

Abstract
Among the modes of transmission available to the cytomegalovirus (CMV) is sexual transmission, primarily via semen. Both male-to-female (M-F) and male-to-male (M-M) sexual transmission significantly contribute toward the spread of CMV infections in the global population. Semen plays an important role in carrying the viral particle that invades the vaginal or rectal mucosa, thereby initiating viral replication. Both semen and seminal plasma (SP) can enhance HIV-1 infection in cell culture, and two amyloid fibrils, semen-derived enhancer of viral infection (SEVI) and amyloids derived from the semenogelins (SEM amyloids), have been identified as seminal factors sufficient to enhance HIV-1 infection (J. Munch et al., Cell 131:1059–1071, 2007; N. R. Roan et al., Cell Host Microbe 10:541–550, 2011; F. Arnold et al., J. Virol. 86:1244–1249, 2012). Whether SP, SEVI, or SEM amyloids can enhance other viral infections has not been extensively examined. In this study, we found that SP, SEVI, and SEM amyloids strongly enhance both human CMV (HCMV) and murine CMV infection in cell culture. SEVI and SEM amyloids increased infection rates by >10-fold, as determined by both flow cytometry and fluorescence microscopy. Viral replication was increased by 50- to 100-fold. Moreover, viral growth curve assays showed that SP, SEVI, and SEM amyloids sped up the kinetics of CMV replication such that the virus reached its replicative peak more quickly. Finally, we discovered that SEM amyloids and SEVI counteracted the effect of anti-gH in protecting against CMV infection. Collectively, the data suggest that semen enhances CMV infection through interactions between semen amyloid fibrils and viral particles, and these interactions may prevent HCMV from being neutralized by anti-gH antibody.
INTRODUCTION
Topical microbicides that prevent sexual transmission of viruses could significantly reduce sexually transmitted diseases. People who are infected with human cytomegalovirus (HCMV) can shed the virus in their body fluids, including semen (1, 2). HCMV replicates in the genital tract, is sexually transmitted, and is highly prevalent worldwide (3). Viral load in semen is directly related to the transmission of HCMV from male-to-male (M-M) and from male-to-female (M-F) (1). In the United States, ca. 30 to 50% of women have never been infected with HCMV. About 1 to 4% of previously uninfected women are infected with HCMV during pregnancy. Upon infection, about one-third of pregnant women will pass HCMV to their fetuses or infants (4, 5). HCMV can cause birth defects, making it a significant public health problem (6). In addition, HCMV infection causes life-threatening diseases in immunocompromised hosts, such as individuals with HIV/AIDS, and is associated with HIV disease progression in both treated and untreated individuals (7, 8).
No effective drugs against CMV-mediated diseases in infants are available, and no vaccine is effective in preventing CMV infection. For these reasons, fresh approaches for developing microbicides effective against CMV could have important benefits for the health of both adults and infants. Identifying risk factors for the transmission of CMV during sexual intercourse and understanding how semen is involved in the transmission of CMV are important elements in the development of innovative strategies against CMV infection, especially in terms of designing nontoxic, effective topical microbicides against the virus. Although it is apparent that semen is an important carrier of HCMV, the effects of semen on CMV transmission remain unknown.
Semen contains proteolytic cleavage products of prostatic acid phosphatase (PAP) and semenogelin (SEM) that form amyloid fibrils in semen. The PAP-derived amyloids were named semen-derived enhancer of viral infection (SEVI) and were the first semen amyloids shown to enhance HIV infection (9). A subsequently identified second set of peptides that form HIV-enhancing amyloid fibrils are derived from SEM and referred to as SEM amyloids (10). Whether other sexually transmitted viral infections can be enhanced by seminal plasma (SP) or semen amyloids has remained largely unexplored.
In the present study, we discovered that SP, SEVI, and SEM amyloids can enhance both HCMV and murine CMV (MCMV) infection of permissive cells. We also observed that the fibrils can interact directly with viral particles and protect viruses from being neutralized by antibodies against glycoprotein H (gH).
MATERIALS AND METHODS
Tissue culture and viruses.
NIH 3T3 (from the American Type Culture Collection [ATCC]), U-251 MG, and MRC-5 (ATCC no. CCL171) cells, permissive to infection by MCMV and HCMV, respectively, were maintained in Dulbecco Modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 1% penicillin-streptomycin.
MCMVE5gfp was generated from the BACmid Sm3fr (11) by tagging green fluorescent protein (GFP) to the C terminus of IE3 (end of exon 5) (12). HCMVgfpSVH was made by tagging GFP to the N terminus of IE1 and IE2 using bacterial artificial chromosome (BAC) (13) techniques. Briefly, we inserted galK between the first and second amino acid codons of the MIE gene. Then, the galK was replaced with the open reading frame of GFP so that GFP and the MIE genes were fused in frame. The BAC DNA was sequenced and confirmed to be correct and transfected into MRC-5 cells to produce the HCMVgfpSVH virus.
Reagents.
SEVI was synthesized by the genomic and the proteomics core laboratories at the University of Pittsburgh. SEM amyloids (amino acid residues 49 to 107) (10) were synthesized by Celtek Peptides. Synthetic peptides were dissolved in phosphate-buffered saline (PBS) or serum-free MEM and agitated overnight at 1,400 rpm at 37°C using an Eppendorf Thermomixer, as described previously (9). Aβ(1-42) amyloids (catalog no. A9810) were purchased from Sigma-Aldrich (St. Louis, MO). Semen samples were pooled from 10 different individuals. To generate seminal plasma, the pooled semen samples were centrifuged at 700 × g for 5 min, and the supernatant was divided into aliquots and stored at −80°C and until use. The process of obtaining and using of semen samples and the informed consent forms were fully reviewed and approved by the committee on human research at Ponce School of Medicine and Health Sciences before the initiation of the study (IRB number 081106-YY).
Antibodies.
