

Abstract
Viral entry by herpes simplex virus (HSV) is executed and tightly regulated by four glycoproteins. While several viral glycoproteins can mediate viral adhesion to host cells, only binding of gD to cellular receptor can activate core fusion proteins gB and gH/gL to execute membrane fusion and viral entry. Atomic structures of gD bound to receptor indicate that the C terminus of the gD ectodomain must be displaced before receptor can bind to gD, but it is unclear which conformational changes in gD activate membrane fusion. We rationally designed mutations in gD to displace the C terminus and observe if fusion could be activated without receptor binding. Using a cell-based fusion assay, we found that gD V231W induced cell-cell fusion in the absence of receptor. Using recombinant gD V231W protein, we observed binding to conformationally sensitive antibodies or HSV receptor and concluded that there were changes proximal to the receptor binding interface, while the tertiary structure of gD V231W was similar to that of wild-type gD. We used a biosensor to analyze the kinetics of receptor binding and the extent to which the C terminus blocks binding to receptor. We found that the C terminus of gD V231W was enriched in the open or displaced conformation, indicating a mechanism for its function. We conclude that gD V231W triggers fusion through displacement of its C terminus and that this motion is indicative of how gD links receptor binding to exposure of interfaces on gD that activate fusion via gH/gL and gB.
INTRODUCTION
Entry of herpes simplex virus (HSV) into host cells is a critical step in the viral replication cycle. Four viral glycoproteins are indispensable for HSV entry into host cells (1, 2). gB and the heterodimer gH/gL form the core fusion machinery that is conserved across herpesviruses. gD is a fourth essential glycoprotein specific to alphaherpesviruses (1). In HSV, membrane fusion occurs only after gD binds receptor, which is an essential step in viral entry. Receptor-bound gD then activates gH/gL (3), which interacts with gB to induce membrane fusion (4).
The role of gD in viral entry can be decomposed into two functions that are postulated to occur on distinct faces of gD, receptor binding and activation of gH/gL and gB for fusion (5). Several cellular receptors that can bind gD and initiate membrane fusion have been described. gD receptors include HVEM (6), which is found on lymphocytes (7), and nectin-1 (8), which is a component of adherens junctions in epithelial tissue (9). While HVEM and nectin-1 are the primary gD receptors (10), other receptors have been identified, including 3-O-sulfated heparan, which can be utilized by HSV-1 (11), and nectin-2, which can be utilized by HSV-2 (12). On cells lacking gD receptor, gD does not activate gH/gL and gB and HSV does not enter these cells, yet these cells become susceptible to HSV entry if they are made to express gD receptor (6). The specificity of gD for receptor plays an important role in the tropism of HSV, and chimeric viruses have been retargeted by inserting alternate receptor binding domains within gD (13–15). Cocrystal structures of gD bound to HVEM (16) and gD bound to nectin-1 (17) indicate that both receptors bind to the same face of gD. A comparison of receptor-bound structures of gD to crystal structures of gD alone indicates a key structural difference involved in receptor binding. When gD is not bound to receptor, the C terminus of the gD ectodomain sits on top of the receptor binding region of gD (18). After gD binds to either HVEM or nectin-1, receptor occupies the site previously occupied by the gD C terminus. The C terminus (residues 255 to 316) is disordered in the receptor-bound cocrystal structure, after being displaced by receptor. Therefore, the C terminus itself has been proposed to play an important role in triggering membrane fusion (5, 18–20).
The second role of gD is interacting with gH/gL to activate fusion (3, 4). Antibodies MC2 and MC5 block the ability of gD to activate fusion, without interfering with receptor binding. These antibodies map to a face of gD opposite of the receptor binding region (5). Near the postulated epitopes of MC2 and MC5, the C terminus is connected to the core immunoglobulin (Ig)-like fold of gD, implicating the C terminus as a link between receptor binding and activation of gH/gL and gB. Disulfide bonds that lock the C terminus in place near the MC2 and MC5 epitopes have been engineered into gD, and these mutations inhibit activation of fusion, without blocking receptor binding (20). While the MC2 and MC5 epitopes are conformational and involve residues attached to the core Ig-like fold of gD, segments of the C terminus near these epitopes have also been implicated as being important for activation of fusion.
