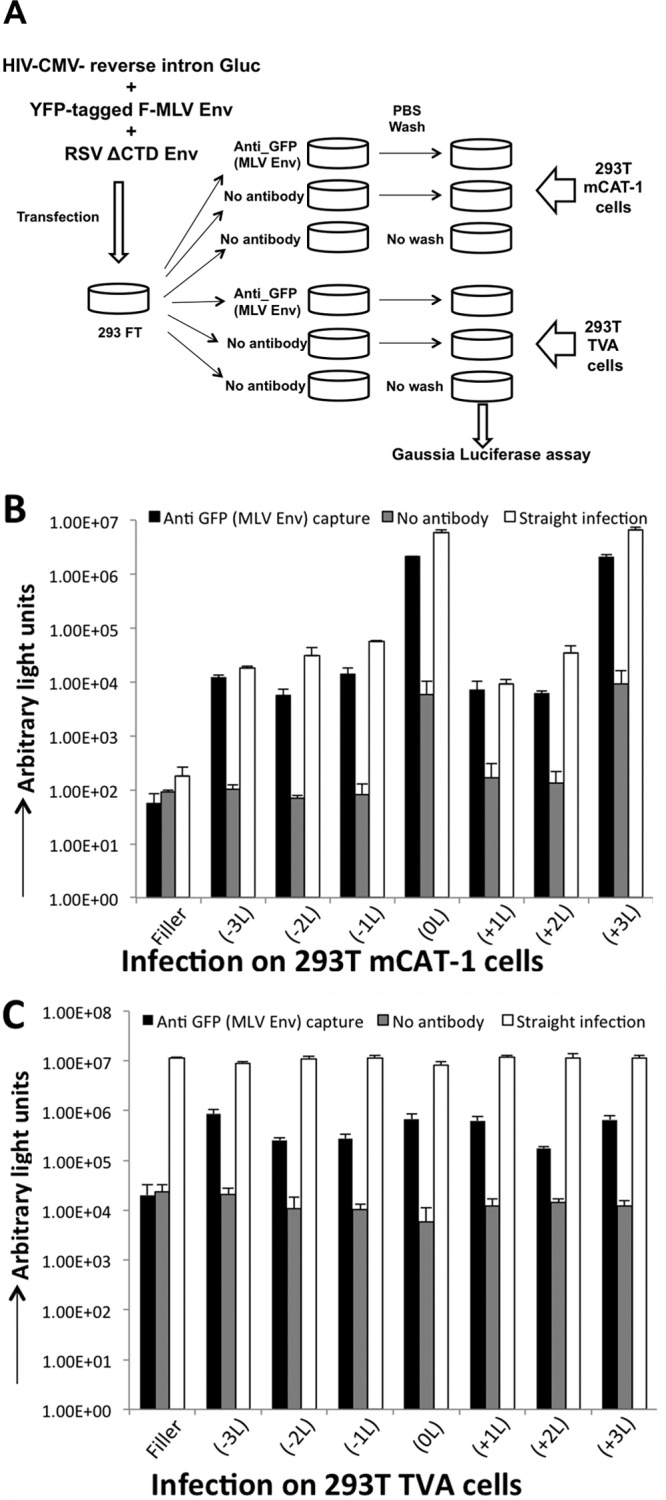
Viral capture of mutant Envs. (A) Schematic of the virus capture assay. Env-defective provirus used in the experiment carries a gene that encodes the Gluc reporter with an intron within the reporter ORF in the reverse orientation, and this ensures reporter signal from a bona fide infection, but not from the transfection. (B and C) Infectivity of viral particles pseudotyped with the indicated mutant F-MLV Envs and RSV Env ΔCTD on cells expressing the mCAT-1 receptor (B) or on cells expressing the TVA receptor (C). Target cells are added to viral particles captured by anti-GFP (F-MLV Env) antibody, no antibody, or no wash/straight infection. Data shown are averages of 3 experiments. Error bars indicate the SD in the experiments.
