

Abstract
Chronic hepatitis B virus (HBV) infection is a major cause of hepatocellular carcinoma (HCC) worldwide. The pre-S2 mutant large HBV surface protein (Δ2 LHBS), which contains an in-frame deletion of approximately 17 amino acids in LHBS, is highly associated with risks and prognoses of HBV-induced HCC. It was previously reported that Δ2 LHBS interacts with the Jun activation domain-binding protein 1 (JAB1), a zinc metalloprotease. This promotes the degradation of the cell cycle regulator p27Kip1 and is believed to be the major mechanism for Δ2 LHBS-induced HCC. In this study, it was found that the interaction between JAB1 and Δ2 LHBS is facilitated by divalent metal Zn2+ ions. The binding of JAB1 to Δ2 LHBS requires the JAB1/CSN5 MPN metalloenzyme (JAMM) motif and residue H138 that binds to Zn2+ ions in JAB1. Isothermal titration calorimetry showed that Δ2 LHBS binds directly to Zn2+ ions in a two-site binding mode. Residues H71 and H116 in Δ2 LHBS, which also contact Zn2+ ions, are also indispensable for Δ2 LHBS-mediated p27Kip1 degradation in human HuH7 cells. These results suggest that developing drugs that interrupt interactions between Δ2 LHBS and JAB1 can be used to mitigate Δ2 LHBS-associated risks for HCC.
INTRODUCTION
Chronic hepatitis B virus (HBV) infection is the most important cause of hepatocellular carcinoma (HCC) worldwide. HBV causes necroinflammatory liver disease of variable duration and severity. A major portion of the viral hepatitis progresses into liver cirrhosis and dysplasia and ultimately into HCC. HBV surface antigen (HBsAg), the major component comprising the viral envelope, is the main serum and tissue marker for the viral infection status (1). HBsAg causes sustained hepatic inflammation and injury in the chronic phase of HBV infection and is therefore highly associated with HCC incidence.
The HBS gene contains three in-frame gene segments: pre-S1, pre-S2, and major (or small) S. Using different start codons but sharing the same C terminus, the viral surface proteins include large, middle, and major protein products. The major HBsAg composes the majority of the viral envelope, whereas the middle and large HBS (MHBS and LHBS), usually much less expressed, are minor envelope proteins. LHBS and MHBS also facilitate secretion of the major HBS out of the host cell. In the chronic phase of HBV infection, the viral genome often integrates into the host chromosome and the viral replication is downregulated (2, 3). In this phase, LHBS is expressed predominantly among various viral surface proteins (3). There also emerges the pre-S2 mutant LHBS (Δ2 LHBS) that is truncated of approximately 17 amino acids (aa) in the N terminus of the pre-S2 region of the protein and often also contains a point mutation in the start codon of the region, which leads to a dramatic decrease in the synthesis of MHBS (4, 5) and potentially affects DNA polymerase activity due to overlap of the surface and polymerase genes in the viral genome. We previously (5–7) found that Δ2 LHBS contributed to the histological morphology of the type II ground glass hepatocyte (GGH) preneoplastic lesions, which was characterized by the marginal HBS staining pattern and proliferation in clusters as hepatic nodules. We recently (8) also found that the type II GGH harboring Δ2 LHBS was a biomarker for tumor recurrence and worse survival of HCC patients after hepatectomy surgery. Therefore, Δ2 LHBS is highly associated with risks and prognoses of HBV-induced HCC (9, 10).
We previously (6) reported that Δ2 LHBS accumulates in endoplasmic reticulum (ER), which induces strong ER stress as well as the associated signaling pathways. Through ER stress, Δ2 LHBS induces oxidative stress, DNA damage, and mutagenesis, all of which cause genomic instability (11, 12). It also induces the overexpression of cell cycle regulator cyclin A and causes cell cycle progression in the presence of DNA lesions (13). We recently (14) found that Δ2 LHBS directly interacts with c-Jun activation domain-binding protein 1 (JAB1) and subsequently causes hyperphosphorylation of the tumor suppressor retinoblastoma and, consequently, G1- to S-phase cell cycle progression.
JAB1 is a key subunit of the COP9 signalosome (CSN) and acts as a multifunctional protein associated with the signaling pathway, cell cycle regulation, and development. JAB1 is oncogenic because it promotes cell proliferation by increasing transcription of activator protein 1 (AP-1) and stimulates cell cycle progression by increasing the degradation of the cyclin-dependent kinase inhibitor p27Kip1 (15, 16). Therefore, this is believed to be an important mechanism for the Δ2 LHBS-induced carcinogenic process. JAB1 interacts with a number of cellular proteins such as psoriasin, protease-activated receptor 2 (PAR-2) and p27Kip1 (17, 18). The MRP1-PAD1-N-terminal (MPN) domain spanning the middle region of JAB1 is the usual interactive domain. The MPN domain consists of a JAB1/MPN/Mov34 metalloenzyme (JAMM) motif, which presents with a deneddylation/isopeptidase activity (19–21). The JAMM motif displays a conserved sequence of His-X-His-X10-Asp motif accompanied by an upstream conserved glutamate (22). As a metalloprotease, JAB1 relies on metals for its enzymatic activities; however, it was not clear whether metals are also important for JAB1 interaction with proteins or whether they modulate JAB1 protein conformation for complex formation. In the present study we sought to characterize the potential involvement of metals in the binding between JAB1 and Δ2 LHBS, as well as to identify the protein domains involved in the interaction between them.
MATERIALS AND METHODS
DNA manipulation.
The pre-S1 and pre-S2 regions of the Δ2 LHBS were PCR amplified and cloned into the Escherichia coli protein expression vector pET21b. The pHBV1.3 plasmid, which contained the whole HBV genome, was generously supplied by Chungming Chang at National Health Research Institutes in Taiwan (23). The plasmid pHBV1.3/Δ2 LHBS, which contained Δ2 instead of the wild-type HBS gene, was constructed by deleting nucleotides (nt) 4 to 57 of the HBV genome. Internal deletion of aa 61 to 119 of Δ2 LHBS was achieved by overlapping PCRs for the HBV genome DNA regions spanning nt 2854 to 3033 and nt 3211 to 155 and then cloned into pET21b. The Δ2 LHBS H28Y, H71Y, H119Y, and H71Y H119Y point mutants were constructed by using a QuikChange site-directed mutagenesis kit (Stratagene). The PCR primers used are listed in Table 1. The JAB1 gene was PCR amplified and cloned into pGEX-5X-1 glutathione S-transferase (GST) fusion plasmid vector (GE Healthcare). The various deletion mutants of JAB1 were constructed using overlapping PCRs and then cloned into pGEX-5X-1 (Table 1). The JAB1 H138Y point mutant was constructed by using the site-directed mutagenesis kit mentioned above (Table 1).
Table 1.
