

Abstract
In order to maintain the gas exchange function of the lung following influenza virus infection, a delicate orchestration of positive and negative regulatory pathways must be maintained to attain viral eradication while minimizing local inflammation. The programmed death receptor 1 ligand/programmed death receptor 1 (PDL-1/PD-1) pathway plays an important immunoregulatory role, particularly in the context of T cell function. Here, we have shown that influenza virus infection of primary airway epithelial cells strongly enhances PDL-1 expression and does so in an alpha interferon receptor (IFNAR) signaling-dependent manner. PD-1 is expressed primarily on effector T cells in the lung, compared to effector memory and central memory cells, and shortly after influenza virus infection, an increased number of PD-1+ T cells are recruited to the airways. Using in vitro cocultures of airway epithelial cells and T cells and in vivo models of influenza virus infection, we have demonstrated that blockade of airway epithelial PDL-1 improves CD8 T cell function, defined by increased production of gamma interferon (IFN-γ) and granzyme B and expression of CD107ab. Furthermore, PDL-1 blockade in the airways served to accelerate influenza virus clearance and enhance infection recovery. Our findings suggest that local manipulation of the PDL-1/PD-1 axis in the airways may represent a therapeutic alternative during acute influenza virus infection.
INTRODUCTION
The respiratory tract is constantly exposed to a broad range of foreign antigens due to respiration. The airway epithelium lining the respiratory tract not only represents the first barrier of defense against respiratory pathogens but is often the primary target of infection (1). Consequently, apart from a critical role in gas exchange function, airway epithelial cells additionally participate in host defense mechanisms by producing cytokines and chemokines (2–8). Since exacerbated responses by airway epithelial cells in response to innocuous antigens or pathogens can prove detrimental to lung function, airway epithelial cells have also evolved to regulate pulmonary homeostasis by multiple mechanisms, including, among others, expression of CD200 (9), MUC-1 (10), and surfactant proteins (11) and reduced expression of adaptor molecules (12–14).
Interaction of programmed cell death ligand 1 (PDL-1) with its receptor, programmed death 1 (PD-1), a member of the B7 family of signaling molecules, has been specifically linked to negative regulation of T cell immune responses (15, 16). Upon activation, PD-1 inhibits T cell receptor signaling by blocking phosphatidylinositol 3-kinase (PI3k)/Akt activation (17). During chronic viral infection and cancer, PD-1 expression leads to T cell dysfunction and exhaustion (15, 16, 18). Conversely, blockade of the PD-1/PDL-1 axis has been shown to either enhance T cell function or reduce the viral burden in a number of chronic viral infections (18–21). Our understanding of the role of the PD-1/PDL-1 pathway during acute viral infection, however, is just beginning to unfold. PD-1 expression is rapidly downregulated during acute lymphocytic choriomeningitis virus (LCMV) infection (22) but is expressed on functional CD8 T cells in other acute viral infections (23, 24). PD-1 has also been shown to regulate the development of T cell memory during vaccinia virus infection (25). Fairly recently, it has been shown that a number of acute respiratory viral infections, including human metapneumovirus, respiratory syncytial virus, and influenza virus, impair CD8 T cell function through PD-1 (26) (27). Although previous studies have shown that lack of PD-1 function due to genetic mutation or to systemic blockade prevented CD8 T cell inhibition and enhanced viral control, the specific role that the airway epithelium plays during PDL-1/PD-1 regulation of pulmonary T cell function has not been fully elucidated.
Our study builds on the observations by Erickson et al. (27) to further show that PDL-1 is constitutively expressed in airway epithelial cells and its expression is upregulated by influenza virus and dependent on interferon alpha receptor (IFNAR) signaling. Coculture studies with airway epithelial cells and T cells in the presence of PDL-1 blocking antibody revealed that PDL-1 expression on airway epithelial cells repressed T cell function. Additionally, intranasal blockade of PDL-1 in influenza virus-infected mice resulted in increased levels of cytotoxic proteins and cytokines in the airways, enhanced CD8 T cell function, and reduced viral load. Our findings suggest that PDL-1 expression by airway epithelial cells during the early phase of the recall response to influenza virus infection represents a mechanism for regulating lung immune homeostasis and reveal a promising strategy to modulate local epithelial-T cell interaction in the airways.
MATERIALS AND METHODS
Mice.
C57BL/6J, BALB/c, and IFNAR−/− mice (BALB/c background) were obtained from The Jackson Laboratory (Bar Harbor, ME) or Harlan (Indianapolis, IN) or were bred at The Research Institute at Nationwide Children's Hospital. Mice were housed in BL2 containment under pathogen-free conditions. The Institutional Animal Care and Use Committee at The Research Institute at Nationwide Children's Hospital approved all of the animal studies described in this study.
Cell culture.
Primary mouse tracheal epithelial cell (TEC) cultures were obtained from tracheas resected from 3- to 10-week-old wild-type BALB/c and IFNAR−/− mice. Tracheal cell isolation and culture were performed as previously described (28). In short, tracheas were collected and incubated with 1.5 mg/ml pronase and 0.5 mg/ml crude pancreatic DNase I. Nonadherent cells were collected and seeded onto 24-mm coated membranes with 50 μg/ml type I rat tail collagen solution. Cells were incubated for 10 to 14 days postharvest and then allowed to differentiate by incubation under an air-liquid interface for an additional 14 days before use.
Virus infection.
Influenza A viruses Udorn, WSN, PR8, and X31 were grown in eggs, and virus titers were determined by an immunofluorescence focus assay. Male mice (6 to 12 weeks of age) were anesthetized with 2,2,2,-tribromoethanol and intranasally inoculated with 3,000 50% egg infective doses (EID50) influenza virus X31 and challenged with 1 × 102 PFU/mouse PR8 in a 30-μl volume. Organs were harvested on the indicated days posttreatment. Groups of three to six animals were used for each data point. For the analysis of PDL-1 transcript, TEC cultures derived from BALB/c and IFNAR−/− mice were exposed to 2 × 105 PFU WSN for 2 h or mock inoculated and harvested 24 h postinfection. Human airway epithelial (HAE) cultures were exposed to 2 × 105 PFU influenza A virus Udorn strain or mock treated for 2 h and harvested after 24 h of incubation as appropriate (28).
Immunofluorescence.
Influenza virus-infected HAE cultures maintained on Snapwell inserts were fixed in 10% paraformaldehyde and then washed and stored in 70% ethanol until paraffin embedded. Six-micrometer sections of HAE cultures or human trachea were deparaffinized for immunofluorescence. Following 30′ antigen retrieval in sodium citrate (pH 6), tissues were incubated in 0.2% Triton X-100 for 15 min and blocked with 5% goat serum for 1 h. PDL-1 was visualized using rabbit polyclonal antibody to CD274 (Abcam, Cambridge, MA) and Texas-red goat anti-rabbit (Vector Laboratories, Burlingame, CA). Control sections were stained with rabbit immunoglobulin or with secondary antibody only. Tissues were mounted with Prolong Gold antifade reagent containing 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen/Molecular Probes). Immunofluorescent images were viewed on an Olympus BX61 microscope using a 40× objective and captured using a Hamamatsu ORCA-GR digital camera in concert with the Slidebook version 4.2 software program (Intelligent Imaging Innovations, Inc., Denver, CO).
