

Abstract
Reovirus nonstructural protein σ1s is implicated in cell cycle arrest at the G2/M boundary and induction of apoptosis. However, the contribution of σ1s to these effects in an otherwise isogenic viral background has not been defined. To evaluate the role of σ1s in cell cycle arrest and apoptosis, we used reverse genetics to generate a σ1s-null reovirus. Following infection with wild-type virus, we observed an increase in the percentage of cells in G2/M, whereas the proportion of cells in G2/M following infection with the σ1s-null mutant was unaffected. Similarly, we found that the wild-type virus induced substantially greater levels of apoptosis than the σ1s-null mutant. These data indicate that σ1s is required for both reovirus-induced cell cycle arrest and apoptosis. To define sequences in σ1s that mediate these effects, we engineered viruses encoding C-terminal σ1s truncations by introducing stop codons in the σ1s open reading frame. We also generated viruses in which charged residues near the σ1s amino terminus were replaced individually or as a cluster with nonpolar residues. Analysis of these mutants revealed that amino acids 1 to 59 and the amino-terminal basic cluster are required for induction of both cell cycle arrest and apoptosis. Remarkably, viruses that fail to induce cell cycle arrest and apoptosis also are attenuated in vivo. Thus, identical sequences in σ1s are required for reovirus-induced cell cycle arrest, apoptosis, and pathogenesis. Collectively, these findings provide evidence that the σ1s-mediated properties are genetically linked and suggest that these effects are mechanistically related.
INTRODUCTION
Apoptosis is a critical host response to viral infection. Induction of apoptotic cell death limits production of viral progeny from infected cells (1) and provides signals that activate adaptive immune responses (2–4). Although thought to be initiated as a protective measure for the host, apoptosis also causes the pathology associated with many viral diseases. Defining viral and cellular determinants that govern virus-induced apoptosis is of fundamental importance to an understanding of viral pathogenesis.
Mammalian orthoreoviruses (reoviruses) are highly tractable models for studies of viral replication and pathogenesis (5). Reoviruses are nonenveloped, icosahedral viruses with segmented double-stranded RNA (dsRNA) genomes. The different reovirus serotypes display differences in cell tropism (6, 7), mechanism of viral dissemination (8), apoptosis induction (9), and central nervous system (CNS) disease (5). In newborn mice, serotype 3 (T3) reoviruses infect neurons and cause apoptosis that leads to lethal encephalitis (8, 10–12). T3 reoviruses also induce high levels of apoptosis in cultured cells (13). Serotype 1 (T1) reoviruses infect ependymal cells and cause ependymitis and hydrocephalus (11, 12). However, T1 reoviruses induce markedly less apoptosis than T3 reoviruses in cell culture (13). Differences in apoptosis induction segregate with the viral S1 gene segment (9), which encodes attachment protein σ1 and nonstructural protein σ1s (14–17), and the M2 gene segment (9, 18), which encodes outer-capsid protein μ1 (14, 15). Fragments of the μ1 protein generated during virus entry activate intrinsic apoptotic pathways (19–23). However, it is not known how attachment protein σ1 or nonstructural protein σ1s contributes to reovirus apoptosis.
Protein σ1s is a 14-kDa nonstructural protein encoded by the viral S1 gene segment (16, 24, 25). The σ1s open reading frame (ORF) completely overlaps the σ1 coding sequence; however, σ1s lies in a different reading frame (16, 24–27). Little amino acid sequence identity exists in the σ1s proteins from different reovirus serotypes (24, 27). The only feature of the σ1s protein that is conserved across the serotypes is a cluster of positively charged amino acids near the amino terminus (24, 27). For T3 reovirus, this cluster is hypothesized to function as a nuclear localization signal (28).
The σ1s protein has been implicated in reovirus-induced cell cycle arrest at the G2/M boundary (29, 30) and may function in reovirus neurovirulence by influencing apoptosis in the murine CNS (31). However, interpreting these studies of σ1s function is complicated because the σ1s-null mutant virus used in previous experiments is not isogenic to the parental strain from which it was derived (32). Serial passage of a T3 field isolate strain, T3C84, in cell culture yielded a variant, T3C84-MA, which due to introduction of a premature stop codon after the seventh amino acid in the σ1s ORF does not express σ1s (32). Following infection with T3C84, the percentage of cells in the G2/M phase of the cell cycle is increased compared to that after mock infection. In contrast, after infection with T3C84-MA, the percentage of cells in G2/M is similar to that of uninfected controls (30). Although these results suggest a role for σ1s in reovirus-induced cell cycle arrest, the viruses used for these studies are not isogenic. Unlike the parental virus, T3C84-MA harbors, in addition to the mutation affecting σ1s expression, a second mutation in the S1 gene that confers binding of σ1 to cell surface sialic acid (33). Importantly, the capacity to bind sialic acid enhances reovirus-induced apoptosis (34) and virulence in vivo (35). Moreover, there may be additional mutations in T3C84-MA acquired during serial passage that influence these phenotypes. Using σ1s-deficient viruses generated by reverse genetics, we found that T1 and T3 reoviruses require σ1s to disseminate within an infected host using hematogenous pathways (36, 37). However, the mechanism by which σ1s promotes spread via the blood is not known.
In this study, we used wild-type and σ1s-null T3 reoviruses generated by reverse genetics to determine whether σ1s is required for reovirus-induced cell cycle arrest and apoptosis; these viruses are isogenic except for σ1s expression. We found that the σ1s-null mutant failed to cause cell cycle arrest and induced lower levels of apoptosis than the wild-type virus. Using a panel of mutant viruses, we identified σ1s residues 1 to 59 and a cluster of basic amino acids near the amino terminus as essential for both effects. Mutants defective for cell cycle arrest and apoptosis also are attenuated in vivo. Collectively, these findings suggest that cell cycle arrest, apoptosis, and reovirus virulence are mechanistically linked.
MATERIALS AND METHODS
Cell lines.
L929 cells were maintained in Joklik's minimal essential medium (SMEM; Lonza) supplemented to contain 5% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 25 ng/ml amphotericin B. HeLa cells were maintained in Dulbecco's MEM (DMEM; Gibco) supplemented to contain 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 25 ng/ml amphotericin B (Invitrogen). HCT-116 cells were maintained in McCoy's 5a medium (Gibco) supplemented to contain 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 25 ng/ml amphotericin B (Invitrogen).
Preparation of murine cortical neuron cultures.
Primary cultures of mouse cortical neurons were established using cerebral cortices of C57/BL6 embryos at embryonic day 15 (E15) (38). Fetuses were decapitated, their brains were removed, and their cortical lobes were dissected and submerged in Hanks' balanced salt solution (Gibco) on ice. Cortices were incubated in 0.6 mg/ml trypsin solution at room temperature for 30 min, washed twice, and manually dissociated twice with a Pasteur pipette. Viable cells were plated at a density of 2.75 × 105 cells/ml in 24-well plates (Corning) or on glass coverslips (BD Biosciences) placed in 24-well plates. Wells were treated prior to plating with a 10-μg/ml poly-d-lysine solution (BD Biosciences) and a 1.64-μg/ml laminin solution (BD Biosciences). Cultures were incubated for the first 24 h in neurobasal medium (Gibco) supplemented to contain 10% FBS (Gibco), 0.6 mM l-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cultures were thereafter maintained in neurobasal medium supplemented to contain 1× B27 (Gibco), 50 units/ml penicillin, and 50 μg/ml streptomycin. One-half of the medium was replaced every 3 to 4 days. Neurons were allowed to mature for 7 days prior to use.
Viruses.
Recombinant reoviruses were generated using plasmid-based reverse genetics (39). Monolayers of L929 cells at approximately 90% confluence (3 × 106 cells) in 60-mm dishes (Corning) were infected with rDIs-T7 pol at a multiplicity of infection (MOI) of ∼0.5 50% tissue culture infective dose (TCID50)/cell. At 1 h postinfection, cells were cotransfected with 9 plasmid constructs representing cloned gene segments from the strain type 3 Dearing (T3D) genome—pT7-L1T3D (2 μg), pT7-L2T3D (2 μg), pT7-L3T3D (2 μg), pT7-M1T3D (1.75 μg), pT7-M2T3D (1.75 μg), pT7-M3T3D (1.75 μg), pT7-S2T3D (1.5 μg), pT7-S3T3D (1.5 μg), and pT7-S4T3D (1.5 μg)—in combination with 2 μg of pBacT7-S1T3D, pBacT7-S1T3D σ1s-null, pBacT7-S1T3D σ1s-S41Stop, pBacT7-S1T3D σ1s-L60Stop, pBacT7-S1T3D σ1s-L80Stop, pBacT7-S1T3D σ1s-L97stop, pBacT7-S1T3D σ1s-R14L, pBacT7-S1T3D σ1s-R14L/R15L, or pBacT7-S1T3D σ1s-R14L/R15L/R19L. For each transfection, 3 μl of TransIT-LT1 transfection reagent (Mirus) was used per μg of plasmid DNA. Following 5 days of incubation, recombinant virus was isolated from transfected cells by plaque purification using monolayers of L929 cells (40). For generation of σ1s mutant viruses, pBacT7-S1T3D was altered by QuikChange (Stratagene) site-directed mutagenesis. Sequences of mutant viruses were confirmed using S1 gene cDNAs prepared from viral RNA extracted from purified virions subjected to OneStep reverse transcriptase PCR (RT-PCR) (Qiagen) with S1-specific primers. Changes introduced into the σ1s ORF did not alter the σ1 protein sequence. Primer sequences are available from the corresponding author upon request. PCR products were analyzed following electrophoresis in Tris-borate-EDTA agarose gels or purified and subjected directly to sequence analysis. The presence of a noncoding signature mutation in the L1 gene of viruses generated by plasmid-based rescue was confirmed using RT-PCR and L1-specific primers (39).
