

Abstract
Alpha interferon (IFN-α) is an essential component of innate antiviral immunity and of treatment regimens for chronic hepatitis C virus (HCV) infection. Resistance to IFN might be important for HCV persistence and failure of IFN-based therapies. Evidence for HCV genetic correlates of IFN resistance is limited. Experimental studies were hampered by lack of HCV culture systems. Using genotype (strain) 1a(H77) and 3a(S52) Core-NS2 JFH1-based recombinants, we aimed at identifying viral correlates of IFN-α resistance in vitro. Long-term culture with IFN-α2b in Huh7.5 cells resulted in viral spread with acquisition of putative escape mutations in HCV structural and nonstructural proteins. Reverse genetic studies showed that primarily amino acid changes I348T in 1a(H77) E1 and F345V/V414A in 3a(S52) E1/E2 increased viral fitness. Single-cycle assays revealed that I348T and F345V/V414A enhanced viral entry and release, respectively. In assays allowing viral spread, these mutations conferred a level of IFN-α resistance exceeding the observed fitness effect. The identified mutations acted in a subtype-specific manner but were not found in genotype 1a and 3a patients, who failed IFN-α therapy. Studies with HCV recombinants with different degrees of culture adaptation confirmed the correlation between viral fitness and IFN-α resistance. In conclusion, in vitro escape experiments led to identification of HCV envelope mutations resulting in increased viral fitness and conferring IFN-α resistance. While we established a close link between viral fitness and IFN-α resistance, identified mutations acted via different mechanisms and appeared to be relatively specific to the infecting virus, possibly explaining difficulties in identifying signature mutations for IFN resistance.
INTRODUCTION
Hepatitis C virus (HCV) infection is a major public health burden (1). Chronic infection increases the risk of developing liver cirrhosis and hepatocellular carcinoma, being the main indication for liver transplantation (2). HCV is a small, enveloped virus with a 9.6-kb single-stranded RNA genome of positive polarity, with 5′ and 3′ untranslated regions and one long open reading frame (ORF) encoding a polyprotein, which is cleaved into structural proteins (Core, E1, and E2) and nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (2). HCV shows significant genetic heterogeneity; therefore, it has been classified in 7 major genotypes and numerous subtypes, differing in approximately 30 and 20% of their sequences, respectively (3).
Recombinant alpha interferon (IFN-α) was first used in 1986 for treatment of chronic non-A/non-B hepatitis (4). Subsequently, a combination of pegylated IFN-α and ribavirin has become the standard of care, resulting in sustained viral response in approximately 50% of treated patients (5). Despite the recent approval of two HCV protease inhibitors for inclusion in treatment regimens for chronic HCV genotype 1 infection, pegylated IFN-α/ribavirin remains the basis of current therapeutic regimens. In addition, induction of interferons is an important innate immune defense mechanism during HCV infection (6).
HCV has apparently developed mechanisms to escape from innate immunity, leading to viral persistence, and from IFN-α-based therapy, leading to treatment failure. Several HCV proteins, specifically Core, E2, NS3/NS4A protease, and NS5A, were reported to interfere with host innate immunity (reviewed in reference 7). The identification of viral and host factors allowing prediction of therapy outcome has been the subject of intensive investigations. Various host factors, such as sex, age, race, and stage of liver fibrosis, were found to influence therapy outcome (8). Recently, certain variations near the IL28B gene were described to correlate with treatment-induced and spontaneous viral clearance (9, 10). Of the viral factors, HCV genotype, viral load, and complexity of HCV quasispecies were associated with treatment outcome (8, 11, 12). Primarily based on analysis of HCV sequences of patients with favorable and nonfavorable treatment outcome, sequence variations in several HCV proteins, mainly Core, E2, and NS5A, were suggested as viral genetic correlates of IFN-α resistance (7, 8). However, consistent identification of signature mutations remains elusive. Also, experimental approaches were limited by lack of suitable HCV cell culture systems allowing for reverse genetic studies. Subgenomic replicons, only recapitulating viral replication, were used to promote in vitro viral escape from IFN in order to identify viral resistance mutations. However, in the majority of studies, no IFN-resistant replicons could be identified and resistance was found to be conferred by cellular and not viral factors (13–18). In 2005, the first HCV cell culture system resulting in production of infectious HCV particles (HCVcc) was developed. Based on the genotype 2a isolate JFH1 and Huh7 human hepatoma cells or derived cell lines with increased permissiveness to HCV infection, this system allowed studies in the context of the complete viral life cycle (19–21). Following this discovery, JFH1-based recombinants expressing genotype-specific genome regions were developed (22, 23). At the outset of this study, JFH1-based recombinants with Core-NS2 of the 7 major HCV genotypes that are able to efficiently infect Huh7.5 hepatoma cells were available (24, 25).
The aim of this study was to identify HCV genomic correlates of IFN-α resistance. Thus, we aimed at inducing viral escape by treatment of cultures infected with genotype 1a and 3a Core-NS2 recombinants with IFN-α2b to identify and characterize mutations conferring resistance to IFN-α. A better understanding of HCV IFN-α resistance mechanisms is of great interest for studies of virus-host interactions. Furthermore, it might contribute to understanding of viral persistence and treatment failure in HCV-infected patients.
MATERIALS AND METHODS
Plasmids.
We used previously described inter- and intragenotypic HCV recombinants with untranslated regions and NS3-NS5B of JFH1 and Core-NS2 of the following genotypes (isolates), with cell culture adaptive amino acid changes as indicated: 1a(H77) with V787A and Q1247L (25); 1a(TN) with R1408W (26); 1a(DH6) with V157A, I414T, Y444H, V787A, S905C, and Q1247L (26); 1b(J4) with F886L and Q1496L (24); 1b(DH1) with F886L and Q1496L (26); 2a(J6) (21); 3a(S52) with I787S and K1398Q (24); and 3a(DBN) with W838R and K1398Q (26) (throughout, nucleotide and amino acid positions are given as absolute H77 [GenBank accession number AF009606] reference numbers [3]). To test the influence of the degree of culture adaptation on IFN-α resistance, the following additional Core-NS2 recombinants were used: genotype 1a isolate H77 recombinants with either Q1247L or R1408W (25) and genotype 3a isolate S52 recombinants with either Y788C and Q1496L or K1398Q (27). Plasmids used to generate HCV pseudoparticles (HCVpp) were a gift from F.-L. Cosset: phCMV-Gag-Pol murine leukemia virus (MLV) (28), MLV-luc (28), and phCMV-ires (29). To generate phCMV-E1E2 3a(S52), which was used for production of 3a(S52) HCVpp, an amplicon encoding the C-terminal 60 amino acids of Core and the entire E1 and E2 proteins was amplified from a plasmid encoding 3a(S52)I787S,K1398Q (24) using PfuI (Stratagene) with primers HCVpp-S52_F (5′-AATGATATCGACCTCATGGGGTACATCCCG-3′) and HCVpp-S52_R (5′-ATTGATATCTTATGCTTCTGCTTGTGATACC-3′), introducing EcoRV restriction sites at the 5′ and 3′ ends and a stop codon at the 3′ end. The EcoRV restriction site naturally present in 3a(S52) E2 was removed by introduction of the synonymous nucleotide change T1901C. The resulting amplicon was introduced into phCMV-ires using EcoRV (Fermentas). Generation of vector phCMV-E1E2 1a(H77), used for production of 1a(H77) HCVpp, was described previously (30). Mutations were introduced in plasmids using PfuI for fusion PCR- and restriction enzyme-based cloning.
Culturing, transfection, infection, and evaluation of Huh7.5 cell cultures.
Overall, culturing of Huh7.5 hepatoma cells (31) was done as previously described (27). One day before transfection or infection, 3.5 × 105 cells were plated per well in 6-well plates. For transfections, HCV RNA transcripts were generated by in vitro transcription as previously described (27), and then 2.5 μg HCV RNA transcripts was incubated with 5 μl Lipofectamine 2000 (Invitrogen) in 500 μl Opti-MEM (Invitrogen) for 20 min at room temperature. Cells were incubated with transfection complexes for 24 h in growth medium and were split as indicated in the figure legends. For infections, cells were inoculated with virus-containing supernatant for 6 h at a multiplicity of infection (MOI) of 0.01 and were split every 2 days, starting from day 1 postinfection. Supernatants were collected each time the cells were split and were stored at −80°C. Viral spread in infected cultures was monitored by estimating the percentage of infected cultured cells following immunostaining, using primary mouse anti-HCV NS5A 9E10 antibody (gift from C. Rice) and secondary antibody Alexa Fluor 594 goat anti-mouse IgG (H+L) (Invitrogen) as described previously (27). The percentage of HCV-positive cells was evaluated by microscopy, assigning values of 0 (no cells infected), 0.1, 1, 5, and 10 to 90% (in steps of 10). Infectivity titers of culture supernatants were determined as focus-forming units (FFU)/ml following infection of replicate cultures with serially diluted supernatant immunostained with primary antibody mouse anti-HCV NS5A 9E10 and secondary antibody ECL anti-mouse immunoglobulin IgG, horseradish-peroxidase-conjugated whole antibody (GE Healthcare Amersham), and automated FFU counting as previously described (26, 27). Extracellular HCV-Core levels were determined on cell culture supernatants using an Architect HCV Ag assay (Abbott Diagnostics) according to the manufacturer's recommendations.
Treatment of cell cultures with IFN-α2b.
For induction of in vitro viral escape, treatment with 500 IU/ml of IFN-α2b (Schering-Plough) was initiated when approximately 60% of the cultured cells were infected with 1a(H77) or 3a(S52). Cultures then were treated at the indicated days with 500 IU/ml IFN-α2b to maintain between 1 and 30% HCV antigen-positive cultured cells (Fig. 1). For transfection cultures, following 24 h of incubation with transfection complexes, cells were split and maintained in growth medium containing IFN-α2b at the indicated concentrations. Medium with IFN-α2b was added when the cells were split, as indicated in the figure legends. For infection cultures, after 6 h of infection, treatment with 5 IU/ml IFN-α2b was initiated and was repeated on days 1 and 3 postinfection, when the cells were split. From day 5 postinfection, medium containing 20 IU/ml IFN-α2b was added to the cultured cells every 2 days, when the cells were split. IFN-α2b concentrations used did not have cytotoxic effects (24). For treated cultures, percent residual infectivity or percent residual Core was calculated by relating supernatant infectivity titers or Core levels to the peak infectivity titers or Core levels of nontreated cultures transfected with the same recombinant.
Fig 1.

Treatment with IFN-α allowed long-term culture of HCV genotype 1a(H77) and 3a(S52) Core-NS2 recombinants and resulted in selection of putative viral escape mutations. Virus stocks of genotype (isolate) 1a(H77) (A) and 3a(S52) (B) Core-NS2 recombinants were used to infect naive Huh7.5 cells (MOI of 0.003) (24). On day 6 postinfection, when 60% of the cultured cells were expressing HCV NS5A as determined by immunostaining, treatment with IFN-α2b was initiated by applying 500 IU/ml three times at 6-h intervals. Cultures then were treated with 500 IU/ml of IFN-α2b (red arrows) in variable intervals in order to maintain 1 to 30% HCV NS5A-positive cells. The presence of IFN-α in culture medium is represented by red bars. To avoid viral eradication, cells were cultured without IFN-α when <1% infected cells were recorded (green bars). Black arrows indicate days at which supernatant for direct ORF sequencing was obtained. At viral breakthrough, 1a(H77) (C) and 3a(S52) (D) viruses had acquired several coding and noncoding mutations, indicated by red and gray arrows, respectively. For 3a(S52), mutations that were common to both of the sequenced samples are shown. For reverse genetic studies, identified coding mutations were introduced individually or in combination in 1a(H77) and 3a(S52), as indicated by brackets; names of resulting mutants are given.
Single-cycle production assay.
To determine intracellular and extracellular HCV-Core levels as well as intracellular and extracellular infectivity titers, 4 × 105 CD81-deficient S29 cells (32) were plated per well of a 6-well plate. After 24 h of incubation, cells were transfected in replicates with HCV RNA as described above; however, growth medium was exchanged with Opti-MEM during a 4-h transfection incubation, leading to increased transfection efficiency. To determine intracellular HCV Core levels at 4 and 48 h posttransfection, cells of one replicate well were trypsinized. At 4 h posttransfection, all harvested cells, and at 48 h posttransfection 25% of the harvested cells, were centrifuged at 1,000 × g for 5 min at 4°C, washed in cold phosphate-buffered saline (PBS), and lysed in cold radioimmunoprecipitation assay (RIPA) buffer (Thermo Scientific) supplemented with protease inhibitor cocktail set III (Calbiochem). Cell lysates were clarified at 20,000 × g for 15 min at 4°C before HCV-Core levels were measured using an Ortho HCV antigen enzyme-linked immunosorbent assay (ELISA) kit (Ortho Clinical Diagnostics) according to the manufacturer's instructions. To determine intracellular infectivity titers, the remaining 75% of cells harvested at 48 h posttransfection were centrifuged at 1,000 × g for 5 min at 4°C and resuspended in 100 μl of growth medium. To release intracellular HCV particles, cells were subjected to four freeze-thaw cycles using liquid nitrogen and a 37°C water bath; supernatant containing the intracellular virus population was clarified by two centrifugations at 1,500 × g for 5 min at 4°C. For determination of extracellular Core levels and infectivity titers, supernatants were collected 48 h after transfection as described above. When specified, S29 cells were treated with the indicated concentrations of IFN-α2b after the 4-h transfection period until termination of the assay 48 h posttransfection.