RNA extraction, reverse transcription, and real-time PCR.
RNA isolation from infected HAE cultures and TEC cultures was achieved by direct lysis of cells with TRIzol reagent (Invitrogen) and application of the resulting aqueous phase to Qiagen RNeasy minicolumns. Total RNA harvested from triplicate (TEC) or quadruplicate (HAE) cultures was processed and analyzed as described in reference 28. One microgram RNA extracted from infected lungs or cell cultures using a combination of TRIzol (Invitrogen) and/or the RNeasy minikit, (Qiagen) was also used for cDNA synthesis (high-capacity reverse transcription [RT] kit; Applied Biosystems), followed by quantitative PCR performed using Sybr green (Applied Biosystems) and primers for human or mouse PDL-1, the Flu NP gene, and human or mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Data were analyzed using the Prism 7500 SDS software program. Mock-infected groups served as the calibrator sample, and all samples were normalized to the endogenous control, GAPDH.
TEC/T lymphocyte cocultures.
TEC cultures derived from BALB/c and IFNAR−/− mice were cocultured with splenic T lymphocytes at a 1:1 ratio (TECs/T cells). T lymphocytes were isolated from spleens of naive BALB/c mice by negative selection. TECs seeded in 24-well plates were exposed to 1 PFU/cell influenza virus WSN for 2 h. Fresh medium was added after the removal of inoculum, and panned T lymphocytes (purity, >95%) were added 24 h postinfection. Blocking antibodies to PDL-1 (clone MIH5; eBioscience) or control antibody (ChromePure rat IgG; Jackson Immuno Research) were added 3 h before the addition of T lymphocytes such that 10 μg blocking antibody was added to 2.5 × 105 TECs.
In vivo antibody blockade.
Mice initially infected with influenza virus X31 were challenged 30 to 40 days later with influenza virus PR8 as described above and either intraperitoneally injected with 200 μg of functional-grade anti-mouse PDL-1 or 200 μg IgG or Hanks buffered salt solution (HBSS) or were intranasally treated with 150 μg anti-PDL-1 or control antibody administered successively over a 3-day time period in 50-μg (50-μl) doses. Sham-treated mice received the same volume of HBSS in lieu of antibody.
Plaque assay.
Lung homogenates collected in 0.5 ml DMEM plus 0.1% bovine serum albumin (BSA) were used for viral titer determination. Plaque assays were performed by exposing 80 to 90% confluent MDCK cells grown in 12-well plates to 100-μl quantities of 10-fold serial dilutions of lung homogenates for 2 h at 37°C. The inoculated monolayers were covered with DMEM containing 0.8% methylcellulose, l-glutamine, 0.1% BSA, antibiotics, and 1 μg/ml l-1-tosylamide-2-phenylethyl chloromethyl ketone (TPCK) trypsin (Sigma, St. Louis, MO) and incubated for 3 days at 37°C. After incubation, the monolayers were permeabilized with 0.2% Triton X-100. Virus-infected cells were immunostained using a monoclonal antibody that reacts with the influenza A virus nucleoprotein (Cal Bioreagents, San Mateo, CA), followed by a 1-h incubation with anti-mouse IgG peroxidase conjugate (Sigma, St. Louis, MO) and a 10- to 30-min incubation with True Blue substrate (KPL, Gaithersburg, MD). Quantification was performed by counting plaques using a dissecting microscope, and counts were expressed as log10 PFU per milliliter.
Flow cytometry. (i) Epithelial cells.
TECs were trypsinized to obtain a cell suspension and then stained and fixed with paraformaldehyde. Analysis of cell surface expression of PDL-1 on TECs was achieved using phycoerythrin (PE)-rat anti-mouse CD274 (BD Pharmingen, Franklin Lakes, NJ) and the BD LSRII flow cytometer with FlowJo software (TreeStar Inc., Ashland, OR). Results were expressed as median fluorescence intensities (MFI) after subtracting the MFI of control cells stained with the appropriate isotype control antibodies (BD Pharmingen).
(ii) T lymphocytes.
Single-cell suspensions were obtained from the bronchoalveolar lavage (BAL) fluid, lung, mesenteric lymph nodes (MLN), and spleen. Red blood cells (RBCs) were lysed, and the number of cells per organ was determined. Cells were blocked with Fc-block (CD16/32) and washed and stained with a combination of NP366–374/Db tetramer and antibodies against CD3, CD4, CD8, CD44, CD62L, CD43 activation-associated glycoform, CD107ab, and PD-1 or combined with intracellular staining for cytokines/cytotoxic proteins: gamma interferon (IFN-γ), interleukin 2 (IL-2), perforin, and granzyme B (eBioscience). Flow cytometry data were acquired on a BD LSR II instrument (BD Biosciences) and analyzed using FlowJo software. Gates were set using negative controls and isotype controls.
ELISA for IFN-γ and granzyme B.
Concentrations of IFN-γ and granzyme B were measured in BAL sample supernatants and cocultures with commercially available enzyme-linked immunosorbent assay (ELISA) kits (Ready-SET-Go! kit; eBioscience). All assays were carried out according to the manufacturer's instructions, and 100 μl from each sample was assayed in duplicate. The lower limits of detection for these assays were 15 pg/ml for IFN-γ and 40 pg/ml (granzyme B). For statistical analysis, samples with optical density readings below the limit of the standard curve of the assay were assigned a value half that of the detection level.
Statistical analysis.
t tests were used to compare both the percentage of cells positive for a given antigen of interest and the mean relative fluorescence intensity (MRFI) determined by flow cytometry analysis. Two-sample homoscedastic Student t tests were used for single comparisons in which a P value less than 0.05 was considered statistically significant. Results are representative of at least 2 to 3 separate experiments using 3 to 6 mice per group and are expressed as means ± standard deviations (SD). One-way analysis of variance (ANOVA) was used to determine statistical significance when comparing three or more independent groups (particularly in cases in which antibody treatment was compared to mock, Flu, and IgG control treatments). When ANOVA demonstrated that the differences among means were statistically significant (P < 0.05), the Tukey posttest was used to correct for multiple comparisons and to generate multiplicity-adjusted P values (Prism GraphPad, version 6).
RESULTS
PDL-1 is constitutively expressed in airway epithelial cells, and influenza virus infection and alpha/beta interferon (IFN-α/β) signaling regulate its expression.