PCR primers used in this study
| Gene | Primer |
|
|---|---|---|
| Orientationa | Sequence (5′–3′) | |
| H28Y | F | GGGATTCTTTCCCGATTATCAGTTGGACCCTGC |
| R | GCAGGGTCCAACTGATAATCGGGAAAGAATCCC | |
| H71Y | F | CAGGGCTCACCCCTCCATACGGCGCTATTTTGGGG |
| R | CCCCAAAATACCGCCGTATGGAGGGGTGAGCCCTG | |
| H116Y | F | CTCTCCACCTCTAAGAGACAGTTATCCTCAGGCCATACAGTGG |
| R | CCACTGTATGGCCTGAGGATAACTGTCTCTTAGAGGTGGTGGAGAG | |
| JAB1ΔJAMM | F | GCTACAAAAAAGTAATGGTGATCCTTAGTCC |
| R | ATTACTTTTTTGTAGCAGTGGTGATTGATCC | |
| JAB1Δ1-52 | F | GAATTCGCAAGTACTGCAAAATC |
| JAB1Δ251-334 | R | CTCGAGCAACGTATTCACCCAGTA |
| H138Y | F | GCAATCGGGTGGTATTATAGCCACCCTGGCTATG |
| R | CATAGCCAGGGTGGCTATAATACCACCCGATTGC | |
F, forward; R, reverse.
RT-PCR.
Human hepatoma HuH7 cells transfected with pHBV1.3 and pHBV1.3/Δ2 LHBS were used for RNA extraction according to a protocol described previously (11). The reverse transcription-PCR (RT-PCR) protocol was also described elsewhere (14). The PCR primers used for the various HBV genes were as follows: HBs, forward (5′-ATGGGAGGTTGGTCATCAAAACCTC-3′) and reverse (5′-GTTCGTCACAGGGTCCCCAGTCCTC-3′), and HBc, forward (5′-TTCAAGCCTCCAAGCTG-3′) and reverse (5′-CCGTAAAGTTTCCCACCTTAT-3′).
Expression and purification of Δ2 LHBS and its mutant proteins.
E. coli BL21 Rosetta (DE3) strain cells carrying the Δ2 LHBS and its mutant genes were grown in Luria-Bertani medium as previously described (24). To induce gene expression, cells were treated with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 8 h at 25°C or, alternatively, for 6 h at 37°C and then centrifuged at 3,000 × g for 10 min. The cell pellets were resuspended in lysis buffer (20 mM potassium phosphate [pH 8.0], 1 mM phenylmethylsulfonyl fluoride, 500 mM NaCl) and then broken open using ultrasonication. The cell lysates were centrifuged at 24,000 × g for 20 min at 4°C to remove debris, and then the clear supernatant was loaded onto a column containing Ni-nitrilotriacetic acid (Ni-NTA) resins (GE Healthcare) to bind to the Δ2 LHBS and its mutant proteins, C-terminal-tagged with 6×His. After the lysates had been extensively washed with lysis buffer containing imidazole in concentrations of 20 to 80 mM, the 6×His-tagged proteins were eluted using a solution containing 20 mM potassium phosphate (pH 8.0), 500 mM NaCl, and 500 mM imidazole and then analyzed by using SDS-PAGE. The purified proteins were dialyzed against the dialysis buffer (20 mM Tris-HCl [pH 8.5], 100 mM NaCl, 0.5 mM MgCl2, 0.1 mM EDTA, 10% glycerol). To remove the proteins and ions that had nonspecifically bound to the Ni-NTA resins, proteins were passed through a column (DEAE Sepharose FF; GE Healthcare) and quantified using the Bradford method (Bio-Rad) and SDS-PAGE (24).
Expression and purification of the GST fusion proteins.
JAB1 and its mutant genes, fused to GST, were transformed into E. coli BL21 Rosetta (DE3) cells and then grown to mid-log phase. Protein expression was induced using 0.1 mM IPTG for 8 h at 25°C. The GST fusion proteins were purified using a protocol previously described (25, 26). Briefly, cells were pelleted and then resuspended in phosphate-buffered saline (pH 7.4) containing 5% N-lauroylsarcosine and 1% (vol/vol) Triton X-100. The cells were broken by using a sonicator. After the cells had been centrifuged at 24,000 × g at 4°C for 20 min to remove the cell debris, the supernatant was loaded onto a column containing glutathione-agarose beads (GE Healthcare). The beads in the column were extensively washed with phosphate-buffered saline (PBS; pH 7.4) and then eluted with a buffer containing 50 mM Tris-HCl (pH 8.0) and 20 mM reduced glutathione.
In vitro GST pulldown assays.
JAB1 and its mutants, fused to GST, and the Δ2 LHBS and its mutants, 50 pmol each, were mixed with 30 μl of glutathione-agarose beads and then incubated in assay buffer (150 mM NaCl, 20 mM HEPES [pH 8.0], 0.1% Tween 20, 10% glycerol, 1× protease inhibitor cocktail [Roche], and various concentrations of ZnCl2) for 4 h at 4°C. After the incubation, the beads were extensively washed with assay buffer. The bound proteins were analyzed by using Western blotting with rabbit antibody that recognizes LHBS or mouse antibody that recognizes 6×His.
Coimmunoprecipitation assays.
For in vitro assays using E. coli recombinant proteins, JAB1 and its mutants fused to GST, as well as Δ2 LHBS and its mutants, 50 pmol each, were incubated overnight at 4°C in assay buffer with protein G-beads (GE Healthcare) and rabbit anti-LHBS antibody or mouse anti-JAB1 antibody. The next day, the beads were extensively washed with assay buffer. The proteins bound to beads were analyzed using SDS-PAGE and then by using Western blotting with antibodies that recognize JAB1 (Becton Dickinson Biosciences), LHBS, or 6×His. For coimmunoprecipitation assays using cell lysates of human hepatocytes, we followed a protocol previously described (14). Cell lysates of the hepatoma HuH7 cells stably expressing the wild-type and Δ2 LHBS or its mutants were mixed with the coimmunoprecipitation solution in the presence of 20 μM ZnCl2 and then analyzed by Western blotting. To detect the binding of JAB1 with Δ2 LHBS in the context of whole HBV genome, HuH7 cells were transfected with pHBV1.3 or pHBV1.3/Δ2LHBS then applied to the coimmunoprecipitation assays following the protocol described above. The antibodies used were mouse monoclonal anti-hemagglutinin (Sigma-Aldrich), anti-JAB1 and anti-p27Kip1 (Santa Cruz Biotechnology), anti-LHBS (clone 7H11, generously provided by C. Zhang, Xiamen University, China), and rabbit anti-HBS and anti-GST antibodies (Abcam).
Immunofluorescence assays.
HuH7 cells transfected with the wild-type and Δ2 LHBS or its H71Y/H116Y double mutant genes were grown on glass coverslips. After harvest, cells were fixed with 4% formaldehyde then washed with PBS (pH 7.4). The cells were then incubated with mouse anti-JAB1 (BD Biosciences) and rabbit anti-HA antibodies (Covance) for 1 h at room temperature. The secondary monoclonal anti-mouse antibody conjugated with Alexa 488 and anti-rabbit antibody conjugated with Alexa 594 fluorescent dyes (Molecular Probes) were then incubated with cells for 1 h. After the reactions, the coverslips were mounted with Prolong Gold Anti-Fade with DAPI (Invitrogen) and then observed by Leica LSM510 META confocal microscopy. The relative levels of JAB1 colocalized with LHBS in the same cells were quantified by using the FUJI signal quantification software.
Protein binding analysis using surface plasmon resonance.