HCV pseudoparticle production and infection assay.
In general, the assay was carried out as described previously (30). In brief, 293T human embryo kidney cells (HEK) (a gift from F.-L. Cosset) plated in 10-cm dishes were transfected during 6 h with expression vectors encoding (i) HCV-E1E2 glycoproteins of 1a(H77) or 3a(S52) [phCMV-E1E2 1a(H77) or phCMV-E1E2 3a(S52); 4 μg], (ii) the firefly luciferase reporter gene (MLV-Luc; 8 μg); and (iii) the MLV-Gag-Pol packaging construct (phCMV-Gag-Pol MLV; 8 μg) using calcium phosphate (CalPhos mammalian transfection kit; Clontech Laboratories, Inc.) by following the manufacturer's instructions. Supernatants containing HCVpp were collected 48 h later and directly used for infection of 1.5 × 104 Huh7.5 cells that had been plated the previous day on each well of a 96-well plate. Per condition tested, 8 wells were infected for 16 to 24 h using 100 μl of HCVpp-containing supernatant supplemented with 4 μg/ml of Polybrene (Sigma-Aldrich). Seventy-two h postinfection, cells were lysed and luciferase activity was measured as relative light units (RLU) using a FLUOstar Optima luminometer (BMG Labtech) after addition of firefly luciferase substrate (luciferase assay system; Promega). For analysis of incorporation of MLV-capsid E1 and E2 into purified HCVpp by Western blotting, samples were prepared by ultracentrifugation of 9 ml viral supernatant through a 20% sucrose cushion using a Beckman SW-41 rotor mounted in a Beckman XL-70 ultracentrifuge at 107,000 × gmax (where gmax is the maximum force of gravity) for 3 h at 4°C. Pellets containing HCVpp were resuspended in 100 μl of PBS after overnight incubation at 4°C, and stored at −80°C. When specified, cells were pretreated with the indicated concentrations of IFN-α2b 24 h before and during infection with HCVpp.
Western blot analysis.
Total protein concentration of ultracentrifugation-purified HCVpp was determined by bicinchoninic acid (BCA) assay (Pierce). Proteins were loaded onto a precast Novex Bis-Tris polyacrylamide gel (Invitrogen) in the presence of reducing agent. After separation, proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane (Hybond-P membrane; 0.45-μm pore size; GE Health Care/Amersham) and incubated overnight at 4°C with the following specific primary antibodies: mouse anti-HCV E1 (A4 clone; gift from S. U. Emerson) (33), mouse anti-HCV E2 (H52 clone; gift from J. Dubuisson) (33), and mouse anti-MLV Gag (gift from J. Dubuisson) (34). Membranes then were incubated for 1 h at room temperature with secondary horseradish peroxidase-conjugated antibody (goat anti-mouse or anti-rabbit IgG [H+L], peroxidase conjugated; Pierce). Proteins were revealed by enhanced chemiluminescence detection (SuperSignal West Femto maximum sensitivity substrate; Pierce).
HCV sequence analysis.
For plasmid preparations (Qiagen Plasmid Maxi Kit; Qiagen), the complete HCV sequence was confirmed (Macrogen).
For cell culture-derived HCV, procedures for RNA extraction, reverse transcription of viral RNA, first-round PCR from cDNA, and second-round nested PCR were described previously (27). Primers used for 1a(H77) are described in reference 25, and those for 3a(S52) are described in reference 27.
Serum from patients chronically infected with HCV genotype 1a or 3a who failed to respond to combination therapy with IFN-α/ribavirin were obtained from Aalborg University Hospital. RNA was extracted using the High Pure viral nucleic acid kit (Roche). Reverse transcription was carried out with random hexamers (TAG Copenhagen) and SuperScript II (Invitrogen) under the following conditions: 25°C for 10 min, 42°C for 1 h, and 48°C for 30 min. The resulting cDNA was treated with RNase H (Invitrogen) and RNase T (Fermentas) at 37°C for 20 min. E1 and E2 were amplified by nested PCR, generating 4 overlapping amplicons. For first-round PCR, 2.5 U of AmpliTaq Gold with buffer II (Invitrogen), 0.24 mM deoxynucleoside triphosphate (dNTP), 4 mM MgCl2, and 0.2 μM reverse and forward primer (TAG Copenhagen), respectively, were used. Primer sequences are shown in Table 1. Cycling conditions were 95°C for 5 min, followed by 35 cycles of 94°C for 1 min, 50°C for 1.5 min, and 72°C for 3 min, followed by 72°C for 6 min. Ten μl of first-round PCR product was used as the template for the nested second-round PCR, carried out as described above, except that 5 U of AmpliTaq Gold was used and 40 cycles were run. Second-round PCR products were column purified using a MinElute PCR purification kit (Qiagen).
Table 1.
Primers used for PCR amplification of E1-E2 of HCV from genotype 1a- or 3a-infected patients
| Genotype and amplicon | Primer name | Sequence (5′ → 3′) |
|---|---|---|
| 1a | ||
| First-round PCR | ||
| Amplicon 1 | Out C E1 F | GCAACAGGGAACCTTCCTGGTTGCTC |
| Out C E1 R | CGTAGGGGACCAGTTCATCATCAT | |
| Amplicon 2 | 1aA2_Fout809 | CAYATCGAYCTGCTYGTCGG |
| 1aA2_Rout1282 | GGCHGTNCTRTTGATGTGCCA | |
| Amplicon 3 | 1aA3_Fout1057 | GGYGCYCACTGGGGAGTCCT |
| 1aA3_Rout1975 | AGYTCRGWCCTGTCCCTGTC | |
| Amplicon 4 | gt1abA4_E2Fout | GTKGTRGTGGGRACGACCGA |
| gt1abA3_p7Rout | YAACGCCAGCAGGAGCAGGAG | |
| Second-round nested PCR | ||
| Amplicon 1 | IN C E1 F | AACCTTCCTGGTTGCTCTTTCTCTAT |
| IN C E1 R | GTTCATCATCATATCCCATGCCAT | |
| Amplicon 2 | 1aA2_Fin855 | TAYGTGGGGGAYYTRTGCGGG |
| 1aA2_Rin1276 | GTNCTRTTGATGTGCCARCTGCC | |
| Amplicon 3 | gt1abA3_E1Fin | TCCATGGTGGGGAACTGGGC |
| 1aA3_Rin1952 | CCCYCCYACRTACATCCTGA | |
| Amplicon 4 | gt1abA4_E2Fin | TGGTTCGGYTGYACVTGGATGAA |
| gt1abA3_p7Rin | CAGAAGAACACRAGGAAGGA | |
| 3a | ||
| First-round PCR | ||
| Amplicon 1 | Out C E1 F | GCAACAGGGAACCTTCCTGGTTGCTC |
| Out C E1 R | CGTAGGGGACCAGTTCATCATCAT | |
| Amplicon 2 | 3a-2-Out F 1200 | GGCCGTCTTYCTYGTGGGACAAGC |
| 3a-2-Out R 1744 | GTGATGGGCTTGCAGCTGCTGAG | |
| Amplicon 3 | 3a-3-Out F 1595 | GGTCAACACCAATGGCTCGTGGCA |
| 3a-3-Out R 2233 | CATGGGTAATGCCAAAGCCGGTA | |
| Amplicon 4 | 3a-4-Out F 2111 | CTTCTGCCCCACCGACTGCTTCAG |
| 3a-4-Out R 2651 | AGCAGCGACGGCGTTCAGCGTGA | |
| Second-round nested PCR | ||
| Amplicon 1 | In C E1 F | AACCTTCCTGGTTGCTCTTTCTCTAT |
| In C E1 R | GTTCATCATCATATCCCATGCCAT | |
| Amplicon 2 | 3a-2-In F 1209 | TTYCTYGTGGGACAAGCCTTCAC |
| 3a-2-ln R 1717 | TGAGGACATCCAGTAGAGTTGAA | |
| Amplicon 3 | 3a-3-In F 1604 | CAATGGCTCGTGGCACATCAACAG |
| 3a-3-In R 2219 | AAGCCGGTATGGATAGTCGACCA | |
| Amplicon 4 | 3a-4-In F 2121 | ACCGACTGCTTCAGGAAACATCCT |
| 3a-4-In R 2639 | GTTCAGCGTGACAAGGTTCTCCAA |
Generated amplicons were directly sequenced (Macrogen). Sequence analysis was done with Sequencher (Gene Codes). Alignments were done using Sequencher and BioEdit freeware. Cloning plans were developed using Vector NTI (Invitrogen). HCV sequences and number tools for determination of H77 reference numbers were from the European and Los Alamos HCV databases (35–37). Ready-made alignments of genotype 1a and 3a HCV sequences were from the Los Alamos 2008 web alignment.
RESULTS
In vitro selection of putative viral escape mutations in HCVcc under treatment with IFN-α.
Huh7.5 cells infected with 1a(H77) or 3a(S52) were treated in various intervals with 500 IU/ml IFN-α2b (Fig. 1A and B). 1a(H77) spread to the majority of cultured cells by day 129 posttreatment following an extended period of continuous treatment, indicating that viral escape might have occurred (Fig. 1A). 3a(S52) spread to the majority of cultured cells on day 122, and a high percentage of infected cells was maintained despite continuous treatment until day 161 (Fig. 1B).
Direct sequence analysis of the complete ORF of viral genomes recovered from culture supernatant obtained at day 129 revealed that 1a(H77) had acquired 5 nucleotide changes coding for I348T in E1, H932D in NS2, T2076A and M2259V in NS5A, and S2788A in NS5B; one noncoding NS3 mutation was also identified (Fig. 1C and Table 2).
Table 2.
Coding nucleotide changes acquired by 1a(H77) at viral breakthrough under IFN-α treatmente
| HCV gene | Nucleotide positiona |
Nucleotide identitiyb |
Amino acid positionc |
Amino acid changed | |||
|---|---|---|---|---|---|---|---|
| 1a(H77) plasmid | H77 absolute reference no. | 1a(H77) plasmid | Escape day 129 | 1a(H77) polyprotein | H77 absolute reference no. | ||
| E1 | 1383 | 1384 | T | C | 348 | 348 | I→T |
| NS2 | 3134 | 3135 | C | G | 932 | 932 | H→D |
| NS5A | 6566 | 6567 | A | G | 2076 | 2076 | T→A |
| NS5A | 7103 | 7116 | A | G | 2255 | 2259 | M→V |
| NS5B | 8756 | 8703 | T | G | 2806 | 2788 | S→A |
Nucleotide positions are numbered according to the HCV sequence of the 1a(H77) plasmid and with H77(GenBank accession number AF009606) absolute reference numbers (3).
Nucleotide identity at the respective position of the plasmid or of viral genomes recovered from infected cell cultures. Coding mutations are shown. Positions were included at which nucleotide changes were detected at least as 50/50 quasispecies. In addition, the noncoding mutation G4514A was found.
Amino acid positions are numbered according to the HCV sequence of the 1a(H77) polyprotein and with H77 (GenBank accession number AF009606) absolute reference numbers (3).
Amino acid changes encoded by the given nucleotide changes are indicated.
Viral genomes for sequencing of the complete ORF were from supernatant obtained from the primary escape experiment at day 129 posttreatment (Fig. 1A).
For 3a(S52), on days 144 and 161, we identified 8 common mutations coding for F345V in E1, V414A in E2, I765V in p7, H837Y in NS2, T2076A, E2381G and M2388T in NS5A, and K2471R in NS5B, as well as 9 common noncoding mutations; 1 coding mutation specific to day 144 as well as 7 coding and 7 noncoding mutations specific to day 161 were also identified (Fig. 1D and Table 3).
Table 3.