An F-MLV Env construct with YFP engineered into the proline-rich region was used for this assay because we have found that the YFP tag in F-MLV Env works well for viral capture with a polyclonal anti-GFP antibody. Each of the mutants was engineered with YFP and assayed to see whether their phenotype was altered by the presence of YFP. For this assay, an Env-defective HIV-1 provirus containing Gluc with a reverse intron (HIV-CMV-iGluc) was used (42, 43). As expected, infectivity on cells with the receptor for F-MLV Env (293T mCAT-1) was highest with F-MLV Env 0L and the +3L mutant (Fig. 3B, straight infection). With this assay, it was apparent that some infectivity occurred with all of the Env mutants, but the infectivities of −1L, −2L, −3L, +1L, and +2L mutants were all reduced about 100-fold compared to those of the 0L and the +3L mutant. This infectivity with the YFP-tagged mutants followed the same pattern as the untagged Envs (compare Fig. 2A and Fig. 3B, white bars). Next we tested whether the viral particles could be captured with the GFP antibody. Virus was bound to the plate and washed prior to infection as outlined in Fig. 3A. Gluc signal equivalent to that from the straight infection was observed with each of the mutants in 293T mCAT-1 cells, and the signal in each case was greatly diminished in controls with no antibody. Note that the infectivity in the control wells with no antibody containing the 0L and +3L Envs appeared high compared to the poorly functioning glycoproteins, but this signal was still 100-fold lower than the equivalent wells that did contain antibody. Next we tested infectivity on cells susceptible to infection with the RSV Env to determine whether any of the mutant glycoproteins were incorporated and capable of facilitating viral capture. Direct transduction of 293T TVA cells with the viral supernatant showed the presence of infectious viral particles in the supernatant from all transfections. Infectivity by virus capture against the YFP tag followed by infection on 293T TVA cells was comparable in all samples containing the YFP-tagged F-MLV Env or a mutant thereof. This experiment confirms that F-MLV Env leucine mutants are all incorporated into similar levels into viral particles.
Addition/deletion of leucines in MSD of F-MLV glycoprotein affects fusogenicity of Env.
Because the leucine insertions and deletions did not affect incorporation into viral particles, we questioned why they were not functional. To understand the infectivity phenotype with these mutants, we tested the fusogenicity of each mutant Env. Because gammaretroviral Env proteins are fusogenically inactive prior to cleavage of the R-peptide by the retroviral protease, each of the Env mutants was engineered to have the R-peptide removed, and the fusogenicity of each mutant (with or without the R-peptide) was assayed. Briefly, each mutant Env was transfected along with a tet-off expression construct into 293FT cells. Twenty-four hours posttransfection, 100,000 of the transfected cells were cocultured with 100,000 293T mCAT-1 cells expressing TRE-driven Gluc (see Materials and Methods). If Env is fusogenic, the transfected cells and the receptor-expressing cells fuse, resulting in a tet-off-dependent induction of Gluc. The extent of induction of the reporter relative to the ΔR (0L) Env indicates the fusogenicity of the Env. The mutant full-length (FL) Envs, regardless of mutations in the MSD, were found to be nonfusogenic by this assay, with the exception of +3L Env. The +3L Env is constitutively fusogenically active despite the presence of the R-peptide (Fig. 4A). Upon deletion of the R-peptide, several of the Env mutants displayed significant fusogenic activity. Because several of the ΔR mutants were at least partially fusion capable, we tested this panel of mutants for the ability to form infectious particles. With the exception of the wild type and +3L mutants, the leucine mutants remained noninfectious despite having the R-peptide deleted (Fig. 4B). Finally, we tested whether there are additional sequences upstream of the R-peptide that act as negative regulators of glycoprotein function by engineering the leucine mutants with the majority of their cytoplasmic tail deleted (Δ25 mutants). Unlike mutants with a FL or partial (ΔR) CTD, most of the mutants with most of their cytoplasmic tail deleted (Δ25) were able to produce infectious particles (compare Fig. 4B and C and Fig. 2A). Thus, the infectivity of these mutants is restricted by their CTDs. The mutant −2L Δ25 Env did not produce a high level of infectious particles compared to the other five mutants. This may relate to the level of the mature Env with this mutant as seen in the case of −2L FL Env (Fig. 2C). In agreement with previous studies, truncation of the CTD in the 0L Env causes some loss in the infectious particle output (Fig. 4C) (21, 56). Having thus implicated the CTD in this observed phenotype, we wanted to determine the sequences in the membrane-proximal region of the CTD responsible for modulating infectivity in the MSD mutants.
Fig 4.