Protein binding affinities of Δ2 LHBS and its mutants, and GST-JAB1 and its mutants, were analyzed using surface plasmon resonance (SPR) spectrometry (Biacore 3000; GE Healthcare) according to a previously described protocol (27). The binding buffer, which contained 10 mM HEPES (pH 7.4), 150 mM NaCl, and 0.005% Tween 20, was incubated in 0.4% Chelex 100 (Bio-Rad) overnight at 4°C to chelate divalent ions. To detect zinc ion-dependent binding between Δ2 LHBS and JAB1, ZnCl2 in various concentrations was added to the metal-chelated buffer. GST-JAB1 was immobilized using an amine coupling kit with a CM4 sensor chip (GE Healthcare). After the immobilization, Δ2 LHBS or its mutants in various concentrations was injected into the sensor chip at 20 μl/min for 4 min at 25°C to detect the Δ2 LHBS-JAB1 association. After that, dissociation between the two proteins was monitored for 4 min. The sensor chip surface was regenerated using a pulse injection of 1 M NaCl and 50 mM EDTA (pH 7.4) after each use. Kinetic constants for the association and dissociation of Δ2 LHBS and JAB1 were then calculated (BIAevaluation software; GE Healthcare).
ITC analysis.
After elution from the Ni-NTA column, the Δ2 LHBS and its H71Y/H116Y double mutant were dialyzed overnight against a buffer containing 20 mM Tris-HCl (pH 8.5), 100 mM NaCl, 5 mM EDTA, and 10% glycerol. A second dialysis step was included to reduce the ionic strength of the buffer against 20 mM Tris-HCl (pH 8.5) and 10% glycerol for an additional 4 h. The low-salt-containing proteins were passed through a Resource Q column (GE HealthCare) and then through a Superdex 75 column (GE HealthCare) for further purification. The purified proteins were concentrated and dialyzed overnight against the buffer containing 50 mM HEPES (pH 6.5) and 50 mM NaCl. To ensure saturation for binding studies, ZnCl2 was dissolved in the same buffer, with a molar ratio of 10:1 to 20:1 for ZnCl2 to the dialyzed proteins. Protein concentration was determined by absorbance measurement using a molar extinction coefficient of 27,500 M−1 cm−1 for Δ2 LHBS and the H71Y/H116Y double mutant, respectively. Both ZnCl2 solution and proteins were degassed for 5 min before titrations. Isothermal titration calorimetry (ITC) experiments were done at 20°C in a MicroCal (Northampton, MA) VP-ITC microcalorimeter. A Zn2+ ion-binding study was performed in the reaction chamber with 1.8 ml of 83.6 μM Δ2 LHBS and 1672 μM ZnCl2 in the titration syringe using an injection increment of 6 μl. The Δ2 LHBS H71Y/H116Y double mutant was also titrated in a similar fashion using 1.8 ml of 81.3 μM in the reaction chamber and 813 μM ZnCl2 in a syringe by using an injection volume of 10-μl. The titration intervals were every 3 min with a stirring rate was 307 min−1. The binding data were analyzed (Origin7.0; OriginLab Corp.). Two sets of binding sites were used as a model to obtain the best fitting parameters.
RESULTS
Divalent ion-mediated interaction between JAB1 and Δ2 LHBS.
We tested binding affinities between JAB1 and Δ2 LHBS E. coli recombinant proteins using GST pulldown assays. Their interaction was found greatly increased by Zn2+ and mildly increased by Ni2+ ions (20 μM each) but not by the same concentration of Co2+, Mn2+, Mg2+, or Ca2+ ions (Fig. 1A). In the presence of EDTA, a divalent cation chelator, the Zn2+ ion-mediated interaction was abolished, which indicated that it was a metal ion-mediated reaction (Fig. 1A). The results of the coimmunoprecipitation assays also showed that JAB1 and Δ2 LHBS E. coli recombinant proteins interact in a Zn2+ ion-dependent manner (Fig. 1B).
Fig 1.
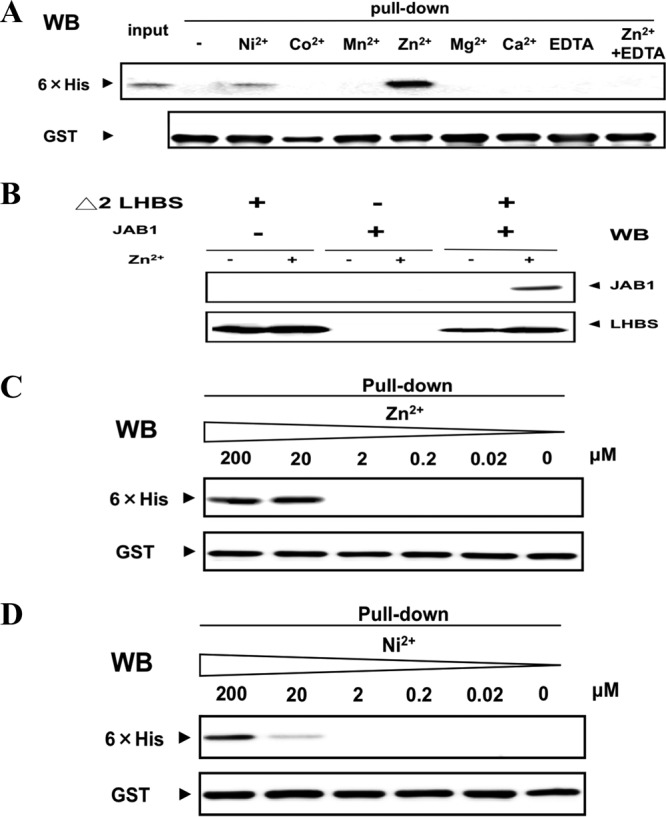
Metal ion-dependent protein binding of Δ2 LHBS protein with JAB1. (A) interactions between the recombinant Δ2 LHBS (50 pM) and JAB1 fused to GST (50 pM) in the presence of various metal ions (20 μM), detected using in vitro GST pulldown assays. The pull-down fractions were probed, using Western blotting (WB), for 6×His tags fused to Δ2 LHBS GST and 6×His with rabbit anti-GST and mouse anti-6×His antibodies, respectively. EDTA (5 mM) was added to the binding solution to chelate the divalent metal ions. (B) Coimmunoprecipitation assays were performed to detect the Zn2+ ion-dependent protein binding of Δ2 LHBS and JAB1 E. coli recombinant proteins. A rabbit anti-LHBS antibody was used to immunoprecipitate Δ2 LHBS in the presence or absence of Zn2+ ions (20 μM). JAB1 in the immunoprecipitant was detected by using Western blotting (WB) with a mouse anti-JAB1 antibody. (C and D) Titrations of the Zn2+ (C) and Ni2+ (D) ions in the binding of Δ2 LHBS protein with JAB1, detected using in vitro GST pulldown assays, followed by Western blotting with antibodies that recognize 6×His (top panel) and GST (bottom panel).
To detect the ion dependencies of the JAB1-Δ2 LHBS binding, Zn2+ and Ni2+ ions in various concentrations were used for the GST pulldown experiments. A dose of Zn2+ ions as low as 20 μM stimulated interaction between the two proteins, and a dose of 200 μM Ni2+ ions was required for an equivalent stimulatory effect (Fig. 1C and D). This suggests that Zn2+ ions increase binding affinities between JAB1 and Δ2 LHBS more efficiently than Ni2+ ions do. The binding affinities between JAB1 and Δ2 LHBS were measured using SPR spectrometry in the presence of 20 μM Zn2+ and 20 μM Ni2+ ions (Fig. 2). The calculated Kd values in the presence of 20 μM Zn2+ and Ni2+ ions were 80 nM and 18.2 μM, respectively (Table 2). These data demonstrated that Zn2+ and, to a lesser extent, Ni2+ ions promote the protein interaction between JAB1 and Δ2 LHBS.