The antibodies used for Western blotting (WB) and immunofluorescence (IF) are listed below. A monoclonal antibody against tubulin (T-9026) was purchased from Sigma-Aldrich (1:1,000 for WB); polyclonal antibodies against GFP (sc-8334) and monoclonal antibodies against HCMV pp65 (3A12), gB (2F12), and gH (monoclonal antibody 0861) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA; 1:1,000 for WB to probe GFP); monoclonal antibodies against MCMV IE1 and E1 were provided by Stipan Jonjic (Croatia; 1:50 for IF and 1:200 for WB) (14). The monoclonal antibodies against HCMV pp65, MCMV gB, m25, and gH were generous gifts from J. D. Shanley (University of Connecticut; 1:250 for WB) (15, 16). The monoclonal antibody against HCMV IE1/2 (MAB810) was purchased from Sigma-Aldrich. The rabbit antibody against HCMV UL112/113 was a gift from J. H. Ahn (Korea University, Seoul, Korea).
Purification of MCMV and HCMV.
MCMV and HCMV were amplified in NIH 3T3 and MRC-5 cells, respectively. The viral supernatant was centrifuged at 8,000 × g for 20 min to remove cell debris. The clarified medium was transferred into SW27/28 Ultraclear centrifuge tubes that were underlain with 7 ml of 20% sorbitol buffer (20% d-sorbitol, 50 mM Tris-HCl [pH 7.2], and 1 mM MgCl) and centrifuged at 55,000 × g for 1 h. The purified viral pellet was resuspended in PBS.
Viral entry assay.
HCMV or MCMV was incubated with MEM (medium control) or with SEVI or SEM amyloid at 37°C for 1 h. The mixtures were then used to infect MRC-5 or NIH 3T3 cells after the cells had reached 80% confluence. Six hours later, the cells were fixed and immunostained for pp65 (HCMV tegument protein) or m25 (tegument protein of MCMV). Total cells and pp65-positive (or m25-positive) cells were counted by flow cytometry. Mock-infected cells treated with SEVI and SEM amyloids were used to control for autofluorescence.
To assess the effect of the amyloids after viral entry, MRC-5 cells or NIH 3T3 cells were infected with HCMV or MCMV for 2 h and washed three times with MEM to remove surface-associated viral particles. The cells were then exposed to MEM (buffer control) or to SEVI or SEM amyloids. After 24 h, whole-cell lysates were collected for WB analyses to detect IE1 and IE2 (for HCMV) or to detect IE1 and IE3 (for MCMV).
Amyloid fibril-virus binding assay.
Amyloid fibrils are structures that can be pelleted by low-speed centrifugation (e.g., 1,000 rpm on a table centrifuge) (17, 18). Purified viral particles (HCMV or MCMV) were incubated with 50 μg of SEVI or SEM amyloid fibrils at 37°C for 1 h. Controls for the experiment included virus in the absence of amyloids and virus incubated with 50 μg of Aβ(1-42) amyloids/ml (Sigma, catalog no. A9810). The samples were then centrifuged at 1,000 rpm for 5 min on a tabletop centrifuge (Eppendorf, model 5424). The pellets were washed twice with serum-free MEM and resuspended in PBS. The resuspended pellets were mixed with the same volume of 2× Laemmli buffer. After a 5-min heat treatment at 95°C, the proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by WB, as described below.
Immunoblot analysis.
Proteins were separated by SDS-PAGE (10 to 20 μg loaded per lane), transferred to nitrocellulose membranes (Amersham, Inc., Piscataway, NJ), and blocked with 5% nonfat milk for 60 min at room temperature. The membranes were incubated overnight at 4°C with primary antibody, followed by incubation with a horseradish peroxidase-coupled secondary antibody and detection with enhanced chemiluminescence (Pierce, Rockford, IL), according to standard methods. Membranes were stripped with stripping buffer (100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl [pH 6.8]), washed with PBS–0.1% Tween 20, and used to detect additional proteins.
Cell visualization by confocal microscopy.
The viruses used in the present study express GFP-fused immediate-early (IE) proteins (GFP-IE3 for MCMV and GFP-IE1 for HCMV). To visualize GFP-expressing cells, the cells were seeded on coverslips, infected for 12 h, washed twice with PBS, fixed in 1% paraformaldehyde for 10 min at room temperature, washed two more times with PBS, and then stained with DAPI. Cells were examined at ×10 magnification with a Leica TCS SPII confocal laser scanning system equipped with a water-cooled argon-krypton laser. Two channels (DAPI [(4′,6′-diamidino-2-phenylindole] and GFP) were recorded sequentially. DAPI staining was used to show the total number of cells recorded.
Flow cytometry.
To quantify the infection efficiency, cells were infected for 12 h with GFP reporter virus, treated with trypsin, and resuspended in PBS. The cells were then analyzed by a FACSCalibur system with two lasers and four channels (BD Biosciences) to detect total cell numbers and cells with GFP fluorescence. Mock-infected cells with or without treatment with amyloids served as autofluorescence controls.
PFU assay.
Virus titers were determined by the plaque assay, essentially as described previously (12), but with some slight modifications. Supernatants containing serially diluted viral particles were added to confluent NIH 3T3 (MCMV) or MRC-5 (HCMV) monolayers in six-well plates. After adsorption for 2 h, the medium was removed, and the cells were washed twice with serum-free DMEM and overlaid with phenol-free DMEM containing 5% FCS, 0.5% low-melting-point agarose (Gibco), and 1% penicillin-streptomycin. The numbers of plaques that were stained red were counted and are reported as PFU/ml. Mean PFU were determined after averaging the numbers of PFU from different dilutions. A Student t test was used to statistically analyze differences between the groups; a P value lower than 0.005 was used as the threshold for a significant difference.
RESULTS
SEVI and SEM amyloids increase CMV infection rates.
Herpesviruses are commonly detected in semen samples, with CMV being the most prevalent virus. SEVI and SEM amyloids have been demonstrated to enhance HIV infection (9, 10). To determine whether these amyloids can also enhance CMV infection, we used HCMVgfpSVH and MCMVE5gfp (12, 19), both of which express MIE proteins fused to GFP, thereby allowing infection to be assessed by flow cytometry (Fig. 1) and fluorescence microscopy (Fig. 2). Infections were carried out in permissive cells (MRC-5 for HCMVgfpSVH and NIH 3T3 for MCMVE5gfp).
Fig 1.