Purified reovirus virions were generated using second- or third-passage L929 cell lysate stocks of twice-plaque-purified reovirus as described previously (41). Viral particles were Freon extracted from infected cell lysates, layered onto 1.2- to 1.4-g/cm3 CsCl gradients, and centrifuged at 62,000 × g for 18 h. Bands corresponding to virions (1.36 g/cm3) (42) were collected and dialyzed in virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.4]). The concentration of reovirus virions in purified preparations was determined from an equivalence to one optical density (OD) unit at 260 nm (2.1 × 1012 virions) (42). Viral titer was determined by plaque assay using L929 cells (40).
Virus replication assays.
L929 cells (5 × 104 cells/well) seeded in 24-well plates were adsorbed in triplicate with reovirus strains at an MOI of 1 PFU/cell at room temperature for 1 h in serum-free medium, washed once with phosphate-buffered saline (PBS), and incubated in serum-containing medium for various intervals. Cells were frozen and thawed twice prior to determination of viral titer by plaque assay using L929 cells (40).
Flow cytometry.
L929 cells (106 cells/well) seeded in 6-well plates were adsorbed with reovirus strains at various MOIs at room temperature for 1 h. At various intervals postinfection, cells were trypsinized, transferred to microcentrifuge tubes, washed twice with PBS, and fixed in 70% ethanol at 4°C overnight. Cells were washed twice with PBS and stained with Krishan's stain, containing 3.8 mM trisodium citrate (Sigma), 70 μM propidium iodide (Sigma), 0.01% Nonidet P-40 (Sigma), and 0.01 mg of RNase A (Boehringer Mannheim) per ml (43). Cellular DNA content was quantified using a Coulter Epics XL flow cytometer (Beckman-Coulter). Alignment of the instrument was verified daily using DNA check beads (Coulter). Peak versus integral gating was used to exclude doublet events from the analysis. Data were collected for 10,000 events. Cell cycle modeling was accomplished using the Flow-Jo program (Verity Software House).
Quantification of apoptosis by AO staining.
L929, HeLa, or HCT-116 cells (5 × 104 cells/well) seeded in 24-well plates were adsorbed with reovirus strains at various MOIs at room temperature for 1 h. After 48 h of incubation, the percentage of apoptotic cells was determined using acridine orange (AO) staining as described previously (13). For each experiment, >200 cells were counted, and the percentage of cells exhibiting condensed chromatin was determined by epi-illumination fluorescence microscopy using a fluorescein filter set (Zeiss).
Assessment of caspase 3/7 activity.
HeLa cells (2 × 105 cells/well) seeded in 24-well plates were adsorbed with reovirus strains at an MOI of 100 PFU/cell at room temperature for 1 h. Cells were frozen 24 h postinfection, and caspase 3/7 activity in thawed lysates containing 5 × 103 cell equivalents was quantified using the Caspase-Glo-3/7 assay system (Promega) according to the manufacturer's instructions.
Assessment of reovirus infectivity by indirect immunofluorescence.
Monolayers of L929, HeLa, or HCT-116 cells (2 × 105 cells/well) seeded in 24-well plates were adsorbed with reovirus strains at an MOI of 1 PFU/cell at room temperature for 1 h. Inocula were removed, cells were washed once with PBS, and fresh medium was added. Cells were incubated at 37°C for 24 h, washed once with PBS, and fixed with ice-cold methanol for 30 min at 4°C. Cells were washed twice with PBS prior to being blocked with 5% bovine serum albumin (BSA) in PBS at room temperature for 15 min. Cells were incubated with polyclonal reovirus-specific serum diluted 1:500 in PBS containing 0.5% Triton X-100 at room temperature for 1 h. Primary antibody was removed, and cells were washed twice with PBS and incubated with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) and Alexa Fluor 488-labeled goat anti-rabbit fluorescent secondary antibody (Invitrogen), both diluted 1:1,000 in PBS containing 0.5% Triton X-100, at room temperature for 1 h. Cells were washed twice with PBS, and infected cells were visualized by fluorescence microscopy. Results are expressed as the mean percentage of infected cells present in three ×10-magnification fields of view.
Assessment of σ1s expression by confocal microscopy.
Monolayers of HeLa cells (105 cells/well) grown on coverslips in 24-well plates were adsorbed with reovirus at an MOI of 10 PFU/cell at room temperature for 1 h. Following removal of the inoculum, cells were washed with PBS and incubated in complete medium at 37°C for 18 h to permit completion of a single cycle of viral replication. Monolayers were fixed with 1 ml of methanol at 20°C for at least 30 min, washed twice with PBS, and blocked with 0.1% gelatin, 0.1% Tween 20 (Sigma), and 20% normal goat serum (Vector Laboratories) in PBS. For detection of σ1s, cells were washed with PBS and incubated with mouse monoclonal anti-σ1s antibody 2F4 (44) at a dilution of 1:500 in PBS with 0.1% gelatin, 2% normal goat serum, and 0.1% Tween 20. For detection of reovirus proteins, cells were washed once with PBS and stained with polyclonal rabbit antireovirus serum at a 1:1,000 dilution in PBS–0.5% Triton X-100 at room temperature for 1 h. Monolayers were washed twice with PBS–0.5% Triton X-100 and incubated with a 1:1,000 dilution of Alexa 488- or Alexa 546-labeled anti-rabbit or anti-mouse IgG (Invitrogen), respectively. Monolayers were washed with PBS, and infected cells were visualized by confocal microscopy using a Ti-U microscope with a 60×/1.4-numerical-aperture (NA) oil immersion objective (Nikon). Images were analyzed using NIS-Elements software (Nikon) and normalized for pixel intensity, brightness, and contrast.
Infection of mice.
C57BL/6J mice were obtained from Jackson Laboratory. Newborn mice weighing 1.5 to 2 g were inoculated intramuscularly with purified reovirus diluted in PBS. Intramuscular inoculations (10 μl) were delivered into the left hind limb using a Hamilton syringe and 30-gauge needle. Mice were monitored for weight loss and symptoms of disease for 21 days postinoculation and euthanized when found to be moribund (defined by rapid or shallow breathing, lethargy, or paralysis). Death was not used as an endpoint in these experiments. Animal husbandry and experimental procedures were performed in accordance with Public Health Service policy and approved by the Vanderbilt University School of Medicine Institutional Animal Care and Use Committee.
Statistical analysis.
Means of results from triplicate samples were compared using an unpaired Student t test. Differences in viral virulence were determined using a log-rank test. Statistical analyses were performed using Prism software (GraphPad Software, Inc.). P values of <0.05 were considered to be statistically significant.
RESULTS
The σ1s protein is required for reovirus-induced cell cycle arrest.
To determine whether σ1s is required for reovirus-induced cell cycle arrest, L929 cells were mock infected or infected with rsT3D or rsT3D σ1s-null at an MOI of 100 PFU/cell. At 24 h postinfection, cellular DNA was stained with propidium iodide and quantified by flow cytometry (Fig. 1A). We observed a >2-fold increase in the percentage of cells with 4N DNA content (representing cells in G2 or mitosis) following infection with rsT3D relative to that in mock-infected cells. In contrast, rsT3D σ1s-null did not alter the proportion of cells in the G2 and M phases of the cell cycle. Infection of HeLa cells (Fig. 1B) or HCT116 cells (Fig. 1C) with rsT3D σ1s-null also did not alter the percentage of cells in G2/M. To ensure that differences in the capacities of rsT3D and rsT3D σ1s-null to induce cell cycle arrest do not reflect a difference in infectivity between the two viruses, we quantified the number of infected cells 24 h after inoculation of L929, HeLa, and HCT-116 cells. No difference in infectivity of rsT3D and rsT3D σ1s-null was observed in any of the cell lines tested (Fig. 1D). This result is consistent with our previous finding that rsT3D and rsT3D σ1s-null produce comparable yields of viral progeny in cultured cells (36). Thus, failure of rsT3D σ1s-null to induce cell cycle arrest is not attributable to a defect in infectivity or replication. These results indicate that σ1s is required for reovirus-induced cell cycle arrest.
Fig 1.

The σ1s protein is required for reovirus-induced cell cycle arrest. (A to C) L929 (A), HeLa (B), or HCT-116 (C) cells were mock infected or infected with rsT3D or rsT3D σ1s-null at an MOI of 100 PFU/cell. At 24 h postinfection, cells were stained with propidium iodide, and cellular DNA content was quantified by flow cytometry. Results are expressed as the mean percentage of cells with a 4N DNA content, which represents cells in the G2 or M phases of the cell cycle, for three independent experiments. (D) L929, HeLa, or HCT116 cells were inoculated with rsT3D or rsT3D σ1s-null at an MOI of 1 PFU/cell. At 24 h postinoculation, infected cells were quantified by indirect immunofluorescence. Results are expressed as the mean percentage of infected cells in a ×10-magnification field of view for three independent samples. Error bars indicate standard deviations (SD). *, P < 0.05 (determined by Student's t test in a comparison with rsT3D-infected cells).
The σ1s protein is required for reovirus-induced apoptosis.
To determine whether σ1s is required for reovirus-induced apoptosis, L929 cells were mock infected or infected with rsT3D or rsT3D σ1s-null at an MOI of 100 PFU/cell. Apoptotic nuclei were quantified by AO staining at 48 h postinfection (Fig. 2A). We found that rsT3D induced apoptosis in nearly 100% of cells. Although rsT3D σ1s-null induced more apoptosis than was observed in mock-infected cells, the level of apoptosis was substantially lower than that following infection with rsT3D. Even when the infectious dose of rsT3D σ1s-null was 10-fold higher than a dose of rsT3D that elicits apoptosis in nearly 100% of cells (1,000 PFU/cell for rsT3D σ1s-null versus 100 PFU/cell for rsT3D), the capacity of rsT3D σ1s-null to induce apoptosis was reduced (Fig. 2E). Concordantly, rsT3D σ1s-null also induced less caspase 3/7 activity than rsT3D (Fig. 2F). These findings are not restricted to cell type, as rsT3D σ1s-null elicited lower levels of apoptosis than rsT3D in HeLa cells (Fig. 2B), HCT116 cells (Fig. 2C), and primary cultures of murine cortical neurons (Fig. 2D). Together, these findings indicate that σ1s expression significantly potentiates the capacity of reovirus to induce apoptotic cell death.