The epithelial surfaces of the lung play an important role in regulating local immunity and homeostasis. In an initial attempt to analyze the role of regulatory factors critical to infection of the respiratory tract, we established polarized, well-differentiated human primary airway epithelial cell cultures (HAEs) and exposed them to influenza A virus. HEA cultures differentiate to form a pseudostratified mucociliary cell layer and effectively mimic the airway epithelial environment. A comparison of cluster of differentiation molecules following gene expression microarray-based screening revealed that one gene in particular, the PDL-1 gene, was drastically upregulated in HAE cultures 24 h post-exposure to influenza virus (data not shown). Our analysis showed that PDL-1 expression was enhanced 83.9-fold 24 h post-exposure to influenza virus compared to that of mock-treated HAEs (Fig. 1A). quantitative RT-PCR (qRT-PCR) analysis corroborated the above findings, showing a 54.3-fold increase in PDL-1 expression in HAEs following influenza virus exposure (Fig. 1B). Additionally, immunofluorescent staining of human tracheal sections showed that PDL-1 was not only constitutively expressed in airway epithelial cells but, perhaps more important, that the staining was prominent on the apical surface (Fig. 1C). To determine if influenza virus infection affected PDL-1 expression at the protein level, we performed immunofluorescent analysis of PDL-1 expression on HAE cultures infected or not with influenza virus. As we observed in human trachea epithelium, PDL-1 was constitutively expressed in control HAEs (Fig. 1D, upper panel). However, compared with mock-treated cultures, PDL-1 staining increased in intensity in HAEs 24 h post-exposure to influenza virus (Fig. 1D, lower panel). Altogether, these data indicate that PDL-1 is constitutively expressed by the human airway epithelium and that influenza virus infection increases the expression of PDL-1 in airway epithelial cells within 24 h of infection.
Fig 1.

PDL-1 is constitutively expressed in the human airway epithelium and is upregulated by influenza infection in HAEs. (A) mRNA expression of PDL-1 in mock treated HAE cultures or in HAE cultures 24 h post-exposure to 2 × 105 PFU influenza virus (Udorn strain). (B) Relative mRNA levels of PDL-1 in influenza-virus-infected HAE cells, validated by qRT-PCR. (C) Immunofluorescence analysis of PDL-1 expression (red) in a paraffin-embedded human trachea section. The box inset represents a high-magnification detail of the apical side of the epithelium. (D) Immunofluorescence analysis of PDL-1 expression (red) in paraffin embedded HAE cultures either mock treated (upper panel) or infected with influenza virus (lower panel). In all immunofluorescent images, the nucleus is counterstained with DAPI (blue). Error bars indicate SD (∗∗∗, P ≤ 0.0005).
To further analyze regulation of PDL-1 expression on airway epithelial cells, we generated primary murine tracheal epithelial cell cultures (TECs). As we observed with the HAEs, exposure of TECs to influenza virus resulted in a significant increase (27.6-fold) in PDL-1 mRNA expression by 24 h (Fig. 2A). Since alpha interferon receptor (IFNAR) signaling is critical for the antiviral response of the airway epithelium (28), we also analyzed the role of IFNAR on the regulation of PD-1L expression in airway epithelial cells during the response to influenza virus. The data show that there was a 21.1-fold difference in PDL-1 mRNA expression when influenza virus-infected IFNAR−/− TECs were compared to wild-type TECs, indicating that PDL-1 mRNA expression is dependent on signaling. We next corroborated these findings at the protein level by fluorescence-activated cell sorting (FACS) analysis of PDL-1 cell surface expression. The data show that influenza virus infection enhanced the cell surface expression of PDL-1 on TECs but that PDL-1 expression on IFNAR-deficient TEC cultures was not different from isotype control staining (Fig. 2B and C). Altogether, these data indicate that the influenza virus-induced increase in PDL-1 expression on epithelial cells is IFNAR signaling dependent.
Fig 2.
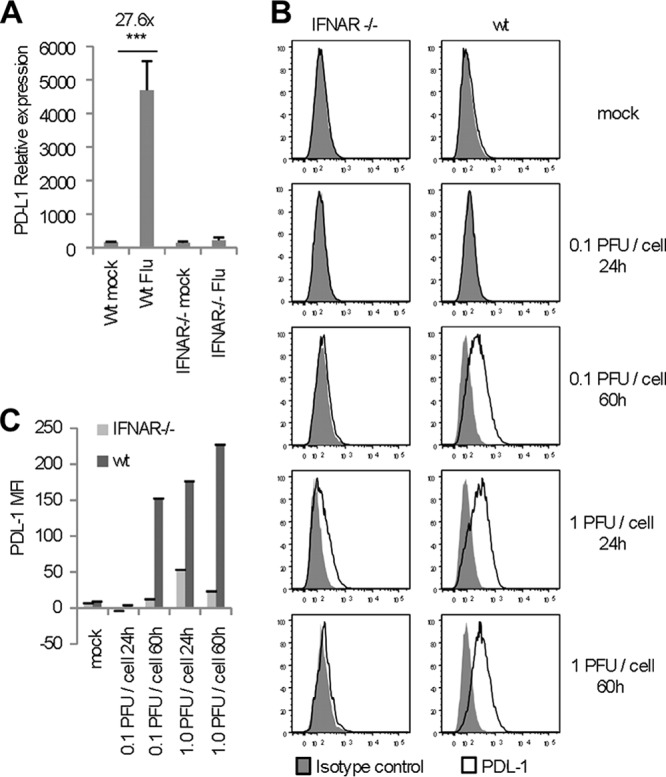
IFNAR signaling regulates PDL-1 expression in TEC cultures. (A) PDL-1 mRNA expression analyzed in TEC cultures derived from BALB/c (wt) and IFNAR−/− mice 24 h after exposure (or not) to 2 × 105 PFU influenza virus (WSN). (B and C) Cell surface expression of PDL-1 on wt or IFNAR−/− TEC cultures infected with 0.1 PFU/cell or 1.0 PFU/cell influenza virus 24 and 60 h postinfection. (B) Representative histograms. (C) Median fluorescent intensity (MFI), in which values used for the bar graph represent the MFI of PDL-1 minus the MFI of the isotype control. All panels are representative of 2 or 3 separate experiments in which TEC cultures were performed in triplicate. Error bars indicate SD (∗∗∗, P ≤ 0.0005).
PD-1+ T cells are present in the lung early during influenza virus infection.