Flow cytometry to detect viral infection rate. (A) MCMV infection in NIH 3T3 cells. NIH 3T3 cells were cultured in a six-well plate. When the cells were 80% confluent, they were mock infected or infected with MCMVE5gfp (12) at an MOI of 0.1. Virus was preincubated with 0, 2, 5, or 10 μg of SEM amyloids or SEVI/ml for 1 h at 37°C before infection. The cells were fixed at 16 h postinfection and measured by flow cytometry to detect the percentage of GFP-positive (infected) cells. The percentage of infected cells is shown in the lower right-hand corner of each plot. (B) HCMV infection in MRC-5 cells. MRC-5 cells were mock infected or infected with HCMVgfpSVH at an MOI of 0.1 for 16 h. Prior to infection, the virus was incubated with 0, 2, 5, or 10 μg of SEM amyloids or SEVI/ml at 37°C for 1 h. Infection rates were detected by flow cytometry, as described in panel A, and the percentage of infected cells is shown in the lower right-hand corner of each plot. Mock-infected cells in the absence or presence of 10 μg of SEM/ml or 10 μg of SEVI/ml were used to control for autofluorescence. These experiments were performed in duplicate, with one set of the results reported in the figure. The results are representative of one of three independent experiments.
Fig 2.

Visualization of HCMV infection in U-251 MG cells. (A to C) The epithelial cell-like cell line U-251 MG was infected with HCMVgfpSVH at an MOI of 0.5 for 16 h in the absence of amyloids (A1 to A3) or after pretreatment with either 10 μg of SEM amyloids/ml (B1 to B3) or 10 μg of SEVI/ml (C1 to C3). The cells were fixed with 1% paraformaldehyde and stained with DAPI. The slides were observed under a fluorescence microscope (×10 amplification lens), and pictures were taken to show infected cells (GFP, A1 to C1) and total cells (DAPI, A2 to C2). The merged pictures are shown in A3 to C3. The percentages of GFP-positive (infected) cells are shown in the upper right-hand corners of A1 to C1. Scale bars, 20 μm. (D) Imaging experiments were performed three independent times. We counted a total of 1,000 cells for each experiment and averaged the percentages of GFP-positive cells. The averages and standard errors are shown in this panel. Statistically significant differences are indicated at the top (P < 0.005).
For MCMVE5gfp infection of NIH 3T3, we treated the virus in the presence of titrations of SEVI or SEM amyloids at 37°C for 1 h and then added the pretreated virus to NIH 3T3 cells (multiplicity of infection [MOI] = 0.1). At 16 h after infection, the cells were washed, treated with trypsin, fixed, and quantitated by flow cytometry. Mock-infected cells with or without amyloid treatment served as autofluorescence controls. As can be seen in Fig. 1A, SEVI and SEM amyloids both strongly enhance MCMV infection in a dose-dependent manner. The percentages of MCMVE5gfp-infected NIH 3T3 cells are indicated in the lower right-hand corners. In the absence of amyloid treatment, the MCMV infection rate was 2.42%. As the amount of SEM amyloids was increased from 2 to 10 μg/ml, infection rates increased from 7.8 to 29.2%. Similarly, increasing SEVI levels from 2 to 10 μg/ml increased infection rates from 6.23 to 23.5%.
For HCMV infection of MRC-5 cells, cell-free virus was treated at 37°C for 1 h with different titrations of SEVI or SEM amyloids (Fig. 1B). Pretreated HCMV was then added to MRC-5 cells in a six-well plate. Sixteen hours later, cells were treated with trypsin and fixed with 1% paraformaldehyde, and the total cells and GFP-positive cells were quantified. As can be seen in Fig. 1B, the basal infection rate in the absence of SEVI or SEM amyloid was 1.68% and increased to 18.2% after treatment with 10 μg of SEM amyloids/ml and to 18.5% after treatment with 10 μg of SEVI/ml.
SEVI and SEM amyloids enhance HCMV infection of U-251 MG epithelial cell-like cells.
HCMV can infect epithelial cells in vivo (8). In order to determine whether SEVI or SEM amyloid can enhance CMV infection of epithelial cell-like cells, we next examined HCMV infection of U-251 MG cells in the absence or presence of semen amyloids. U-251 MG cells seeded on coverslips were infected with HCMVgfpSVH at an MOI of 0.5 for 16 h and then fixed with 1% paraformaldehyde. After a PBS wash, the cells were stained with DAPI to identify nuclei. The coverslips were transferred onto slides and then visualized using confocal microscopy. CMV-infected cells were identified by GFP expression, whereas DAPI was used to quantify the total number of cells. Consistent with the flow cytometry data, HCMV infection was drastically enhanced by SEVI and SEM amyloids (Fig. 2). Whereas basal infection levels were low in the absence of semen amyloids (Fig. 2A), infection was substantially increased in the presence of either SEM amyloids (Fig. 2B) or SEVI amyloids (Fig. 2C). The percentages of GFP-positive cells are shown in the upper right-hand corners of plots in Fig. 2A1 to C1. A total of 1,000 cells were counted in each experiment to obtain the percentage of GFP-positive cells, and the numbers were averaged from three independent experiments. As shown in Fig. 2D, the differences between the virus-only group and the SEVI- or SEM amyloid-treated groups were significant (P < 0.005).
Viral protein production is enhanced by SP, SEVI, and SEM amyloids.
To determine whether the enhancement of CMV infection mediated by semen amyloid fibrils results in increased viral protein production, we infected NIH 3T3 or MRC-5 cells with MCMV or HCMV, respectively, and detected viral protein production by WB. Four groups of cells are shown in Fig. 3: (i) virus infection alone (that is, virus not exposed to SP or amyloids), (ii) virus treated with SP at a dilution of 1:1,000, (iii) virus treated with SEM amyloids (5 μg/ml), and (iv) virus treated with SEVI (5 μg/ml). At 24 h after infection, whole-cell lysates were fractionated by SDS-PAGE and analyzed by WB with anti-CMV antibodies (Fig. 3). The immunoblotted viral proteins include (i) the IE proteins IE1 and IE3-GFP for MCMV and IE1 and IE2 for HCMV (the bands above IE1 and IE2 are SUMOylated IE1 or IE2) and (ii) the very early proteins M112-113 (for MCMV) and UL112-113 (for HCMV). Viral protein production was significantly increased by SP, SEM amyloids, and SEVI. At the doses tested, SP had the lowest viral-enhancing effect on viral protein production. This, however, was probably caused by the low concentrations of SP used in these studies, chosen since high concentrations of SP can lead to marked cytotoxicity (20). To quantify the relative increases in viral protein production by SP, SEM, and SEVI, we compared the density of the IE3 bands for MCMV (or the IE1 bands for HCMV) relative to virus alone and normalized all signals to tubulin. The fold increases in IE3 levels for MCMV (left) and IE1 for HCMV (right) are indicated. As shown below the corresponding Western blots, SEVI was superior to SEM amyloids in enhancing expression of both IE3 (MCMV, left) and IE1 (HCMV, right).