Fig 2.
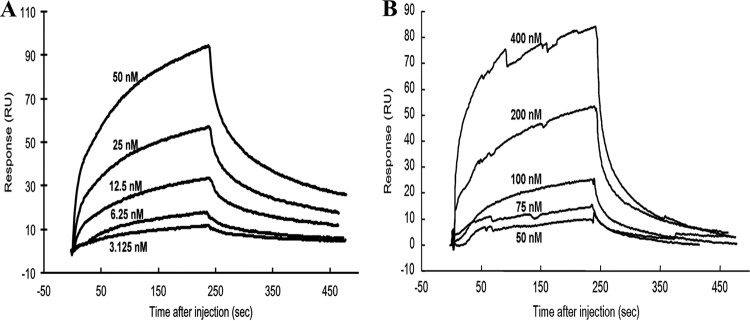
Binding analysis of Δ2 LHBS with JAB1 in the presence of different metal ions. (A) Surface plasmon resonance analysis to measure the binding affinities of Δ2 LHBS with JAB1 in the presence of 20 μM Zn2+ (A) and Ni2+ ions (B). Δ2 LHBS in the indicated concentrations was injected onto the sensor chip on which JAB1 had been immobilized. Reactions of the protein association and dissociation were monitored for 4 min. RU, response unit.
Table 2.
Kinetic constants of protein interaction between JAB1 and the full-length and Δ61-119 Δ2 LHBS in the presence of Zn2+ and Ni2+ ions
| Δ2 LHBS | Metal ion (20 μM) | Mean ± SEM |
||
|---|---|---|---|---|
| kon (M−1 s−1) | koff (s−1) | KD (M) | ||
| Full length | Ni2+ | (5.92 ± 0.14) × 102 | (1.08 ± 0.09) × 10−2 | (1.82 ± 0.10) × 10−5 |
| Zn2+ | (1.44 ± 0.07) × 105 | (1.17 ± 0.03) × 10−2 | (8.09 ± 0.60) × 10−8 | |
| Δ61-119 | Zn2+ | (8.32 ± 0.87) × 103 | (8.95 ± 0.70) × 10−3 | (1.08 ± 0.11) × 10−6 |
Involvement of the JAB1 JAMM/MPN+ domain in binding to Δ2 LHBS.
Serial deletion mutants of JAB1, fused to GST, were constructed to study the essential region for its binding to Δ2 LHBS (Fig. 3A). The Δ2 LHBS and various GST-JAB1 mutant proteins were purified to near homogeneity (Fig. 3B, bottom panel) and then used for the GST pulldown assays. The results showed that because of the deletion of the JAMM/MPN+ domain, JAB1 lost its ability to bind with Δ2 LHBS (Fig. 3B, top panel), which suggested that the JAMM/MPN+ domain, known to interact with several different proteins, is involved in the protein binding to Δ2 LHBS.
Fig 3.

Requirement of JAMM domain of JAB1 for its binding with Δ2 LHBS in the presence of Zn2+ ions. (A) Schematic representations showing the partial deletion constructs of JAB1 used in the present study. The wild-type (FL) and mutant JAB1 proteins (Δ1-52, Δ251-334, and ΔJAMM) were N-terminal fused to GST. (B) GST pulldown assays to detect the binding of Δ2 LHBS to JAB1 in the presence of (2 μM, + Zn2+) or absence of Zn2+ ions, followed with Western blotting (WB) with antibodies that recognize GST (top panel) and LHBS (middle panel). The bottom panel shows the input amounts of the JAB1 proteins to the GST pull-down reactions, detected by using Coomassie blue staining. (C) Protein binding of Δ2 LHBS to JAB1 and its H138Y mutant in the presence of Zn2+ ions, detected by using GST pulldown assays, followed by WB to detect LHBS (top panel) and GST (bottom panel).
Histidine is prone to bind to Zn2+ ions. To test whether histidine mediates the binding between JAB1 and Δ2 LHBS, the H138 residue in the JAMM/MPN+ domain was mutated to tyrosine for analysis. The results of the GST pulldown assays found that the JAB1 H138Y mutant lost most of its binding activity to Δ2 LHBS (Fig. 3C), which suggested that JAB1 H138 residue was important for the Zn2+ ion-mediated JAB1-Δ2 LHBS binding.
Involvement of Δ2 LHBS H71 and H116 residues in binding to JAB1.
We recently found that the region from aa 61 to 119 in Δ2 LHBS was involved in its interaction with JAB1 in in vitro-cultured human cells (28). Here, the GST pulldown assays using the E. coli recombinant proteins showed that the Δ2 LHBS deletion mutant, truncated from aa 61 to 119 (Δ2 LHBS/Δ61-119), lost its ability to bind with JAB1 (Fig. 4A). The binding affinities between JAB1 and Δ2 LHBS/Δ61-119 were measured using SPR spectrometry in the presence of 20 μM Zn2+ ions. The calculated KD (equilibrium dissociation constant) value was 1.08 μM, ∼16 times higher than that between JAB1 and Δ2 LHBS (Fig. 4B and Table 2). This indicates that the region from aa 61 to 119 in Δ2 LHBS is required for its binding with JAB1.
Fig 4.
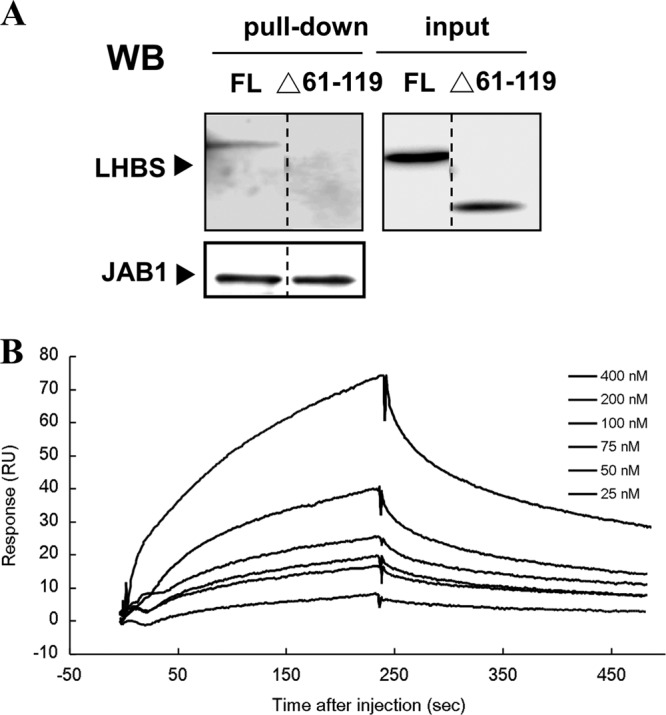
Requirement of the region from aa 61 to 119 of Δ2 LHBS for its binding to JAB1. (A) GST pulldown assays to detect the binding of JAB1 to Δ2 LHBS (FL) and its Δ61-119 mutant (Δ61-119), followed by Western blotting (WB) with antibodies that recognize LHBS and JAB1. The FL and Δ61-119 lanes were originally run in one gel but not next to each other. To show this, a dotted dividing line was placed between two lanes. (C) Surface plasmon resonance analysis to measure binding affinities of GST-JAB1 with various concentrations of the Δ2 LHBS Δ61-119 mutant in the presence of 20 μM Zn2+ ions. The kinetic constants of protein interaction are shown at the bottom of the curve analysis.