Coding nucleotide changes acquired by 3a(S52) at viral breakthrough under IFN-αe
| HCV gene | Nucleotide positiona |
Nucleotide identitiyb |
Amino acid positionc |
Amino acid changed | ||||
|---|---|---|---|---|---|---|---|---|
| 3a(S52) plasmid | H77 absolute reference no. | 3a(S52) plasmid | Escape day 144 | Escape day 161 | 3a(S52) polyprotein | H77 absolute reference no. | ||
| E1 | 1373 | 1374 | T | G | G | 345 | 345 | F→V |
| E2 | 1581 | 1582 | T | C | C | 414 | 414 | V→A |
| E2 | 2067 | 2062 | A | • | G | 576 | 574 | E→G |
| E2 | 2079 | 2066h | A | • | G | 580 | 575c | E→G |
| p7 | 2651 | 2634 | A | G | G | 771 | 765 | I→V |
| NS2 | 2867 | 2850 | C | C/T | T | 843 | 837 | H→Y |
| NS2 | 3329 | 3312 | G | • | A | 997 | 991 | A→T |
| NS5A | 6396 | 6379 | A | • | G | 2019 | 2013 | K→R |
| NS5A | 6584 | 6567 | A | A/g | A/G | 2082 | 2076 | T→A |
| NS5A | 6800 | 6783 | T | T/C | • | 2154 | 2148 | C→R |
| NS5A | 7020 | 7003 | C | • | C/T | 2227 | 2221 | T→I |
| NS5A | 7121 | 7116 | A | • | A/G | 2261 | 2259 | M→V |
| NS5A | 7500 | 7483 | A | A/G | G | 2387 | 2381 | E→G |
| NS5A | 7521 | 7504 | T | T/C | C | 2394 | 2388 | M→T |
| NS5B | 7824 | 7753 | A | G | G | 2495 | 2471 | K→R |
| NS5B | 8045 | 7974 | G | • | G/A | 2569 | 2545 | D→N |
Nucleotide positions are numbered according to the HCV sequence of the 3a(S52) plasmid and with H77 (GenBank accession number AF009606) absolute reference numbers (3).
Nucleotide identity at the respective position of the plasmid or of viral genomes recovered from infected cell cultures. Coding mutations are shown. Positions at which nucleotide changes were detected as at least 50/50 quasispecies in at least one ORF were included. In addition, on day 144 the following noncoding mutations were found: T2375T/G, A3449a/G, T3699T/C, A4763A/G, T5261t/C, A5327C, G5504g/A, T8192T/C, and A8576A/G. On day 161, the following noncoding mutations were found: A440G, T2375G, A3449G, T3699C, A4763A/G, T5261T/C, A5327C, G5504G/A, G6623G/A, A6749A/G, A6752A/G, C7685C/T, A7895A/G, T8192t/C, A8501A/G, and A8576A/G. Dots indicate the identity to the original plasmid sequence.
Amino acid positions are numbered according to the HCV sequence of the 3a(S52) polyprotein and with H77 (GenBank accession number AF009606) absolute reference numbers (3).
Amino acid changes encoded by the given nucleotide changes are indicated.
Viral genomes for sequencing of the complete ORF were from supernatants obtained from the primary escape experiment at days 144 and 161 posttreatment (Fig. 1B).
Primarily the I348T change in E1 of 1a(H77) increased viral fitness and conferred IFN-α resistance.
We first transfected Huh7.5 cells with RNA transcripts of 1a(H77) recombinants containing (i) the 5 identified coding mutations, designated 1a(H77)5mut; (ii) the 2 mutations located in 1a(H77) Core-NS2, 1a(H77)I348T,H932D; and (iii) the 3 mutations located in NS3-NS5B, 1a(H77)NS5A/B (Fig. 1C and Table 2). Without IFN-α treatment, all recombinants infected ≥80% of cultured cells by days 3 and 5 (Fig. 2A). Compared to the original 1a(H77) and the 1a(H77)NS5A/B recombinants, 1a(H77)I348T,H932D and 1a(H77)5mut had ∼0.5 log10 higher supernatant HCV infectivity titers, reaching up to 4.3 log10 FFU/ml (Fig. 2A, left). Under treatment with 20 IU/ml IFN-α, spread of the original 1a(H77) was inhibited, with ≤20% of infected cells on days 3 to 9; similar spread kinetics were observed for 1a(H77)NS5A/B (Fig. 2B, left). In contrast, 1a(H77)I348T,H932D and 1a(H77)5mut showed only slightly delayed spread kinetics, infecting ≥80% of cultured cells on day 5. These differences were reflected by 1a(H77)I348T,H932D and 1a(H77)5mut reaching up to 3.8 and 4.1 log10 FFU/ml, respectively, on day 9, while no infectivity was detected for 1a(H77) and 1a(H77)NS5A/B (Fig. 2B, left). The 1a(H77)I348T,H932D and 1a(H77)5mut viruses showed at least 0.6 to 1.7 log10 higher infectivity titers than 1a(H77). Thus, differences in spread kinetics and infectivity titers were greater in IFN-α-treated than in nontreated cultures (Fig. 2A and B). When calculating percent residual infectivity for treated cultures, under IFN-α treatment, 1a(H77) and 1a(H77)NS5A/B showed <10% residual infectivity, whereas 1a(H77)I348T,H932D and 1a(H77)5mut showed up to 37 and 61% residual infectivity, respectively (Fig. 2C, left). This suggested that among the 5 tested mutations, primarily I348T and H932D increased viral fitness and conferred IFN-α resistance, even though combination with NS5A/B mutations led to slight enhancement of the observed phenotype.
Fig 2.

I348T in E1 increased infectivity titers of 1a(H77) and conferred IFN-α resistance. In vitro HCV RNA transcripts of the indicated recombinants were transfected into replicate Huh7.5 cultures. From day 1 posttransfection, cultures were split every second day. (A and B) The percentage of infected cultured cells (% inf) was monitored by immunostaining for HCV NS5A and is indicated above the graph at the indicated day posttransfection. ND, the percentage was not determined. Supernatant HCV infectivity titers are shown as means from 3 replicates with standard errors of the means (SEM). The lower limit of detection in the experiments shown was up to 2.4 log10 FFU/ml, indicated by the y axis breaks. (A) Cultures were monitored without IFN-α treatment until viruses had spread to almost all cultured cells. (B) Replicate cultures were treated with 20 IU/ml of IFN-α2b on day 1 posttransfection and subsequently each time the cells were split. Cultures were monitored until the fittest virus had spread to almost all cultured cells. 1a(H77)I348T and 1a(H77)H932D only spread to a maximum of 60 and 40% of cells, respectively, followed by a decrease in percent infected cells. (C) Percent residual infectivity was determined by relating infectivity titers of treated cultures to peak infectivity titers of nontreated cultures infected with the same recombinant.
We next tested 1a(H77)I348T and 1a(H77)H932D (Fig. 2, right). Similar to 1a(H77)I348T,H932D, without treatment, 1a(H77)I348T infectivity titers were 0.6 to 0.7 log10 higher than those of 1a(H77) on days 3 and 5 (Fig. 2A, right). Under treatment, spread of 1a(H77) was inhibited with no infectivity detectable, while 1a(H77)I348T spread to 60% of cultured cells with 3.8 log10 FFU/ml and 37% residual infectivity on day 11 (Fig. 2B and C); similar findings were obtained in 5 independent transfections. Without IFN-α, 1a(H77)H932D showed slightly increased infectivity titers compared to those of 1a(H77) (Fig. 2A, right). In the presence of IFN-α, 1a(H77)H932D spread to 40% of cells with 3.1 log10 FFU/ml and 14% residual infectivity on day 11 (Fig. 2B and C).
When determining HCV core levels in culture supernatants, differences were most apparent for IFN-α-treated cultures (Fig. 3). Thus, 1a(H77)I348T, 1a(H77)I348T,H932D, and 1a(H77)5mut had peak Core levels of ≥4.6 log10 fmol/liter, while 1a(H77) had Core levels of ≤3.8 log10 fmol/liter (Fig. 3B). 1a(H77)NS5A/B had Core levels similar to those of 1a(H77), while 1a(H77)H932D had slightly higher levels. Further, 1a(H77)I348T, 1a(H77)I348T,H932D, and 1a(H77)5mut showed up to 31% residual Core, while 1a(H77) and 1a(H77)NS5A/B showed ≤6% residual Core; 1a(H77)H932D showed up to 13% residual Core (Fig. 3C).
Fig 3.

Differences in supernatant Core levels confirmed that I348T conferred IFN-α resistance to 1a(H77). Core levels were measured in supernatants obtained in the transfection cultures shown in Fig. 2, which were nontreated (A) or treated with IFN-α (B), as described in the Fig. 2 legend and in Materials and Methods. The lower limit of detection was 2.5 log10 fmol/liter, indicated by the y axis breaks. (C) Percent residual Core level was calculated by relating Core level of treated cultures to the peak Core level of nontreated cultures infected with the same recombinant.
The 1a(H77)I348T, 1a(H77)5mut, and 1a(H77) viruses responded in a concentration-dependent manner to IFN-α treatment (Fig. 4). Previously observed differences in spread kinetics and infectivity titers (Fig. 2B) were greatest using 20 or 100 IU/ml IFN-α. To investigate significance of the observed differences in percent residual infectivity as shown in Fig. 4C, we carried out statistical analysis as previously described (38). Median 50% effective concentrations (EC50) with 95% confidence intervals (CI) were 0.22 (0.16 to 0.29), 5.0 (3.6 to 7.0), and 15.5 (13.8 to 17.5) IU/ml for 1a(H77), 1a(H77)I348T, and 1a(H77)5mut, respectively. Median fold EC50 differences (with 95% CI) were 23.2 (14.8 to 36.5) for 1a(H77) versus 1a(H77)I348T (P < 0.0001) and 72.1 (52.3 to 99.3) for 1a(H77) versus 1a(H77)5mut (P < 0.0001). Median EC75 (with 95% CI) were 2.8 (1.9 to 4.1), 31.8 (21.2 to 47.9), and 43.9 (37.3 to 51.6) IU/ml for 1a(H77), 1a(H77)I348T, and 1a(H77)5mut, respectively. Median fold EC75 differences (with 95% CI) were 11.4 (6.5 to 20.0) for 1a(H77) versus 1a(H77)I348T (P < 0.0001) and 15.7 (10.3 to 23.9) for 1a(H77) versus 1a(H77)5mut (P < 0.0001). Finally, median EC90 (with 95% CI) were 36.4 (19.2 to 69.1), 202.3 (104.3 to 392.6), and 123.9 (95.3 to 161.0) IU/ml for 1a(H77), 1a(H77)I348T, and 1a(H77)5mut, respectively. Median fold EC90 differences (with 95% CI) were 5.6 (2.2 to 14.0) for 1a(H77) versus 1a(H77)I348T (P = 0.0003) and 3.4 (1.7 to 6.8) for 1a(H77) versus 1a(H77)5mut (P = 0.0005). Thus, the interferon concentrations required to inhibit HCV strain H77 infection at EC50, EC75, and EC90 were statistically significantly higher for the mutant viruses than for the corresponding original virus, indicating interferon resistance.
Fig 4.
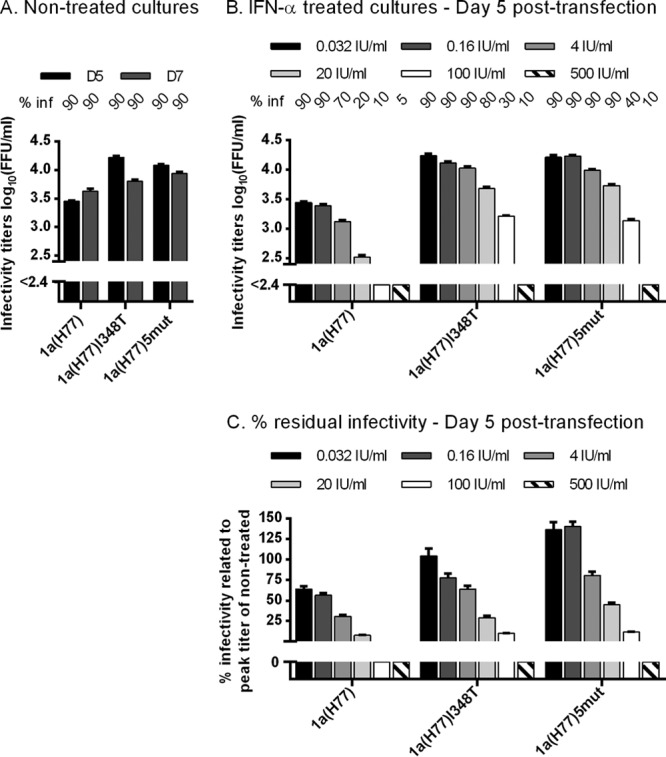
1a(H77) with the I348T substitution showed resistance to several concentrations of IFN-α. In vitro HCV RNA transcripts of the indicated recombinants were transfected into replicate Huh7.5 cultures. From day 1 posttransfection, cultures were split every second day. (A and B) The percentage of infected cultured cells (% inf) was monitored by immunostaining for HCV NS5A and is indicated above the graph at the indicated day posttransfection. Supernatant infectivity titers are means from 3 replicate determinations of 3 replicate cultures with SEM. The lower limit of detection in the experiments shown was up to 2.4 log10 FFU/ml, indicated by y axis breaks. (A) Cultures were monitored without IFN-α treatment until day 7 posttransfection. (B) Replicate cultures were treated with a 5-fold serial dilution of IFN-α2b (0.032 to 500 IU/ml, excluding 0.8 IU/ml); each concentration was tested in triplicate cultures. Treatment was initiated on day 1 posttransfection and administered each time the cells were split. Presented infectivity titers were from day 5 posttransfection; similar results were obtained at day 7. (C) Percent residual infectivity was determined by relating infectivity titers of treated cultures to peak infectivity titers of nontreated cultures infected with the same recombinant.