Fig 3.
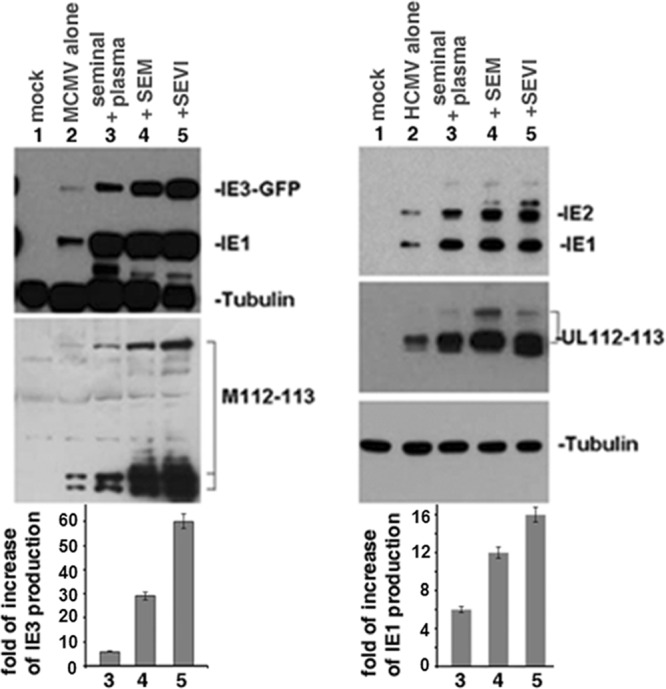
Western blot assay to detect viral protein production. (Left) NIH 3T3 cells were mock infected or infected with either MCMVE5gfp alone (MOI = 0.1) or MCMVE5gfp treated with SP, SEM amyloids, or SEVI for 24 h. Whole-cell lysates were prepared for Western blot with antibodies against GFP (IE3-GFP), IE1, M112-113, and tubulin (as loading control). (Right) MRC-5 cells were mock infected or infected either with HCMV alone or HCMV treated with SP, SEM amyloids, or SEVI; all infections were carried out for 24 h at an MOI of 0.1. Whole-cell lysates were prepared for Western blot with antibodies against IE1, IE2, UL112-113, and tubulin. By comparing the density of the IE3 bands of MCMV or the IE1 bands of HCMV with that of the virus alone (with all intensities normalized to the tubulin control), the fold increase of IE3 levels for MCMV (left) and that of IE1 for HCMV (right) were calculated (Quantity One 4.5.0 software; Bio-Rad Laboratories, Richmond, CA). Normalized IE3 (MCMV, left) or IE1 (HCMV, right) levels are shown below the corresponding Western blots.
SEVI and SEM amyloids enhance CMV entry.
To determine whether the enhancement of CMV infection by the amyloids occurs via increasing viral entry, we performed a viral entry assay. We incubated HCMV or MCMV with 10 μg of SEVI or SEM amyloid/ml at 37°C for 1 h. Viral mixtures were then used to infect MRC-5 or NIH 3T3 cells at an MOI of 0.1 for 6 h. The cells were fixed and examined for tegument protein expression (pp65 for HCMV and m25 for MCMV) by flow cytometry (Fig. 4A). Because tegument proteins enter cells along with viral particles and their de novo production occurs later than 24 h postinfection, they can be used as biomarkers for viral entry. Amyloid-treated cells in the absence of virus were used as controls for the autofluorescence. As shown in Fig. 4A, both SEVI and SEM amyloids increased the percentage of cells that are positive with tegument proteins upon infection, suggesting that these amyloids increased CMV entry into target cells.
Fig 4.
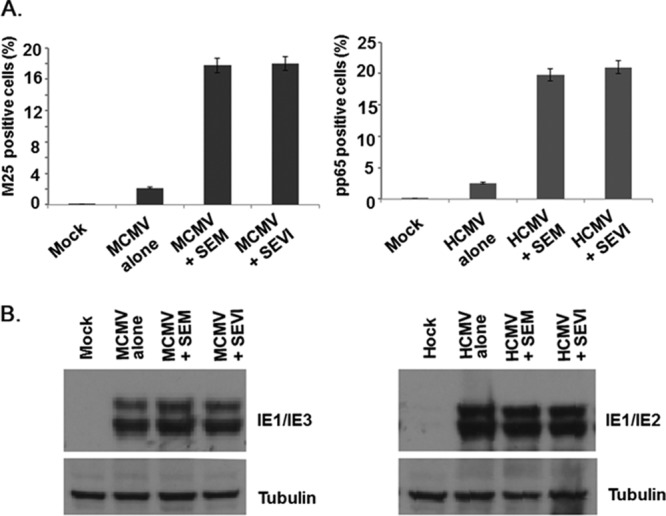
Viral entry assay. (A) MCMV and HCMV infection rate. HCMV or MCMV was incubated with 10 μg of SEVI or SEM amyloids/ml or MEM (as a buffer control) at 37°C for 1 h. Pretreated virions were used to infect MRC-5 or NIH 3T3 cells (MOI = 0.1). Six hours later, the cells were fixed with 1% paraformaldehyde and immunostained for pp65 (HCMV tegument protein, right) or m25 (tegument protein of MCMV, left). The total cells and pp65-positive (or m25-positive) cells were counted by flow cytometry. Mock-infected cells treated with SEVI and SEM amyloids were used to control for autofluorescence. Based on statistical analysis, the average values (± standard deviations) of duplicate measurements from three independent experiments that yielded similar results were determined. (B) MRC-5 cells or NIH 3T3 cells were infected with HCMV or MCMV for 2 h and washed three times with MEM. The cells were then treated with MEM, with SEVI, or with SEM amyloids. After 24 h, whole-cell lysates were collected for Western blot analysis to assess for IE1 and IE2 (for HCMV, right) or IE1 and IE3 (for MCMV, left) expression.