In order for PDL-1 to be functional on airway epithelial cells, contact with cells expressing its receptor is necessary. Therefore, we investigated whether or not a significant population of PD-1-expressing cells was available in the respiratory tract. In order to do this, cohorts of mice were intranasally infected with influenza A virus, and cell populations in the bronchoalveolar lavage (BAL) fluid and lung parenchyma were analyzed at different time points after infection. The data show that PD-1 was expressed in 10 to 40% of the CD4 and CD8 T cells present in the airways and lung parenchyma of naive mice and that there were not significant changes in the frequency of PD-1-expressing T cells during the first week after influenza virus infection (Fig. 3A and B). The analysis of T cell numbers shows that a small population of PD-1-expressing CD4 and CD8 T cells was present in the lung parenchyma (∼1,000 cells), while the number of PD-1+ T cells in the airways was very low (10 CD8 T cells and 100 CD4 T cells) (Fig. 3C and D). However, we observed a significant increase in the absolute number of PD-1+ CD4 and CD8 T cells present in the airways by day 3 after influenza virus infection (Fig. 3C). Additionally, the number of PD-1+ T cells in the lung parenchyma also increased over time, though the differences were not statistically significant (Fig. 3D). Although the number of PD1-expressing T cells returned to baseline levels in the lung parenchyma by day 12 postinfection, greater numbers of PD-1+ CD4 and CD8 T cells remained in the airways of influenza virus-infected mice up to 1 month after infection. To more precisely determine the expression pattern of PD-1 on different populations of influenza virus-specific effector and memory T cells, influenza virus memory mice (40 or more days after inoculation with influenza virus) were intranasally stimulated with poly(I·C) to induce nonspecific activation of T cells in the respiratory tract. We found that PD-1 was expressed on effector CD8 T cells but was absent from the cell surface of effector memory and central-memory CD8 T cells as defined by the expression of L-selectin and CD43 activation-associated glycoform (Fig. 4). This pattern of expression was found on influenza virus NP366-374-specific CD8 T cells as well as NP tetramer-negative CD8 T cells. Altogether, these data indicate that the majority of influenza virus-specific effector CD8 T cells present in the lung express PD-1.
Fig 3.

Influenza virus infection induces early, nonspecific recruitment of PD-1+ effector T cells into the lung airways. Cohorts of naive C57BL/6 mice were infected with influenza virus X31, and the T cell phenotype was assessed at the indicated time points. Representative dot plots show the frequency of PD-1+ CD44+ T cells present in the CD4 and CD8 subpopulations of the BAL fluid (A) or lung parenchyma (B) at the indicated time points after infection. Bar diagrams show the numbers of antigen-experienced PD1+ CD4 and PD-1+ CD8 T cells present in the BAL fluid (C) or lung parenchyma (D). Data are representative of 3 independent experiments with 3 mice per group; error bars indicate SD (∗, P ≤ 0.05; ∗∗, P ≤ 0.005).
Fig 4.

PD-1 is mostly expressed on effector CD8 T cells. Cohorts of mice previously infected with influenza virus X31 received intranasal administration of 50 μg poly(I·C). (A) Representative dot plots and histograms show PD-1 expression on effector and memory T cell subsets within NP tetramer-positive and -negative CD8 T cell populations in the lung parenchyma 3 days poststimulation. Bar diagrams show the MFI and frequency of PD-1+ effector (CD43+ CD62L−), effector-memory (CD43− CD62L−), and central-memory (CD43− CD62L+) CD8 T cells residing in the NP366–374/Db tetramer-positive subset (B) or in the tetramer-negative subset (C). Data are representative of 2 independent experiments with 3 mice per group. Error bars indicate SD (∗∗∗, P ≤ 0.0005).
PDL-1 expression on airway epithelial cells controls CD8 T cell function.
To determine if PDL-1 expression on the airway epithelium modulated the function of T cells, we used a memory system that allowed us to analyze the function of virus-specific T cells. We incubated T cells isolated from the spleens of day 30 to 40 influenza virus-infected mice with primary, polarized TECs in the presence or absence of PDL-1-blocking antibodies. TECs were first exposed to influenza virus for 24 h to induce the upregulation of PDL-1 on the epithelial cell surface and then cocultured with T cells for an additional 36 h. T cell function was assessed by measuring the levels of granzyme B secreted into the culture medium. As shown in Fig. 5, granzyme B was undetectable in uninfected cocultures regardless of the presence or not of anti-PDL-1 antibodies. However, in the presence of influenza virus, granzyme B was released into the culture medium, and PDL-1 blockade resulted in significantly higher levels of granzyme B in culture supernatants. These data indicate that PDL-1 expressed on airway epithelial cells can inhibit the cytotoxic activity of T cells through its interaction with PD-1 and that this process can be reversed in vitro by blockade of PDL-1/PD-1 interaction. To address the functional role of PDL-1/PD-1 interactions in vivo during influenza virus infection, we performed PDL-1 blockade studies and monitored parameters of T cell function in the airways. For these studies, we used influenza virus memory mice to ensure that an elevated number of virus-specific T cells were present in the respiratory tract very early after challenge. The memory mice were intranasally challenged with a heterosubtypic influenza virus (PR8) infection and injected with PDL-1 blocking antibody or with isotype control antibody, and the levels of granzyme B and of IFN-γ were assayed in BAL fluid 3 days after viral challenge. As shown in Fig. 6, intraperitoneal administration of anti-PDL-1 blocking antibody resulted in a significant increase in the concentrations of granzyme B and IFN-γ in the airways compared to results for mice treated with control IgG.
Fig 5.
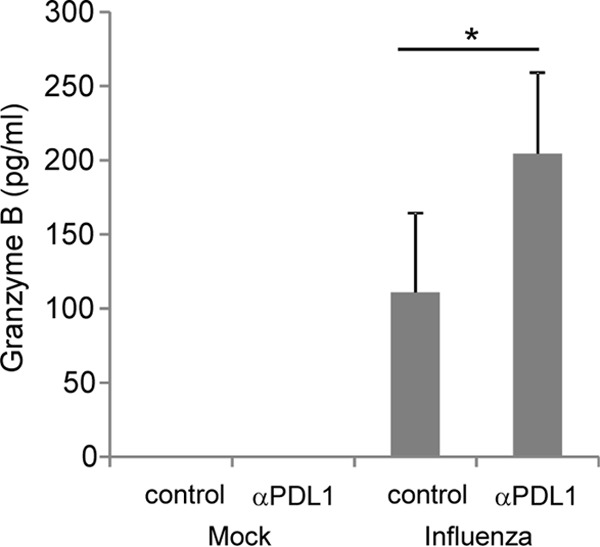
PDL-1 blockade on airway epithelial cells enhances CD8 T cell function. T cells isolated from the spleens of influenza virus-infected mice were cocultured with TEC cells infected or not with influenza virus WSN in the presence of anti-PDL-1 blocking antibody or control IgG antibody. Thirty-six hours after coculture, supernatants were harvested for the determination of granzyme B secretion by ELISA. Data are combined from 2 separate experiments employing triplicate wells per group. Error bars indicate SD (∗, P ≤ 0.05).
Fig 6.