Finally, we demonstrated that differences observed in transfection experiments were also found during viral infection (Fig. 5). Neither additional dominant mutations nor reversions of engineered mutations were identified when we analyzed the complete ORF of viral genomes from supernatant derived at the peak of infection of nontreated or treated first-passage cultures.
Fig 5.
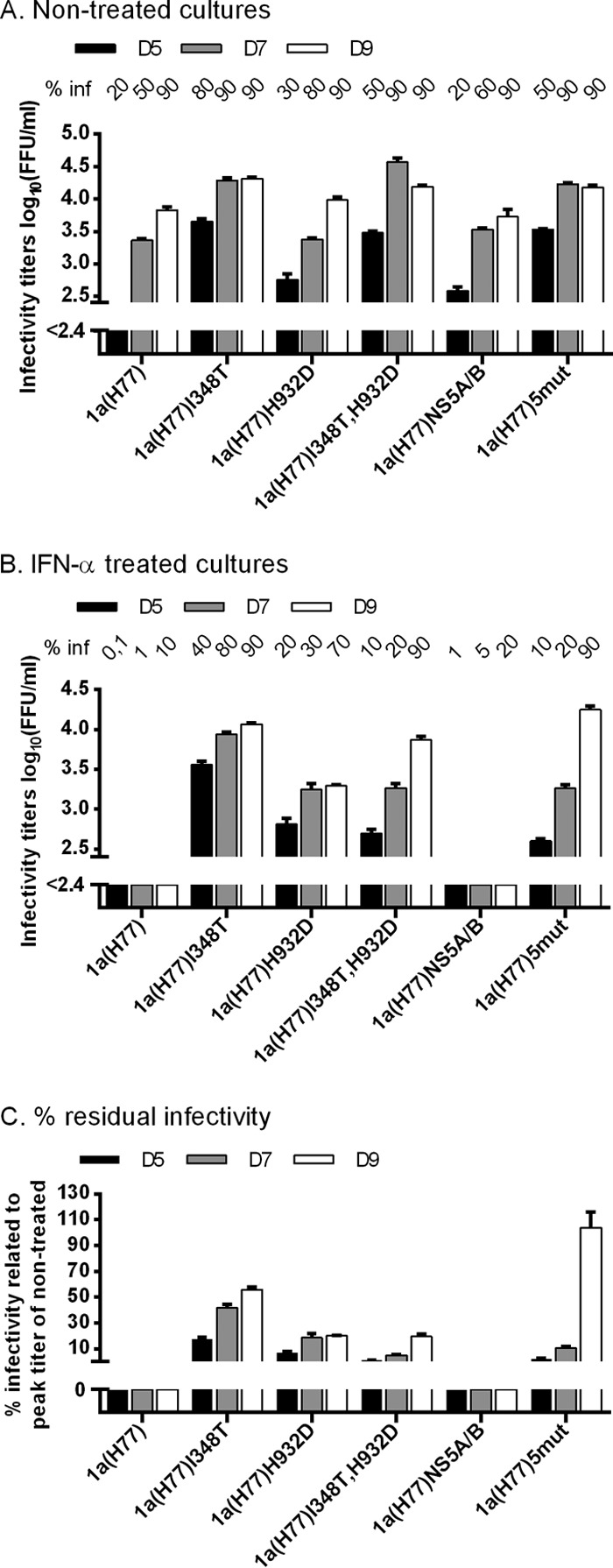
I348T in E1 increased infectivity titers of 1a(H77) and conferred IFN-α resistance in a kinetic first-passage infection experiment. Replicate Huh7.5 cultures were infected at an MOI of 0.01 with 1a(H77), 1a(H77)I348T, 1a(H77)I348T,H932D, 1a(H77)NS5A/B, and 1a(H77)5mut contained in supernatants derived from nontreated transfection cultures at the peak of infection shown in Fig. 2A. From day 1 postinfection, cultures were split every second day. (A and B) The percentage of infected cultured cells (% inf) was monitored by immunostaining for HCV NS5A and is indicated above the graph at the indicated day postinfection. Supernatant HCV infectivity titers are shown as means of 3 replicates with SEM. The lower limit of detection in the experiments shown was up to 2.4 log10 FFU/ml, indicated by the y axis breaks. (A) Cultures were monitored without IFN-α until day 9, when all viruses had spread to the entire cell culture. (B) After 6 h of infection, replicate cultures were treated with 5 IU/ml IFN-α. Treatment with 5 IU/ml IFN-α was repeated on days 1 and 3 postinfection. On days 5, 7, and 9 postinfection, cell cultures were treated with 20 IU/ml IFN-α. The experiment was terminated when the most resistant viruses had spread to the entire culture. (C) Percent residual infectivity was determined by relating infectivity titers of treated cultures to peak infectivity titers of nontreated cultures infected with the same recombinant.
Overall, among the 5 coding mutations identified in 1a(H77) under long-term IFN-α treatment at viral breakthrough, it appeared to be primarily I348T that increased viral infectivity and conferred IFN-α resistance.
Primarily the F345V change in E1 in combination with V414A in E2 of 3a(S52) increased viral fitness and conferred IFN-α resistance.
We next examined the 3a(S52) coding mutations identified in the primary escape experiment (Fig. 1D and Table 3). We transfected Huh7.5 cells with recombinants containing (i) the 8 identified coding mutations common to the two escape samples analyzed, 3a(S52)8mut; (ii) the 4 mutations located in 3a(S52) Core-NS2, 3a(S52)F345V,V414A,I765V,H837Y; and (iii) the 4 mutations located in NS3-NS5B, 3a(S52)NS5A/B. Without IFN-α, 3a(S52)F345V,V414A,I765V,H837Y and 3a(S52)8mut showed slightly accelerated spread kinetics and higher infectivity titers than 3a(S52) and 3a(S52)NS5A/B, reaching up to 4.7 log10 FFU/ml (Fig. 6A, left). Under IFN-α treatment, 3a(S52)F345V,V414A,I765V,H837Y and 3a(S52)8mut spread to 90% of cultured cells by day 5, whereas 3a(S52) and 3a(S52)NS5A/B remained below 50% for 7 days (Fig. 6B, left). While 3a(S52) infectivity titers were below the detection limit, 3a(S52)F345V,V414A,I765V,H837Y and 3a(S52)8mut reached up to 4.4 and 4.6 log10 FFU/ml, respectively (Fig. 6B, left), corresponding to 43 and 61% residual infectivity (Fig. 6C). Thus, mutations in the 3a(S52) Core-NS2 region apparently increased viral infectivity and conferred IFN-α resistance. Similar studies with 3a(S52) recombinants containing Core-NS2 mutations singly or in combination indicated that the phenotype observed for 3a(S52)F345V,V414A,I765V,H837Y was primarily caused by F345V in E1 in combination with V414A in E2 (Fig. 6, middle and right).
Fig 6.

F345V mutation in combination with V414A increased infectivity titers of 3a(S52) and conferred IFN-α resistance. In vitro HCV RNA transcripts of the indicated recombinants were transfected into replicate Huh7.5 cultures. From day 1 posttransfection, cultures were split every second day. (A and B) The percentage of infected cultured cells (% inf) was monitored by immunostaining for HCV NS5A and is indicated above the graph at the indicated day posttransfection. ND, the percentage was not determined. Supernatant infectivity titers are shown as means from 3 replicates with SEM. The lower limit of detection in the experiments shown was up to 2.4 log10 FFU/ml, indicated by the y axis breaks. (A) Cultures were monitored without IFN-α treatment until viruses had spread to almost all cultured cells. (B) Replicate cultures were treated with 20 IU/ml of IFN-α2b on day 1 posttransfection and subsequently each time the cells were split. Cultures were monitored until the fittest virus had spread to almost all cells. (C) Percent residual infectivity was determined by relating infectivity titers of treated cultures to peak infectivity titers of nontreated cultures infected with the same recombinant.
For IFN-α-treated cultures of the different 3a(S52) recombinants, differences in HCV Core levels reflected differences observed in spread kinetics and infectivity titers (data not shown). Also, differences observed in transfections were found during viral infection (data not shown). Neither additional dominant mutations nor reversions of engineered mutations were identified when analyzing the complete ORF of viral genomes from first-passage supernatant derived at the peak of infection of nontreated or treated cell cultures.
Overall, of the 8 coding mutations identified in 3a(S52) escape cultures, it appeared that primarily F345V in E1 but also V414A in E2 conferred increased fitness and IFN-α resistance to 3a(S52).
Effect of 348T identified in 1a(H77) and 345V identified in 3a(S52) on other HCV isolates, subtypes, and genotypes.
We first introduced 348T into JFH1-based recombinants with Core-NS2 of genotype 1a isolates TN and DH6 (26), genotype 1b isolates J4 and DH1 (24, 26), and genotype 3a isolates S52 and DBN (24, 26). Without IFN-α, original as well as mutant genotype 1a and 3a recombinants showed efficient spread in cell culture, with infectivity titers of 1a(TN)I348T being slightly higher than those of 1a(TN) (Fig. 7A). Introduction of 348T impaired fitness of 1b(J4) but not 1b(DH1) (Fig. 7A). Under IFN-α, introduction of 348T resulted in a slight increase in titers and residual infectivity for 1a(TN) and 1a(DH6) but not for 1b and 3a recombinants (Fig. 7B and C).
Fig 7.

E1 348T increased infectivity titers and conferred IFN-α resistance to another 1a isolate. In vitro HCV RNA transcripts of the indicated recombinants were transfected into replicate Huh7.5 cultures. From day 1 posttransfection, cultures were split every second day. (A and B) The percentage of infected cultured cells (% inf) was monitored by immunostaining for HCV NS5A and is indicated above the graph at the indicated day posttransfection. Supernatant infectivity titers are shown as means from 3 replicates with SEM. The lower limit of detection in the experiments shown was up to 2.4 log10 FFU/ml, indicated by y axis breaks. (A) Cultures were monitored without IFN-α treatment until viruses had spread to almost all cultured cells or until the percentage of infected cells decreased. (B) Replicate cultures were treated with 20 IU/ml of IFN-α2b on day 1 posttransfection and subsequently each time the cells were split. Cultures were monitored until the fittest virus had spread to almost all cultured cells. (C) Percent residual infectivity was determined by relating infectivity titers of treated cultures to peak infectivity titers of nontreated cultures infected with the same recombinant.
We next tested 3a(DBN), 1a(H77), and 1a(TN) mutants with 345V; 1b(J4) and 1b(DH1) have V at amino acid 345. Without IFN-α treatment, 345V increased infectivity of 3a(DBN) but not 1a(TN) while impairing fitness of 1a(H77) (Fig. 8A). Under IFN-α treatment, 345V increased titers and residual infectivity for 3a(DBN) but not for the 1a recombinants (Fig. 8B and C). Thus, 348T and 345V apparently increased infectivity titers and conferred IFN-α resistance in a subtype-specific manner.
Fig 8.
E1 345V increased infectivity titers and conferred IFN-α resistance to another 3a isolate. In vitro HCV RNA transcripts of the indicated recombinants were transfected into replicate Huh7.5 cultures. From day 1 posttransfection, cultures were split every second day. (A and B) The percentage of infected cultured cells (% inf) was monitored by immunostaining for HCV NS5A and is indicated above the graph at the indicated day posttransfection. Supernatant infectivity titers are shown as means from 3 replicates with SEM. The lower limit of detection in the experiments shown was up to 2.4 log10 FFU/ml, indicated by y axis breaks. (A) Cultures were monitored without IFN-α treatment until viruses had spread to almost all cultured cells or until the percentage of infected cells decreased. (B) Replicate cultures were treated with 20 IU/ml of IFN-α2b on day 1 posttransfection and subsequently each time the cells were split. Cultures were monitored until the fittest virus had spread to almost all cells. (C) Percent residual infectivity was determined by relating infectivity titers of treated cultures to peak infectivity titers of nontreated cultures infected with the same recombinant.
I348T identified in 1a(H77) and F345V identified in 3a(S52) affect different steps of the viral life cycle.