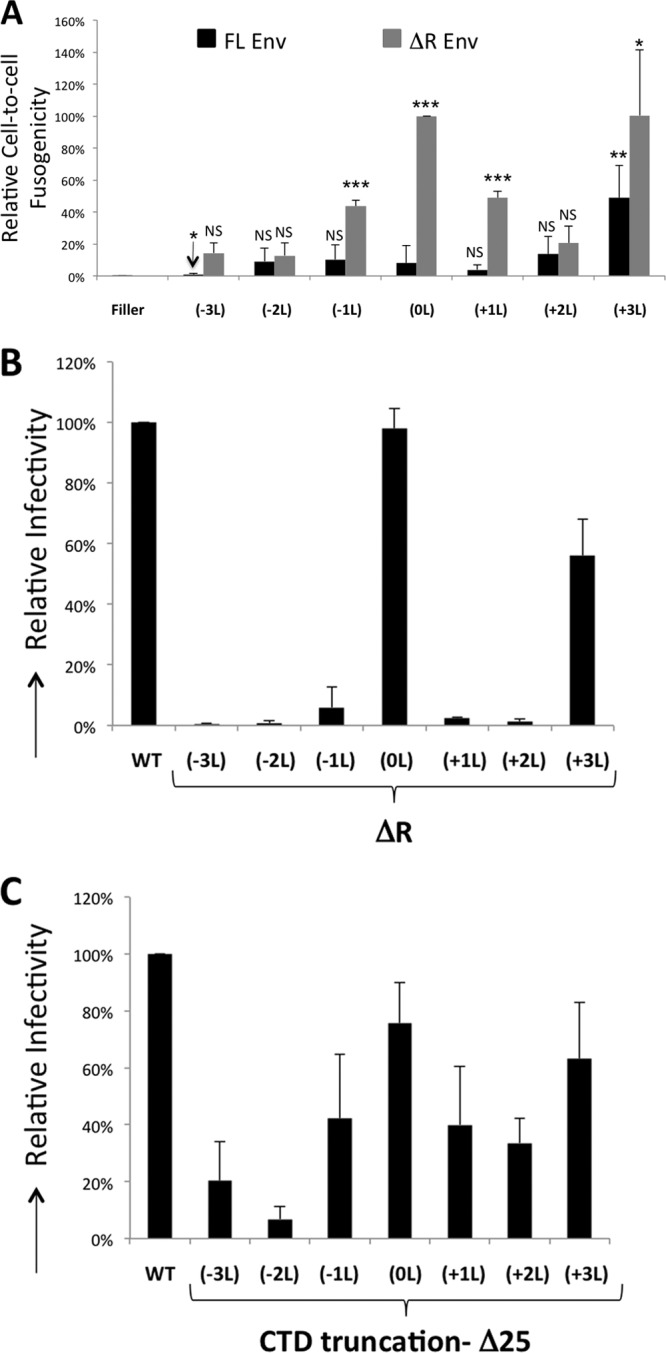
Fusogenicity of Env. (A) Cell-to-cell fusogenicity of the indicated F-MLV FL and ΔR Env mutants in the absence of GagPol relative to F-MLV Env ΔR. The fusogenicity of each mutant was compared with that of FL Env (0L) (NS, not significant [P > 0.1]; ∗, P < 0.1; ∗∗, P < 0.05; ∗∗∗, P < 0.01). The fusogenicity of FL Env (−3L) was statistically less than that of FL +0. (B) Infectivity of FL Env and ΔR Envs with the indicated MSD mutations, relative to WT Env infectivity, is shown. (C) Infectivity of WT Env and Δ25 Envs with the indicated mutations in the MSD relative to WT Env infectivity. The averages of three independent experiments are shown. Error bars indicate the SD within the experiments.
Sequences in the CTD contributing to infectivity with MSD mutations in Env.
While removal of most of the CTD (Δ25) restored infectivity to most of the MSD mutants, removal of the R-peptide did not. This suggests that the sequence just proximal to the MSD can modulate functionality. MLV Env is trimeric in the ectodomain and also is modeled to be trimeric in the CTD prior to R-peptide cleavage (11, 29). A recent study by Löving et al. (29) demonstrated that the membrane-proximal ectodomain of MLV Env is tightly packed prior to R-peptide cleavage. These pieces of data led us to hypothesize that there are protein contacts in both the glycoprotein ectodomain and CTD that contribute to Env trimerization and that glycoprotein function is inhibited if these two trimer interfaces are not synchronous with each other.
To identify the minimum portion of the CTD that negatively regulated fusion of the MSD mutants, we created truncations in the 0L and +1L Env. These Envs are processed efficiently in the cell; they are incorporated into viral particles and are not fusogenic with FL CTD. However, whereas the +0L Env can produce infectious particles regardless of its CTD, the +1 Env mutant can produce infectious particles only if its CTD is deleted. Exploiting the singular difference in the infectivity with these two Envs, we explored the sequences required for modulation of infectivity. Initially, truncations in the CTD removing 21, 22, 23, or 24 amino acids were made in the 0L and +1L Envs. The infectivity of HIV-1 particles with these Envs showed that a minimum of 23 C-terminal amino acids in the CTD of the +1L Env have to be removed to restore infectious particle production (Fig. 5A). Immunoblotting analysis of the FL, Δ21, and Δ23 versions of the 0L and +1L Env showed that the Envs are produced and processed to similar extents in cells and are also incorporated normally into viral particles (Fig. 5A).