To identify which histidine residues in the region from aa 61 to 119 in Δ2 LHBS were required for the protein binding with JAB1, we mutated H71 and H116, the two conserved histidines in the region (Fig. 5A). The results of the GST pulldown assays showed that both of the Δ2 LHBS H71Y and H116Y mutants dramatically lost their ability to bind with JAB1, and the H71Y/H116Y double mutant showed no detectable level of its binding to JAB1; however, the binding of the H28Y mutation outside the region from aa 61 to 119 with JAB1 was not affected (Fig. 5B). The binding affinities detected using SPR spectrometry showed that the calculated KD values between JAB1 and Δ2 LHBS and its mutants H71Y and H116Y, and the H71Y/H116Y double mutant, were approximately 0.1, 0.3, 0.7, and 2.0 μM, respectively (Table 3). The H71Y/H116Y double mutant had a KD value 19 times higher than that of Δ2 LHBS, which indicated that both the H71 and H116 residues were important in the protein binding with JAB1 and that they work synergistically in the binding process.
Fig 5.

Requirement of the H71 and H116 residues of Δ2 LHBS for its binding to JAB1. (A) Sequence alignment to compare the pre-S1 region from aa 61 to 119 of LHBS in various HBV genotypes. The GenBank accession number (gi) for each protein sequence is shown following the genotype. The conserved and identical amino acid residues among various genotypes are shaded. The conserved H71 and H116 residues are shaded and marked. (B) GST pulldown assays to detect the binding of GST-JAB1 to Δ2 LHBS and its mutants H28Y, H71Y, and H116Y and to the H71Y/H116Y double mutant, followed by Western blotting (WB) with antibodies that recognize 6×His (top panel) and GST (middle panel). The bottom panel shows the input amounts of Δ2 LHBS and its mutant proteins for the GST pull-down analysis.
Table 3.
Binding affinities of Δ2 LHBS and its mutants to JAB1 in the presence of Zn2+ ions
| Δ2 LHBS or mutant | Mean KD (M) ± SEM |
|---|---|
| Δ2 LHBS | (8.09 ± 0.60) × 10−8 |
| Δ2 LHBS/Δ61-119 | (1.08 ± 0.11) × 10−6 |
| Δ2 LHBS/H71Y,H116Y | (1.96 ± 0.77) × 10−6 |
| Δ2 LHBS/H71Y | (2.98 ± 0.31) × 10−7 |
| Δ2 LHBS/H116Y | (6.54 ± 0.15) × 10− |
Zn2+ ion-dependent JAB1-Δ2 LHBS association in human cell lysates.
We previously found that Δ2 LHBS accumulated in ER lumen, where it interacted with JAB1 (14). Because Δ2 LHBS H71 and H116 and JAB1 H138 residues were required for the interaction between the two proteins expressed in E. coli, we hypothesized that in human cells the JAB1-Δ2 LHBS binding is facilitated by zinc in physiological concentrations, known to be a few hundred micromolar in a typical eukaryotic cell (29, 30). By coimmunoprecipitation analyses in the presence of 5 to 20 μM Zn2+ ions, we confirmed the binding of JAB1 with Δ2 LHBS in cell lysates of the human hepatoma HuH7 cells carrying Δ2 but not the wild-type LHBS expressed alone (Fig. 6A) or in the context of the whole HBV genome (Fig. 6B and C). This finding suggested that JAB1 was associated with Δ2 LHBS expressed from the viral genome in the host hepatocytes. This interaction was promoted by Zn2+ ions in a dose-dependent manner, because it increased, along with the elevations of zinc concentrations, and was nearly completely abolished with the addition of the metal chelator EDTA (Fig. 6C). By coimmunofluorescence and confocal microcopy analyses, we also found JAB1 colocalized with Δ2 LHBS much more than with wild type and the H71Y/H116Y double mutant (Fig. 6D), a finding which suggested that Zn2+ ions are involved in the specific interaction between JAB1 and Δ2 LHBS in hepatocytes, probably in the ER lumen (data not shown), where Δ2 LHBS has been reported to reside (6). Interestingly, we also found that the Δ2 LHBS H71Y and H116Y mutants and the H71Y/H116Y double mutant had significantly weaker protein associations with JAB1 than did Δ2 LHBS (Fig. 6E). Our previous studies (14, 28) showed that binding of JAB1 to Δ2 LHBS caused degradation of the cyclin-dependent kinase inhibitor p27Kip1, which resulted in G1- to S-phase cell cycle progression. Here, we also found that indeed all three histidine mutants no longer caused p27Kip1 decrease as Δ2 LHBS did (Fig. 6F). Taken together, these data indicated that Zn2+ ions facilitate the binding of Δ2 LHBS with JAB1 and that H71 and H116 residues are the two key residues in the protein for this binding, which induced p27Kip1 degradation in human HuH7 cells.
Fig 6.

Zn2+ ion-dependent JAB1-Δ2 LHBS association in human cell lysates. (A) The wild-type (W) and Δ2 LHBS stably expressed in HuH7 cells were immunoprecipitated in the presence of 20 μM Zn2+ ions with an antibody that recognizes hemagglutinin (HA) tagged to LHBS. The immunoprecipitants were probed, using Western blotting (WB), with antibodies that recognize HA and JAB1. V, cells containing the gene expression vector pQCXIP. (B) Expression of HBs and HBc RNA and proteins in HuH7 cells transfected with pHBV1.3 (W), pHBV1.3/Δ2LHBS (Δ2), or the vector control (V) by RT-PCR, Northern blotting (with digoxigenin-labeled HBx gene as a probe), and Western blotting. In Western blotting, the HBS proteins were detected with two different antibodies: the total HBS and LHBS alone were detected using the antibodies recognizing the major S and the pre-S1 regions, respectively. (C) Coimmunoprecipitation assays were performed to detect the association of JAB1 with the wild-type and Δ2 LHBS in HuH7 cells transfected with pHBV1.3 (W), pHBV1.3/Δ2LHBS (Δ2), or the vector control (V) in the presence of 5, 10, and 20 μM Zn2+ ions and EDTA (1 mM). The mouse monoclonal antibody recognizing the pre-S1 region of LHBS was used to immunoprecipitate the LHBS-associated protein complex, which was then analyzed by Western blotting (WB). The levels of JAB1 immunoprecipitated with LHBS from at least three experiments were quantified, as shown as a bar chart below the WB images. (D) Coimmunofluorescence assays were performed to detect the localizations of JAB1 with the wild-type (WT) and Δ2 LHBS (Δ2) or its H71Y/H116Y double mutant (Δ2 H71Y/H116Y). The bar chart below the cell images indicates the relative JAB1 levels colocalized with LHBS, detected in at least three independent experiments. *, P < 0.05. (E) Coimmunoprecipitation assays were performed to detect the association of JAB1 with Δ2 LHBS and its H71Y and H116Y mutants and the H71Y/H116Y double mutant, using the same method as described for panel A. (F) Levels of p27Kip1 affected by the wild-type and Δ2 LHBS and its respective mutants, detected using WB. HA and JAB1, WB shows the levels of LHBS-HA and endogenous JAB1. Tubulin was the loading control.
Direct binding of Δ2 LHBS to Zn2+ ions (ITC analysis).