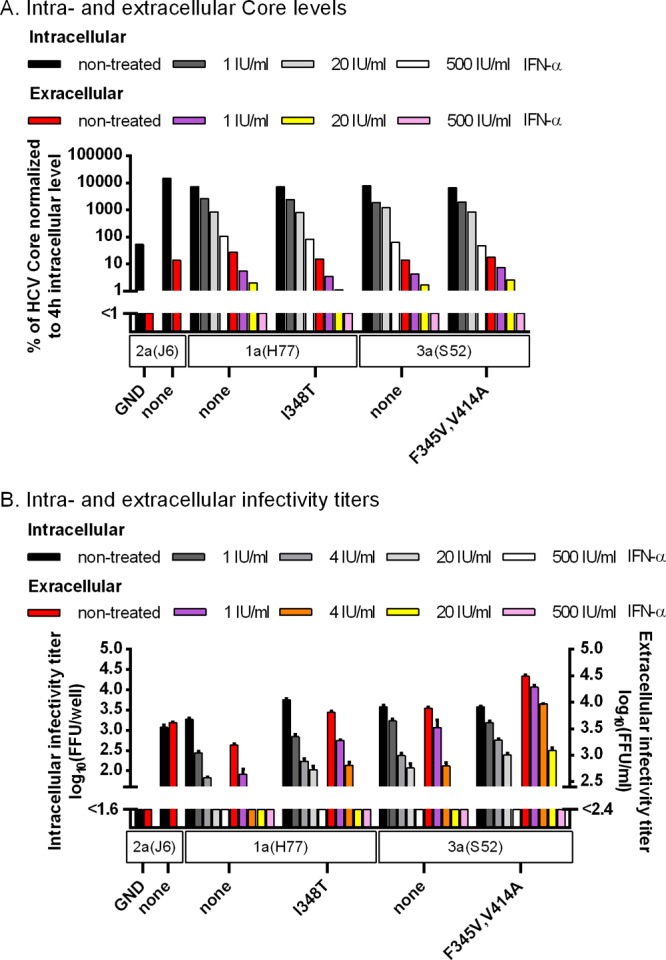
To explore the effect of the identified mutations on the viral life cycle, we first did transfection experiments in CD81-deficient S29 cells, precluding HCV entry (32). For 1a(H77) and 3a(S52) recombinants, no major differences in intracellular or extracellular Core levels were observed, indicating that neither 348T nor 345V influenced viral replication/translation or the number of viral particles released (Fig. 9A). For 1a(H77)I348T and 1a(H77)5mut, intracellular and extracellular infectivity titers were slightly increased compared to those of 1a(H77) (Fig. 9B), with the ratio of intra- to extracellular titers being similar for all 3 recombinants. Since infectivity titers were determined in Huh7.5 cells allowing viral entry, I348T might merely increase entry efficacy (as confirmed below). In contrast, 3a(S52) mutants, particularly 3a(S52)F345V,V414A and 3a(S52)8mut, showed increased extracellular infectivity titers compared to 3a(S52), while intracellular infectivity titers were similar (Fig. 9B). Thus, these mutants showed an increased ratio between extra- and intracellular titers, suggesting that envelope mutations enhance release of infectious viral particles. IFN-α treatment decreased intracellular as well as extracellular Core levels and infectivity titers for tested 1a(H77) and 3a(S52) recombinants in a similar, concentration-dependent manner, indicating an effect of IFN-α on viral replication and/or translation (Fig. 10).
Fig 9.

Envelope mutations identified in 1a(H77) and 3a(S52) affected different steps of the viral life cycle, as determined by a single-cycle production assay. CD81-deficient S29 cell cultures were transfected in duplicate with HCV RNA transcripts of 1a(H77) and 3a(S52) recombinants along with positive [2a(J6)] and negative [2a(J6)GND] controls. (A) After 4 h, cells of one replicate culture were collected and lysed, and intracellular HCV Core levels were measured as described in Materials and Methods. Forty-eight hours posttransfection, supernatant and cells of the replicate cultures were collected; intra- and extracellular Core levels were measured and normalized for transfection efficiency to the 4-h intracellular Core level. Core values are single determinations from one of three independent experiments. (B) In addition, for the cultures harvested 48 h posttransfection, intra- and extracellular infectivity titers were determined in Huh7.5 cells. Infectivity titers are means from 3 replicates of 3 independent experiments ± SEM. The lower limit of detection in the experiments shown was up to 1.8 log10 FFU/well for intracellular infectivity titers and up to 2.5 log10 FFU/ml for extracellular infectivity titers, indicated by y axis breaks.
Fig 10.
In a single-cycle production assay, IFN-α showed a similar effect on replication/translation of 1a(H77) and 3a(S52) original and mutant viruses. CD81-deficient S29 cell cultures were transfected in replicates with HCV RNA transcripts of 1a(H77) and 3a(S52) recombinants along with positive [2a(J6)] and negative [2a(J6)GND] controls. (A) After 4 h, cells of one replicate culture were collected and lysed, and intracellular HCV Core levels were measured as described in Materials and Methods. The other replicate cultures were maintained untreated or treated with the indicated concentrations of IFN-α2b. Forty-eight hours posttransfection, supernatant and cells of these replicate cultures were collected; intra- and extracellular Core levels were measured and normalized for transfection efficiency to the 4-h intracellular Core level. Core values are single determinations. (B) In addition, for the cultures harvested 48 h posttransfection, intra- and extracellular infectivity titers were determined in Huh7.5 cells. Infectivity titers are shown as means from 3 replicates with SEM. The lower limit of detection in the experiments shown was up to 1.6 log10 FFU/well for intracellular infectivity titers and up to 2.4 log10 FFU/ml for extracellular infectivity titers (indicated by y axis breaks).
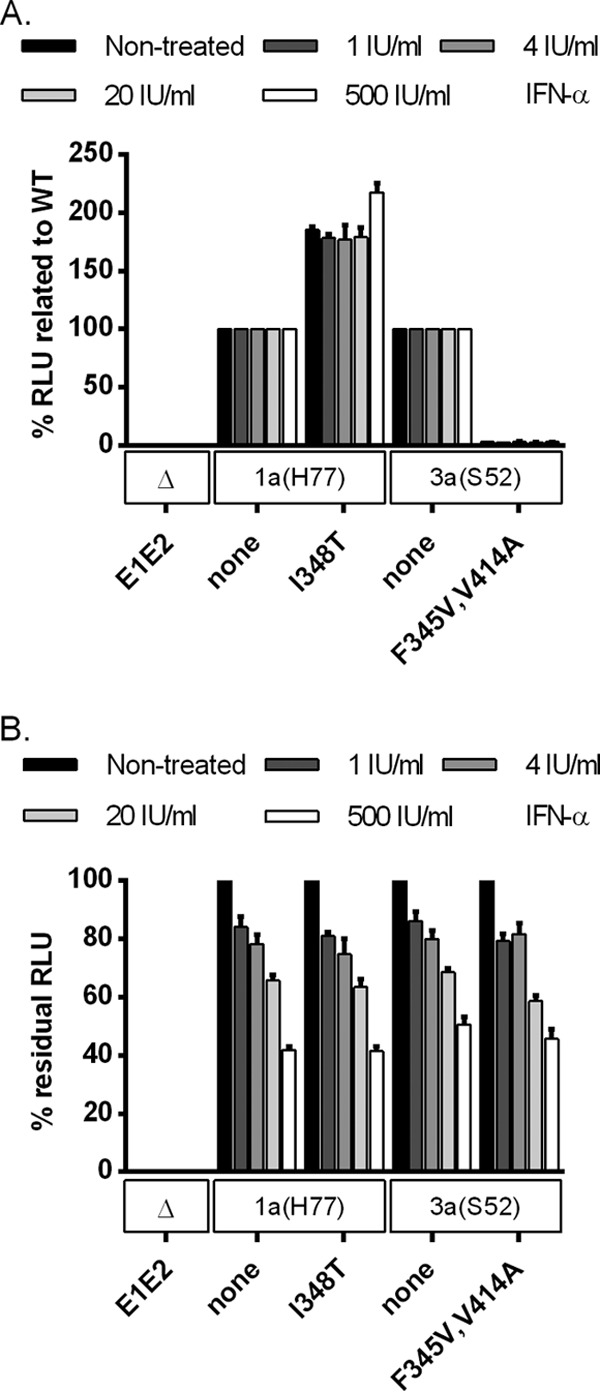
We then carried out experiments with HCVpp to investigate the effect of the identified envelope mutations on viral entry. Compared to 1a(H77) HCVpp, entry efficacy of 1a(H77)I348T HCVpp was approximately 2- to 5-fold increased (Fig. 11A and B and 12A). In contrast, compared to 3a(S52) HCVpp, entry efficacy of 3a(S52)F345V, 3a(S52)V414A, and 3a(S52)F345V,V414A HCVpp was decreased (Fig. 11C and D and 12A). IFN-α treatment decreased entry efficacy for tested 1a(H77) and 3a(S52) HCVpp in a similar, concentration-dependent manner (Fig. 12B).
Fig 11.
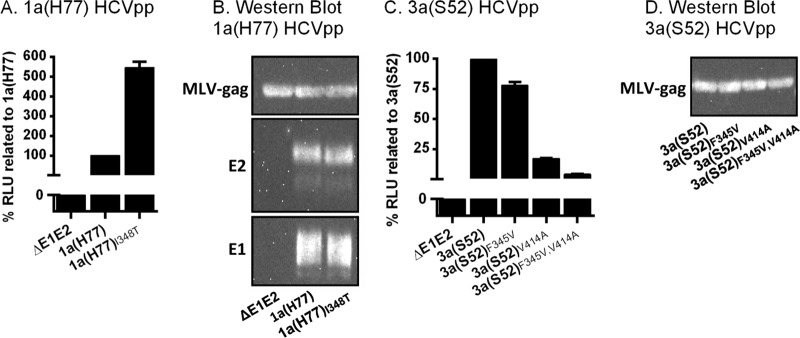
Impact of envelope mutations identified in 1a(H77) and 3a(S52) on HCVpp entry efficacy. (A and C) Huh7.5 cells were inoculated with HCVpp not harboring (ΔE1E2) or harboring 1a(H77) (A) or 3a(S52) (C) envelope protein with or without putative envelope resistance mutations as indicated. Seventy-two hours postinoculation, luciferase activity was measured. RLU of cultures inoculated with HCVpp with envelope mutations were related to RLU of cultures inoculated with HCVpp without envelope mutations and expressed as percent entry efficacy. (B) Western blot detection of E1, E2, and MLV-gag proteins in 1a(H77) HCVpp, enriched from HCVpp containing supernatant by ultracentrifugation. Data indicate similar expression of these 3 proteins for HCVpp of 1a(H77) and 1a(H77)I348T. (D) Western blot detection of MLV-gag protein in 3a(S52) HCVpp, enriched from HCVpp containing supernatant by ultracentrifugation. Data indicate similar expression of this protein for the 4 different 3a(S52) HCVpp. No antibodies were available for detection of 3a(S52) E1 and E2. The presented results are from 1 of 2 representative experiments.
Fig 12.
IFN-α showed a similar effect on entry of 1a(H77) and 3a(S52) original and mutant HCVpp. (A and B) Replicate Huh7.5 cell cultures were inoculated with HCVpp not harboring (ΔE1E2) or harboring 1a(H77) or 3a(S52) envelope protein with or without putative envelope resistance mutations as indicated. IFN-α treatment was carried out as described in Materials and Methods. Seventy-two hours postinoculation, luciferase activity was measured. (A) For each IFN-α2b concentration, RLU of cultures inoculated with mutated HCVpp were related to RLU of cultures inoculated with HCVpp without envelope mutations (WT). (B) Percent residual RLU was calculated by relating RLU of treated cultures to RLU of nontreated cultures infected with the same recombinant. Western blots indicated similar expression of E1, E2, and MLV-gag proteins in 1a(H77) HCVpp with and without envelope mutations that were enriched from HCVpp containing supernatant by ultracentrifugation. Further, similar expression of MLV-gag proteins in 3a(S52) HCVpp with and without envelope mutations was found.
Overall, these functional studies suggested that 348T increased 1a(H77) entry capacity, while 3a(S52) envelope mutations most likely promoted viral release. In these single-cycle assays, the effect of IFN-α treatment on 1a and 3a original versus mutant viruses was similar.
The degree of cell culture adaptation correlated with sensitivity to IFN-α.
To further investigate the correlation between viral fitness and IFN-α resistance, we carried out experiments with genotype 1a (isolate H77) and genotype 3a (isolate S52) Core-NS2 recombinants, showing different degrees of cell culture adaptation (24, 25, 27). Genotype 1a recombinants with culture adaptive mutation Q1247L or R1408W were previously reported to be less adapted than 1a(H77) with culture adaptive mutations V787A and Q1247L (25). Also, genotype 3a recombinants with culture adaptive mutations Y788C and Q1496L or K1398Q were previously reported to be less adapted than 3a(S52) with culture adaptive mutations I787S and K1398Q (24, 27). These less adapted recombinants showed greater sensitivity to IFN-α treatment than recombinants with greater fitness (Fig. 13).
Fig 13.