Fig 5.

Characterization of +1 F-MLV Env. (A) Truncations of CTD and their sequences in the C terminus are shown. Amino acids that are unchanged compared to those shown in the FL Env are indicated by dashes, and an asterisk indicates a stop codon in the ORF. Infectivity and expression of WT and +1L Env are shown. (B) Scheme of double alanine mutants and their infectivity is shown. A boldface letter at a particular position represents mutation of the amino acid in that position. (C) Scheme of single alanine mutations in the context of WT Δ21 Env and +1L Δ21 Env. Infectivity is normalized to that of Δ21 WT Env. Data shown are the averages of at least 3 experiments. Error bars indicate the SD in the experiments.
To test whether the amino acid sequences N terminal to position 21 were involved in the modified infectivity, amino acids between positions 21 and 29 were mutated to alanine as sequential double mutants or single point mutants in the context of the Env Δ21 mutants. Mutation of F28, either as a part of a double substitution or as a point substitution, reduces infectivity drastically to those of 0L Env Δ21 and +1L Env Δ21. Mutations of amino acids R24 and D25 restored infectious particle production to +1L Env Δ21, while other mutations in this section of eight amino acids did not change its phenotype (Fig. 5B). Mutation of R24 or D25 alone has a less significant effect (Fig. 5C). In the case of all other mutations, 0L Env is not affected in its capacity to produce infectious pseudotyped HIV-1 particles (Fig. 5B and C). To test whether the RD24, 25AA mutation was sufficient to restore Env function to the +1 Env in the context of the FL CTD, the mutation was recreated in the FL 0L and +1L Envs and tested for infectivity (Fig. 5B). Mutation of these two residues was not sufficient to restore infectivity in the context of the FL CTD. In fact, mutation of these residues also drastically reduced infectivity of the +0 FL Env. Thus, these two residues are important contributors to Env regulation but are not the only residues required to negatively regulate Env function.
Insertions into the HIV-1 Env MSD modulate infectivity independent of the CTD.
With F-MLV Env, fusion regulation is achieved by sequences in the CTD that are cleaved by the viral protease. While HIV-1 Env is activated for fusion upon HIV-1 protease activation and viral maturation, there is no proteolytic cleavage of the Env itself (57, 58). To understand whether this infectivity phenotype was restricted to Envs processed by protease for fusion activation, we examined HIV-1 Env. Leucine addition mutations in the MSD of HIV-1 Env were created, and the infectivity was assayed in each case. Similar to F-MLV Env, addition of one or two leucines in the MSD reduced infectivity relative to the infectivity of the WT and +3L HIV-1 Env (Fig. 6). Further, the CTD of each of the leucine addition mutants in HIV-1 Env was deleted to understand the role of the CTD in this phenotype for a nonproteolytic-fusion-controlled Env. The HIV-1 Env CTD truncation mutants behaved similar to their FL counterparts, indicating that although MSD hydrophobicity may affect the function of Env, the CTD is not required for this modulation (Fig. 6).
Fig 6.
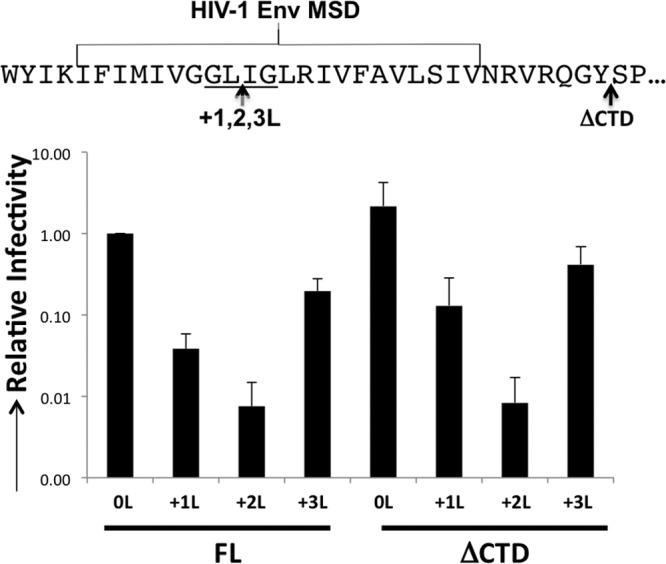
Leucine insertions into the HIV-1 MSD. (Top) HIV-1 MSD illustrating the locations of insertion and truncation. The conserved GLIG motif is underlined. (Bottom) Infectivity of HIV-1 Env MSD mutations with or without the CTD. Infectivity is normalized to that of WT HIV-1 Env. Data shown are the averages of at least 3 experiments. Error bars indicate the SD in the experiments.