Fig 2.

The σ1s protein enhances reovirus-induced apoptosis. (A to D) L929 (A), HeLa (B), or HCT-116 (C) cells or mouse primary cortical neurons (D) were mock infected or infected with rsT3D or rsT3D σ1s-null at an MOI of 100 PFU/cell. At 48 h postinfection, the percentage of apoptotic nuclei was determined following AO staining. (E) L929 cells were infected with rsT3D or rsT3D σ1s-null at the indicated MOIs. At 48 h postinfection, the percentage of apoptotic nuclei was determined following AO staining. Results are expressed as the mean percentage of apoptotic nuclei for three independent experiments. Error bars indicate SD. (F) HeLa cells were mock infected or infected with rsT3D or rsT3D σ1s-null at an MOI of 100 PFU/cell. Caspase 3/7 activity was quantified at 24 h postinfection. Results are expressed as the mean caspase 3/7 activity relative to that in mock-infected cells for three independent experiments. Error bars indicate SD. *, P < 0.0001 (determined by Student's t test in a comparison with rsT3D-infected cells).
Cell cycle arrest precedes apoptosis following reovirus infection.
To define the kinetics of cell cycle arrest and apoptosis during reovirus infection, L929 cells were mock infected or infected with rsT3D at an MOI of 100 PFU/cell. Cell cycle status was assessed (Fig. 3A), and apoptotic cells were quantified (Fig. 3B) at 12-h intervals over a 48-h period. Cell cycle progression was not affected by rsT3D until 24 h postinfection. At this time point, the percentage of cells with a 4N DNA content was approximately twice that detected following mock infection. Although no differences in the percentages of cells in G2/M were detected between mock- and rsT3D-infected cells at 36 or 48 h postinfection, cell cycle modeling at those times is imprecise due to cytopathic effects associated with late stages of viral infection. Apoptosis was not detected in rsT3D-infected cells until 36 h postinfection, and levels of apoptosis were increased at 48 h. These data indicate that cell cycle arrest occurs prior to the induction of apoptosis during reovirus infection.
Fig 3.
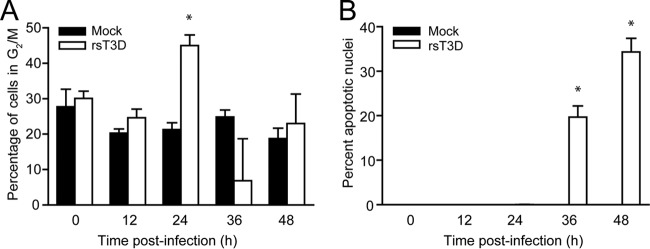
Cell cycle arrest precedes apoptosis during reovirus infection. (A) L929 cells were either mock infected or infected with rsT3D at an MOI of 100 PFU/cell. At the indicated times postinfection, cells were stained with propidium iodide, and cellular DNA content was quantified by flow cytometry. Results are expressed as the mean percentage of cells with a 4N DNA content for three independent experiments. Error bars indicate SD. *, P < 0.05 by Student's t test in comparison to mock infection. (B) HeLa cells were either mock infected or infected with rsT3D at an MOI of 100 PFU/cell. At the indicated times postinfection, the percentage of apoptotic nuclei was determined following AO staining. Results are expressed as the mean percentage of apoptotic nuclei from three independent experiments. Error bars indicate SD. *, P < 0.0001 (determined by Student's t test in a comparison with mock-infected cells).
Identical sequences in σ1s mediate cell cycle arrest and apoptosis.
To define sequences in σ1s required for cell cycle arrest and apoptosis, we used reverse genetics to generate reoviruses expressing truncated σ1s proteins. Stop codons were inserted into the σ1s ORF at amino acid positions 41, 60, 80, and 97 to yield the following viruses: rsT3D σ1s 1–40, rsT3D σ1s 1–59, rsT3D σ1s 1–79, and rsT3D σ1s 1–96. Stop codon insertion and the absence of other S1 gene mutations were confirmed by direct sequencing of viral RNA (data not shown). To determine whether truncating the σ1s protein affects reovirus replication in cell culture, we quantified viral yields following infection of L929 cells (Fig. 4A). Yields of infectious progeny for each mutant were equivalent to those produced by rsT3D. Moreover, immunoblot analysis of infected cell lysates revealed no differences in viral protein levels between rsT3D and the truncation mutants (Fig. 4B). These findings indicate that truncating the σ1s protein does not affect viral gene expression or replication in cultured cells.
Fig 4.
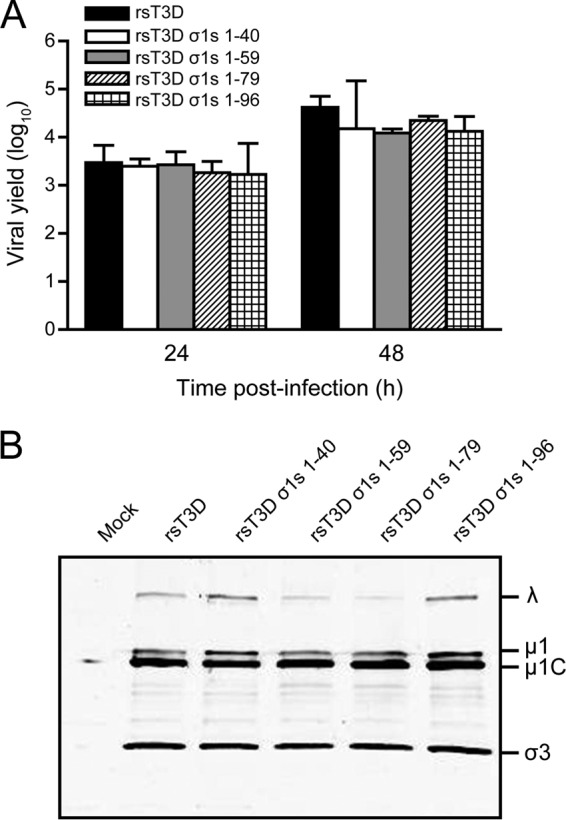
Truncation of the σ1s protein does not alter reovirus replication in cell culture. (A) L929 cells were infected with rsT3D, rsT3D σ1s 1–40, rsT3D σ1s 1–59, rsT3D σ1s 1–79, or rsT3D σ1s 1–96 at an MOI of 1 PFU/cell. Titers of virus in cell lysates were determined by plaque assay at 24 and 48 h postinfection. Results are expressed as the mean viral yield for triplicate samples. Error bars indicate SD. (B) L929 cells were mock infected or infected with rsT3D, rsT3D σ1s 1–40, rsT3D σ1s 1–59, rsT3D σ1s 1–79, or rsT3D σ1s 1–96 at an MOI of 100 PFU/cell. Whole-cell lysates were prepared from infected cells at 48 h postinfection and resolved by SDS-polyacrylamide gel electrophoresis. Reovirus proteins were detected by immunoblotting using a reovirus-specific polyclonal antiserum. Reovirus proteins are labeled on the right.
To identify sequences in σ1s required for reovirus-induced cell cycle arrest, L929 cells were mock infected or infected with rsT3D, rsT3D σ1s-null, or each of the σ1s truncation mutants at an MOI of 100 PFU/cell. We used L929 cells for cell cycle analysis because experiments using these cells provided the largest dynamic range of the three cell lines tested. At 24 h postinfection, cellular DNA was stained with propidium iodide and quantified by flow cytometry (Fig. 5A). We found that the percentage of cells in G2/M was increased following infection with rsT3D σ1s 1–59, rsT3D σ1s 1–79, and rsT3D σ1s 1–96 but not to the levels achieved by rsT3D. In contrast, rsT3D σ1s 1–40 did not alter the percentage of cells in G2/M relative to mock infection. These findings indicate that amino acids 1 to 59 in σ1s are required for reovirus-induced cell cycle arrest. Residues 60 to 120 in σ1s are not essential for this property, but these sequences may enhance the capacity of the protein to cause cell cycle dysregulation.
Fig 5.