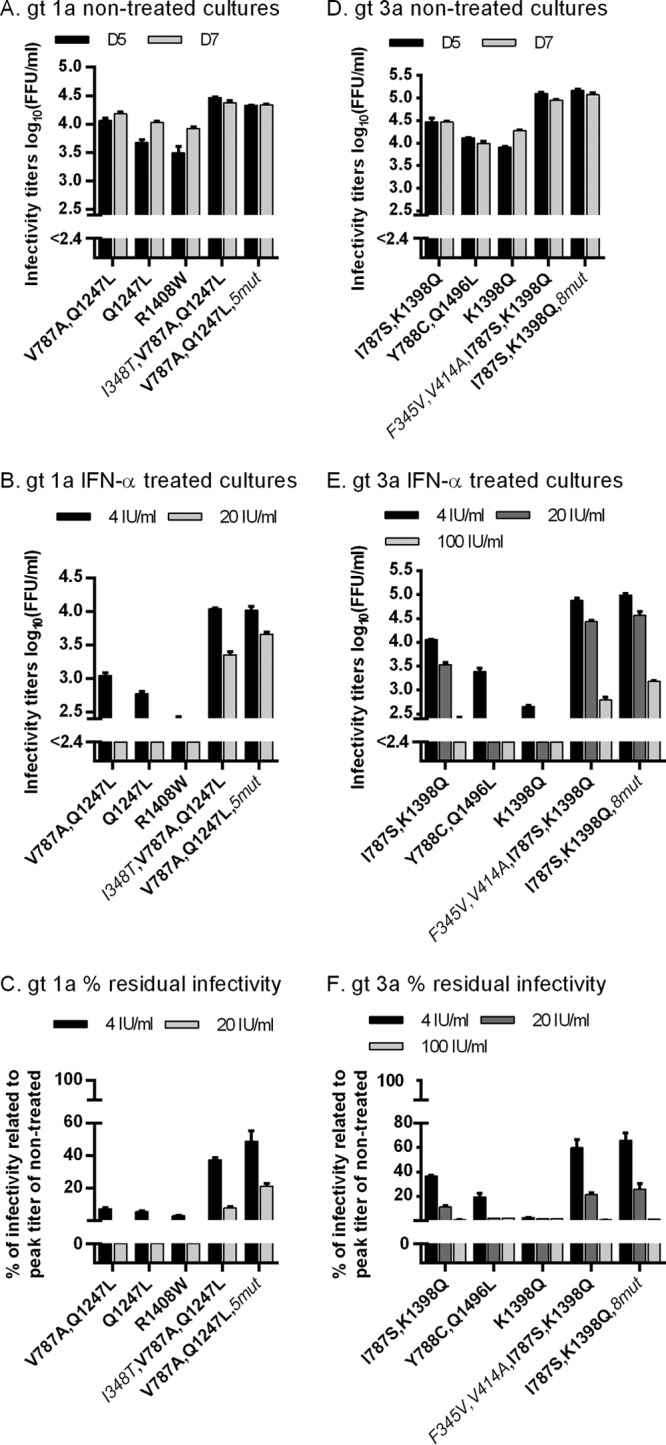
Degree of cell culture adaptation correlated with resistance to IFN-α. In vitro HCV RNA transcripts of the indicated recombinants were transfected into replicate Huh7.5 cultures. Genotype (gt) 1a isolate H77 recombinants had the following mutations: cell culture adaptive mutations V787A and Q1247L, resulting in the 1a(H77) recombinant that is used throughout this study; cell culture-adaptive mutation Q1247L; cell culture adaptive mutation R1408W; cell culture adaptive mutations V787A and Q1247L as well as putative IFN-α escape mutation I348T (the latter is in italics); and cell culture adaptive mutations V787A and Q1247L, as well as 5 putative IFN-α escape mutations, I348T, H923D, T2076A, M2259V, and S2788A (the latter are in italics). Genotype 3a isolate S52 recombinants had the following mutations: cell culture adaptive mutations I787S and K1398Q, resulting in the 3a(S52) recombinant used throughout this study; cell culture adaptive mutations Y788C and Q1496L; cell culture adaptive mutation K1398Q; cell culture adaptive mutations I787S and K1398Q as well as putative IFN-α escape mutations F345V and V414A (the latter are in italics); cell culture adaptive mutations I787S and K1398Q as well as 8 putative IFN-α escape mutations, F345V, V414A, I765V, H837Y, T2076A, E2381G, M2388T, and K2471R (the latter are in italics). From day 1 posttransfection, cultures were split every second day. For genotype 1a (A and B) and 3a (D and E) recombinants, supernatant infectivity titers are shown as means from 3 replicates with SEM. The lower limit of detection in the experiments shown was up to 2.4 log10 FFU/ml, indicated by y axis breaks. Cultures infected with genotype 1a (A) or 3a (D) recombinants were monitored without IFN-α treatment until viruses had spread to almost all cultured cells or until the percentage of infected cells decreased. Replicate cultures infected with genotype 1a (B) or 3a (E) recombinants were treated with the indicated concentrations of IFN-α on day 1 posttransfection and subsequently each time the cells were split. Cultures were monitored until the fittest virus had spread to almost all cells. Values shown are from day 5 posttransfection. Percent residual infectivity was determined by relating infectivity titers of treated cultures to peak infectivity titers of nontreated cultures infected with the same genotype 1a (C) or 3a (F) recombinant.
Analysis of positions 345, 348, and 414 in patients chronically infected with HCV genotypes 1a and 3a who failed to respond to IFN-α.
We amplified the complete envelope sequence of 8 genotype 1a-infected and 6 genotype 3a-infected patients who had failed to respond to IFN-α. We did not detect 345V, 348T, or 414A in any of these patients. Further, at these amino acid positions, amino acid residues typically found for HCV genotypes 1a and 3a were detected. Thus, at position 345 all analyzed genotype 1a patients had L, found in ∼80% of genotype 1a sequences in the 2008 web alignment in the Los Alamos database; all analyzed genotype 3a patients had F, which is found in all genotype 3a sequences. At position 348, all analyzed 1a and 3a patients had I, which is found in ∼95% of 1a and ∼60% of 3a sequences. Finally, at position 414, analyzed 1a patients had I or T (7 or 1 patient, respectively), which is found in ∼75 or <1% of 1a sequences, respectively; analyzed 3a patients had V or I (4 or 2 patients, respectively), which is found in ∼85 or 15% of 3a sequences, respectively.
DISCUSSION
Treatment with IFN-α allowed long-term culture of 1a(H77) and 3a(S52) Core-NS2 JFH1-based recombinants, resulting in viral escape. Reverse genetic studies of mutations present at viral breakthrough showed that primarily the identified envelope mutations increased viral fitness and conferred resistance to IFN-α, with resistance exceeding the observed effect on fitness. We confirmed a close link between viral fitness and IFN-α resistance by studying recombinants with various degrees of cell culture adaptation. For 1a(H77), I348T increased viral entry efficacy, while for 3a(S52), F345V and V414A apparently increased efficacy of release of infectious viral particles. The effects caused by these envelope mutations were apparently subtype specific. Given the importance of IFN for the innate antiviral immune system and treatment of chronic HCV infection, this study has important implications for our understanding of viral persistence and treatment failure.
Identification of viral genetic correlates of resistance to IFN-α has been a main research focus. However, experimental approaches were impaired by the lack of in vitro cell culture systems mimicking the entire HCV life cycle. Replicons only recapitulate viral replication and depend on selectable markers, resulting in selection of not only viral but also cellular phenotypes (31). In most replicon studies that aimed at inducing escape from interferon, no resistant cell clones could be established, or analysis of resistant cell clones showed that cellular factors were responsible for the observed phenotype (13–18). Results by Noguchi et al. indicated that accumulation of various mutations in NS3 and NS5A contributed to IFN resistance; however, no reverse genetic studies were done to identify the effect of specific mutations (39). Interestingly, here we found that HCVcc envelope mutations increasing efficacy of viral entry and release conferred IFN resistance. These genome regions and aspects of the viral life cycle could not be studied in subgenomic replicons, highlighting the importance of the availability of full viral life cycle cell culture systems for studies of interferon resistance of HCV. Earlier this year, a first approach to induce IFN resistance in a full viral life cycle genotype 2a culture system was reported (40). By serial passage in Huh7.5 cells in the presence of up to 10 IU/ml IFN-α2b but also in the absence of IFN-α2b, Perales et al. were able to select viral populations showing a certain degree of IFN-α resistance (40). Multiple mutations were observed in viruses passaged with or without IFN-α. Reverse genetic studies were limited to 3 mutations in E1, E2, and NS2, different from mutations found in our study, which were found to confer resistance to comparatively low concentrations (∼1 IU/ml) of IFN-α2b. It should be noted that in this prior study a reporter virus was used; reporter viruses might be characterized by comparatively high genetic instability (41).
Using reverse genetic studies, we revealed that mutations identified in the 1a(H77) and 3a(S52) envelope proteins primarily mediated increased fitness and conferred resistance to IFN-α. Mutations identified in other genes appeared to only slightly contribute to the observed phenotype (Fig. 2 to 6); alternatively, they might be spontaneously occurring genetic variations that were coselected during treatment. I348T in E1 of 1a(H77) and F345V/V414A in E1/E2 of 3a(S52) conferred resistance to IFN-α at concentrations of 20 to 100 IU/ml (Fig. 2 to 8). For 1a(H77), at 500 IU/ml, the concentration for which viral breakthrough had occurred (Fig. 1), the resistant phenotype was not observed (Fig. 4). Thus, changes of the host cell phenotype might have contributed to the higher resistance observed in primary escape cultures compared to cell cultures infected with recombinants with engineered resistance mutations. However, we demonstrated by Western blotting that interferon-stimulated genes (ISG), MxA, ISG15, protein kinase R (PKR), and phosphorylated PKR, known to have antiviral effects, were induced in 1a(H77)-infected Huh7.5 cells undergoing repetitive treatments with IFN-α throughout a 49-day period (data not shown). Also, compared to the replicon system, changes of the cell phenotype are less likely to occur in the culture system used in this study because of the absence of selective pressure by the use of antibiotics (2).
Typically, efficient infectious culture systems employ Huh7-derived cell lines, such as Huh7.5 cells, because of their increased permissiveness to HCV infection (31). Huh7 and Huh7.5 cells have a partially impaired cell innate immunity (42–44). Nevertheless, initiation of treatment with IFN-α, which is part of the IFN type I pathway, resulted in a decline in the percentage of HCV-infected cells followed by on-treatment virologic suppression for approximately 4 months, until viral breakthrough occurred (Fig. 1). Thus, Huh7.5 cells were responsive to IFN-α treatment. In addition, removal of IFN-α from culture medium, occasionally required to avoid viral eradication from infected cultures, resulted in an increase in the percentage of HCV-infected cells, indicating that IFN-α was responsible for the control of viral infection observed (Fig. 1). Supporting this observation, we and others previously showed that IFN-α treatment suppressed viral infection in Huh7.5 cells (24, 38, 40, 45–47). In addition, in cells from a typical transfection experiment, as shown in Fig. 2, 24 h after IFN-α treatment on day 5 posttransfection, PKR phosphorylation was observed in 1a(H77)-infected and in noninfected Huh7.5 cells in a concentration-dependent manner (data not shown). Finally, Huh7.5 cells expressed MxA, ISG15, PKR, and phosphorylated PKR upon repetitive stimulation with IFN-α for 49 days (data not shown). These observations confirm that Huh7.5 cells were responsive to IFN-α treatment.
Previously, we developed a high-throughput short-term assay to study the effect of antivirals on HCV infection 48 h after treatment (38, 46). Of note, in this assay concentration-response curves for treatment of 1a(H77) and 1a(H77)5mut with IFN-α were similar (data not shown). In the long-term assays carried out in this study, the greatest differences between original and mutant viruses were observed from day 4 of treatment (day 5 posttransfection) (Fig. 2 to 8). Of note, also in S29 cell and HCVpp assays only mimicking defined steps of the viral life cycle and precluding viral spread, the identified envelope mutations did not confer resistance to IFN-α (Fig. 10 and 12). This is in contrast to results in long-term Huh7.5 cell assays, where the identified mutations resulted in clear differences in residual infectivity when 20 IU/ml IFN-α was applied (Fig. 2 to 8) and in statistically significant differences in EC50, EC75, and EC90 when various concentrations of IFN-α were applied (Fig. 4C). Thus, the observed IFN-α-resistant phenotype apparently depended on viral spread.
For 1a(H77), primarily I348T in E1 increased viral fitness, apparent by an ∼0.5 log10 increase in infectivity titers (Fig. 2A). Slightly smaller differences between mutant and original 1a(H77) viruses were found when using supernatant Core levels as the readout, probably due to the absence of an additional infection cycle in this assay (Fig. 3A). In contrast, under IFN-α, 1a(H77)I348T showed 1.4 log10 greater infectivity titers and 1.2 log10 greater Core levels than 1a(H77) on day 11 posttransfection (Fig. 2B and 3B). For 3a(S52), primarily combination of F345V in E1 and V414A in E2 mediated similar effects (Fig. 6). Thus, the effect of identified envelope mutations on viral resistance exceeded the effect on viral fitness. Also, IFN-α concentrations required to inhibit HCV infection by 50, 75, and 90% were statistically significantly higher for the 1a(H77)I348T and 1a(H77)5mut mutants than for the original 1a(H77) virus (Fig. 4C). However, several observations suggest a positive correlation between viral fitness and IFN resistance. First, Sumpter et al. found that several replicon adaptive mutations in NS3, NS4B, NS5A, and NS5B selected without IFN pressure, increased replicon fitness, and conferred resistance to IFN-α (48). Second, specific replicon adaptive mutations were associated with a slower decrease in HCV RNA concentration during IFN-based therapy in patients (49). Third, viability of HCVcc typically depended on acquisition of cell culture adaptive mutations selected by serial viral passage (23–27, 30, 38, 41, 46, 47, 50–56). Mutations at positions 345, 348, and 414 were previously acquired by intergenotypic HCV recombinants and might contribute to cell culture adaptation (24, 26, 41, 56, 57). Fourth, studies of genotype 1a and 3a recombinants with different degrees of cell culture adaptation (24, 25, 27) revealed that increased viral fitness was associated with IFN-α resistance (Fig. 13). Finally, Perales et al. found that control virus populations serially passaged without IFN-α acquired multiple putative cell culture adaptive mutations and showed a certain level of IFN-α resistance (40). For the escape experiments done in our study (Fig. 1) such controls were not feasible, because viruses in nontreated control cultures rapidly spread to the majority of culture cells, followed by cell death and decline in the percentage of infected cells, as usually observed for efficient HCVcc (24, 27). In summary, the discussed observations suggest a close link between viral fitness and resistance to IFN-α.