We hypothesized that displacement of the C terminus of the gD ectodomain, either by receptor binding or by mutation, would be sufficient for gD to activate gH/gL and gB for membrane fusion. For wild-type (WT) gD, the C terminus must first vacate the receptor binding site before receptor may bind; therefore, the conformation of the gD C terminus must be naturally dynamic. As receptor binds to gD, it prevents the C terminus from rebinding, stabilizing the dissociated C terminus. The gD-receptor complex can then activate core fusion proteins gH/gL and gB. While the structures of gD alone or bound to receptor are known, it is not known which aspects of these structures are deterministic in activation of fusion. Using a transfection-based cell-cell fusion assay, protein expression levels may be high enough that even without receptor, the transient dissociation of the C terminus from WT gD may encounter core fusion proteins and trigger fusion. Indeed, when cells that do not express HSV receptor are transfected with gD, gH, gL, and gB, they undergo a small amount of fusion that we call the basal level of fusion. In contrast, no fusion is observed at all when any one of the viral glycoproteins is omitted (3, 21, 22). Because activation of gD is first in the series of events that lead to fusion (3, 4), we infer that gD is the origin of this increased cell-cell fusion activity. Therefore, we interpreted the basal level of cell-cell fusion activity with WT proteins to be due to transient activation of gD, and we postulated that we could enrich this form of gD by rationally designed mutagenesis.
We designed mutations in gD that were predicted to displace the C terminus from the receptor binding site. According to our hypothesis, displacement of the gD C terminus would reveal binding interfaces that interact with and activate gH/gL and gB. Using a cell-cell fusion assay that allowed us to study individual gD mutations in isolation, we found one such mutation, V231W, which increased fusion on cells that do not express HSV receptor. Using recombinant protein, we found that gD V231W had changes in antigenic structure at the receptor binding interface and a reduced affinity for nectin-1. Finally, we used a biosensor to deduce that the C terminus of gD V231W was enriched in the open or displaced conformation in comparison to that of the WT protein. The displacement of the C terminus correlates with the increase in fusion by gD V231W in the absence of receptor. We conclude that displacement of the C terminus is the structural mechanism that links receptor binding to activation of gH/gL and gB for fusion.
MATERIALS AND METHODS
Site-directed mutagenesis.
HSV-1 (KOS) gD mutants A12W and V231W were generated by QuikChange site-directed mutagenesis, using the pCAGGS gD expression vector pPEP99 (23) as the template. The primers used to create A12W were CTCAAGATGTGGGACCCCAATCGCTTTCGCGGCAAAGACC-5′ and GGGGTCCCACATCTTGAGAGAGGCATCCGCCAAGGCATATTTG-3′. The primers used to create V231W were CGCACCTGGGCCGTATACAGCTTGAAGATCGCCGGGTG-5′ and CGGCCCAGGTGCGCTGGTTCTCGGGGATGAAGCGG-3′. Sequence-validated clones were named pJG975 (A12W) and pJG976 (V231W). For baculovirus production, pVT-BAC plasmid pCP265, which contains gD with an N-terminal secretion signal and a stop codon that truncates the protein at position 306 (gD 306t), was used as the template for QuikChange site-directed mutagenesis. Primers containing the V231W mutation were used to mutagenize gD, creating pVT-BAC plasmid pJG977.
Cell-cell fusion.