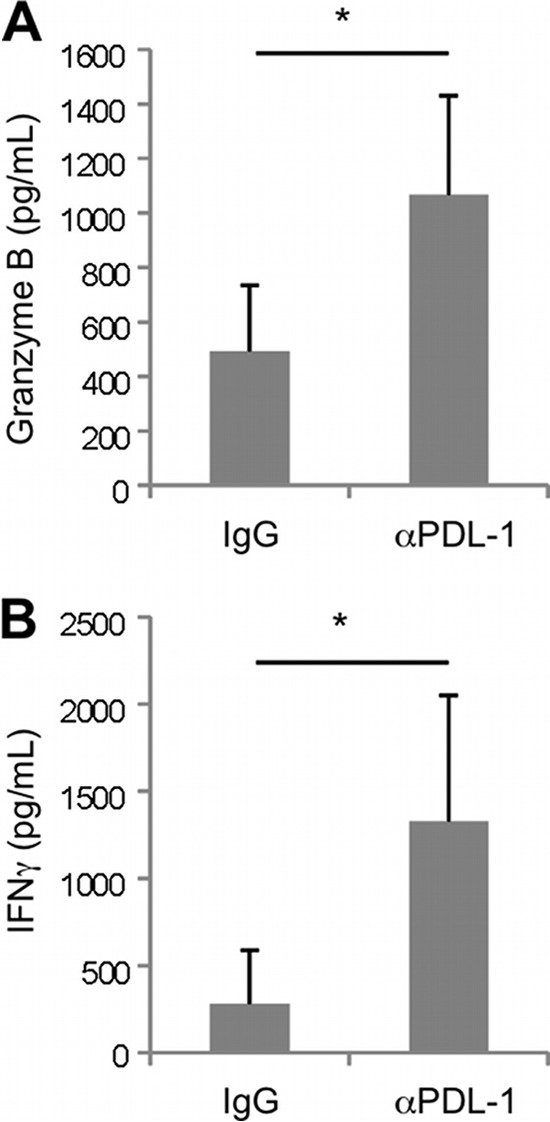
PDL-1 blockade increases the concentration of granzyme B and IFN-γ in the airways during recall. Influenza virus memory mice were secondarily challenged with heterosubtypic influenza virus PR8 and intraperitoneally treated with anti-mouse PDL-1 blocking antibody or rat IgG control. Three days postinfection, BAL fluid samples were assayed for the presence of granzyme B (A) or gamma interferonγ (B) by ELISA. Data are representative of 3 independent experiments (3 mice per group). Error bars indicate SD (∗, P ≤ 0.05).
Although these in vivo experiments demonstrated that PDL-1/PD-1 interaction regulates the effector functions during influenza virus infection, they did not specifically address the role of the airway epithelium in this process. This is important because the PDL-1/PD-1 pathway has been shown to impact the priming of T cell responses (29–34). Thus, in order to elucidate the role of the PDL-1/PD-1 axis in regulating intraepithelial T cell function, we administered anti-PDL-1 blocking antibodies intranasally to induce blockade in the airways. Preliminary studies demonstrated that intranasal administration of antibodies had a specific local effect in the airways but not in the lung parenchyma, draining lymph nodes, or spleen (data not shown). The results show that intranasal administration of anti-PDL-1 blocking antibodies resulted in significantly higher concentrations of granzyme B and IFN-γ in the airways 3 days after influenza virus infection (Fig. 7A and B). By day 5 after influenza virus infection, the levels of granzyme B and IFN-γ in mice treated with anti-PDL1 antibodies were comparable to those of nonchallenged mice (Fig. 7 C and D). These results indicate that intranasal PDL-1 blockade can enhance and accelerate the cytotoxic function of immune cells in the airways.
Fig 7.
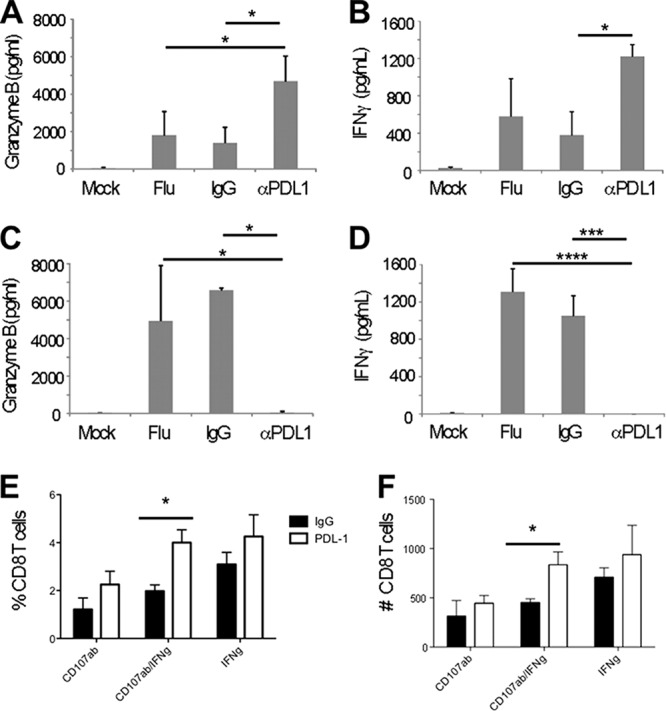
Intranasal PDL-1 blockade enhances the secretion of granzyme B and IFN-γ in the airways during influenza virus recall. Influenza virus memory mice were challenged with influenza virus PR8 (or mock challenged with HBSS) and treated intranasally with anti-mouse PDL-1 blocking antibody or IgG control or HBSS (Flu). ELISA was used to determine the levels of granzyme B or IFN-γ present in the BAL fluid on days 3 and 5 post-PDL-1 blockade (A to D). One-way ANOVA was used to compare the differences between means, and when these differences were significant (P ≤ 0.05), the Tukey test was used to correct for multiple comparisons and to generate the multiplicity-adjusted P values shown. (A) Granzyme B, day 3 (ANOVA; ∗∗, P < 0.01). (B) IFN-γ, day 3 (ANOVA; ∗∗, P < 0.01). (C) Granzyme B, day 5 (ANOVA; ∗∗, P < 0.01). (D) IFN-γ, day 5 (ANOVA; ∗∗∗∗, P < 0.0001). (E) Frequency of IFN-γ and CD107ab-expressing NP366–374-specific CD8 T cells in lung. (F) Number of IFN-γ- and CD107ab-expressing NP366–374-specific CD8 T cells in lung. Data are representative of 2 to 4 independent experiments (3 mice per group). Error bars indicate SD (∗, P ≤ 0.05; ∗∗∗, P ≤ 0.0005; ∗∗∗∗, P ≤ 0.0001).
We next analyzed the effect of intranasal PDL-1 blockade on CD8 T cell function. Influenza virus-challenged mice treated with anti-PDL-1 antibodies had a statistically significant higher frequency and absolute number of IFN-γ CD107ab-doubly positive cells than mice treated with control antibody or mock treated (Fig. 7E and F). The percentage of IFN-γ and of CD107ab-singly positive CD8 T cells was also higher in mice treated with anti-PDL-1 antibodies, but the differences were not statistically significant. We did not observe any differences in CD8 T cell function in the draining lymph nodes or the spleen between mice treated or not with anti-PDL-1 antibodies (data not shown). Altogether, our data indicate that preventing the PDL-1/PD-1 interaction between the airway epithelia and T cells results in enhanced IFN-γ production and cytolytic function by influenza virus-specific CD8 T cells.
PDL-1 blockade in the airways reduces influenza virus load and ameliorates disease.