To provide further support for the notion that the amyloids increased CMV infection at the stage of viral entry, we tested whether the amyloids could increase viral gene expression if added to the cell culture postentry. We infected MRC-5 or NIH 3T3 cells with HCMV or MCMV at an MOI of 0.1 for 2 h, and then washed the cells three times with MEM to remove surface-associated virions. A final concentration of 10 μg of SEVI or SEM amyloids/ml was then added to the cells. Whole-cell lysates were collected at 24 h postinfection and assessed for viral protein expression (IE1 and IE2 for HCMV; IE1 and IE3 for MCMV). As shown in Fig. 4B, neither SEVI nor SEM amyloids enhanced viral gene expression when they were added after infection had been established. These experimental results further confirm that SEVI and SEM amyloids enhance CMV infection by increasing viral entry.
Viral replication is accelerated and enhanced by SP, SEVI, and SEM amyloids.
We then examined whether viral replication can be enhanced by SP and semen components. Five groups of cultures were set up and infected with virus that was preincubated at 37°C for 1 h with the following: (i) mock treatment (CMV alone, no SEM or SEVI), (ii) SP (1:1,000), (iii) SEM amyloids (5 μg/ml), (iv) SEVI (5 μg/ml), and (v) 5 μg of SEM/ml plus 5 μg of SEVI/ml. Infection of both NIH 3T3 and MRC-5 cells were carried out at an MOI of 0.1. Media were collected every day for 6 days (for MCMV) or 9 days (for HCMV) to monitor CMV replication by PFU counts (Fig. 5). Collected viral samples were freeze-thawed three times to release the viral particles and centrifuged to pellet cellular debris. Supernatants were assayed for viral production in NIH 3T3 (for MCMV) and MRC-5 (for HCMV) cells. As shown in Fig. 5, both MCMV (lower) and HCMV (upper) infections were strongly enhanced by a 1:1,000 dilution of SP (20-fold for MCMV and 10-fold for HCMV), 5 μg of SEM amyloids/ml (100-fold for MCMV and 50-fold for HCMV), and 5 μg of SEVI/ml (50-fold for both viruses). The less-pronounced effect on viral replication by SP might be caused by the lower amount of SEVI and SEM amyloids in the diluted SP. According to some estimations, the concentration of SEVI in semen is ∼35 μg/ml (9) and that of SEM amyloids 90 to 3,000 μg/ml (10), suggesting that the diluted SP used in this experiment contained only 0.035 μg of SEVI/ml and 0.09 to 3 μg of SEM amyloid/ml. High concentrations of SP were not used in this experiment since such concentrations have been shown to be toxic to cell culture when used for prolonged periods (20). We also tested whether SEM amyloid and SEVI could have a synergistic effect on enhancing CMV infection. Combining SEVI and SEM amyloids led to an additive, and not a synergistic, effect on enhancing HIV infection.
Fig 5.
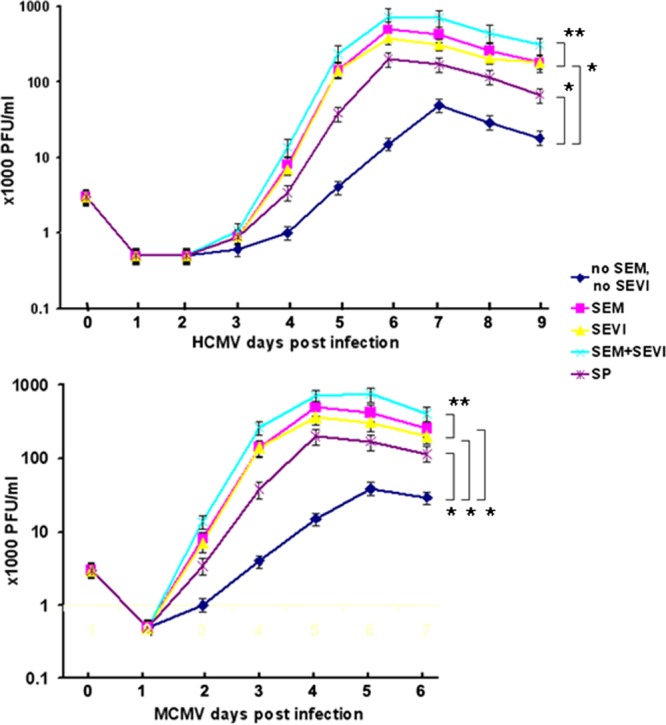
Viral growth curve assay. (Upper panel) HCMV infection of MRC-5 cells. MRC-5 cells were infected with virus alone or with virus treated with SP (1:1,000 dilution), SEM amyloids (5 μg/ml), SEVI (5 μg/ml), or SEM amyloids (5 μg/ml) plus SEVI (5 μg/ml) at an MOI of 0.1 for 24 h (as indicated). Each day for a period of 9 days, one well of each culture was collected (along with medium) and stored at −80°C. The collected cells (with medium) were freeze-thawed for three cycles and then centrifuged at 8,000 × g for 20 min to remove the cellular debris. To perform a PFU assay, a 25-μl supernatant of each sample was used to infect the MRC-5 cells in triplicates. (Lower panel) MCMV infection in NIH 3T3 cells. The procedure is the same as that used for HCMV infection of MRC-5 cells, except that the infection period was 6 days. A Student t test was used to statistically analyze the difference between the groups versus virus alone (*, P < 0.001) and that of the groups of SEM+SEVI versus SEM or SEVI (**, P < 0.005).
SEVI and SEM amyloids interact with CMV viral particles.
One mechanism by which semen amyloids enhance viral infection is promoting the binding of virions (17, 18) to cellular targets. SEVI and SEM amyloids directly interact with HIV particles to form fibril-virus complexes (9, 10, 17). We next sought to determine whether SEVI and SEM amyloids could also directly interact with CMV particles. To answer the question, we performed a viral binding assay. CMV viral particles (5 × 107 particles/ml) were incubated (i) in the absence of added factors, (ii) with 50 μg of Aβ(1-42) amyloids/ml, (iii) with 50 μg of SEVI/ml, or (iv) with 50 μg of SEM amyloid fibrils/ml. All treatments were carried out at 37°C for 1 h. The mixtures were then centrifuged at 700 × g for 5 min, and the resulting pellets were washed twice with serum-free MEM. Pellets were then lysed in gel loading buffer and assessed by Western blotting. As demonstrated in Fig. 6, viral particles incubated with only buffer or with Aβ amyloids failed to be pelleted; in contrast, SEM and SEVI fibrils efficiently pelleted viral particles after low-speed centrifugation. Only viral surface proteins (gB and gH) and tegument protein (m25 of MCMV and pp71 of HCMV) were pulled down, whereas the nonstructural protein IE1, which is in infected cells but not present in the viral particle, was not pelleted. These results suggest that CMV particles directly bind both SEVI and SEM amyloids.