The transfection-based cell-cell fusion assay of Turner et al. (24) was modified for use with B78H1 cells, which do not express HSV receptor (25). B78H1 cells were selected because they lack HSV receptor and do not permit HSV entry but can be made competent for virus entry by transfection of HSV receptor HVEM or nectin-1. Derived cell lines A10 and C10 are stably transfected with HVEM and nectin-1, respectively (25). When B78H1 cells are transfected with HSV gD, gH, gL, gB, and HSV receptor, they form large and stable syncytia, but cell-cell fusion rarely occurs when any of these proteins is not present (3, 21). B78H1 cells or cells of derived lineage C10 or A10 (2.25 ×104) were plated in a 48-well plate, grown overnight, and then transfected with 1.2 μl Fugene6 reagent (Promega), using 200 ng of DNA (50 ng each gD, gB, gH, and gL). Cells cultured under conditions with HSV receptor were fixed at 24 h posttransfection, while cells cultured under conditions without HSV receptor were fixed at 36 h posttransfection. Cells were fixed with 3% paraformaldehyde and stained with Giemsa stain, and then microscope fields were photographed at a ×10 magnification for counting of the cells. The number of nuclei of cells containing 4 or more nuclei were summed over the field using ImageJ software (26) with the cell counter plug-in and averaged across multiple fields, a procedure analogous to cell counting with a hemacytometer. Four fields of cells containing a total of 3,600 cells per construct were counted for each construct. The error bars reflect the variance across each of the four fields of counted cells. WT glycoproteins expressed in B78H1 cells (which have no HSV receptor) produced 127 nuclei in syncytia for the set of experiments reported. Vector alone produced 8 nuclei in syncytia, although these were likely to be false positives due to cell overlap, representing the errors inherent to manual counting. gD V231W produced 448 nuclei in syncytia, but the number is presented as a percentage of that for the WT to highlight differences between gD constructs. The reported results are representative of those for other independently performed sets of experiments. For B78H1 cells, these results were also found to be representative for varied growth times following transfection (24 to 48 h) and varied total amounts of DNA per transfection (150 to 240 μg/ml) distributed evenly among the four glycoproteins.
Split luciferase assay.
The split luciferase assay was done as previously described (27, 28) with several modifications. Briefly, 5 × 104 B78H1 effector cells were seeded on a 96-well plate treated for cell culture. C10 or B78H1 target cells (2 × 105) were seeded on 24-well plates. On the next day, effector cells were transfected with a mix containing 4 μl of Lipofectamine 2000, 375 ng gB, 125 ng of each plasmid (gD, gH, gL), and Rluc81-7 in a final volume of 250 μl in Opti-MEM medium. This master mix was divided over 3 wells. Target cells were transfected with 250 ng of Rluc88-11 plasmid per well. At 24 h posttransfection, the effector cells were preincubated with EnduRen substrate diluted 1:1,000 in fusion medium (Dulbecco modified Eagle medium without phenol red supplemented with 50 mM HEPES and 5% fetal bovine serum). Target cells were then detached with EDTA, washed with fusion medium, and spun for 5 min at 4°C at low speed. Cells were resuspended in fusion medium and transferred over the effector cells containing fusion medium and substrate. The final volume in each 96 well was 60 μl. Luciferase activity (formation of the luminescent product) was monitored at the time intervals specified below with a BioTek plate reader set at 37°C.
Cell surface expression CELISA.
Cell-based competitive enzyme-linked immunosorbent assay (CELISA) was performed as described previously (20), using B78H1 cells to mirror the conditions of the fusion assay. B78H1 cells (2 ×104) were plated in wells of a 96-well plate, grown overnight, and then transfected with Fugene6 using 25 ng gD expression plasmid and 75 ng pUC19 vector carrier DNA. At 36 h posttransfection, cells were moved to 4°C and then incubated for 1 h at 4°C with polyclonal gD antibody R7 (29) in 5% serum. Cells were rinsed three times in phosphate-buffered saline (PBS) and then fixed with 3% paraformaldehyde. Cells were rinsed, blocked with 5% serum for 1 h at 23°C, and incubated with horseradish peroxidase (HRP)-conjugated antirabbit antibody for 1 h at 23°C. Finally, cells were rinsed three times with PBS, and then the amount of bound antibody was quantified by adding ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid)] substrate (Rockland Immunochemicals, Gilbertsville, PA) and recording the optical density at 405 nm. Similar results were obtained if cells were fixed first and then blocked and incubated with primary antibody, before continuing as described above.