To clarify the nature of Zn2+ ion binding to Δ2 LHBS, we directly measured Zn2+ ion binding to Δ2 LHBS and its H71Y/H116Y double mutant variant using ITC analysis. There were two Zn2+ binding sites on the Δ2 LHBS. The KD for one binding site (site 1) was 81.32 nM, which represents the strong binding site, which produced an exothermic reaction (ΔH = −1,555 cal/mol), whereas the second metal-binding site (site 2) had a KD of 64.54 μM, representing weak binding, which produced an endothermic reaction (ΔH = +2,774 cal/mol) (Table 4). These two different isothermal patterns suggested that the Δ2 LHBS protein contains two types of bound Zn2+ ions: one strong and the other weak. In contrast, when Zn2+ ions were titrated into the H71Y/H116Y double mutant of Δ2 LHBS, the exothermic reaction was significantly reduced or in some cases eliminated, and mostly the endothermic reaction was observed. The weak endothermic binding reaction remained (ΔH = +3,813 cal/mol) and produced a KD of 17.73 μM. Representative binding isotherms for both the Δ2 LHBS and its double mutant reacting with Zn2+ ion are shown in Fig. 7. These ITC results demonstrated that Δ2 LHBS alone can bind with Zn2+ ions and with high affinity. The amino acid side chains for H71 and H116 residues would appear to maximize the binding of Zn2+ ions to the Δ2 LHBS protein.
Table 4.
Binding affinities and isothermal properties of Zn2+ ions to Δ2 LHBS and its H71Y/H116Y double mutant (ITC) analysis
| Δ2 LHBS or mutant | Site 1 |
Site 2 |
||
|---|---|---|---|---|
| KD (nM) | ΔH (cal/mol) | KD (nM) | ΔH (cal/mol) | |
| Δ2 LHBS | 81 | –1,555 | 64 | +2,774 |
| Δ2 LHBS/H71Y,H116Y | NDa | ND | 17 | +3,813 |
ND, not determined.
Fig 7.

ITC analysis of ZnCl2 binding to Δ2 LHBS and its H71Y/H116Y double mutant. The raw heat change data for each injection from ITC analysis of Zn2+ binding to Δ2 LHBS (A) and Zn2+ binding to the H71Y/H116Y double mutant of Δ2 LHBS (B) are presented.
DISCUSSION
We found that Zn2+ ions mediated an interaction between Δ2 LHBS and JAB1 in vitro and in vivo. Among all of the other divalent ions tested, the Ni2+ ion, but not others, promoted a mild or higher binding between Δ2 LHBS and JAB1, which indicated that the binding is highly preferentially activated by Zn2+ and, to a lesser degree, Ni2+ ions. The protein-protein binding affinity in the presence of the Zn2+ ion was relatively high (KD = 80 nM), which indicates that this binding was strong. The interactive domain on each protein was also identified. The histidine residues essential for Zn2+ ion-mediated protein-protein binding were found on both Δ2 LHBS and JAB1, which suggested that they both directly bind to the Zn2+ ion. ITC analysis also showed that Δ2 LHBS directly binds to the Zn2+ ion at two sites; the binding at one site was strong, which indicated that Δ2 LHBS binds to the Zn2+ ions with high affinity and independently of JAB1. The Zn2+ ion is known to induce the structural reorganization of some proteins and affect their intra- and intermolecular binding affinities (31, 32). Therefore, the Zn2+ ion might have acted as a metal ligand to modulate conformations of both proteins and stimulate formation of the complex. The residues involved in the binding were essential for Δ2 LHBS-induced functional activation of JAB1: p27Kip1 degradation in human cells, which indicates that the Zn2+ ion-mediated Δ2 LHBS-JAB1 interaction plays a physiological role in Δ2 LHBS-induced cellular effects. Previous studies have reported that the total zinc concentrations in most eukaryotic cells are a few hundred micromolar (29, 30). In addition, the vast majority of cytosolic zinc is buffered by the numerous metal-binding proteins. Given the highly dynamic association of zinc with metalloproteins, the physiological concentration of the free form of Zn2+ ions in human cells is almost impossible to be precisely defined. Thus, in most of the experiments performed here we examined the JAB1-Δ2 LHBS binding in the presence of 20 μM Zn2+ ions to elaborate the effect of zinc to it. Given that this Zn2+ ion concentration was much lower than the physiological total zinc amount in a normal cell, it was believed to be able to provide relevant information for the physiological role of Zn2+ ions in cells.
We also found that the JAB1 JAMM domain, which contains a putative metal-binding motif (EXnHS/THX7SXXD), was essential for its binding to Δ2 LHBS (22). H138 residue on the motif was essential for this binding activity, which supports the notion that the Zn2+ ion's binding to the JAB1 JAMM motif is crucial for the interaction between the two proteins. Thus far, the X-ray structure of the archaebacterial protein AfJAMM, a eukaryotic JAMM ortholog, has been solved, and it has been shown (22) that the Zn2+ ion-binding site of AfJAMM is located in a furrow formed by the convex surface of the β2-β4 sheet and α2 helix. The catalytic Zn2+ ion has a tetrahedral coordination sphere with ligands provided by H67 and H69 on β4, D80 on α2, and a water molecule. Interestingly, the human JAB1 H138 residue, essential for Δ2 LHBS binding, is equivalent to the AfJAMM H67 residue, a ligand for the Zn2+ ion (22). Therefore, the Zn2+ ion's binding to the JAB1 H138 residue might have induced a metal ion-dependent conformational change of the protein and made it accessible to Δ2 LHBS. Our preliminary data found that the Zn2+ ion also greatly increased the binding of JAB1 to MIF1 (data not shown), which is known to form a complex with JAB1, suggesting that the Zn2+ ion is important for maintaining JAB1 structure for the formation of the complex associated with it.
In the present study the direct binding of Zn2+ ions with Δ2 LHBS protein was clearly demonstrated using ITC analysis. The binding pattern fits into a two-site binding model, with one site binding to Zn2+ ions much more strongly than the other site (81 nM versus 64 μM). Interestingly, the H71 and H116 residues are required for the stronger binding rather than the weaker one, which implies that Zn2+ ions bind to these two residues to facilitate the protein conformation of Δ2 LHBS, in E. coli and probably also human cells, the natural HBV host in physiological concentrations of Zn2+ ions. Thus, how the Zn2+ ion regulates the Δ2 LHBS-induced pro-oncogenic effects, such as distorting cell cycle checkpoints and inducing oxidative DNA damage, is an important question to be addressed (11, 13, 14). Also, the physiological role of the second, i.e., weak, Zn2+ ion-binding to Δ2 LHBS remains to be clarified in order to gain a complete picture of the Zn2+ binding properties of the protein and the carcinogenic pathways associated with it.
The Zn2+ ion was reported (33) to be involved in viral particle assembly through tetrahedral coordination with viral proteins. It was found to trigger conformational change and oligomerization of HBV capsid protein. It was also found that micromolar concentrations of Zn2+ ions are sufficient to initiate the assembly of HBV capsid protein, whereas other divalent cations elicited assembly only at millimolar concentrations, which suggests a specific and selective binding of Zn2+ ions. Zn2+ ion-induced HBV assembly was found to be an allosterically regulated process: ligand binding at one site influenced binding at other sites, which indicated that binding was associated with conformational change and that binding of the ligand altered the biological activity of assembly (33). In addition to the Zn2+ ion, which showed a strong stimulatory effect for HBV assembly, the Ni2+ ion, another divalent ion with preferential tetrahedral coordination, showed a moderate effect (33). This was consistent with our finding that the Ni2+ ion presented with a moderate stimulating effect for Δ2 LHBS-JAB1 binding. Therefore, the metal tetrahedral coordination property is believed to be involved in the binding of the two proteins.