Fig 6.
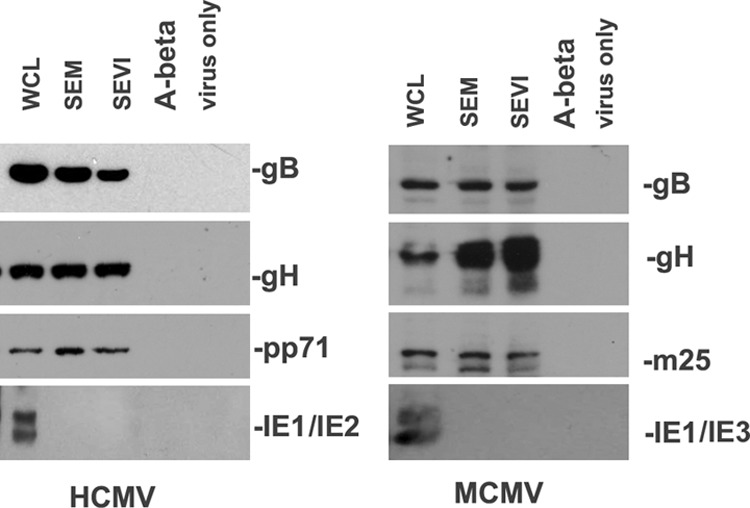
Semen amyloids bind CMV. (Left panel) A total of 1 ml of HCMV (107/ml) was incubated with SEVI or SEM amyloid fibrils at 50 μg/ml for 1 h at 37°C. Controls include virus alone and virus incubated with 50 μg of Aβ(1-42) amyloids/ml. The mixtures were centrifuged, and the pellet was washed twice with MEM. The pellet was lysed in 2× Laemmli buffer and blotted with anti-IE1 (nonstructural protein), anti-gH (envelope protein), anti-gB (envelope protein), and anti-pp71 (tegument protein) antibodies. Whole-cell lysates (WCL) of HCMV-infected NIH 3T3 cells were used as input controls for the Western blot. (Right panel) Similar conditions, but using MCMV instead of HCMV. The tegument protein detected for MCMV was m25.
SEVI and SEM amyloids protect viruses from being neutralized by anti-gH antibodies.
Several antibodies against CMV glycoproteins have been tested for their ability to neutralize CMV. The major targets of the neutralizing antibody response to HCMV are the glycoproteins gB, gM/gN, and gH/gL/gO, all of which mediate entry into host cells (21). In our laboratory, we have two monoclonal antibodies against gB and gH. We found that only the anti-gH antibodies significantly neutralized CMV; in contrast, the anti-gB monoclonal antibody had little neutralizing effect on either MCMV or HCMV infection (data not shown).
The physical interaction between SEVI and SEM amyloids with CMV particles prompted us to speculate that the amyloids might overcome the effects of antibody-mediated neutralization. To test this conjecture, we examined whether the amyloids protected against neutralization by anti-gH (22). We set up the following five treatment groups (Fig. 7): (i) virus only (HCMV or MCMV), (ii) virus incubated with anti-gH antibody for 30 min, (iii) virus incubated with anti-gH antibody for 30 min and then with SEVI or SEM amyloids for another 30 min, (iv) virus incubated with SEVI or SEM amyloids for 30 min and then with anti-gH antibody for another 30 min, and (v) virus incubated with only SEVI or SEM amyloids for 30 min. Infection rates were determined by flow cytometry. The results showed that adding either SEVI or SEM amyloids before the addition of anti-gH significantly reduced the neutralizing effects of the antibody on HCMV and MCMV. Therefore, we postulate that SEM amyloids and SEVI not only enhance CMV infection but might also protect the virus from antibody-mediated neutralization.
Fig 7.

Semen amyloids overcome the antiviral effects of anti-CMV neutralizing antibodies. Five groups were set up as follows: MCMV or HCMV was (i) mock treated, (ii) treated with anti-gH antibody, (iii) treated with anti-gH antibody and then SEVI or SEM amyloids, (iv) treated with SEVI or SEM amyloids and then anti-gH antibody, or (v) treated with SEVI or SEM amyloids. Based on the statistical analysis, the average values (± standard deviations) of duplicate measurements from three independent experiments that yielded similar results were determined. Values were averaged from three independent experiments performed at different times. A Student t test was used to statistically analyze the difference between the groups. *, P < 0.005; **, P = 0.024; ***, P = 0.016.
DISCUSSION
Clinical significance of sexually transmitted CMV.
Although the majority of herpes simplex virus and CMV infections are clinically mild or asymptomatic, CMV infection in two populations can result in serious disease and mortality. First, primary infection during the fetal or perinatal period can be neurologically devastating, even fatal, for the unborn child (23, 24). A CMV-negative woman who becomes infected with CMV during pregnancy has a high probability of transmitting CMV to her fetus (24). CMV causes birth defects (6) and therefore poses a significant public health problem. The precise reasons for the increased severity of the disease early in life are not clear and may involve aspects of immune defense. The severity of CMV-caused diseases is directly influenced by viral load (6). Significantly, one method of CMV acquisition by pregnant women is via sexual intercourse (6). The second population at high risk for CMV infection consists of immunocompromised individuals, including AIDS and organ transplantation patients (6). Many cases of HIV infection are associated with CMV infection, mainly at the stage when the CD4+ T cell count is <50 cell/mm3 (25). Over three-fourths of HIV-infected men who have sex with men have at least one herpesvirus detected in their semen, with CMV being the most prevalent of these (3, 13, 26). CMV infection in AIDS patients can cause myelitis, retinitis, encephalitis, and hepatitis. CMV myelitis is often associated with CMV infection in the peripheral nervous system, whereas patients with CMV encephalitis always have high titers of CMV in the cerebrospinal fluid (25). Although nonsexual routes of CMV transmission are common, sexual transmission also contributes toward the global spread of CMV, with semen being the primary vehicle for both male-to-female (M-F) and male-to-male (M-M) transmission (1, 6). Understanding the molecular details underlying semen-mediated CMV infections will therefore aid in the development of anti-CMV strategies for minimizing or eliminating M-M and M-F sexual transmission of CMV, which can decrease overall CMV transmission rates.