We next analyzed the consequences of intranasal PDL-1 blockade on influenza virus control on days 3 and 5 following a secondary viral challenge. The viral loads were measured in lung homogenates using both qRT-PCR and infectious plaque assays. The data show that PDL-1 blockade resulted in a slight reduction of influenza virus nucleoprotein (NP) transcripts and of infectious viral titers by day 3 postinfection (Fig. 8A). However, by day 5 postinfection, NP transcript levels and infectious virus titers were below the limit of detection in the anti-PDL-1 treated group (Fig. 8B). Conversely, untreated or control IgG-treated mice had significantly greater levels of expression of NP RNA in the lungs as measured by qRT-PCR and several orders of magnitude (102 to 105) higher viral titers measured by plaque assay. We also subjected each mouse from each group to whole-body plethysmography on day 5 post-infection with influenza virus. Enhanced pause (Penh) values in influenza virus memory mice exposed to a heterosubtypic influenza virus challenge but treated with anti-PDL-1 registered values equivalent to those for influenza virus memory mice that did not receive the secondary influenza challenge. Conversely, Penh values were significantly elevated in influenza virus memory mice that were exposed to a heterosubtypic influenza virus challenge but were left untreated or treated with control anti-IgG (data not shown). Last, to determine the effect of intranasal anti-PDL-1 treatment on acute respiratory disease, we monitored weight loss. We observed that mice treated with anti-PDL-1 antibodies during influenza virus challenge did not present any significant weight loss between day 0 and day 5 of treatment, while untreated or control IgG-treated mice had a significant decrease in weight loss (Fig. 9), suggesting that intranasal anti-PDL-1 treatment can influence acute respiratory disease severity. Altogether, our data indicate that blockade of PDL-1/PD-1 interaction in the airways during influenza virus infection can result in enhanced viral control and disease recovery.
Fig 8.
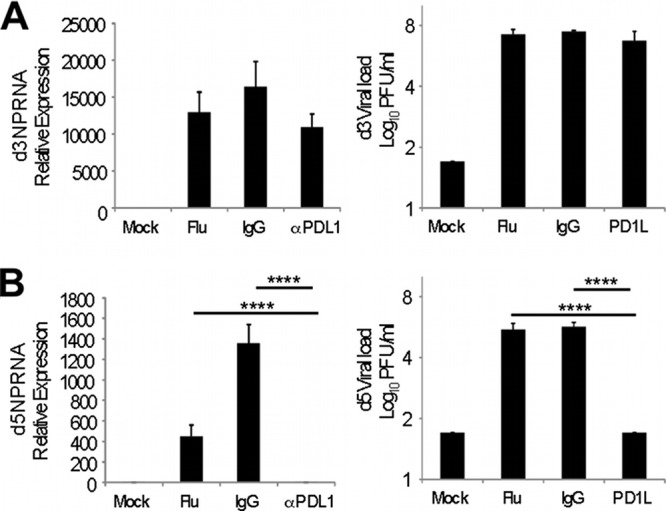
Reduced viral load in anti-PDL-1-treated mice. Influenza virus memory mice (30 to 40 days post-intranasal inoculation with the influenza virus X-31 strain) were intranasally challenged with 1 × 102 PFU/ml influenza virus PR8 (or mock challenged with HBSS) and treated with intranasal instillations of anti-mouse PDL-1 blocking antibody, rat IgG, or HBSS (Flu). On days 3 and 5 after initiation of PDL-1 blockade, the influenza virus load in lung homogenates was determined by qRT-PCR for the NP transcript (left panels) and by plaque assay (right panels). (A) Day 3. (B) Day 5. Values for plaque assay represent the means (± SD) of RSV log10 PFU/ml, where 1.7 log10 PFU/ml represents the limit of detection. Comparisons were made by one-way ANOVA (∗∗∗∗, P < 0.0001 for all panels), and the Tukey posttest was used to correct for multiple comparisons and to generate the multiplicity-adjusted P values shown. Data are representative of at least 2 independent experiments (3 mice per group). Error bars indicate SD (∗∗∗∗, P ≤ 0.0001).
Fig 9.
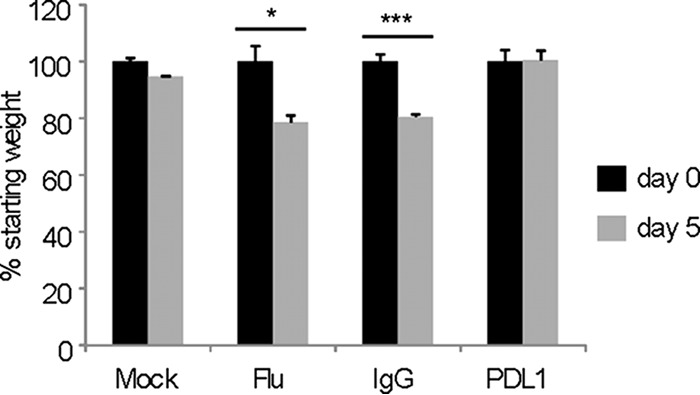
Intranasal PDL-1 blockade prevents influenza-associated weight loss. Percent weight loss in mice inoculated and treated as for Fig. 8 was determined by dividing the average starting weight of each group (day 0, preinoculation) by the average weight of each treatment group on day 5 postinfection. Data are representative of 3 independent experiments with 3 mice per group each. Error bars indicate SD (∗, P ≤ 0.05; ∗∗∗, P ≤ 0.0005).
DISCUSSION
In this report, we showed that influenza virus infection of primary airway epithelial cells strongly enhances PDL-1 expression and does so in an IFNAR signaling-dependent manner. Most important, we observed that intranasal administration of anti-PDL-1 blocking antibodies to induce local blockade of PDL-1 in the airways improved T cell function in the acute phase of infection as defined by increased production of IFN-γ, granzyme B, and cell surface mobilization of CD107ab. PDL-1 blockade in the airways resulted in accelerated influenza virus clearance and enhanced infection recovery as measured by weight loss. These data indicate that at the onset of influenza virus infection, the PD-1/PDL-1 axis is available to limit effector T cell function in the airways.
Our findings showing that PDL-1 was constitutively expressed on airway epithelial cells, prompted us to hypothesize that the PDL-1/PD-1 axis could be regulating local lung immune responses as a means to maintain the integrity of the epithelial lining of the respiratory tract during infection. The epithelial surfaces of the lung have been shown to express molecules such as CD200 among others to regulate the function of immune cells and maintain lung homeostasis (9). The observation that expression of PDL-1 in airway epithelial cells is controlled by viral infection and IFNAR signaling suggests that the PDL-1/PD-1 pathway plays an important role during inflammatory responses in the lung. Innate interferons are critical for the response of airway epithelial cells to respiratory viral infection (28). These observations indicate that interferon signaling also contributes to the regulation of inflammatory homeostasis in the lung by enhancing the expression of PDL-1 in airway epithelial cells, adding another layer of complexity to the regulatory pathways operated by interferon-stimulated genes. Our findings with airway epithelial cells are in accordance with observations in other systems. Mice lacking Toll-like receptor 3 (TLR3) had reduced PDL-1 expression (35), and a major downstream product of TLR3 signaling, IFN-β, has been shown to enhance PDL-1 expression on monocytes, dendritic cells. and hippocampal cultures (35, 36). Furthermore, synthetic double-stranded RNA (dsRNA) has been shown to trigger PDL-1 expression on epithelial cells (37, 38). Taken together, the above findings suggest that PDL-1 expression in the periphery during the effector phase of the response to virus infection likely represents a generalized host regulatory response to viral infection rather than a virus-specific immunoevasion strategy.