Using yeast two-hybrid and in vivo coimmunoprecipitation approaches, we recently found (28) that Δ2 LHBS, but not the wild-type, interacted with JAB1 through the region from aa 61 to 119, the C-terminal region of the pre-S1 domain. Here we dug down to identify the key histidine residues in this region required for the binding and found that the H71 and H116 residues in this region were indeed the indispensable ones. The rationale for the specific binding of Δ2 LHBS to JAB1 through this domain remains to be clarified. Thus, we hypothesize that the 17-residue deletion in the pre-S2 domain of Δ2 LHBS causes a protein structural change, one which likely allows the domain spanning from aa 61 to 119 to be presented at the surface of the molecule and becomes accessible for protein-protein interaction. Our preliminary data showed that the wild-type and Δ2 LHBS E. coli recombinant proteins presented with different solubilities in the same solvent (data not shown), which implied that they were in distinct conformation; however, their structural characterizations must be investigated before a conclusion can be drawn.
JAB1 is a zinc-associated metalloprotease with isopeptidase activity that presents with deubiquitination and deneddylation properties (19–21). The substrate protein catalyzed by the isopeptidase activity of JAB1 is not yet clear. Thus, the finding that its association with Δ2 LHBS was stimulated by Zn2+ ions has raised the possibility that activation of its protease promotes the assembly of the protein complex. There are many examples of this type of protein interacting with and inducing conformational changes of their substrates (34–39). Interestingly, two putative ubiquitination sites were identified in Δ2 LHBS, which suggested the hypothesis that Δ2 LHBS undergoes deubiquitination processes catalyzed by JAB1. Further experiments to identify the ubiquitination sites on the LHBS are under way to explore this mechanism.
In summary, we identified here a Zn2+ ion-mediated protein interaction between Δ2 LHBS and JAB1. This is the first report of binding analysis for the formation of the Δ2 LHBS-associated protein complex, showing protein-protein communication between an oncogenic viral protein and a host factor in viral infection. Potential approaches to disrupt this complex using zinc-binding inhibitors such as matrix metalloprotease inhibitors, commonly used in cancer treatments, should greatly reduce the effect of Δ2 LHBS on p27Kip1 stability and ameliorate the oncogenic process contributed by Δ2 LHBS.
ACKNOWLEDGMENTS
We thank Scott C. Tso and R. Max Wynn at the University of Texas Southwestern Medical Center for their great help with the ITC analyses and providing valuable advices to this study.
This study was financially supported by grant NSC101-2320-B-006-031-MY3 from the Taiwan National Science Council (to W.H.), grant DOH102-TD-B-111-002 from the Taiwan Multidisciplinary Center of Excellence for Clinical Trials and Research (to W.H.), grant D102-22004 from the Center of Infectious Disease and Signaling Research (to W.-J.C. and W.H.) in National Cheng Kung University, Tainan, Taiwan, and grants DK26758 and DK62306 from the National Institutes of Health and grant I-1286 from the Welch Foundation (to D.T.C.). This research was also, in part, supported by the Headquarters of University Advancement at the National Cheng Kung University (to W.-J.C. and W.H.), which is sponsored by the Ministry of Education, Taiwan.
Footnotes
Published ahead of print 18 September 2013
REFERENCES
- 1.Gudat F, Bianchi L, Sonnabend W, Thiel G, Aenishaenslin W, Stalder GA. 1975. Pattern of core and surface expression in liver tissue reflects state of specific immune response in hepatitis B. Lab. Invest. 32:1–9 [PubMed] [Google Scholar]
- 2.Shafritz DA, Kew MC. 1981. Identification of integrated hepatitis B virus DNA sequences in human hepatocellular carcinomas. Hepatology 1:1–8 [DOI] [PubMed] [Google Scholar]
- 3.Ibarra MZ, Mora I, Bartolome J, Porres JC, Carreno V. 1989. Detection of proteins encoded by the pre-S region of hepatitis B virus in the sera of HBsAg carriers: relation to viral replication. Liver 9:153–158 [DOI] [PubMed] [Google Scholar]
- 4.Fan YF, Lu CC, Chang YC, Chang TT, Lin PW, Lei HY, Su IJ. 2000. Identification of a pre-S2 mutant in hepatocytes expressing a novel marginal pattern of surface antigen in advanced disease of chronic hepatitis B virus infection. J. Gastroenterol. Hepatol. 15:519–528 [DOI] [PubMed] [Google Scholar]
- 5.Fan YF, Lu CC, Chen WC, Yao WJ, Wang HC, Chang TT, Lei HY, Shiau AL, Su IJ. 2001. Prevalence and significance of hepatitis B virus (HBV) pre-S mutants in serum and liver at different replicative stages of chronic HBS infection. Hepatology 33:277–286 [DOI] [PubMed] [Google Scholar]
- 6.Wang HC, Wu HC, Chen CF, Lei HY, Su IJ. 2003. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am. J. Pathol. 163:2441–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su IJ, Wang HC, Wu HC, Huang W. 2008. Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J. Gastroenterol. Hepatol. 23:1169–1174 [DOI] [PubMed] [Google Scholar]
- 8.Tsai HW, Lin YJ, Lin PW, Wu HC, Hsu KH, Yen CJ, Chan SH, Huang W, Su IJ. 2011. A clustered ground-glass hepatocyte pattern represents a new prognostic marker for the recurrence of hepatocellular carcinoma after surgery. Cancer 117:2951–2960 [DOI] [PubMed] [Google Scholar]
- 9.Shen FC, Su IJ, Wu HC, Hsieh YH, Yao WJ, Young KC, Chang TC, Hsieh HC, Tsai HN, Huang W. 2009. A Pre-S Gene Chip to detect the pre-S deletions in the hepatitis B virus large surface antigen as a predictive marker for hepatoma risk in the chronic HBV carriers. J. Biomed. Sci. 16:84–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen BF, Liu CJ, Jow GM, Chen PJ, Kao JH, Chen DS. 2006. High prevalence and mapping of pre-S deletion in hepatitis B virus carriers with progressive liver diseases. Gastroenterology 130:1153–1168 [DOI] [PubMed] [Google Scholar]
- 11.Hsieh YH, Su IJ, Wang HC, Chang WW, Lei HY, Lai MD, Chang WT, Huang W. 2004. The pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 25:2023–2032 [DOI] [PubMed] [Google Scholar]
- 12.Hsieh YH, Hsu JL, Su IJ, Huang W. 2011. Genomic instability caused by hepatitis B virus: into the hepatoma inferno. Front. Biosci. 17:2586–2597 [DOI] [PubMed] [Google Scholar]
- 13.Wang HC, Chang WT, Chang WW, Huang W, Lei HY, Lai MD, Fausto N, Su IJ. 2005. Upregulation of cyclin A and nodular proliferation of hepatocytes induced by a pre-S2 deletion mutant in chronic HBV infection. Hepatology 41:761–770 [DOI] [PubMed] [Google Scholar]