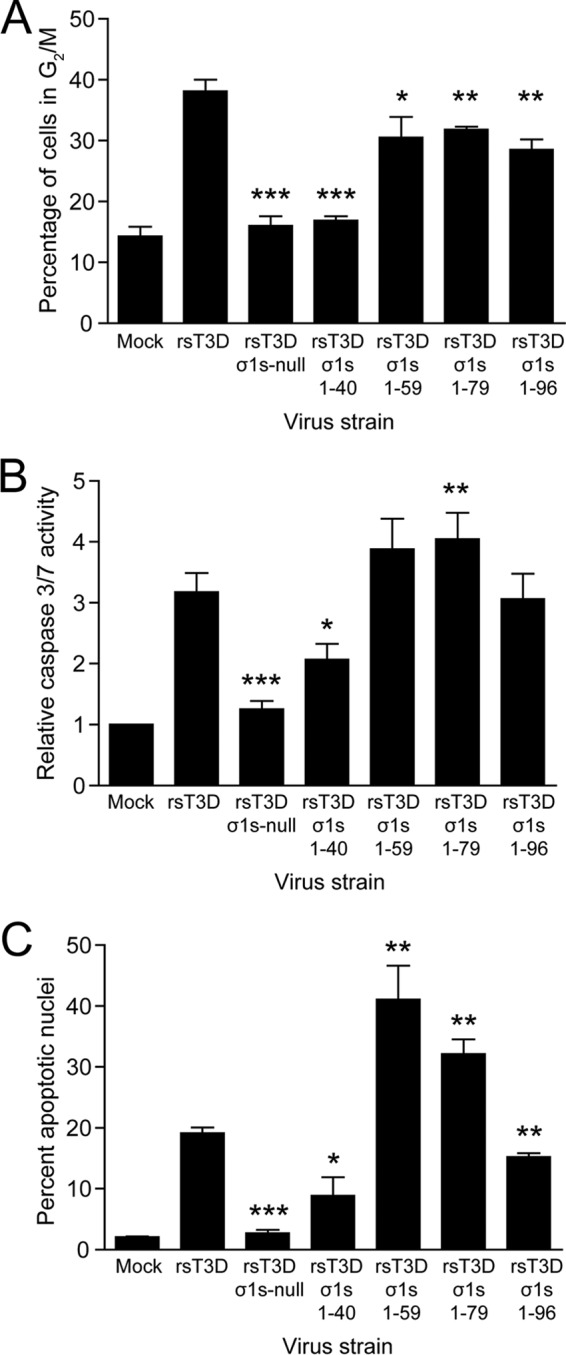
Amino acids 1 to 59 of σ1s are required for reovirus-induced cell cycle arrest and apoptosis. (A) L929 cells were mock infected or infected with rsT3D, rsT3D σ1s-null, rsT3D σ1s 1–40, rsT3D σ1s 1–59, rsT3D σ1s 1–79, or rsT3D σ1s 1–96 at an MOI of 100 PFU/cell. At 24 h postinfection, cells were stained with propidium iodide, and cellular DNA content was quantified by flow cytometry. Results are expressed as the mean percentage of cells with a 4N DNA content from three independent experiments. Error bars indicate SD. *, P < 0.05; **, P < 0.005; ***, P < 0.0001 (as determined by Student's t test in comparison to rsT3D). (B and C) HeLa cells were mock infected or infected with rsT3D, rsT3D σ1s-null, rsT3D σ1s 1–40, rsT3D σ1s 1–59, rsT3D σ1s 1–79, or rsT3D σ1s 1–96 at an MOI of 100 PFU/cell. Caspase 3/7 activity was quantified at 24 h postinfection (B), and the percentages of apoptotic nuclei were determined following AO staining at 48 h postinfection (C). Shown are mean values from three independent experiments. Error bars indicate SD. *, P < 0.01; **, P < 0.005; ***, P < 0.0001 (as determined by Student's t test in a comparison with rsT3D-infected cells).
To identify sequences in σ1s required for apoptosis induction, HeLa cells were mock infected or infected with rsT3D, rsT3D σ1s-null, or each of the σ1s truncation viruses at an MOI of 100 PFU/cell. In parallel cultures, apoptosis induction was assessed by quantifying caspase 3/7 activity or determining the percentage of apoptotic cells using AO staining (Fig. 5B and C). We assessed apoptosis in HeLa cells because experiments using these cells provided the largest dynamic range of the three cell lines tested. By both measures, rsT3D σ1s 1–96 induced levels of apoptosis comparable to those elicited by rsT3D. Strikingly, levels of apoptosis were markedly higher following infection with rsT3D σ1s 1–59 and rsT3D σ1s 1–79 than with rsT3D. In contrast, rsT3D σ1s 1–40 induced significantly less apoptosis than did the wild-type virus. These results indicate that σ1s residues 1 to 59 are required for apoptosis induction and suggest that σ1s amino acids 60 to 96 function as a regulatory domain that negatively modulates the capacity of σ1s to promote apoptosis. Together, these findings indicate that the same amino acids in σ1s are required for reovirus-induced cell cycle arrest and apoptosis induction, suggesting that these properties are mechanistically linked.
The σ1s amino-terminal basic cluster is required for reovirus-induced cell cycle arrest and apoptosis.
To determine whether the σ1s amino-terminal basic cluster is required for reovirus-induced cell cycle arrest and apoptosis, we used reverse genetics to generate three mutant viruses in which arginine residues at positions 14, 15, and 19 in the basic cluster (R14RSRRRLK21) were replaced individually or in combination with leucine to yield the following viruses: rsT3D σ1s R14L, rsT3D σ1s R14L/R15L, and rsT3D σ1s R14L/R15L/R19L. In each case, the arginine-to-leucine substitution preserved the coding sequence of the overlapping σ1 ORF. Arginine 17, arginine 18, and lysine 21 cannot be altered without changing the σ1 coding sequence. The arginine-to-leucine substitutions and absence of other S1 gene mutations were confirmed by direct sequencing of viral RNA (data not shown). To test whether altering the σ1s basic cluster affects viral replication, we quantified viral yields following infection of L929 cells (Fig. 6A). Yields of infectious progeny for each mutant were equivalent to those produced by rsT3D. This finding indicates that the σ1s basic cluster is not required for reovirus replication in cultured cells. In addition, no differences in viral protein levels were detected between rsT3D and the σ1s basic cluster mutants, indicating that viral gene expression is not affected by altering the charge in this region of σ1s (Fig. 6B). Thus, basic residues at some positions in the σ1s amino-terminal basic cluster are dispensable for viral gene expression and replication in cultured cells.
Fig 6.
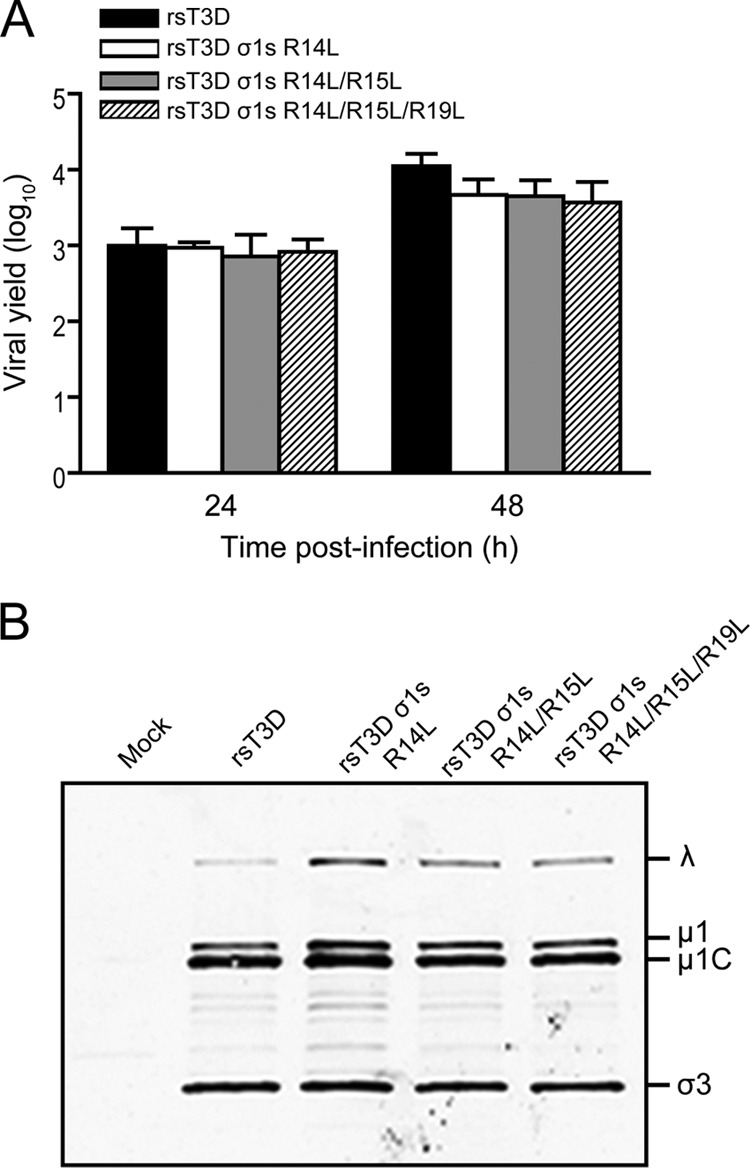
The σ1s N-terminal basic cluster is not required for reovirus replication in cell culture. (A) L929 cells were infected with rsT3D, rsT3D σ1s R14L, rsT3D σ1s R14L/R15L, or rsT3D σ1s R14L/R15L/R19L at an MOI of 1 PFU/cell. Titers of virus in cell lysates were determined by plaque assay at 24 and 48 h postinfection. Results are expressed as the mean viral yields from triplicate samples. Error bars indicate SD. (B) L929 cells were mock infected or infected with rsT3D, rsT3D σ1s R14L, rsT3D σ1s R14L/R15L, or rsT3D σ1s R14L/R15L/R19L at an MOI of 100 PFU/cell. Whole-cell lysates were prepared from infected cells at 48 h postinfection and resolved by SDS-polyacrylamide gel electrophoresis. Reovirus proteins were detected by immunoblotting using a reovirus-specific polyclonal antiserum. Reovirus proteins are labeled on the right.
To determine whether the σ1s amino-terminal basic cluster is required for reovirus-induced cell cycle arrest, L929 cells were mock infected or infected with rsT3D, rsT3D σ1s-null, rsT3D σ1s R14L, rsT3D σ1s R14L/R15L, or rsT3D σ1s R14L/R15L/R19L at an MOI of 100 PFU/cell. At 24 h postinfection, cellular DNA was stained with propidium iodide and quantified by flow cytometry (Fig. 7A). The percentage of cells in G2/M was significantly lower following infection with all three basic cluster mutants than with rsT3D. These data indicate that the σ1s amino-terminal basic cluster is required for reovirus-induced cell cycle arrest.
Fig 7.