Baculovirus protein production.
gD V231W recombinant protein was produced using a baculovirus expression system, following previously established methods for gD 306t (30). To generate baculovirus expressing a soluble truncation of gD-1 V231W, Sf9 insect cells were transfected using Cellfectin II reagent with 500 ng BaculoGold DNA (BD) and 500 ng pJG977 (gD-1 V231W 306t in pVT-BAC) according to the recommended BaculoGold protocol. Resulting virus was plaque purified twice and amplified. Expression of gD was confirmed by Western blotting of the virus supernatant. One liter of Sf9 cells, grown to a density of 4 ×106 cells/ml, was infected with gD V231W baculovirus at a multiplicity of 4 PFU/cell. Virus was grown until Sf9 cell viability dropped to 50%, at which point the culture supernatant was harvested by centrifugation at 2,000 × g for 30 min. The culture supernatant was filtered with a 0.45-μm-pore-size filter and then dialyzed into PBS. An immunoaffinity column, consisting of a bed volume of 120 ml DL6 monoclonal antibody (MAb)-conjugated resin, was prepared by washing with 3 cycles of alternating between 100 mM sodium acetate, pH 4, and 50 mM sodium borate, pH 8, for 3 column volumes (CV) each. Cell culture supernatant containing gD V231W was loaded onto the column and then washed with 5 CV of 10 mM Tris, pH 7.2 and 500 mM sodium chloride (TS buffer). gD V231W was eluted with 3 CV of 100 mM ethanolamine, pH 11, followed by 5 CV of TS buffer. Eluted fractions of gD V231W protein were immediately neutralized using Tris hydrochloride. Protein samples were concentrated, dialyzed into PBS, and concentrated again to a final concentration of 2.7 mg/ml, yielding approximately 10 mg of protein.
Western blotting and MAb ELISA.
gD was resolved for Western blotting by SDS-PAGE using a 10% Tris-glycine gel, with a notable modification that reducing agent was omitted from the sample preparation, and the sample was loaded directly on the gel, without heating (31). After transfer to a nitrocellulose membrane and blocking, the blot was probed with primary antibodies overnight at 4°C and then washed and incubated with horseradish peroxidase-conjugated secondary antibody and, finally, washed and visualized by chemiluminescence.
ELISA for MAb binding was performed with 10 μg/ml gD protein in PBS supplemented with 0.2% Tween 20 (PBST) that was adhered to a 96-well ELISA plate at 4°C overnight. Wells were blocked with 5% milk–PBST (PBS with 5% nonfat dry milk and 0.2% Tween 20), rinsed three times with PBS, and then incubated with 12 μg/ml MAb in 1% milk–PBST at 23°C for 2 h. Wells were rinsed three times, and HRP-conjugated secondary antibody in 1% milk–PBST was added for 1 h at 23°C. Wells were again rinsed three times, and then ABTS substrate was added. The optical density at 405 nm, indicating the amount of monoclonal antibody captured by the gD construct, was recorded.
Receptor binding ELISA.
HSV receptor was adhered to an ELISA plate, using 5 μg/ml HVEM truncated at residue 200 (200t) (32) or 10 μg/ml nectin-1 truncated at residue 245 (245t) (33), at 4°C overnight. The plate was blocked with 5% milk–PBST and rinsed three times with PBS. gD dilutions were made in 1% milk–PBST and incubated on the ELISA plate for 2 h. The gD solution was removed, and the plate was washed three times. Bound gD was detected with R7 polyclonal gD antibody as a primary antibody, rinsed in 1% milk–PBST three times, and then incubated with HRP-conjugated secondary antibody. The plate was washed three times, and then ABTS substrate was added. The optical density at 405 nm, indicating the total amount of gD bound to the ELISA plate, was recorded.
Biosensor analysis.