- 14.Hsieh YH, Su IJ, Wang HC, Tsai JH, Huang YJ, Chang WW, Lai MD, Lei HY, Huang W. 2007. Hepatitis B virus pre-S2 mutant surface antigen induces degradation of cyclin-dependent kinase inhibitor p27Kip1 through c-Jun activation domain-binding protein 1. Mol. Cancer Res. 5:1063–1072 [DOI] [PubMed] [Google Scholar]
- 15.Tomoda K, Kubota Y, Arata Y, Mori S, Maeda M, Tanaka T, Yoshida M, Yoneda-Kato N, Kato JY. 2002. The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. J. Biol. Chem. 277:2302–2310 [DOI] [PubMed] [Google Scholar]
- 16.Chamovitz DA, Segal D. 2001. JAB1/CSN5 and the COP9 signalosome: a complex situation. EMBO Rep. 2:96–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emberley ED, Niu Y, Leygue E, Tomes L, Gietz RD, Murphy LC, Watson PH. 2003. Psoriasin interacts with Jab1 and influences breast cancer progression. Cancer Res. 63:1954–1961 [PubMed] [Google Scholar]
- 18.Luo W, Wang Y, Hanck T, Stricker R, Reiser G. 2006. Jab1, a novel protease-activated receptor-2 (PAR-2)-interacting protein, is involved in PAR-2-induced activation of activator protein-1. J. Biol. Chem. 281:7927–7936 [DOI] [PubMed] [Google Scholar]
- 19.Tran HJ, Allen MD, Löwe J, Bycroft M. 2003. Structure of the Jab1/MPN domain and its implications for proteasome function. Biochemistry 42:11460–11465 [DOI] [PubMed] [Google Scholar]
- 20.Bellare R, Kutach AK, Rines AK, Guthrie C, Sontheimer EJ. 2006. Ubiquitin binding by a variant Jab1/MPN domain in the essential pre-mRNA splicing factor Prp8p. RNA 12:292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cope GA, Suh GSB, Aravind L, Schwarz SE, Zipursky SL, Koonin EV, Deshaies RJ. 2002. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science 298:608–611 [DOI] [PubMed] [Google Scholar]
- 22.Ambroggio XI, Rees DC, Deshaies RJ. 2004. JAMM: a metalloprotease-like zinc site in the proteasome and signalosome. PLoS Biol. 2:E2. 10.1371/journal.pbio.0020002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chou YC, Jeng KS, Chen ML, Liu HH, Liu TL, Chen YL, Liu YC, Hu CP, Chang C. 2005. Evaluation of transcriptional efficiency of hepatitis B virus covalently closed circular DNA by reverse transcription-PCR combined with the restriction enzyme digestion method. J. Virol. 79:1813–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu JL, Chen HC, Peng HL, Chang HY. 2008. Characterization of the histidine-containing phosphotransfer protein B-mediated multistep phosphorelay system in Pseudomonas aeruginosa PAO1. J. Biol. Chem. 283:9933–9944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oh W, Yang MR, Lee EW, Park KM, Pyo S, Yang JS, Lee HW, Song J. 2006. Jab1 mediates cytoplasmic localization and degradation of West Nile virus capsid protein. J. Biol. Chem. 281:30166–30174 [DOI] [PubMed] [Google Scholar]
- 26.Tao H, Liu W, Simmons BN, Harris HK, Cox TC, Massiah MA. 2010. Purifying natively folded proteins from inclusion bodies using Sarkosyl, Triton X-100, and CHAPS. Biotechniques 48:61–64 [DOI] [PubMed] [Google Scholar]
- 27.Kato N, Okayama T, Isawa H, Yuda M, Chinzei Y, Iwanaga S. 2005. Contribution of the N-terminal and C-terminal domains of haemaphysalin to inhibition of activation of plasma kallikrein-kinin system. J. Biochem. 138:225–235 [DOI] [PubMed] [Google Scholar]
- 28.Hsieh YH, Su IJ, Yen CJ, Tsai TF, Tsai HW, Tsai HN, Huang YJ, Chen YY, Ai YL, Kao LY, Hsieh WC, Wu HC, Huang W. 2013. Histone deacetylase inhibitor suberoylanilide hydroxamic acid suppresses the pro-oncogenic effects induced by hepatitis B virus pre-S2 mutant oncoprotein and represents a potential chemopreventive agent in high-risk chronic HBV patients. Carcinogenesis. 34:475–485 [DOI] [PubMed] [Google Scholar]
- 29.Maret W. 2009. Molecular aspects of human cellular zinc homeostasis: redox control of zinc potentials and zinc signals. Biometals 22:149–157 [DOI] [PubMed] [Google Scholar]
- 30.Maret W. 2012. New perspectives of zinc coordination environments in proteins. J. Inorg. Biochem. 111:110–116 [DOI] [PubMed] [Google Scholar]
- 31.Mekmouche Y, Coppel Y, Hochgrafe K, Guilloreau L, Talmard C, Mazarguil H, Faller P. 2005. Characterization of the ZnII binding to the peptide amyloid-β1-16 linked to Alzheimer's disease. Chembiochem 6:1663–1671 [DOI] [PubMed] [Google Scholar]
- 32.Ilari A, Alaleona F, Petrarca P, Battistoni A, Chiancone E. 2011. The X-ray structure of the zinc transporter ZnuA from Salmonella enterica discloses a unique triad of zinc-coordinating histidines. J. Mol. Biol. 409:630–641 [DOI] [PubMed] [Google Scholar]
- 33.Stray SJ, Ceres P, Zlotnick A. 2004. Zinc ions trigger conformational change and oligomerization of hepatitis B virus capsid protein. Biochemistry 43:9989–9998 [DOI] [PubMed] [Google Scholar]
- 34.Dueber EC, Schoeffler AJ, Lingel A, Elliott JM, Fedorova AV, Giannetti AM, Zobel K, Maurer B, Varfolomeev E, Wu P, Wallweber HJ, Hymowitz SG, Deshayes K, Vucic D, Fairbrother WJ. 2011. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science 334:376–380 [DOI] [PubMed] [Google Scholar]
- 35.Song AX, Zhou CJ, Peng Y, Gao XC, Zhou ZR, Fu QS, Hong J, Lin DH, Hu HY. 2010. Structural transformation of the tandem ubiquitin-interacting motifs in ataxin-3 and their cooperative interactions with ubiquitin chains. PLoS One 5:e13202. 10.1371/journal.pone.0013202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prager K, Wang-Eckhardt L, Fluhrer R, Killick R, Barth E, Hampel H, Haass C, Walter J. 2007. A structural switch of presenilin 1 by glycogen synthase kinase 3β-mediated phosphorylation regulates the interaction with beta-catenin and its nuclear signaling. J. Biol. Chem. 282:14083–14093 [DOI] [PubMed] [Google Scholar]
- 37.Raaf J, Brunstein E, Issinger OG, Niefind K. 2008. The interaction of CK2alpha and CK2beta, the subunits of protein kinase CK2, requires CK2beta in a preformed conformation and is enthalpically driven. Protein Sci. 17:2180–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Norris NC, Bingham RJ, Harris G, Speakman A, Jones RP, Leech A, Turkenburg JP, Potts JR. 2011. Structural and functional analysis of the tandem β-zipper interaction of a streptococcal protein with human fibronectin. J. Biol. Chem. 286:38311–38320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shim JY, Bertalovitz AC, Kendall DA. 2011. Identification of essential cannabinoid-binding domains: structural insights into early dynamic events in receptor activation. J. Biol. Chem. 286:33422–33435 [DOI] [PMC free article] [PubMed] [Google Scholar]