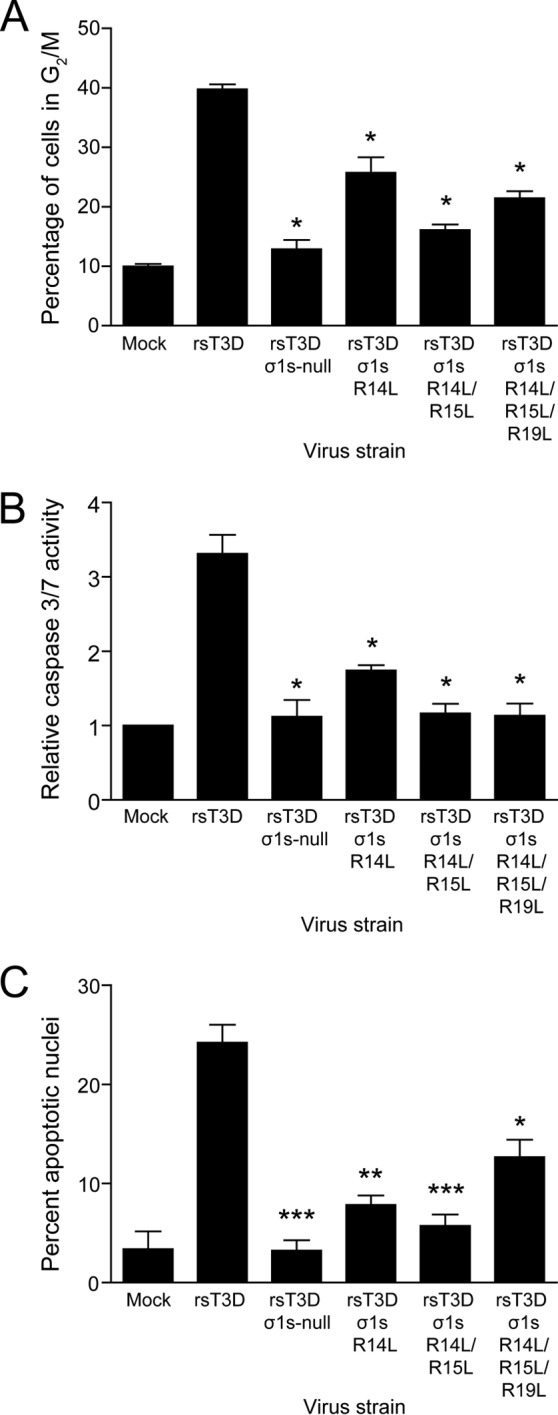
The σ1s N-terminal basic cluster is required for reovirus-induced cell cycle arrest and apoptosis. (A) L929 cells were mock infected or infected with rsT3D, rsT3D σ1s-null, rsT3D σ1s R14L, rsT3D σ1s R14L/R15L, or rsT3D σ1s R14L/R15L/R19L at an MOI of 100 PFU/cell. At 24 h postinfection, cells were stained with propidium iodide, and cellular DNA content was quantified by flow cytometry. Results are expressed as the mean percentage of cells with a 4N DNA content from three independent experiments. Error bars indicate SD. *, P < 0.05 (as determined by Student's t test in a comparison with rsT3D-infected cells). (B and C) HeLa cells were mock infected or infected with rsT3D, rsT3D σ1s-null, rsT3D σ1s R14L, rsT3D σ1s R14L/R15L, or rsT3D σ1s R14L/R15L/R19L at an MOI of 1,000 PFU/cell. Caspase 3/7 activity was quantified at 24 h postinfection (B) and the percentage of apoptotic nuclei was determined following AO staining at 48 h postinfection (C). Shown are mean values from three independent experiments. Error bars indicate SD. *, P < 0.05; **, P < 0.005; ***, P < 0.001 (as determined by Student's t test in a comparison with rsT3D-infected cells).
To determine whether the σ1s amino-terminal basic cluster is required for apoptosis induction, HeLa cells were mock infected or infected with rsT3D, rsT3D σ1s-null, rsT3D σ1s R14L, rsT3D σ1s R14L/R15L, or rsT3D σ1s R14L/R15L/R19L at an MOI of 100 PFU/cell. In parallel cultures, apoptosis induction was assessed by quantifying caspase 3/7 activity (Fig. 7B) or determining the percentage of apoptotic cells using AO staining (Fig. 7C). By both techniques, each σ1s basic cluster mutant was impaired in apoptosis induction compared with rsT3D, indicating that the σ1s basic cluster is required for reovirus-induced apoptosis. We conclude that the σ1s amino-terminal basic cluster is an essential sequence region for reovirus-induced cell cycle arrest and apoptosis, which provides further evidence that these effects are linked.
Changes in the amino-terminal basic cluster do not affect σ1s nuclear translocation.
To test whether altering the σ1s amino-terminal basic cluster affects the capacity of the protein to enter the nucleus, HeLa cells were mock infected or infected with rsT3D, rsT3D σ1s R14L, rsT3D σ1s R14L/R15L, or rsT3D σ1s R14L/R15L/R19L. At 24 h postinfection, the intracellular distribution of σ1s was assessed by indirect immunofluorescence (Fig. 8). The σ1s protein of wild-type rsT3D was observed in the nucleus and cytoplasm, consistent with results of previous reports (28, 32, 36). The σ1s proteins of all three basic cluster mutant viruses were similarly distributed at the time point tested. These data indicate that mutations in the amino-terminal basic cluster do not alter σ1s nuclear translocation and suggest that the basic cluster is not required for nuclear localization.
Fig 8.

Mutations in the σ1s amino-terminal basic cluster do not affect nuclear translocation of the protein. Cos-7 cells were either mock infected or infected with rsT3D, rsT3D σ1s R14L, rsT3D σ1s R14L/R15L, or rsT3D σ1s R14L/R15L/R19L at an MOI of 10 PFU/cell. Following incubation for 24 h, cells were fixed and stained with a T3D σ1s-specific monoclonal antibody or a reovirus-specific polyclonal antiserum. Nuclei were stained with DAPI.
Sequences in σ1s essential for cell cycle arrest and apoptosis induction are required for reovirus virulence.
To determine whether sequences in σ1s required for cell cycle arrest and apoptosis induction also are required for reovirus virulence, we inoculated newborn C57BL/6 mice in the left hind limb muscle with 106 PFU of rsT3D, rsT3D σ1s-null, rsT3D σ1s 1–40, rsT3D σ1s 1–59, or rsT3D σ1s R14L/R15L (Fig. 9). Mice were monitored for 21 days for signs of disease and euthanized when moribund. Hind limb inoculations were performed because reovirus strain T3D replicates poorly in the gastrointestinal tract and fails to disseminate systemically from that site (45–47). All mice inoculated with rsT3D succumbed to infection. The median survival time (MST) for animals infected with rsT3D was 9 days (Table 1). In contrast, 50% of mice survived infection with rsT3D σ1s-null (MST = 18 days). Similarly, 55% of mice survived infection with rsT3D σ1sR14L/R15L, suggesting that the σ1s amino-terminal basic cluster is required for reovirus virulence. The rsT3D σ1s 1–59 mutant was slightly attenuated compared with rsT3D; 19% of mice survived infection (MST = 12 days). However, the rsT3D σ1s 1–40 mutant was substantially attenuated, with a survival rate of 88%. These results indicate that σ1s residues 1 to 59 are essential for full reovirus virulence. Collectively, these findings indicate that sequences in σ1s required for cell cycle arrest and apoptosis induction also are required for reovirus pathogenesis, which suggests a mechanistic link between cell cycle dysregulation, apoptosis induction, and disease.
Fig 9.
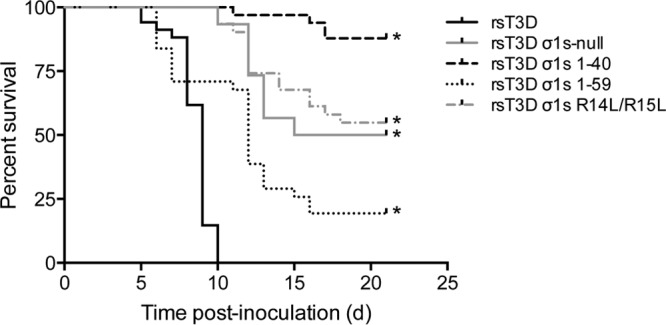
The σ1s amino-terminal basic cluster and amino acids 1 to 59 are required for full reovirus neurovirulence. Newborn C57BL/6 mice were inoculated in the left hind limb with 106 PFU of rsT3D, rsT3D σ1s-null, rsT3D σ1s 1–40, rsT3D σ1s 1–59, or rsT3D σ1s R14L/R15L. Mice were monitored for illness for 21 days (d) and euthanized when moribund. *, P < 0.001 (as determined by log-rank test in a comparison with rsT3D-infected cells).
Table 1.
Survival statistics following intramuscular inoculationa
| Virus strain | No. of animals inoculated | No. of eventsb | % survival | Median survival time (days) |
|---|---|---|---|---|
| rsT3D | 34 | 34 | 0 | 9 |
| rsT3D σ1s-null | 30 | 15 | 50 | 18 |
| rsT3D σ1s 1–40 | 33 | 4 | 88 | >21 |
| rsT3D σ1s 1–59 | 31 | 25 | 19 | 12 |
| rsT3D σ1s R14L/R15L | 31 | 14 | 55 | >21 |
Newborn C57BL/6 mice were inoculated intramuscularly in the left hind limb with 106 PFU of the indicated strains. Mice were monitored for signs of disease for 21 days postinoculation and euthanized when moribund.
Number of mice that did not survive the 21-day observation period.
DISCUSSION
Nonstructural protein σ1s is required for hematogenous reovirus dissemination (36, 37). However, mechanisms by which σ1s promotes systemic spread have not been determined. Previous studies using a σ1s-deficient reovirus strain suggest that σ1s inhibits cell cycle progression at the G2/M boundary (29) and influences reovirus-induced apoptosis (31). However, interpreting these studies is complicated by the use of viruses that are not isogenic with respect to σ1s expression. In this study, we used isogenic viruses generated by reverse genetics to determine whether σ1s mediates reovirus cell cycle arrest and apoptosis. We found that σ1s expression is required for cell cycle arrest, consistent with previous studies. We also found that σ1s enhances reovirus-induced apoptosis. Using a panel of mutant viruses, we identified σ1s amino acids 1 to 59 and the amino-terminal basic cluster as essential for both effects. Finally, we found that mutant viruses that fail to induce cell cycle arrest and apoptosis also are attenuated in vivo. Together, these data provide evidence that σ1s-mediated cell cycle arrest, apoptosis, and virulence are genetically linked and suggest that a common mechanism underlies these effects.