This correlation of fitness and resistance is in contrast to findings for other drugs, such as novel directly acting antivirals. Mutations conferring resistance to this drug class often impair viral fitness (55, 58, 59), which can be rescued by coselection of compensatory mutations (58). Further, after termination of treatment with HCV protease inhibitors, in patients with treatment failure, resistance mutations were shown to revert to the wild-type sequence (60). In contrast, 1a(H77) mutants with I348T as well as 3a(S52) mutants with F345V and V414A did not acquire additional dominant mutations during viral passage with IFN-α. Also, engineered envelope mutations did not revert during viral passage without IFN-α. Thus, in the clinical setting, given a positive impact on viral fitness, mutations selected under IFN-α treatment might be expected to persist after termination of treatment and to spread in human populations.
Even though it is intriguing that Perales et al. also found envelope mutations to confer IFN-α resistance in vitro (40), the specific envelope mutations identified in this study previously were not reported to be associated with IFN-α resistance in vitro or in vivo. Also in this study, the identified envelope mutations were not detected in 8 genotype 1a and 6 genotype 3a patients who had failed treatment with IFN-α/ribavirin. In vitro studies suggested that several HCV proteins counteract response to IFN (reviewed in references 7 and 8). Thus, Core was shown to attenuate IFN-induced JAK/STAT signaling (7). Also, Core showed increased sequence variation in patients with breakthrough during IFN-based therapy (61). Recently, specific Core amino acid changes (at positions 70 and 91) were reported to be associated with IFN resistance for Japanese genotype 1b isolates in vivo (62, 63). These amino acid changes apparently also resulted in increased intracellular but not extracellular HCV RNA levels under IFN-α treatment in the JFH1 HCVcc system (64). Of note, insertion of R70Q and L91M, singly or in combination, into the JFH1-based recombinant with Core-NS2 of the Japanese genotype 1b isolate J4 did not result in IFN-α resistance in assays similar to those shown in Fig. 7 and 8 (data not shown). E2, specifically its C-terminal PKR-eIF2α phosphorylation homology domain (PePHD), was shown to block phosphorylation of PKR, mediating antiviral effects induced by IFN (7, 65). Sequence variation of the PePHD and HVR1 domains of E2 was suggested to influence outcome of IFN-α-based therapy in patients (7, 66). Further, regions flanking PePHD showed sequence variation in breakthrough patients (61). NS5A also was suggested to mediate IFN resistance, possibly interfering with PKR via the NS5A PKR-binding domain containing the IFN sensitivity determining region (ISDR) or interfering with interferon-induced 2′-5′-oligoadenylate synthetase (7). Sequence variation of NS5A ISDR received considerable attention as a determinant of IFN resistance, mainly for Japanese genotype 1b isolates (67, 68). However, other studies, including a study of patients with viral breakthrough during IFN treatment (61), suggested that sequence variation in other NS5A regions is associated with IFN treatment response (7, 8, 69). Of note, previously developed JFH1-based recombinants with 1b(J4) NS5A with and without sensitive-type ISDR mutations (46) responded similarly to IFN-α in experiments similar to those shown in Fig. 7 and 8 (data not shown). Thus, despite major research efforts, it remains difficult to consistently identify signature mutations associated with IFN resistance in vivo and in vitro. In contrast, for novel directly acting antivirals, distinct resistance mutations were identified in vivo as well as in vitro in several independent studies (38, 46, 58, 59). This discrepancy might be explained by the more complicated mode of action of IFN, relying on induction of various cellular effectors. Another explanation could be the proposed link between viral fitness and IFN resistance, with different mutations increasing fitness of different HCV isolates. This might also explain why several determinants of IFN resistance could only be identified in specific populations; thus, resistance mutations in Core and the ISDR were mainly identified in Japanese patients (62, 63, 67, 68). We previously showed that the effect of mutations on HCVcc fitness depended on the sequence context (26). In line with these previous findings, reverse genetic studies carried out here revealed that identified envelope mutations increased fitness and conferred IFN resistance to isolates of the same subtype but not of other subtypes or genotypes (Fig. 7 and 8).
Functional studies showed that I348T increased viral entry efficacy for 1a(H77) HCVpp (Fig. 11). Recently, the ISG interferon-induced transmembrane protein 1 (IFITM1) was shown to accumulate at hepatocyte tight junctions in the liver of HCV-infected patients during IFN therapy and to disrupt HCV entry through its interaction with HCV coreceptor CD81 and occludin (70). Also, a recent study suggested that certain envelope glycoprotein signatures, leading to alteration of host cell entry factor usage, were associated with failure of IFN-based therapy in genotype 1-infected patients (71). Supporting these findings, we found an obvious effect of IFN-α not only on viral replication/translation in S29 cells but also on viral entry in the HCVpp assay (Fig. 10 and 12). Thus, enhanced fitness, mediated by increased entry efficacy, might be a mechanism employed by HCV to escape IFN effector mechanisms. However, this effect might only be apparent in assays allowing for viral spread, since the effect of IFN-α treatment on original versus mutant HCVpp was similar (Fig. 12). Surprisingly, F345V and V414A seemed to impair 3a(S52) HCVpp entry efficacy (Fig. 11). Previous studies highlighted differences between HCVcc and HCVpp (72–74). Thus, 3a(S52) envelope mutations might have a different impact on entry of HCVpp than of HCVcc. However, our data suggest that combination of F345V and V414A promote release of infectious 3a(S52) from the host cell (Fig. 9), suggesting a different mechanism for mutations mediating 3a(S52) IFN resistance. It seems most likely that the fitness-enhancing effect of the 3a(S52) mutations, observed in culture systems mimicking the complete viral life cycle, is caused by enhanced viral release, while their negative effect on viral entry could be caused by the HCVpp system not reflecting the authentic entry process.
In conclusion, long-term treatment with IFN-α led to HCVcc viral escape. Reverse genetic studies revealed that identified mutations in the envelope proteins increased viral fitness, which was apparently mediated by increased efficacy of HCV entry or release. In assays allowing viral spread, fitness-enhancing envelope mutations conferred a level of IFN-α resistance which exceeded the observed fitness effect. In contrast to resistance to directly acting antivirals, IFN resistance apparently is positively linked to viral fitness, which might lead to selection of resistant variants in patient populations over time. Our data indicated that mutations conferring resistance to IFN are relatively specific to the infecting virus. This might explain why researchers have been struggling to clearly define genetic correlates of IFN resistance by sequence analysis of patient populations.
ACKNOWLEDGMENTS
We thank Santseharay Ramirez for sharing protocols and for discussions regarding the HCVpp assay; Troels Scheel for sharing protocols regarding the S29 cell assay; Anna-Louise Sørensen for technical assistance as well as Lubna Gahnem and Lotte Mikkelsen for laboratory assistance; Steen Ladelund for statistical advice; Jens Ole Nielsen and Ove Andersen for support (Copenhagen University Hospital, Hvidovre); Charles Rice (Rockefeller University), François-Loïc Cosset (Université de Lyon), Suzanne U. Emerson (National Institutes of Health), and Jean Dubuisson (Institut Pasteur de Lille) for reagents; and CTL Europe GmbH for customized software.
This study was supported by grants from Copenhagen University Hospital, Hvidovre (S.S. and J.M.G.), Region H Foundation (J.B. and J.M.G.), The Lundbeck Foundation (J.B. and J.M.G.), The Novo Nordisk Foundation (J.B. and J.M.G.), The Danish Council for Independent Research, Medical Science (J.B.), The A. P. Møller and Chastine Mc-Kinney Møller Foundation (J.B. and J.M.G.), The Danish Cancer Society (J.B. and J.M.G.), and a Ph.D. stipend from the Faculty of Health and Medical Sciences, University of Copenhagen (S.S.).
Footnotes
Published ahead of print 18 September 2013
REFERENCES
- 1.Mohd HK, Groeger J, Flaxman AD, Wiersma ST. 2013. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57:1333–1342 [DOI] [PubMed] [Google Scholar]
- 2.Gottwein JM, Bukh J. 2008. Cutting the Gordian knot–development and biological relevance of hepatitis C virus cell culture systems. Adv. Virus Res. 71:51–133 [DOI] [PubMed] [Google Scholar]
- 3.Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin IT, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A. 2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42:962–973 [DOI] [PubMed] [Google Scholar]
- 4.Hoofnagle JH, Mullen KD, Jones DB, Rustgi V, Di BA, Peters M, Waggoner JG, Park Y, Jones EA. 1986. Treatment of chronic non-A, non-B hepatitis with recombinant human alpha interferon. A preliminary report. N. Engl. J. Med. 315:1575–1578 [DOI] [PubMed] [Google Scholar]
- 5.Strader DB, Wright T, Thomas DL, Seeff LB. 2004. Diagnosis, management, and treatment of hepatitis C. Hepatology 39:1147–1171 [DOI] [PubMed] [Google Scholar]
- 6.Heim MH. 2013. Innate immunity and HCV. J. Hepatol. 58:564–574 [DOI] [PubMed] [Google Scholar]
- 7.Wohnsland A, Hofmann WP, Sarrazin C. 2007. Viral determinants of resistance to treatment in patients with hepatitis C. Clin. Microbiol. Rev. 20:23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asselah T, Estrabaud E, Bieche I, Lapalus M, De MS, Vidaud M, Saadoun D, Soumelis V, Marcellin P. 2010. Hepatitis C: viral and host factors associated with non-response to pegylated interferon plus ribavirin. Liver Int. 30:1259–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, Sulkowski M, McHutchison JG, Goldstein DB. 2009. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461:399–401 [DOI] [PubMed] [Google Scholar]
- 10.Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O'Huigin C, Kidd J, Kidd K, Khakoo SI, Alexander G, Goedert JJ, Kirk GD, Donfield SM, Rosen HR, Tobler LH, Busch MP, McHutchison JG, Goldstein DB, Carrington M. 2009. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 461:798–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Estrabaud E, Vidaud M, Marcellin P, Asselah T. 2012. Genomics and HCV infection: progression of fibrosis and treatment response. J. Hepatol. 57:1110–1125 [DOI] [PubMed] [Google Scholar]
- 12.Farci P, Strazzera R, Alter HJ, Farci S, Degioannis D, Coiana A, Peddis G, Usai F, Serra G, Chessa L, Diaz G, Balestrieri A, Purcell RH. 2002. Early changes in hepatitis C viral quasispecies during interferon therapy predict the therapeutic outcome. Proc. Natl. Acad. Sci. U. S. A. 99:3081–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aus dem Siepen M, Oniangue-Ndza C, Wiese M, Ross S, Roggendorf M, Viazov S. 2007. Interferon-alpha and ribavirin resistance of Huh7 cells transfected with HCV subgenomic replicon. Virus Res. 125:109–113 [DOI] [PubMed] [Google Scholar]
- 14.Naka K, Takemoto K, Abe K, Dansako H, Ikeda M, Shimotohno K, Kato N. 2005. Interferon resistance of hepatitis C virus replicon-harbouring cells is caused by functional disruption of type I interferon receptors. J. Gen. Virol. 86:2787–2792 [DOI] [PubMed] [Google Scholar]
- 15.Namba K, Naka K, Dansako H, Nozaki A, Ikeda M, Shiratori Y, Shimotohno K, Kato N. 2004. Establishment of hepatitis C virus replicon cell lines possessing interferon-resistant phenotype. Biochem. Biophys. Res. Commun. 323:299–309 [DOI] [PubMed] [Google Scholar]
- 16.Hazari S, Taylor L, Haque S, Garry RF, Florman S, Luftig R, Regenstein F, Dash S. 2007. Reduced expression of Jak-1 and Tyk-2 proteins leads to interferon resistance in hepatitis C virus replicon. Virol. J. 4:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu H, Nelson DR, Crawford JM, Liu C. 2005. Defective Jak-Stat activation in hepatoma cells is associated with hepatitis C viral IFN-alpha resistance. J. Interferon Cytokine Res. 25:528–539 [DOI] [PubMed] [Google Scholar]
- 18.Guo JT, Bichko VV, Seeger C. 2001. Effect of alpha interferon on the hepatitis C virus replicon. J. Virol. 75:8516–8523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 22.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408–7413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yi M, Ma Y, Yates J, Lemon SM. 2007. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 81:629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gottwein JM, Scheel TK, Jensen TB, Lademann JB, Prentoe JC, Knudsen ML, Hoegh AM, Bukh J. 2009. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49:364–377 [DOI] [PubMed] [Google Scholar]