Semen is an important vector for CMV transmission.
In order to be successfully transmitted and thereby infect genital cells, viruses have to survive in both vaginal and seminal fluids. Recent studies demonstrated that semen enhances the HIV infection of multiple cell types in vitro (9, 10, 17, 27). Two different semen-derived amyloid fibrils were identified as factors sufficient to enhance HIV infection (9, 10). In the present study, we found that SP and semen amyloid fibrils (SEVI and SEM amyloids) enhanced both HCMV and MCMV infection of permissive cells (Fig. 1 and 2). Semen amyloid-mediated enhancement of CMV infection was dose dependent. SP and purified amyloids not only caused higher viral gene expression but also led to increased viral production (Fig. 3 and 5). The enhancement of CMV infection by SP and the amyloids is likely caused by the direct binding and clustering of the virions, since the fibrils can bind CMV (Fig. 6) in a manner similar to the way in which the fibrils bind HIV (17). Overall, semen amyloids increased viral replication 50- to 100-fold, which could have significant implications for semen-mediated transmission of CMV.
SEVI and SEM amyloids interact with CMV and overcome the neutralizing effects of antibodies.
Both neutralizing and non-neutralizing CMV-specific antibodies have been identified (28, 29). The major targets of the neutralizing antibody response to HCMV are glycoproteins gB, gM/gN, and gH/gL/gO, all of which mediate entry into host cells (21, 30). In our assays, monoclonal antibodies against HCMV (gB and gH) and MCMV (gB and gH) were tested for neutralization activity. In the absence of complement, there was no neutralizing effect mediated by anti-gB. In contrast, anti-gH antibodies neutralized viral infection (Fig. 6) in the absence of complement, a finding consistent with previously reported results (31). Interestingly, in the present study we found that the semen amyloids can overcome the neutralizing effects of anti-gH antibodies. When CMV was first incubated with SEVI or SEM amyloids and then with anti-gH antibody, the neutralizing effects were significantly reduced (Fig. 6). These results may provide an explanation for why the antiviral components in vaginal fluid are not effective at blocking CMV infection.
Conclusion and perspectives.
Our results suggest that (i) SEVI and SEM amyloids, as well as SP, enhance CMV infection, (ii) SEVI and SEM amyloids represent novel microbicide targets for CMV infection, and (iii) SEVI and SEM amyloids can prevent neutralization of CMV by antibodies. It is possible that the combination of inhibitors targeting the activities of SEVI and SEM amyloids and anti-gH antibodies may have a synergistic effect in protecting against CMV transmission. It would therefore be worthwhile to test a novel, innovative approach to CMV microbicide development by testing agents that target SEVI, SEM, or both. Our studies point to a potential strategy of inhibiting the semen amyloid-mediated infection of CMV by preventing the formation of amyloids in semen. This approach of targeting host-derived factors is in contrast to traditional microbicidal strategies that target the virus itself. It has been shown that the genital tract of a man infected with HIV-1 is an anatomic compartment that supports CMV replication (26, 32). That being the case, it would be intriguing to investigate the extent to which semen from AIDS patients enhances CMV transmission.
ACKNOWLEDGMENTS
We thank J. Kerry, S. Jonjic, M. Messerle, and J. Shanley for donating reagents. We acknowledge the instrument support provided by the PSM Molecular Biology Core Laboratory. Finally, we thank Bob Ritchie of the Ponce School of Medicine and Health Sciences/RCMI Publications Office (G12 RR003050/8G12MD007579-27) for help with manuscript preparation.
This study was supported by a pilot grant from the National Center for Research Resources (G12 RR003050) and the National Institute on Minority Health and Health Disparities (8G12MD007579-27) from the National Institutes of Health (Q.T.), an American Cancer Society grant (117448-RSG-09-289-01-MPC) (Q.T.), NIH/NCRR U54RR022762 (Q.T.), and the 5K12 DK083021-04 KURe fellowship and a K99/R00 grant 1K99AI104262 (N.R.R.).
Footnotes
Published ahead of print 11 September 2013
REFERENCES
- 1.Fowler KB, Pass RF. 1991. Sexually transmitted diseases in mothers of neonates with congenital cytomegalovirus infection. J. Infect. Dis. 164:259–264 [DOI] [PubMed] [Google Scholar]
- 2.Pereira L, Maidji E, Fisher SJ, McDonagh S, Tabata T. 2007. HCMV persistence in the population: potential transplacental transmission. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 3.Gianella S, Strain MC, Rought SE, Vargas MV, Little SJ, Richman DD, Spina CA, Smith DM. 2012. Associations between virologic and immunologic dynamics in blood and in the male genital tract. J. Virol. 86:1307–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Revello MG, Gerna G. 2002. Diagnosis and management of human cytomegalovirus infection in the mother, fetus, and newborn infant. Clin. Microbiol. Rev. 15:680–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Revello MG, Zavattoni M, Furione M, Lilleri D, Gorini G, Gerna G. 2002. Diagnosis and outcome of preconceptional and periconceptional primary human cytomegalovirus infections. J. Infect. Dis. 186:553–557 [DOI] [PubMed] [Google Scholar]
- 6.Cannon MJ. 2009. Congenital cytomegalovirus (CMV) epidemiology and awareness. J. Clin. Virol. 46(Suppl 4):S6–S10 [DOI] [PubMed] [Google Scholar]
- 7.Deayton JR, Sabin CA, Johnson MA, Emery VC, Wilson P, Griffiths PD. 2004. Importance of cytomegalovirus viraemia in risk of disease progression and death in HIV-infected patients receiving highly active antiretroviral therapy. Lancet 363:2116–2121 [DOI] [PubMed] [Google Scholar]
- 8.Mocarski ES, Jr, Shenk T, Pass RF. 2006. Cytomegaloviruses, 5th ed. Lippincott/The Williams &Wilkins Co, Philadelphia, PA [Google Scholar]