Although our data indicate that σ1s mediates cell cycle arrest and apoptosis in reovirus-infected cells, it is not known how these effects are functionally associated. During cell division, cell cycle arrest and apoptosis act in concert to prevent passing damaged DNA to daughter cells. If DNA is damaged or replication stress is detected, checkpoints are activated to inhibit cell cycle progression. Repair of such damage terminates the arrest and allows the cell cycle to proceed through mitosis. When the damage cannot be repaired, apoptosis is induced to ensure that daughter cells receive only faithfully replicated DNA. We envision three possibilities to explain how σ1s-dependent cell cycle arrest leads to apoptosis during reovirus infection. First, σ1s may trigger cellular pathways that are activated by DNA damage or replication stress. For example, activation of the DNA damage response (DDR) proteins ataxia telangiectasia mutated (ATM) or ATM-Rad3-related (ATR) can induce cell cycle arrest that leads to apoptosis (48). Although activation of ATM and ATR following DNA virus infection is well documented (49), RNA viruses also can activate these pathways. For example, Rift Valley fever virus causes cell cycle arrest in an ATM-dependent manner (50). Avian reovirus induces phosphorylation of ATM that is consistent with DDR activation and causes G2/M arrest (51). However, it is not known whether DDR induction leads to cell cycle arrest or apoptosis in avian-reovirus-infected cells. Second, σ1s may halt cell cycle progression by inhibiting cell cycle regulatory proteins. Several viruses encode proteins that cause cell cycle arrest and apoptosis by blocking the activities of cell cycle control factors. Adenovirus E4orf4 (52) and hepatitis B virus X protein (53) directly engage components of the anaphase-promoting complex (APC), which powers mitotic exit. Cell cycle arrest and apoptosis can result from impaired APC function (54). In reovirus-infected cells, σ1s might inhibit cell cycle progression by interfering with the function of cellular proteins that control the G2-to-M transition, with the resultant stress associated with prolonged arrest in G2 leading to apoptosis. Third, σ1s may indirectly elicit cell cycle arrest and apoptosis by disrupting a vital cellular process that causes cell stress. For example, the σ1s protein is implicated in altering the architecture of the nuclear envelope by disrupting the lamin network, which is integral to nuclear structure and function (28). Disrupting nuclear lamin can activate the DNA replication checkpoint, leading to cell cycle arrest and ultimately apoptosis (55, 56). Identifying cellular proteins that interact with σ1s will likely provide insight into how σ1s functions to cause cell cycle arrest and apoptotic cell death.
If the contribution of σ1s to reovirus apoptosis is dependent on induction of cell cycle arrest, then the proapoptotic activity of σ1s would manifest only in actively dividing cells. A causal relationship between σ1s-dependent cell cycle arrest and apoptosis would explain why rsT3D and rsT3D σ1s-null are comparably virulent following intracranial inoculation of newborn mice (36), even though rsT3D σ1s-null induces apoptosis less efficiently than rsT3D in cultured cells. Unlike most cells in culture, neurons do not divide in vivo following completion of a neural developmental program (57). Consequently, σ1s-mediated proapoptotic signals would not be induced in neurons, and apoptosis would result solely from σ1s-independent mechanisms. In keeping with this idea, reovirus can activate proapoptotic networks that function regardless of whether σ1s is expressed. For example, cleavage fragments of outer-capsid protein μ1 generated during viral disassembly activate intrinsic apoptotic pathways (20, 23). In addition, proinflammatory cytokines secreted in response to reovirus infection may contribute to virus-induced apoptosis by activating death receptors (21, 58–62). Both of these mechanisms are likely induced by rsT3D and rsT3D σ1s-null in the murine CNS. Although we found that rsT3D infection induces more apoptosis in primary cortical neuron cultures than rsT3D σ1s-null, the difference is not statistically significant. The primary cortical neurons were harvested from mice at E15, which is while neurons are undergoing active cell division (57). Moreover, the cells undergo several rounds of cell division once they are placed in culture (J. L. Konopka-Anstadt and T. S. Dermody, unpublished data). Thus, the modest difference in apoptosis in cultured neurons displayed by rsT3D and rsT3D σ1s-null may reflect the fact that the primary cortical neuron cultures were still dividing.
Apoptosis is initiated during binding and entry of reovirus into host cells. Attachment of σ1 to cellular receptors, such as cell surface sialic acid or junctional adhesion molecule A (JAM-A), may provide signals that potentiate apoptosis (63). During reovirus internalization, fragments of outer-capsid protein μ1 generated by endosomal proteases are delivered into the cytoplasm along with the viral core (20, 64). Once in the cytoplasm, the μ1 fragments associate with mitochondria, destabilize the mitochondrial membrane, and facilitate cytochrome c release and loss of mitochondrial membrane potential (22, 23, 65). Because viral transcription and translation occur subsequent to viral entry, proapoptotic signals are initiated before σ1s is expressed. The σ1s protein may enhance apoptosis by modulating signals initiated by entry-related events. Alternatively, σ1s may activate a different arm of the apoptotic program that boosts the apoptotic capacity of reovirus. Both models are consistent with our finding that rsT3D σ1s-null enhances apoptosis but is not absolutely required for this effect (Fig. 2). It is likely that apoptosis in rsT3D σ1s-null-infected cells results from entry-related proapoptotic signaling. It remains to be determined how signals from σ1s integrate with the activities of capsid components known to induce apoptosis.
One of the hallmarks of Reoviridae viruses is that disease develops primarily in neonatal or juvenile hosts (5). At these stages of development, high levels of cell division occur to support rapid growth. If σ1s-dependent cell cycle arrest and apoptosis are triggered in dividing cells, it is possible that the proapoptotic activity of σ1s contributes to the enhanced pathogenesis of reovirus observed in younger hosts. Although rsT3D and rsT3D σ1s-null are comparably virulent following intracranial inoculation, rsT3D σ1s-null is dramatically attenuated relative to rsT3D after hind limb inoculation of newborn mice (36). Attenuation of rsT3D σ1s-null is due to failure of the virus to disseminate systemically via the blood, which impairs viral delivery to the brain and subsequent CNS disease (36). Similarly, a serotype 1 σ1s-null reovirus also fails to spread hematogenously following peroral inoculation (37). We found that mutant viruses that do not induce cell cycle arrest and apoptosis also are attenuated relative to wild-type virus following hind limb inoculation (Fig. 9). These observations raise the possibility that induction of cell cycle arrest and apoptosis following infection at peripheral sites, such as the intestine or hind limb muscle, facilitate hematogenous reovirus dissemination. In at least some circumstances, apoptosis is an immunologically silent process that does not activate immune or inflammatory responses. However, some apoptotic cells secrete a subset of proinflammatory chemokines and cytokines (3) along with soluble factors, such as ATP and UTP, that recruit phagocytes to clear debris from dying cells (2, 4). Following peroral inoculation, reovirus induces apoptosis in intestinal epithelial cells that are taken up by Peyer's patch dendritic cells (66). It is possible that σ1s causes cell cycle arrest and apoptosis in reovirus-infected intestinal epithelial cells and that apoptotic cells filled with progeny virus are engulfed by phagocytic cells, which are in turn responsible for systemic viral dissemination from the site of inoculation.
Reoviruses normally infect their hosts via the intestinal tract, where intestinal epithelial cells undergo continuous replication and shedding. Rapid epithelial cell turnover prevents pathogens from gaining a foothold in the intestine via physiologic turnover of infected cells. Impairing intestinal epithelial cell renewal may slow the process of cell shedding and allow reovirus to persist in the gut of an infected host. Intestinal bacteria, including enteropathogenic and enterohemorrhagic Escherichia coli, Salmonella enterica serovar Typhi, and Shigella dysenteriae encode cyclomodulins that inhibit cell cycle progression (67, 68). Although the function of cyclomodulins has not been defined in vivo, these molecules are hypothesized to prevent renewal of the intestinal epithelium to allow bacterial colonization. In the intestine, reovirus antigen is detected in villus epithelial cells and cells at the base of intestinal crypts (69). Stem cells in intestinal crypts are self-renewing and serve as the source of epithelial cells that line the villi. Inhibiting cell cycle progression in crypt cells would slow epithelial cell migration from the crypt to the villus tips and allow reovirus to persist in the intestine. Following peroral inoculation, titers of the serotype 1 σ1s-null virus in the intestine are lower than those produced by wild-type virus (37). By failing to cause cell cycle arrest in intestinal epithelial cells, the σ1s-null virus would not be anticipated to affect the normal turnover of the intestinal lining, which would explain its diminished capacity to infect the intestine.
Reovirus is one of many RNA viruses that modulate cell cycle progression during infection. Like mammalian reovirus, avian reovirus (51), Borna disease virus (70), hepatitis C virus (71), infectious bronchitis virus (44), and respiratory syncytial virus (72) cause cell cycle arrest in G2. Other RNA viruses, including coronavirus (73), influenza virus (74), and measles virus (75, 76), arrest the cell cycle in G1. Although much is known about the relationship between DNA viruses and the cell cycle, little is understood about how or why RNA viruses dysregulate this vital cellular process. DNA viruses, such as adenovirus or papillomavirus, drive the cell into S phase to create a suitable environment for viral DNA replication (77). Perhaps G2 is the optimal cell cycle stage for reovirus replication. It is noteworthy that the σ1s protein is not required for reovirus replication in cultured cells (32, 36, 37). However, these studies were performed using transformed cells, including HeLa, L929, and MDCK cells. Transformed cells are more metabolically active than the majority of cells in vivo. Because they cycle rapidly, transformed cells enter each stage of the cell cycle more frequently than nontransformed cells. Thus, any benefit gained by halting cell cycle progression in transformed cells may not provide a σ1s-expressing virus with a replication advantage. Because transformed cells divide rapidly, it is possible that they are more susceptible to σ1s-mediated apoptosis. Based on an enhanced capacity to infect and kill cancer cells, reovirus is currently being evaluated in clinical trials as an oncolytic adjunct to conventional chemotherapy (78, 79). If σ1s-mediated effects on cell cycle control are more evident in proliferating cells, then σ1s-dependent cell cycle arrest and apoptosis may be important mechanisms underlying reovirus oncolysis.