- 25.Scheel TK, Gottwein JM, Jensen TB, Prentoe JC, Hoegh AM, Alter HJ, Eugen-Olsen J, Bukh J. 2008. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc. Natl. Acad. Sci. U. S. A. 105:997–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scheel TK, Gottwein JM, Carlsen TH, Li YP, Jensen TB, Spengler U, Weis N, Bukh J. 2011. Efficient culture adaptation of hepatitis C virus recombinants with genotype-specific core-NS2 by using previously identified mutations. J. Virol. 85:2891–2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gottwein JM, Scheel TK, Hoegh AM, Lademann JB, Eugen-Olsen J, Lisby G, Bukh J. 2007. Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology 133:1614–1626 [DOI] [PubMed] [Google Scholar]
- 28.Callens N, Ciczora Y, Bartosch B, Vu-Dac N, Cosset FL, Pawlotsky JM, Penin F, Dubuisson J. 2005. Basic residues in hypervariable region 1 of hepatitis C virus envelope glycoprotein e2 contribute to virus entry. J. Virol. 79:15331–15341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pestka JM, Zeisel MB, Blaser E, Schurmann P, Bartosch B, Cosset FL, Patel AH, Meisel H, Baumert J, Viazov S, Rispeter K, Blum HE, Roggendorf M, Baumert TF. 2007. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. U. S. A. 104:6025–6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carlsen TH, Scheel TK, Ramirez S, Foung SK, Bukh J. 2013. Characterization of hepatitis C virus recombinants with chimeric E1/E2 envelope proteins and identification of single amino acids in the E2 stem region important for entry. J. Virol. 87:1385–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russell RS, Meunier JC, Takikawa S, Faulk K, Engle RE, Bukh J, Purcell RH, Emerson SU. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 105:4370–4375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, Rice CM. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68:6147–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Albecka A, Montserret R, Krey T, Tarr AW, Diesis E, Ball JK, Descamps V, Duverlie G, Rey F, Penin F, Dubuisson J. 2011. Identification of new functional regions in hepatitis C virus envelope glycoprotein E2. J. Virol. 85:1777–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuiken C, Combet C, Bukh J, Shin I, Deleage G, Mizokami M, Richardson R, Sablon E, Yusim K, Pawlotsky JM, Simmonds P. 2006. A comprehensive system for consistent numbering of HCV sequences, proteins and epitopes. Hepatology 44:1355–1361 [DOI] [PubMed] [Google Scholar]
- 36.Combet C, Garnier N, Charavay C, Grando D, Crisan D, Lopez J, Dehne-Garcia A, Geourjon C, Bettler E, Hulo C, Le MP, Bartenschlager R, Diepolder H, Moradpour D, Pawlotsky JM, Rice CM, Trepo C, Penin F, Deleage G. 2007. euHCVdb: the European hepatitis C virus database. Nucleic Acids Res. 35:D363–D366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuiken C, Mizokami M, Deleage G, Yusim K, Penin F, Shin I, Charavay C, Tao N, Crisan D, Grando D, Dalwani A, Geourjon C, Agrawal A, Combet C. 2006. Hepatitis C databases, principles and utility to researchers. Hepatology 43:1157–1165 [DOI] [PubMed] [Google Scholar]
- 38.Gottwein JM, Scheel TK, Jensen TB, Ghanem L, Bukh J. 2011. Differential efficacy of protease inhibitors against HCV genotypes 2a, 3a, 5a, and 6a NS3/4A protease recombinant viruses. Gastroenterology 141:1067–1079 [DOI] [PubMed] [Google Scholar]
- 39.Noguchi T, Otsubaki T, Ando I, Ogura N, Ikeda S, Shimotohno K. 2008. Isolation and gene analysis of interferon alpha-resistant cell clones of the hepatitis C virus subgenome. Virology 375:424–432 [DOI] [PubMed] [Google Scholar]
- 40.Perales C, Beach NM, Gallego I, Soria ME, Quer J, Esteban JI, Rice C, Domingo E, Sheldon J. 2013. Response of hepatitis C virus to long-term passage in the presence of alpha interferon: multiple mutations and a common phenotype. J. Virol. 87:7593–7607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gottwein JM, Jensen TB, Mathiesen CK, Meuleman P, Serre SB, Lademann JB, Ghanem L, Scheel TK, Leroux-Roels G, Bukh J. 2011. Development and application of hepatitis C reporter viruses with genotype 1 to 7 core-nonstructural protein 2 (NS2) expressing fluorescent proteins or luciferase in modified JFH1 NS5A. J. Virol. 85:8913–8928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Jr, Lemon SM. 2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U. S. A. 102:2992–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li K, Chen Z, Kato N, Gale M, Jr, Lemon SM. 2005. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 280:16739–16747 [DOI] [PubMed] [Google Scholar]
- 44.Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li YP, Ramirez S, Gottwein JM, Bukh J. 2011. Non-genotype-specific role of the hepatitis C virus 5′ untranslated region in virus production and in inhibition by interferon. Virology 421:222–234 [DOI] [PubMed] [Google Scholar]
- 46.Scheel TK, Gottwein JM, Mikkelsen LS, Jensen TB, Bukh J. 2011. Recombinant HCV variants with NS5A from genotypes 1–7 have different sensitivities to an NS5A inhibitor but not interferon-alpha. Gastroenterology 140:1032–1042 [DOI] [PubMed] [Google Scholar]
- 47.Li YP, Ramirez S, Jensen SB, Purcell RH, Gottwein JM, Bukh J. 2012. Highly efficient full-length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc. Natl. Acad. Sci. U. S. A. 109:19757–19762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sumpter R, Jr, Wang C, Foy E, Loo YM, Gale M., Jr 2004. Viral evolution and interferon resistance of hepatitis C virus RNA replication in a cell culture model. J. Virol. 78:11591–11604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarrazin C, Mihm U, Herrmann E, Welsch C, Albrecht M, Sarrazin U, Traver S, Lengauer T, Zeuzem S. 2005. Clinical significance of in vitro replication-enhancing mutations of the hepatitis C virus (HCV) replicon in patients with chronic HCV infection. J. Infect. Dis. 192:1710–1719 [DOI] [PubMed] [Google Scholar]
- 50.Zhong J, Gastaminza P, Chung J, Stamataki Z, Isogawa M, Cheng G, McKeating JA, Chisari FV. 2006. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J. Virol. 80:11082–11093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li YP, Ramirez S, Gottwein JM, Scheel TK, Mikkelsen L, Purcell RH, Bukh J. 2012. Robust full-length hepatitis C virus genotype 2a and 2b infectious cultures using mutations identified by a systematic approach applicable to patient strains. Proc. Natl. Acad. Sci. U. S. A. 109:E1101–E1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jensen TB, Gottwein JM, Scheel TK, Hoegh AM, Eugen-Olsen J, Bukh J. 2008. Highly efficient JFH1-based cell-culture system for hepatitis C virus genotype 5a: failure of homologous neutralizing-antibody treatment to control infection. J. Infect. Dis. 198:1756–1765 [DOI] [PubMed] [Google Scholar]
- 53.Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. 2006. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. U. S. A. 103:2310–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Imhof I, Simmonds P. 2010. Development of an intergenotypic hepatitis C virus (HCV) cell culture method to assess antiviral susceptibilities and resistance development of HCV NS3 protease genes from HCV genotypes 1 to 6. J. Virol. 84:4597–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shimakami T, Welsch C, Yamane D, McGivern DR, Yi M, Zeuzem S, Lemon SM. 2011. Protease inhibitor-resistant hepatitis C virus mutants with reduced fitness from impaired production of infectious virus. Gastroenterology 140:667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gottwein JM, Jensen SB, Li YP, Ghanem L, Scheel TK, Serre SB, Mikkelsen L, Bukh J. 2013. Combination treatment with hepatitis C virus protease and NS5A inhibitors is effective against recombinant genotype 1a, 2a, and 3a viruses. Antimicrob. Agents Chemother. 57:1291–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McMullan LK, Grakoui A, Evans MJ, Mihalik K, Puig M, Branch AD, Feinstone SM, Rice CM. 2007. Evidence for a functional RNA element in the hepatitis C virus core gene. Proc. Natl. Acad. Sci. U. S. A. 104:2879–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sarrazin C, Zeuzem S. 2010. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138:447–462 [DOI] [PubMed] [Google Scholar]
- 59.Sarrazin C, Hezode C, Zeuzem S, Pawlotsky JM. 2012. Antiviral strategies in hepatitis C virus infection. J. Hepatol. 56(Suppl 1):S88–S100 [DOI] [PubMed] [Google Scholar]
- 60.Susser S, Vermehren J, Forestier N, Welker MW, Grigorian N, Fuller C, Perner D, Zeuzem S, Sarrazin C. 2011. Analysis of long-term persistence of resistance mutations within the hepatitis C virus NS3 protease after treatment with telaprevir or boceprevir. J. Clin. Virol. 52:321–327 [DOI] [PubMed] [Google Scholar]
- 61.Vuillermoz I, Khattab E, Sablon E, Ottevaere I, Durantel D, Vieux C, Trepo C, Zoulim F. 2004. Genetic variability of hepatitis C virus in chronically infected patients with viral breakthrough during interferon-ribavirin therapy. J. Med. Virol. 74:41–53 [DOI] [PubMed] [Google Scholar]
- 62.Akuta N, Suzuki F, Kawamura Y, Yatsuji H, Sezaki H, Suzuki Y, Hosaka T, Kobayashi M, Kobayashi M, Arase Y, Ikeda K, Kumada H. 2007. Predictors of viral kinetics to peginterferon plus ribavirin combination therapy in Japanese patients infected with hepatitis C virus genotype 1b. J. Med. Virol. 79:1686–1695 [DOI] [PubMed] [Google Scholar]
- 63.Akuta N, Suzuki F, Sezaki H, Suzuki Y, Hosaka T, Someya T, Kobayashi M, Saitoh S, Watahiki S, Sato J, Matsuda M, Kobayashi M, Arase Y, Ikeda K, Kumada H. 2005. Association of amino acid substitution pattern in core protein of hepatitis C virus genotype 1b high viral load and non-virological response to interferon-ribavirin combination therapy. Intervirology 48:372–380 [DOI] [PubMed] [Google Scholar]
- 64.Funaoka Y, Sakamoto N, Suda G, Itsui Y, Nakagawa M, Kakinuma S, Watanabe T, Mishima K, Ueyama M, Onozuka I, Nitta S, Kitazume A, Kiyohashi K, Murakawa M, Azuma S, Tsuchiya K, Watanabe M. 2011. Analysis of interferon signaling by infectious hepatitis C virus clones with substitutions of core amino acids 70 and 91. J. Virol. 85:5986–5994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. 1999. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science 285:107–110 [DOI] [PubMed] [Google Scholar]
- 66.Saludes V, Gonzalez-Candelas F, Planas R, Sola R, Ausina V, Martro E. 2013. Evolutionary dynamics of the E1-E2 viral populations during combination therapy in non-responder patients chronically infected with hepatitis C virus subtype 1b. Infect. Genet. Evol. 13:1–10 [DOI] [PubMed] [Google Scholar]
- 67.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Izumi N, Marumo F, Sato C. 1995. Comparison of full-length sequences of interferon-sensitive and resistant hepatitis C virus 1b. Sensitivity to interferon is conferred by amino acid substitutions in the NS5A region. J. Clin. Investig. 96:224–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Ogura Y, Izumi N, Marumo F, Sato C. 1996. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N. Engl. J. Med. 334:77–81 [DOI] [PubMed] [Google Scholar]
- 69.Yahoo N, Sabahi F, Shahzamani K, Malboobi MA, Jabbari H, Sharifi H, Mousavi-Fard SH, Merat S. 2011. Mutations in the E2 and NS5A regions in patients infected with hepatitis C virus genotype 1a and their correlation with response to treatment. J. Med. Virol. 83:1332–1337 [DOI] [PubMed] [Google Scholar]
- 70.Wilkins C, Woodward J, Lau DT, Barnes A, Joyce M, McFarlane N, McKeating JA, Tyrrell DL, Gale M., Jr 2013. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 57:461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schvoerer E, Moenne-Loccoz R, Murray JM, Velay A, Turek M, Fofana I, Fafi-Kremer S, Erba AC, Habersetzer F, Doffoel M, Gut JP, Donlin MJ, Tavis JE, Zeisel MB, Stoll-Keller F, Baumert TF. 2013. Hepatitis C virus envelope glycoprotein signatures are associated with treatment failure and modulation of viral entry and neutralization. J. Infect. Dis. 207:1306–1315 [DOI] [PubMed] [Google Scholar]
- 72.Helle F, Vieyres G, Elkrief L, Popescu CI, Wychowski C, Descamps V, Castelain S, Roingeard P, Duverlie G, Dubuisson J. 2010. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J. Virol. 84:11905–11915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fraser J, Boo I, Poumbourios P, Drummer HE. 2011. Hepatitis C virus (HCV) envelope glycoproteins E1 and E2 contain reduced cysteine residues essential for virus entry. J. Biol. Chem. 286:31984–31992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV. 2007. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J. Virol. 81:374–383 [DOI] [PMC free article] [PubMed] [Google Scholar]