The PD-1/PDL-1 signaling cascade is a prominent mechanism by which T cell nonresponsiveness occurs in the face of chronic viral infections and cancer. The role of PD-1-expressing T cells during acute infection has remained controversial (39), although recent observations report increased primary T cell responses in the absence of PD-1 signaling (25, 27, 40). Additionally, a recent study showed that the majority of PD-1-expressing T cells in healthy human adults do not exhibit an exhausted phenotype (41). Our data show that PD-1 is primarily expressed on effector T cells in the lung, compared to findings for effector memory and central memory cells, and that shortly after influenza virus infection, an increased number of PD-1+ T cells are recruited to the airways. Zelinskyy et al. (23) also demonstrated that during acute Friend retrovirus infection, the PD-1-expressing CD8 T cells were terminally differentiated cytotoxic effectors. These studies indicate that PD-1 is not a marker of T cell exhaustion per se and suggest a likely scenario during acute infection in which the PD-1/PDL-1 axis modulates inflammation and ultimately serves to prevent tissue damage from activated effector T cells.
Using cocultures of primary airway epithelial cells and T cells and a mouse model of influenza virus infection, we determined that blockade of airway epithelial PDL-1 improves CD8 T cell function, defined by increased production of IFN-γ, granzyme B, and expression of CD107ab. Importantly, therapeutic PDL-1 blockade in the airways accelerated influenza virus clearance and enhanced infection recovery as measured by weight loss. Our results are in accordance with recent findings by Erickson (27) showing that viral acute respiratory infections impair CD8 T cells through PD-1 and that systemic inhibition of PD-1 prevented CD8 T cell impairment. Our study adds that PD-1/PDL-1 regulation of CD8 T cell effector function is mediated locally by the epithelial cells of the lung, is regulated by IFNAR signaling, and can be therapeutically manipulated in the airways by intranasal administration of anti-PDL-1 blocking antibodies. Since PD-1 signaling has been shown to impact dendritic cell maturation, antigen presentation, and T cell priming and expansion (34, 42–44), it is important to differentiate the systemic role of PD-1/PDL-1 during the generation of adaptive immune responses from the specific role at the mucosal site of infection during epithelial-T cell interaction cross talk.
Our findings are in line with those of several studies demonstrating that loss of PDL-1 during acute viral infection can promote viral clearance (45, 46) and that PDL-1 blockade at the onset of infection may serve as a potential therapeutic treatment (27, 35). Clearly, further work is needed to optimize the timing of therapy and to address role of other cell populations expressing PD-1/PDL-1 in the lung. Nevertheless, our findings indicate that local manipulation of the PDL-1/PD-1 pathway in the airways may represent a therapeutic alternative during acute influenza virus infection.
ACKNOWLEDGMENTS
We thank the Biomedical Genomics Core and Flow Cytometry Core Laboratory at the Research Institute and Mark Peeples and Sara Mertz for help with the HAE cultures.
This work was supported in part by NIH grant AI082962 and by The Research Institute.
Footnotes
Published ahead of print 25 September 2013
REFERENCES
- 1.Taubenberger JK, Morens DM. 2008. The pathology of influenza virus infections. Annu. Rev. Pathol. 3:499–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diamond G, Legarda D, Ryan LK. 2000. The innate immune response of the respiratory epithelium. Immunol. Rev. 173:27–38 [DOI] [PubMed] [Google Scholar]
- 3.Bals R, Hiemstra PS. 2004. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur. Respir. J. 23:327–333 [DOI] [PubMed] [Google Scholar]
- 4.Kato A, Schleimer RP. 2007. Beyond inflammation: airway epithelial cells are at the interface of innate and adaptive immunity. Curr. Opin. Immunol. 19:711–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. 2007. Epithelium: at the interface of innate and adaptive immune responses. J. Allergy Clin. Immunol. 120:1279–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaykhiev R, Bals R. 2007. Interactions between epithelial cells and leukocytes in immunity and tissue homeostasis. J. Leukoc. Biol. 82:1–15 [DOI] [PubMed] [Google Scholar]
- 7.Shornick LP, Wells AG, Zhang Y, Patel AC, Huang G, Takami K, Sosa M, Shukla NA, Agapov E, Holtzman MJ. 2008. Airway epithelial versus immune cell Stat1 function for innate defense against respiratory viral Infection. J. Immunol. 180:3319–3328 [DOI] [PubMed] [Google Scholar]
- 8.Humlicek AL, Manzel LJ, Chin CL, Shi L, Excoffon KJ, Winter MC, Shasby DM, Look DC. 2007. Paracellular permeability restricts airway epithelial responses to selectively allow activation by mediators at the basolateral surface. J. Immunol. 178:6395–6403 [DOI] [PubMed] [Google Scholar]
- 9.Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L, Gwyer E, Sedgwick JD, Barclay AN, Hussell T. 2008. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat. Immunol. 9:1074–1083 [DOI] [PubMed] [Google Scholar]
- 10.Ueno K, Koga T, Kato K, Golenbock DT, Gendler SJ, Kai H, Kim KC. 2008. MUC1 mucin is a negative regulator of toll-like receptor signaling. Am. J. Respir. Cell Mol. Biol. 38:263–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamada C, Sano H, Shimizu T, Mitsuzawa H, Nishitani C, Himi T, Kuroki Y. 2006. Surfactant protein A directly interacts with TLR4 and MD-2 and regulates inflammatory cellular response. Importance of supratrimeric oligomerization. J. Biol. Chem. 281:21771–21780 [DOI] [PubMed] [Google Scholar]
- 12.Jia HP, Kline JN, Penisten A, Apicella MA, Gioannini TL, Weiss J, McCray PB., Jr 2004. Endotoxin responsiveness of human airway epithelia is limited by low expression of MD-2. Am. J. Physiol. Lung Cell. Mol. Physiol. 287:L428–L437 [DOI] [PubMed] [Google Scholar]
- 13.Muir A, Soong G, Sokol S, Reddy B, Gomez MI, Van Heeckeren A, Prince A. 2004. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 30:777–783 [DOI] [PubMed] [Google Scholar]
- 14.Greene CM, Carroll TP, Smith SG, Taggart CC, Devaney J, Griffin S, O'Neill SJ, McElvaney NG. 2005. TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J. Immunol. 174:1638–1646 [DOI] [PubMed] [Google Scholar]
- 15.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. 2007. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 27:111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T. 2000. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192:1027–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Francisco LM, Sage PT, Sharpe AH. 2010. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 236:219–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687 [DOI] [PubMed] [Google Scholar]