DISCUSSION
We have shown previously the active recruitment of foreign glycoproteins to HIV-1 budding sites (55). Further studies of protein domains in F-MLV Env required for recruitment to HIV-1 assembly sites showed that the CTD was not required for specific recruitment (40). Analysis of the domains in Gag required for selective recruitment of F-MLV Env to HIV-1 assembly sites found that the MA domain is dispensable (59). Gag is a membrane binding protein that may modify the lipid environment at the viral assembly site (47, 48), so it may recruit Env to viral budding sites through interactions with the MSD. This study evaluated the role of the hydrophobicity of Env MSD in Env recruitment to HIV-1 viral budding sites.
MSD hydrophobicity does not affect Env incorporation.
In contrast to our expectations, the hydrophobicity of the MSD did not appear to play a role in incorporation into viral particles (Fig. 2A and 6). The results of virion biochemical analysis and the viral capture assay show that Env is incorporated into viral particles regardless of the mutations that alter its hydrophobicity (Fig. 2C and 3B). Further, the mutant Envs are incorporated to comparable levels (Fig. 3C). These data showed that the hydrophobicity of the Env glycoprotein is not the major determining factor in its incorporation into viral particles, although the hydrophobicity could affect other stages of Env biogenesis. With the leucine addition/deletion mutants, since Env is present in viral particles, the most likely explanation for its variation in infectivity is loss of fusogenicity. Indeed, a test of cell-to-cell fusogenicity of the different mutant Envs showed that the variation in infectivity mirrored the fusion function of the different Envs (Fig. 4A).
The helical orientation of Env MSD controls Env function.
Evaluation of gp70 and gp85 in cells showed that removal of leucines in the MSD reduced Env processing (Fig. 2C) and was deleterious for Env function (Fig. 4A). The addition of one or two leucines abolished fusogenicity of Env, while the addition of three leucines made the Env constitutively fusogenic (Fig. 4A). These data would be consistent with the helical orientation of the Env. The addition of a single amino acid would twist the orientation of the protein with an alpha helix by approximately 103°. The addition of three leucines would effectively introduce one turn of the alpha helix and hence would bring the protein back to the original orientation. If the CTD forms a trimer that is out of phase with the trimer interface of the ectodomain, it could disrupt the ectodomain interface and cause the protein to be nonfunctional. However, if the CTD is deleted, only the trimer interface in the ectodomain would be present, and therefore, there would not be conflicting trimer interfaces. The observation that the +3L Env is not only functional but is also constitutively fusogenically active (analogous to mutants with a truncated R-peptide) suggests that the CTD trimer is in the correct orientation relative to the ectodomain but perhaps is a weaker trimer that is not able to hold Env in the tight (fusogenically inactive) conformation.
With HIV Env, insertion of single alanines at various positions in the MSD has shown that the spacing between the conserved GXXXG motif and a conserved arginine two amino acids downstream in the MSD is important for Env processing and intracellular localization (60). The site that we introduced leucines in HIV Env was in the middle of this GXXXG motif. The same study introduced a single alanine insertion in the same location as our leucine insertions and did not find that the mutant was defective in fusogenicity, but viral infectivity was not tested. The CTD of F-MLV Env is predicted to be a helix continuous with the MSD, while HIV Env is not predicted to form a continuous helix in its MSD CTD. This may explain the differences in CTD dependence of the infectivity phenotype with the two Envs (Fig. 2 and 6).
Within the membrane-proximal region of the CTD of the gammaretroviral Envs of murine origin, the primary amino acid sequence FVKDRIS is biochemically well conserved (61). With the F-MLV +1 Env, truncation of 23 C-terminal amino acids or truncation of 21 amino acids and mutation of the 24th and 25th amino acids, allows for infectious particle output (Fig. 5). However, mutation of these same two amino acids when the full CTD is present abolishes functionality. Together these data suggest that this membrane-proximal region is a critical modulator of the viral glycoprotein function, presumably through coordination of a cytoplasmic trimer interface.
ACKNOWLEDGMENTS
We thank Walther Mothes, Beatrice Hahn, David Derse, Vineet KewalRamani, John Young, and the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH for reagents.
This work was supported by U.S. Public Health Service grant AI73098.