Biosensor analysis was performed on a Biacore 3000 instrument. For binding to HVEM, monoclonal antibody CW10 (34), which binds HVEM without blocking the interaction with gD, was conjugated to a CM5 Biacore chip and used to capture 700 resonance units (RU) of soluble recombinant HVEM 200t (32) in one flow cell, keeping a second flow cell as a reference for antibody only. Using Biacore running buffer HBS-EP (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% polysorbate 20), gD was injected at 40 μl/min over both flow cells for 30 s with a 2-min dissociation phase. The CM5 chip surface was regenerated with a 3-min pulse of 100 mM glycine, pH 2.5, followed by a 3-min pulse of 100 mM NaHCO3, pH 10, and then a 2-min reequilibration phase in HSB-EP. The cycle was repeated for a gD concentration series consisting of 1 μM, 2 μM, 4 μM, 8 μM, and 10 μM. Using BIAevaluation software, version 4.1, the data were globally fit using a 1:1 Langmuir binding model to fit association and dissociation rates and calculate an equilibrium binding constant. Errors were represented by the standard error reported by the BIAevaluation software. For nectin-1 binding, polyclonal antibody to tetra-His was immobilized on a CM5 Biacore chip. Nectin-1 245t contains a hexahistidine tag, which was used for capture on the biosensor chip. Binding and analysis for nectin-1 were performed as described above for HVEM binding.
RESULTS
The design of mutations to activate fusion was based upon two observations. First, crystal structures indicate that the C terminus of the gD ectodomain undergoes a large change as it is displaced by the binding of gD receptor (18), suggesting a physical linkage between displacement of the C terminus and receptor binding. Second, a point mutation, W294A, dissociates the C terminus from the gD core, yet complemented virus generated with this mutant gD is impaired in infectivity (18). This implies that residues in the C terminus itself have a role in entry. We propose that displacement of the C terminus is the physical mechanism that links receptor binding to exposure of interfaces on gD that activate fusion. To test our hypothesis, we have designed two mutations in gD that were predicted to displace the C terminus. Since residues on the C terminus itself have been proposed to play a role in blocking receptor binding (18) as well as activation of fusion (19), we selected mutations that were outside the C terminus, so as not to inadvertently interfere with the function of the gD C terminus in fusion activation. We predicted that these mutations would activate fusion even when they were not bound to receptor.
Design of mutations.
In order to determine which residues are important for binding of the C terminus to the gD core, we examined the crystal structure of gD without receptor (18). Residue W294 of the C terminus is largely responsible for anchoring the C terminus to the receptor binding face of gD, and it does so by binding in a hydrophobic pocket on the Ig-like core of gD (18). Instead of mutating W294, we asked if we could block binding of the C terminus to the hydrophobic pocket that is bound by W294. The back of the hydrophobic pocket is created by valine residue 231. Thus, if we mutated V231 to a larger residue, such as tryptophan (V231W), it would fill this hydrophobic pocket and block the interaction with the C terminus (Fig. 1B and C). V231 is centrally located on the receptor binding interface of gD, which made it an attractive location to modify. Because V231 is proximal to residues that contact HVEM as well as residues that contact nectin-1, the effect of the mutation on receptor binding was difficult to predict. V231 does not directly contact either of these receptors in the cocrystal structures (16, 17). Still, V231 is in proximity to gD residues arginine 15 and alanine 12, which are part of the N-terminal hairpin that does contact HVEM. An effect on nectin-1 binding is more likely than an effect on HVEM binding, because in the gD–nectin-1 cocrystal structure, gD V231 is only a few angstroms away from nectin-1 residues threonine 131 and glycine 132.
Fig 1.