- 19.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–354 [DOI] [PubMed] [Google Scholar]
- 20.Trautmann L, Janbazian L, Chomont N, Said EA, Wang G, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, Routy JP, Haddad EK, Sekaly RP. 2006. Upregulation of PD-1 expression on HIV-specific CD8 + T cells leads to reversible immune dysfunction. Nat. Med. 12:1198–1202 [DOI] [PubMed] [Google Scholar]
- 21.Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, Ferrari C. 2006. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J. Virol. 80:11398–11403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77:4911–4927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zelinskyy G, Myers L, Dietze KK, Gibbert K, Roggendorf M, Liu J, Lu M, Kraft AR, Teichgraber V, Hasenkrug KJ, Dittmer U. 2011. Virus-specific CD8+ T cells upregulate programmed death-1 expression during acute friend retrovirus infection but are highly cytotoxic and control virus replication. J. Immunol. 187:3730–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowen DG, Shoukry NH, Grakoui A, Fuller MJ, Cawthon AG, Dong C, Hasselschwert DL, Brasky KM, Freeman GJ, Seth NP, Wucherpfennig KW, Houghton M, Walker CM. 2008. Variable patterns of programmed death-1 expression on fully functional memory T cells after spontaneous resolution of hepatitis C virus infection. J. Virol. 82:5109–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allie SR, Zhang W, Fuse S, Usherwood EJ. 2011. Programmed death 1 regulates development of central memory CD8 T cells after acute viral infection. J. Immunol. 186:6280–6286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Telcian AG, Laza-Stanca V, Edwards MR, Harker JA, Wang H, Bartlett NW, Mallia P, Zdrenghea MT, Kebadze T, Coyle AJ, Openshaw PJ, Stanciu LA, Johnston SL. 2011. RSV-induced bronchial epithelial cell PD-L1 expression inhibits CD8+ T cell nonspecific antiviral activity. J. Infect. Dis. 203:85–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erickson JJ, Gilchuk P, Hastings AK, Tollefson SJ, Johnson M, Downing MB, Boyd KL, Johnson JE, Kim AS, Joyce S, Williams JV. 2012. Viral acute lower respiratory infections impair CD8+ T cells through PD-1. J. Clin. Invest. 122:2967–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ioannidis I, McNally B, Willette M, Peeples ME, Chaussabel D, Durbin JE, Ramilo O, Mejias A, Flano E. 2012. Plasticity and virus specificity of the airway epithelial cell immune response during respiratory virus infection. J. Virol. 86:5422–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. 2005. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat. Immunol. 6:280–286 [DOI] [PubMed] [Google Scholar]
- 30.Keir ME, Freeman GJ, Sharpe AH. 2007. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J. Immunol. 179:5064–5070 [DOI] [PubMed] [Google Scholar]
- 31.Martin-Orozco N, Wang YH, Yagita H, Dong C. 2006. Cutting edge: programmed death (PD) ligand-1/PD-1 interaction is required for CD8+ T cell tolerance to tissue antigens. J. Immunol. 177:8291–8295 [DOI] [PubMed] [Google Scholar]
- 32.Wei S, Shreiner AB, Takeshita N, Chen L, Zou W, Chang AE. 2008. Tumor-induced immune suppression of in vivo effector T-cell priming is mediated by the B7-H1/PD-1 axis and transforming growth factor beta. Cancer Res. 68:5432–5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wahl C, Bochtler P, Chen L, Schirmbeck R, Reimann J. 2008. B7-H1 on hepatocytes facilitates priming of specific CD8 T cells but limits the specific recall of primed responses. Gastroenterology 135:980–988 [DOI] [PubMed] [Google Scholar]
- 34.Goldberg MV, Maris CH, Hipkiss EL, Flies AS, Zhen L, Tuder RM, Grosso JF, Harris TJ, Getnet D, Whartenby KA, Brockstedt DG, Dubensky TW, Jr, Chen L, Pardoll DM, Drake CG. 2007. Role of PD-1 and its ligand, B7-H1, in early fate decisions of CD8 T cells. Blood 110:186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lafon M, Megret F, Meuth SG, Simon O, Velandia Romero ML, Lafage M, Chen L, Alexopoulou L, Flavell RA, Prehaud C, Wiendl H. 2008. Detrimental contribution of the immuno-inhibitor B7-H1 to rabies virus encephalitis. J. Immunol. 180:7506–7515 [DOI] [PubMed] [Google Scholar]
- 36.Schreiner B, Mitsdoerffer M, Kieseier BC, Chen L, Hartung HP, Weller M, Wiendl H. 2004. Interferon-beta enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. J. Neuroimmunol. 155:172–182 [DOI] [PubMed] [Google Scholar]
- 37.Heinecke L, Proud D, Sanders S, Schleimer RP, Kim J. 2008. Induction of B7-H1 and B7-DC expression on airway epithelial cells by the Toll-like receptor 3 agonist double-stranded RNA and human rhinovirus infection: In vivo and in vitro studies. J. Allergy Clin. Immunol. 121:1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsuda M, Matsumoto K, Inoue H, Matsumura M, Nakano T, Mori A, Azuma M, Nakanishi Y. 2005. Expression of B7-H1 and B7-DC on the airway epithelium is enhanced by double-stranded RNA. Biochem. Biophys. Res. Commun. 330:263–270 [DOI] [PubMed] [Google Scholar]
- 39.Brown KE, Freeman GJ, Wherry EJ, Sharpe AH. 2010. Role of PD-1 in regulating acute infections. Curr. Opin. Immunol. 22:397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Channappanavar R, Twardy BS, Suvas S. 2012. Blocking of PDL-1 interaction enhances primary and secondary CD8 T cell response to herpes simplex virus-1 infection. PLoS One 7:e39757. 10.1371/journal.pone.0039757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duraiswamy J, Ibegbu CC, Masopust D, Miller JD, Araki K, Doho GH, Tata P, Gupta S, Zilliox MJ, Nakaya HI, Pulendran B, Haining WN, Freeman GJ, Ahmed R. 2011. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J. Immunol. 186:4200–4212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Talay O, Shen CH, Chen L, Chen J. 2009. B7-H1 (PD-L1) on T cells is required for T-cell-mediated conditioning of dendritic cell maturation. Proc. Natl. Acad. Sci. U. S. A. 106:2741–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, Xu H, Ruff W, Broadwater M, Choi IH, Tamada K, Chen L. 2009. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood 113:5811–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rowe JH, Johanns TM, Ertelt JM, Way SS. 2008. PDL-1 blockade impedes T cell expansion and protective immunity primed by attenuated Listeria monocytogenes. J. Immunol. 180:7553–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. 2003. PD-1 inhibits antiviral immunity at the effector phase in the liver. J. Exp. Med. 198:39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kirchberger S, Majdic O, Steinberger P, Bluml S, Pfistershammer K, Zlabinger G, Deszcz L, Kuechler E, Knapp W, Stockl J. 2005. Human rhinoviruses inhibit the accessory function of dendritic cells by inducing sialoadhesin and B7-H1 expression. J. Immunol. 175:1145–1152 [DOI] [PubMed] [Google Scholar]