Footnotes
Published ahead of print 18 September 2013
REFERENCES
- 1.Cronin J, Zhang XY, Reiser J. 2005. Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene Ther. 5:387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson MC. 2011. Mechanisms for Env glycoprotein acquisition by retroviruses. AIDS Res. Hum. Retroviruses 27:239–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mazari PM, Roth MJ. 2013. Library screening and receptor-directed targeting of gammaretroviral vectors. Future Microbiol. 8:107–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palù G, Parolin C, Takeuchi Y, Pizzato M. 2000. Progress with retroviral gene vectors. Rev. Med. Virol. 10:185–202 [DOI] [PubMed] [Google Scholar]
- 5.Arce F, Breckpot K, Collins M, Escors D. 2011. Targeting lentiviral vectors for cancer immunotherapy. Curr. Cancer Ther. Rev. 7:248–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desmaris N, Bosch A, Salaün C, Petit C, Prévost MC, Tordo N, Perrin P, Schwartz O, de Rocquigny H, Heard JM. 2001. Production and neurotropism of lentivirus vectors pseudotyped with lyssavirus envelope glycoproteins. Mol. Ther. 4:149–156 [DOI] [PubMed] [Google Scholar]
- 7.Hachiya A, Sriwiriyanont P, Patel A, Saito N, Ohuchi A, Kitahara T, Takema Y, Tsuboi R, Boissy RE, Visscher MO, Wilson JM, Kobinger GP. 2007. Gene transfer in human skin with different pseudotyped HIV-based vectors. Gene Ther. 14:648–656 [DOI] [PubMed] [Google Scholar]
- 8.Kato S, Kuramochi M, Takasumi K, Kobayashi K, Inoue K, Takahara D, Hitoshi S, Ikenaka K, Shimada T, Takada M, Kobayashi K. 2011. Neuron-specific gene transfer through retrograde transport of lentiviral vector pseudotyped with a novel type of fusion envelope glycoprotein. Hum. Gene Ther. 22:1511–1523 [DOI] [PubMed] [Google Scholar]
- 9.Palomares K, Vigant F, Van Handel B, Pernet O, Chikere K, Hong P, Sherman SP, Patterson M, An DS, Lowry WE, Mikkola HK, Morizono K, Pyle AD, Lee B. 2013. Nipah virus envelope-pseudotyped lentiviruses efficiently target ephrinB2-positive stem cell populations in vitro and bypass the liver sink when administered in vivo. J. Virol. 87:2094–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin F, Neil S, Kupsch J, Maurice M, Cosset F, Collins M. 1999. Retrovirus targeting by tropism restriction to melanoma cells. J. Virol. 73:6923–6929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fass D, Harrison SC, Kim PS. 1996. Retrovirus envelope domain at 1.7 angstrom resolution. Nat. Struct. Biol. 3:465–469 [DOI] [PubMed] [Google Scholar]
- 12.Johnston ER, Radke K. 2000. The SU and TM envelope protein subunits of bovine leukemia virus are linked by disulfide bonds, both in cells and in virions. J. Virol. 74:2930–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leamnson RN, Halpern MS. 1976. Subunit structure of the glycoprotein complex of avian tumor virus. J. Virol. 18:956–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Opstelten DJ, Wallin M, Garoff H. 1998. Moloney murine leukemia virus envelope protein subunits, gp70 and Pr15E, form a stable disulfide-linked complex. J. Virol. 72:6537–6545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pinter A, Fleissner E. 1977. The presence of disulfide-linked gp70-p15(E) complexes in AKR murine leukemia virus. Virology 83:417–422 [DOI] [PubMed] [Google Scholar]
- 16.Pinter A, Lieman-Hurwitz J, Fleissner E. 1978. The nature of the association between the murine leukemia virus envelope proteins. Virology 91:345–351 [DOI] [PubMed] [Google Scholar]
- 17.Henzy JE, Coffin JM. 2013. Betaretroviral envelope subunits are noncovalently associated and restricted to the mammalian class. J. Virol. 87:1937–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowalski M, Potz J, Basiripour L, Dorfman T, Goh WC, Terwilliger E, Dayton A, Rosen C, Haseltine W, Sodroski J. 1987. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science 237:1351–1355 [DOI] [PubMed] [Google Scholar]
- 19.Hunter E. 1997. Viral entry and receptors, p 71–119 In Coffin JM, Hughes SH, Varmus HE. (ed), Retroviruses. Cold Spring Harbor Lab Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 20.Löving R, Li K, Wallin M, Sjöberg M, Garoff H. 2008. R-peptide cleavage potentiates fusion-controlling isomerization of the intersubunit disulfide in Moloney murine leukemia virus Env. J. Virol. 82:2594–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ragheb JA, Anderson WF. 1994. pH-independent murine leukemia virus ecotropic envelope-mediated cell fusion: implications for the role of the R peptide and p12E TM in viral entry. J. Virol. 68:3220–3231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallin M, Ekstrom M, Garoff H. 2004. Isomerization of the intersubunit disulphide-bond in Env controls retrovirus fusion. EMBO J. 23:54–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kubo Y, Tominaga C, Yoshii H, Kamiyama H, Mitani C, Amanuma H, Yamamoto N. 2007. Characterization of R peptide of murine leukemia virus envelope glycoproteins in syncytium formation and entry. Arch. Virol. 152:2169–2182 [DOI] [PubMed] [Google Scholar]