The σ1s protein is one of many viral nonstructural proteins that induce cell cycle arrest and apoptosis. Adenovirus E4orf4 (80, 81), avian reovirus p17 (51), and HIV Vpr (82–86) cause both effects, whereas other viral nonstructural proteins, such as influenza virus PB1-F2 (87), directly induce apoptosis. Apoptotic cell death is an important component of the host response to viral infection. Apoptosis limits viral replication in infected cells and alerts adaptive immune responses to the presence of an invading pathogen. Consequently, viruses use multiple strategies to limit apoptosis, which allows the virus to evade immune detection. It is not clear why viruses encode proteins that actively promote a cellular process specifically designed to combat viral infection. Determining how σ1s modulates host cell cycle progression and apoptosis will enhance an understanding of the molecular and cellular basis of reovirus cell cycle arrest, apoptosis, and dissemination and may provide new knowledge about how and why viral nonstructural proteins from numerous viruses promote these effects. Given the likelihood of significant interplay between σ1s and cell cycle control pathways, enhanced knowledge of σ1s function also may lead to new insights into these highly regulated, interrelated processes that play fundamental roles in development, immunity, and cancer.
ACKNOWLEDGMENTS
The flow cytometry experiments were performed in the Vanderbilt Cytometry Shared Resource. David Cortez (Vanderbilt University) provided HCT-116 cells. We thank Dan Voth for assistance with confocal microscopy.
This work was supported by Public Health Service awards T32 CA09385 (K.W.B.), F32 AI075776 (K.W.B.), K22 AI94079 (K.W.B.), P20 GM103625 (K.W.B.), T32 CA09385 (J.L.K.-A.), F32 AI081486 (J.L.K.-A.), and R37 AI38296 (T.S.D.) and the Elizabeth B. Lamb Center for Pediatric Research. Additional support was provided by Public Health Service awards CA68485 for the Vanderbilt-Ingram Cancer Center and DK20593 for the Vanderbilt Diabetes Research and Training Center.
Footnotes
Published ahead of print 25 September 2013
REFERENCES
- 1.Roulston A, Marcellus RC, Branton PE. 1999. Viruses and apoptosis. Annu. Rev. Microbiol. 53:577–628 [DOI] [PubMed] [Google Scholar]
- 2.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. 2010. Pannexin 1 channels mediate ‘find-me' signal release and membrane permeability during apoptosis. Nature 467:863–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cullen SP, Henry CM, Kearney CJ, Logue SE, Feoktistova M, Tynan GA, Lavelle EC, Leverkus M, Martin SJ. 2013. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol. Cell 49:1034–1048 [DOI] [PubMed] [Google Scholar]
- 4.Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS. 2009. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461:282–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dermody TS, Parker J, Sherry B. 2013. Orthoreovirus. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed, vol 2, p 1304–1346Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 6.Dichter MA, Weiner HL. 1984. Infection of neuronal cell cultures with reovirus mimics in vitro patterns of neurotropism. Ann. Neurol. 16:603–610 [DOI] [PubMed] [Google Scholar]
- 7.Tardieu M, Weiner HL. 1982. Viral receptors on isolated murine and human ependymal cells. Science 215:419–421 [DOI] [PubMed] [Google Scholar]
- 8.Tyler KL, McPhee DA, Fields BN. 1986. Distinct pathways of viral spread in the host determined by reovirus S1 gene segment. Science 233:770–774 [DOI] [PubMed] [Google Scholar]
- 9.Tyler KL, Squier MKT, Brown AL, Pike B, Willis D, Oberhaus SM, Dermody TS, Cohen JJ. 1996. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes. J. Virol. 70:7984–7991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrison LA, Sidman RL, Fields BN. 1991. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc. Natl. Acad. Sci. U. S. A. 88:3852–3856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weiner HL, Drayna D, Averill DR, Jr, Fields BN. 1977. Molecular basis of reovirus virulence: role of the S1 gene. Proc. Natl. Acad. Sci. U. S. A. 74:5744–5748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiner HL, Powers ML, Fields BN. 1980. Absolute linkage of virulence and central nervous system tropism of reoviruses to viral hemagglutinin. J. Infect. Dis. 141:609–616 [DOI] [PubMed] [Google Scholar]
- 13.Tyler KL, Squier MK, Rodgers SE, Schneider SE, Oberhaus SM, Grdina TA, Cohen JJ, Dermody TS. 1995. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein σ1. J. Virol. 69:6972–6979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCrae MA, Joklik WK. 1978. The nature of the polypeptide encoded by each of the ten double-stranded RNA segments of reovirus type 3. Virology 89:578–593 [DOI] [PubMed] [Google Scholar]
- 15.Mustoe TA, Ramig RF, Sharpe AH, Fields BN. 1978. Genetics of reovirus: identification of the dsRNA segments encoding the polypeptides of the μ and σ size classes. Virology 89:594–604 [DOI] [PubMed] [Google Scholar]
- 16.Sarkar G, Pelletier J, Bassel-Duby R, Jayasuriya A, Fields BN, Sonenberg N. 1985. Identification of a new polypeptide coded by reovirus gene S1. J. Virol. 54:720–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiner HL, Ault KA, Fields BN. 1980. Interaction of reovirus with cell surface receptors. I. Murine and human lymphocytes have a receptor for the hemagglutinin of reovirus type 3. J. Immunol. 124:2143–2148 [PubMed] [Google Scholar]
- 18.Danthi P, Hansberger MW, Campbell JA, Forrest JC, Dermody TS. 2006. JAM-A-independent, antibody-mediated uptake of reovirus into cells leads to apoptosis. J. Virol. 80:1261–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coffey CM, Sheh A, Kim IS, Chandran K, Nibert ML, Parker JS. 2006. Reovirus outer capsid protein μ1 induces apoptosis and associates with lipid droplets, endoplasmic reticulum, and mitochondria. J. Virol. 80:8422–8438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danthi P, Coffey CM, Parker JS, Abel TW, Dermody TS. 2008. Independent regulation of reovirus membrane penetration and apoptosis by the μ1 ϕ domain. PLoS Pathog. 4:e1000248. 10.1371/journal.ppat.1000248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Danthi P, Pruijssers AJ, Berger AK, Holm GH, Zinkel SS, Dermody TS. 2010. Bid regulates the pathogenesis of neurotropic reovirus. PLoS Pathog. 6:e1000980. 10.1371/journal.ppat.1000980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JW, Lyi SM, Parrish CR, Parker JS. 2011. A proapoptotic peptide derived from reovirus outer capsid protein μ1 has membrane-destabilizing activity. J. Virol. 85:1507–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wisniewski ML, Werner BG, Hom LG, Anguish LJ, Coffey CM, Parker JS. 2011. Reovirus infection or ectopic expression of outer capsid protein μ1 induces apoptosis independently of the cellular proapoptotic proteins Bax and Bak. J. Virol. 85:296–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cashdollar LW, Chmelo RA, Wiener JR, Joklik WK. 1985. Sequences of the S1 genes of the three serotypes of reovirus. Proc. Natl. Acad. Sci. U. S. A. 82:24–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ernst H, Shatkin AJ. 1985. Reovirus hemagglutinin mRNA codes for two polypeptides in overlapping reading frames. Proc. Natl. Acad. Sci. U. S. A. 82:48–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cenatiempo Y, Twardowski T, Shoeman R, Ernst H, Brot N, Weissbach H, Shatkin AJ. 1984. Two initiation sites detected in the small s1 species of reovirus mRNA by dipeptide synthesis in vitro. Proc. Natl. Acad. Sci. U. S. A. 81:1084–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dermody TS, Nibert ML, Bassel-Duby R, Fields BN. 1990. Sequence diversity in S1 genes and S1 translation products of 11 serotype 3 reovirus strains. J. Virol. 64:4842–4850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoyt CC, Bouchard RJ, Tyler KL. 2004. Novel nuclear herniations induced by nuclear localization of a viral protein. J. Virol. 78:6360–6369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poggioli GJ, Dermody TS, Tyler KL. 2001. Reovirus-induced σ1s-dependent G2/M cell cycle arrest results from inhibition of p34cdc2. J. Virol. 75:7429–7434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poggioli GJ, Keefer CJ, Connolly JL, Dermody TS, Tyler KL. 2000. Reovirus-induced G2/M cell cycle arrest requires σ1s and occurs in the absence of apoptosis. J. Virol. 74:9562–9570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoyt CC, Richardson-Burns SM, Goody RJ, Robinson BA, Debiasi RL, Tyler KL. 2005. Nonstructural protein σ1s is a determinant of reovirus virulence and influences the kinetics and severity of apoptosis induction in the heart and central nervous system. J. Virol. 79:2743–2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodgers SE, Connolly JL, Chappell JD, Dermody TS. 1998. Reovirus growth in cell culture does not require the full complement of viral proteins: identification of a σ1s-null mutant. J. Virol. 72:8597–8604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chappell JD, Gunn VL, Wetzel JD, Baer GS, Dermody TS. 1997. Mutations in type 3 reovirus that determine binding to sialic acid are contained in the fibrous tail domain of viral attachment protein σ1. J. Virol. 71:1834–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Connolly JL, Barton ES, Dermody TS. 2001. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J. Virol. 75:4029–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barton ES, Youree BE, Ebert DH, Forrest JC, Connolly JL, Valyi-Nagy T, Washington K, Wetzel JD, Dermody TS. 2003. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J. Clin. Invest. 111:1823–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boehme KW, Frierson JM, Konopka JL, Kobayashi T, Dermody TS. 2011. The reovirus σ1s protein is a determinant of hematogenous but not neural virus dissemination in mice. J. Virol. 85:11781–11790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boehme KW, Guglielmi KM, Dermody TS. 2009. Reovirus nonstructural protein σ1s is required for establishment of viremia and systemic dissemination. Proc. Natl. Acad. Sci. U. S. A. 106:19986–19991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Antar AAR, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi T, Antar AAR, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Virgin HW, Bassel-Duby IVR, Fields BN, Tyler KL. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J. Virol. 62:4594–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J. Virol. 62:246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith RE, Zweerink HJ, Joklik WK. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791–810 [DOI] [PubMed] [Google Scholar]