- 9.Munch J, Rucker E, Standker L, Adermann K, Goffinet C, Schindler M, Wildum S, Chinnadurai R, Rajan D, Specht A, Gimenez-Gallego G, Sanchez PC, Fowler DM, Koulov A, Kelly JW, Mothes W, Grivel JC, Margolis L, Keppler OT, Forssmann WG, Kirchhoff F. 2007. Semen-derived amyloid fibrils drastically enhance HIV infection. Cell 131:1059–1071 [DOI] [PubMed] [Google Scholar]
- 10.Roan NR, Muller JA, Liu H, Chu S, Arnold F, Sturzel CM, Walther P, Dong M, Witkowska HE, Kirchhoff F, Munch J, Greene WC. 2011. Peptides released by physiological cleavage of semen coagulum proteins form amyloids that enhance HIV infection. Cell Host Microbe 10:541–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner M, Jonjic S, Koszinowski UH, Messerle M. 1999. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 73:7056–7060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez FP, Cosme RS, Tang Q. 2010. Murine cytomegalovirus major immediate-early protein 3 interacts with cellular and viral proteins in viral DNA replication compartments and is important for early gene activation. J. Gen. Virol. 91:2664–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wurm M, Schambach A, Lindemann D, Hanenberg H, Standker L, Forssmann WG, Blasczyk R, Horn PA. 2010. The influence of semen-derived enhancer of virus infection on the efficiency of retroviral gene transfer. J. Gene Med. 12:137–146 [DOI] [PubMed] [Google Scholar]
- 14.Tang Q, Li L, Maul GG. 2005. Mouse cytomegalovirus early M112/113 proteins control the repressive effect of IE3 on the major immediate-early promoter. J. Virol. 79:257–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loh LC, Keeler VD, Shanley JD. 1999. Sequence requirements for the nuclear localization of the murine cytomegalovirus M44 gene product pp50. Virology 259:43–59 [DOI] [PubMed] [Google Scholar]
- 16.Wu CA, Carlson ME, Henry SC, Shanley JD. 1999. The murine cytomegalovirus m25 open reading frame encodes a component of the tegument. Virology 262:265–276 [DOI] [PubMed] [Google Scholar]
- 17.Roan NR, Munch J, Arhel N, Mothes W, Neidleman J, Kobayashi A, Smith-McCune K, Kirchhoff F, Greene WC. 2009. The cationic properties of SEVI underlie its ability to enhance human immunodeficiency virus infection. J. Virol. 83:73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yolamanova M, Meier C, Shaytan AK, Vas V, Bertoncini CW, Arnold F, Zirafi O, Usmani SM, Muller JA, Sauter D, Goffinet C, Palesch D, Walther P, Roan NR, Geiger H, Lunov O, Simmet T, Bohne J, Schrezenmeier H, Schwarz K, Standker L, Forssmann WG, Salvatella X, Khalatur PG, Khokhlov AR, Knowles TP, Weil T, Kirchhoff F, Munch J. 2013. Peptide nanofibrils boost retroviral gene transfer and provide a rapid means for concentrating viruses. Nat. Nanotechnol. 8:130–136 [DOI] [PubMed] [Google Scholar]
- 19.Perez KJ, Martinez FP, Cosme-Cruz R, Perez-Crespo NM, Tang Q. 2013. A short cis-acting motif in the M112-113 promoter region is essential for IE3 to activate M112-113 gene expression and is important for murine cytomegalovirus replication. J. Virol. 87:2639–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim KA, Yolamanova M, Zirafi O, Roan NR, Staendker L, Forssmann WG, Burgener A, Dejucq-Rainsford N, Hahn BH, Shaw GM, Greene WC, Kirchhoff F, Munch J. 2010. Semen-mediated enhancement of HIV infection is donor-dependent and correlates with the levels of SEVI. Retrovirology 7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Compton T, Feire A. 2007. Early events in human cytomegalovirus infection. Chapter 16. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 22.Britt WJ, Vugler L, Butfiloski EJ, Stephens EB. 1990. Cell surface expression of human cytomegalovirus (HCMV) gp55-116 (gB): use of HCMV-recombinant vaccinia virus-infected cells in analysis of the human neutralizing antibody response. J. Virol. 64:1079–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crumpacker CS, Zhang J. 2009. Cytomegalovirus, p 1971–1989 In Mandell GL, Bennett JE, Dolin R. (ed), Principles and practice of infectious diseases, 6th ed. Churchill Livingstone, New York, NY [Google Scholar]
- 24.Walker SP, Palma-Dias R, Wood EM, Shekleton P, Giles ML. 2013. Cytomegalovirus in pregnancy: to screen or not to screen. BMC Pregnancy Childbirth 13:96. 10.1186/1471-2393-13-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffiths P. 2004. Cytomegalovirus infection of the central nervous system. Herpes 11(Suppl 2):95A–104A [PubMed] [Google Scholar]
- 26.Gianella S, Anderson CM, Vargas MV, Richman DD, Little SJ, Morris SR, Smith DM. 2013. Cytomegalovirus DNA in semen and blood is associated with higher levels of proviral HIV DNA. J. Infect. Dis. 207:898–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roan NR, Sowinski S, Munch J, Kirchhoff F, Greene WC. 2010. Aminoquinoline surfen inhibits the action of SEVI (semen-derived enhancer of viral infection). J. Biol. Chem. 285:1861–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maidji E, McDonagh S, Genbacev O, Tabata T, Pereira L. 2006. Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal Fc receptor-mediated transcytosis. Am. J. Pathol. 168:1210–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonagh S, Maidji E, Chang HT, Pereira L. 2006. Patterns of human cytomegalovirus infection in term placentas: a preliminary analysis. J. Clin. Virol. 35:210–215 [DOI] [PubMed] [Google Scholar]
- 30.Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. U. S. A. 102:18153–18158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, Coelingh KL, Britt WJ. 1995. Human cytomegalovirus neutralizing antibody-resistant phenotype is associated with reduced expression of glycoprotein H. J. Virol. 69:6047–6053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gianella S, Mehta SR, Strain MC, Young JA, Vargas MV, Little SJ, Richman DD, Kosakovsky Pond SL, Smith DM. 2012. Impact of seminal cytomegalovirus replication on HIV-1 dynamics between blood and semen. J. Med. Virol. 84:1703–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]