- 24.Li M, Li ZN, Yao Q, Yang C, Steinhauer DA, Compans RW. 2006. Murine leukemia virus R peptide inhibits influenza virus hemagglutinin-induced membrane fusion. J. Virol. 80:6106–6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rein A, Mirro J, Haynes JG, Ernst SM, Nagashima K. 1994. Function of the cytoplasmic domain of a retroviral transmembrane protein: p15E-p2E cleavage activates the membrane fusion capability of the murine leukemia virus Env protein. J. Virol. 68:1773–1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor GM, Sanders DA. 2003. Structural criteria for regulation of membrane fusion and virion incorporation by the murine leukemia virus TM cytoplasmic domain. Virology 312:295–305 [DOI] [PubMed] [Google Scholar]
- 27.Yang C, Compans RW. 1997. Analysis of the murine leukemia virus R peptide: delineation of the molecular determinants which are important for its fusion inhibition activity. J. Virol. 71:8490–8496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henderson LE, Sowder R, Copeland TD, Smythers G, Oroszlan S. 1984. Quantitative separation of murine leukemia virus proteins by reversed-phase high-pressure liquid chromatography reveals newly described gag and env cleavage products. J. Virol. 52:492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Löving R, Wu SR, Sjöberg M, Lindqvist B, Garoff H. 2012. Maturation cleavage of the murine leukemia virus Env precursor separates the transmembrane subunits to prime it for receptor triggering. Proc. Natl. Acad. Sci. U. S. A. 109:7735–7740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gebhardt A, Bosch JV, Ziemiecki A, Friis RR. 1984. Rous sarcoma virus p19 and gp35 can be chemically crosslinked to high molecular weight complexes. An insight into virus assembly. J. Mol. Biol. 174:297–317 [DOI] [PubMed] [Google Scholar]
- 31.Manrique JM, Affranchino JL, González SA. 2008. In vitro binding of simian immunodeficiency virus matrix protein to the cytoplasmic domain of the envelope glycoprotein. Virology 374:273–279 [DOI] [PubMed] [Google Scholar]
- 32.Murakami T, Freed EO. 2000. Genetic evidence for an interaction between human immunodeficiency virus type 1 matrix and alpha-helix 2 of the gp41 cytoplasmic tail. J. Virol. 74:3548–3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cosson P. 1996. Direct interaction between the envelope and matrix proteins of HIV-1. EMBO J. 15:5783–5788 [PMC free article] [PubMed] [Google Scholar]
- 34.Chakrabarti L, Emerman M, Tiollais P, Sonigo P. 1989. The cytoplasmic domain of simian immunodeficiency virus transmembrane protein modulates infectivity. J. Virol. 63:4395–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones DR, Suzuki K, Piller SC. 2002. A 100-amino acid truncation in the cytoplasmic tail of glycoprotein 41 in the reference HIV type 1 strain RF. AIDS Res. Hum. Retroviruses 18:513–517 [DOI] [PubMed] [Google Scholar]
- 36.Kodama T, Burns DP, Kestler HW, III, Daniel MD, Desrosiers RC. 1990. Molecular changes associated with replication of simian immunodeficiency virus in human cells. J. Med. Primatol. 19:431–437 [PubMed] [Google Scholar]
- 37.Kodama T, Wooley DP, Naidu YM, Kestler HW, III, Daniel MD, Li Y, Desrosiers RC. 1989. Significance of premature stop codons in env of simian immunodeficiency virus. J. Virol. 63:4709–4714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimizu H, Hasebe F, Tsuchie H, Morikawa S, Ushijima H, Kitamura T. 1992. Analysis of a human immunodeficiency virus type 1 isolate carrying a truncated transmembrane glycoprotein. Virology 189:534–546 [DOI] [PubMed] [Google Scholar]
- 39.Shimizu H, Morikawa S, Yamaguchi K, Tsuchie H, Hachimori K, Ushijima H, Kitamura T. 1990. Shorter size of transmembrane glycoprotein of an HIV-1 isolate. AIDS 4:575–576 [DOI] [PubMed] [Google Scholar]