- 43.Yao K, Vakharia VN. 1998. Generation of infectious pancreatic necrosis virus from cloned cDNA. J. Virol. 72:8913–8920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dove B, Brooks G, Bicknell K, Wurm T, Hiscox JA. 2006. Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. J. Virol. 80:4147–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bodkin DK, Fields BN. 1989. Growth and survival of reovirus in intestinal tissue: role of the L2 and S1 genes. J. Virol. 63:1188–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bodkin DK, Nibert ML, Fields BN. 1989. Proteolytic digestion of reovirus in the intestinal lumens of neonatal mice. J. Virol. 63:4676–4681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nibert ML, Chappell JD, Dermody TS. 1995. Infectious subvirion particles of reovirus type 3 Dearing exhibit a loss in infectivity and contain a cleaved σ1 protein. J. Virol. 69:5057–5067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sperka T, Wang J, Rudolph KL. 2012. DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 13:579–590 [DOI] [PubMed] [Google Scholar]
- 49.Turnell AS, Grand RJ. 2012. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 93:2076–2097 [DOI] [PubMed] [Google Scholar]
- 50.Baer A, Austin D, Narayanan A, Popova T, Kainulainen M, Bailey C, Kashanchi F, Weber F, Kehn-Hall K. 2012. Induction of DNA damage signaling upon Rift Valley fever virus infection results in cell cycle arrest and increased viral replication. J. Biol. Chem. 287:7399–7410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chulu JL, Huang WR, Wang L, Shih WL, Liu HJ. 2010. Avian reovirus nonstructural protein p17-induced G2/M cell cycle arrest and host cellular protein translation shutoff involve activation of p53-dependent pathways. J. Virol. 84:7683–7694 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Kornitzer D, Sharf R, Kleinberger T. 2001. Adenovirus E4orf4 protein induces PP2A-dependent growth arrest in Saccharomyces cerevisiae and interacts with the anaphase-promoting complex/cyclosome. J. Cell Biol. 154:331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim S, Park SY, Yong H, Famulski JK, Chae S, Lee JH, Kang CM, Saya H, Chan GK, Cho H. 2008. HBV X protein targets hBubR1, which induces dysregulation of the mitotic checkpoint. Oncogene 27:3457–3464 [DOI] [PubMed] [Google Scholar]
- 54.Heilman DW, Green MR, Teodoro JG. 2005. The anaphase promoting complex: a critical target for viral proteins and anti-cancer drugs. Cell Cycle 4:560–563 [PubMed] [Google Scholar]
- 55.Liu Y, Rusinol A, Sinensky M, Wang Y, Zou Y. 2006. DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J. Cell Sci. 119:4644–4649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manju K, Muralikrishna B, Parnaik VK. 2006. Expression of disease-causing lamin A mutants impairs the formation of DNA repair foci. J. Cell Sci. 119:2704–2714 [DOI] [PubMed] [Google Scholar]
- 57.Dehay C, Kennedy H. 2007. Cell-cycle control and cortical development. Nat. Rev. Neurosci. 8:438–450 [DOI] [PubMed] [Google Scholar]
- 58.Clarke P, Beckham JD, Leser JS, Hoyt CC, Tyler KL. 2009. Fas-mediated apoptotic signaling in the mouse brain following reovirus infection. J. Virol. 83:6161–6170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clarke P, Meintzer SM, Gibson S, Widmann C, Garrington TP, Johnson GL, Tyler KL. 2000. Reovirus-induced apoptosis is mediated by TRAIL. J. Virol. 74:8135–8139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clarke P, Meintzer SM, Spalding AC, Johnson GL, Tyler KL. 2001. Caspase 8-dependent sensitization of cancer cells to TRAIL-induced apoptosis following reovirus-infection. Oncogene 20:6910–6919 [DOI] [PubMed] [Google Scholar]
- 61.Kominsky DJ, Bickel RJ, Tyler KL. 2002. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ. 9:926–933 [DOI] [PubMed] [Google Scholar]
- 62.Richardson-Burns SM, Kominsky DJ, Tyler KL. 2002. Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with the activation of death receptors. J. Neurovirol. 8:365–380 [DOI] [PubMed] [Google Scholar]
- 63.Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell F, Nusrat A, Parkos CA, Dermody TS. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441–451 [DOI] [PubMed] [Google Scholar]
- 64.Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS. 2008. Reovirus apoptosis and virulence are regulated by host cell membrane-penetration efficiency. J. Virol. 82:161–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaufer S, Coffey CM, Parker JS. 2012. The cellular chaperone hsc70 is specifically recruited to reovirus viral factories independently of its chaperone function. J. Virol. 86:1079–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fleeton M, Contractor N, Leon F, Wetzel JD, Dermody TS, Kelsall B. 2004. Peyer's patch dendritic cells process viral antigen from apoptotic epithelial cells in the intestine of reovirus-infected mice. J. Exp. Med. 200:235–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jubelin G, Chavez CV, Taieb F, Banfield MJ, Samba-Louaka A, Nobe R, Nougayrede JP, Zumbihl R, Givaudan A, Escoubas JM, Oswald E. 2009. Cycle inhibiting factors (CIFs) are a growing family of functional cyclomodulins present in invertebrate and mammal bacterial pathogens. PLoS One 4:e4855. 10.1371/journal.pone.0004855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oswald E, Nougayrede JP, Taieb F, Sugai M. 2005. Bacterial toxins that modulate host cell-cycle progression. Curr. Opin. Microbiol. 8:83–91 [DOI] [PubMed] [Google Scholar]
- 69.Rubin DH, Kornstein MJ, Anderson AO. 1985. Reovirus serotype 1 intestinal infection: a novel replicative cycle with ileal disease. J. Virol. 53:391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Planz O, Pleschka S, Oesterle K, Berberich-Siebelt F, Ehrhardt C, Stitz L, Ludwig S. 2003. Borna disease virus nucleoprotein interacts with the CDC2-cyclin B1 complex. J. Virol. 77:11186–11192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kannan RP, Hensley LL, Evers LE, Lemon SM, McGivern DR. 2011. Hepatitis C virus infection causes cell cycle arrest at the level of initiation of mitosis. J. Virol. 85:7989–8001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bian T, Gibbs JD, Orvell C, Imani F. 2012. Respiratory syncytial virus matrix protein induces lung epithelial cell cycle arrest through a p53 dependent pathway. PLoS One 7:e38052. 10.1371/journal.pone.0038052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen CJ, Makino S. 2004. Murine coronavirus replication induces cell cycle arrest in G0/G1 phase. J. Virol. 78:5658–5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.He Y, Xu K, Keiner B, Zhou J, Czudai V, Li T, Chen Z, Liu J, Klenk HD, Shu YL, Sun B. 2010. Influenza A virus replication induces cell cycle arrest in G0/G1 phase. J. Virol. 84:12832–12840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McChesney MB, Altman A, Oldstone MB. 1988. Suppression of T lymphocyte function by measles virus is due to cell cycle arrest in G1. J. Immunol. 140:1269–1273 [PubMed] [Google Scholar]
- 76.McChesney MB, Kehrl JH, Valsamakis A, Fauci AS, Oldstone MB. 1987. Measles virus infection of B lymphocytes permits cellular activation but blocks progression through the cell cycle. J. Virol. 61:3441–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dyson N, Howley PM, Munger K, Harlow E. 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243:934–937 [DOI] [PubMed] [Google Scholar]
- 78.Adair RA, Roulstone V, Scott KJ, Morgan R, Nuovo GJ, Fuller M, Beirne D, West EJ, Jennings VA, Rose A, Kyula J, Fraser S, Dave R, Anthoney DA, Merrick A, Prestwich R, Aldouri A, Donnelly O, Pandha H, Coffey M, Selby P, Vile R, Toogood G, Harrington K, Melcher AA. 2012. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci. Transl. Med. 4:138ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karapanagiotou EM, Roulstone V, Twigger K, Ball M, Tanay M, Nutting C, Newbold K, Gore ME, Larkin J, Syrigos KN, Coffey M, Thompson B, Mettinger K, Vile RG, Pandha HS, Hall GD, Melcher AA, Chester J, Harrington KJ. 2012. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin. Cancer Res. 18:2080–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lavoie JN, Nguyen M, Marcellus RC, Branton PE, Shore GC. 1998. E4orf4, a novel adenovirus death factor that induces p53-independent apoptosis by a pathway that is not inhibited by zVAD-fmk. J. Cell Biol. 140:637–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marcellus RC, Lavoie JN, Boivin D, Shore GC, Ketner G, Branton PE. 1998. The early region 4 orf4 protein of human adenovirus type 5 induces p53-independent cell death by apoptosis. J. Virol. 72:7144–7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bartz SR, Rogel ME, Emerman M. 1996. Human immunodeficiency virus type 1 cell cycle control: Vpr is cytostatic and mediates G2 accumulation by a mechanism which differs from DNA damage checkpoint control. J. Virol. 70:2324–2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR. 1995. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69:6705–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jowett JB, Planelles V, Poon B, Shah NP, Chen ML, Chen IS. 1995. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J. Virol. 69:6304–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Re F, Braaten D, Franke EK, Luban J. 1995. Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J. Virol. 69:6859–6864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rogel ME, Wu LI, Emerman M. 1995. The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J. Virol. 69:882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zamarin D, Garcia-Sastre A, Xiao X, Wang R, Palese P. 2005. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 1:e4. 10.1371/journal.ppat.0010004 [DOI] [PMC free article] [PubMed] [Google Scholar]