- 40.Lucas TM, Lyddon TD, Grosse SA, Johnson MC. 2010. Two distinct mechanisms regulate recruitment of murine leukemia virus envelope protein to retroviral assembly sites. Virology 405:548–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kothe DL, Decker JM, Li Y, Weng Z, Bibollet-Ruche F, Zammit KP, Salazar MG, Chen Y, Salazar-Gonzalez JF, Moldoveanu Z, Mestecky J, Gao F, Haynes BF, Shaw GM, Muldoon M, Korber BT, Hahn BH. 2007. Antigenicity and immunogenicity of HIV-1 consensus subtype B envelope glycoproteins. Virology 360:218–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aloia AL, Duffy L, Pak V, Lee KE, Sanchez-Martinez S, Derse D, Heidecker G, Cornetta K, Rein A. 2013. A reporter system for replication-competent gammaretroviruses: the inGluc-MLV-DERSE assay. Gene Ther. 20:169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazurov D, Ilinskaya A, Heidecker G, Lloyd P, Derse D. 2010. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog. 6:e1000788. 10.1371/journal.ppat.1000788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lewis BC, Chinnasamy N, Morgan RA, Varmus HE. 2001. Development of an avian leukosis-sarcoma virus subgroup A pseudotyped lentiviral vector. J. Virol. 75:9339–9344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. 1995. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. U. S. A. 92:7297–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chesebro B, Wehrly K, Nishio J, Perryman S. 1992. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J. Virol. 66:6547–6554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krementsov DN, Rassam P, Margeat E, Roy NH, Schneider-Schaulies J, Milhiet PE, Thali M. 2010. HIV-1 assembly differentially alters dynamics and partitioning of tetraspanins and raft components. Traffic 11:1401–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ono A, Freed EO. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. U. S. A. 98:13925–13930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buchan DW, Ward SM, Lobley AE, Nugent TC, Bryson K, Jones DT. 2010. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 38(Suppl 2):W563–W568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292:195–202 [DOI] [PubMed] [Google Scholar]
- 51.Erlwein O, Buchholz C, Schnierle BS. 2003. The proline-rich region of the ecotropic Moloney murine leukaemia virus envelope protein tolerates the insertion of the green fluorescent protein and allows the generation of replication-competent virus. J. Gen. Virol. 84:369–373 [DOI] [PubMed] [Google Scholar]
- 52.Rothenberg SM, Olsen MN, Laurent LC, Crowley RA, Brown PO. 2001. Comprehensive mutational analysis of the Moloney murine leukemia virus envelope protein. J. Virol. 75:11851–11862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu BW, Lu J, Gallaher TK, Anderson WF, Cannon PM. 2000. Identification of regions in the Moloney murine leukemia virus SU protein that tolerate the insertion of an integrin-binding peptide. Virology 269:7–17 [DOI] [PubMed] [Google Scholar]
- 54.Ng VL, Wood TG, Arlinghaus RB. 1982. Processing of the env gene products of Moloney murine leukaemia virus. J. Gen. Virol. 59:329–343 [DOI] [PubMed] [Google Scholar]
- 55.Jorgenson RL, Vogt VM, Johnson MC. 2009. Foreign glycoproteins can be actively recruited to virus assembly sites during pseudotyping. J. Virol. 83:4060–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Januszeski MM, Cannon PM, Chen D, Rozenberg Y, Anderson WF. 1997. Functional analysis of the cytoplasmic tail of Moloney murine leukemia virus envelope protein. J. Virol. 71:3613–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wyma DJ, Jing J, Shi J, Zhou J, Lineberger JE, Miller MD, Aiken C. 2004. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J. Virol. 78:3429–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wyss S, Dimitrov AS, Baribaud F, Edwards TG, Blumenthal R, Hoxie JA. 2005. Regulation of human immunodeficiency virus type 1 envelope glycoprotein fusion by a membrane-interactive domain in the gp41 cytoplasmic tail. J. Virol. 79:12231–12241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gregory DA, Lyddon TD, Johnson MC. 2013. Multiple Gag domains contribute to selective recruitment of murine leukemia virus (MLV) Env to MLV virions. J. Virol. 87:1518–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyauchi K, Curran AR, Long Y, Kondo N, Iwamoto A, Engelman DM, Matsuda Z. 2010. The membrane-spanning domain of gp41 plays a critical role in intracellular trafficking of the HIV envelope protein. Retrovirology 7:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Janaka SK, Lucas TM, Johnson MC. 2011. Sequences in gibbon ape leukemia virus envelope that confer sensitivity to HIV-1 accessory protein Vpu. J. Virol. 85:11945–11954 [DOI] [PMC free article] [PubMed] [Google Scholar]