Mutations designed to displace the gD C terminus. (A) Structures of gD alone or gD bound to HVEM or nectin-1 depict the receptor binding interface of gD. The C terminus (red) lies across the interface that binds either HVEM or nectin-1. Boxed regions are enlarged in panels B through E. (B) When receptor is not bound, W294 (stick representation) of the C terminus binds in a hydrophobic pocket formed by helix 3 of the gD core (box in panel A). V231 (cyan) is at the back of this pocket. (C) Mutating V231 to tryptophan (purple) was predicted to prevent W294 from binding in the hydrophobic pocket. (D) From the structure of gD bound to HVEM, the N terminus of gD forms a hairpin (green), and HVEM Y23 binds in the same hydrophobic pocket of gD. Residue A12 (cyan) makes up an edge of this hydrophobic pocket. By overlaying the gD N-terminal hairpin of the HVEM bound structure on the gD core of the unbound structure, A12W is positioned near the same hydrophobic pocket bound by W294. (E) Mutating A12 to tryptophan (purple) was predicted to fill this hydrophobic pocket, inducing the N-terminal hairpin to form even when HVEM is not bound. The asterisk in panel B denotes S140. Mutation S140N introduces an N-linked glycan and has been implicated in receptor-independent virus entry.
A second mutation was rationally designed on the basis of the gD-HVEM cocrystal structure. The C terminus of the gD ectodomain is not visible in this structure, but the same hydrophobic pocket on the Ig-like core of gD binds the critical residue Y23 of HVEM, in conjunction with the N-terminal hairpin of gD (16, 35, 36). If the two crystal structures are superimposed, alanine 12 of the N-terminal gD hairpin occupies the same position as W294; therefore, mutating A12 to a tryptophan (A12W) would cause gD to continuously form the N-terminal hairpin of the receptor-bound conformation. Thus, A12W of the N terminus would fill the hydrophobic pocket on the core of gD, even in the absence of HVEM (Fig. 1D and E). A challenge to this approach is that the N terminus is unstructured when HVEM is not bound, and point mutation A12W would need to bring order to the N-terminal hairpin to allow A12W to bind the hydrophobic pocket. Again, this would block binding of the C terminus to this interface, potentially allowing gD to activate fusion without receptor. A12W was likely to affect HVEM binding because it is a component of the N-terminal hairpin that interacts with HVEM, but A12W was unlikely to alter nectin-1 binding because A12 is disordered and not seen in the gD–nectin-1 cocrystal structure. The two gD constructs A12W and V231W would then occupy the hydrophobic pocket on the receptor binding face of gD from opposite sides, with V231W pushing out from the gD core and A12W reaching in from the N terminus.
Cell surface expression.
First, we established if gD proteins containing mutations V231W and A12W were expressed at the cell surface at levels comparable to those expressed by the WT. We measured surface expression with a cell-based ELISA using cells that were fixed but not permeabilized. B78H1 cells transfected with gD constructs were probed with R7 polyclonal antibody to gD. The gD expression levels of both the A12W and V231W mutants were similar to the expression level of WT gD (Fig. 2A). In addition, we included for comparison two previously reported gD mutations, W294A and deletion of residues 290 to 299 (Δ290-299) (18, 37). While these mutations have not been characterized in a cell-cell fusion assay, they are known to reduce the association of the C terminus with the gD core, resulting in an increased affinity to both nectin-1 and HVEM (18, 38). W294A was designed to dissociate the C terminus from the gD core (18). In contrast, gD Δ290-299 was generated by random linker insertion mutagenesis (37), consisting of a deletion at residues 290 to 299, combined with an insertion of five amino acids: GKIFP. Only later was gD Δ290-299 characterized as a high-affinity binder for receptor due to displacement of the C terminus and the loss of residue 294 (38). The expression of gD Δ290-299 was reduced from that of WT gD, but the difference was small. gD W294A was expressed at 60% of the level of WT gD. gD is known to efficiently trigger viral entry, even at low levels of expression (39), so we considered the expression levels of all the constructs to be suitable for further analysis.
Fig 2.

Cell surface expression and cell-cell fusion. (A) Cell surface expression of gD constructs was quantified by measurement of reactivity with R7 polyclonal antibody and normalized to that of the WT. All gD mutants were expressed at levels similar to that for the WT, with the exception of W294A, whose level of expression was reduced 30 to 40%. (B) Cell-cell fusion with C10 cells (expressing nectin-1) transfected with gB, gH, and gL and various gD constructs. All gD constructs have fusion activity similar to that of the WT. (C) Cell-cell fusion with A10 cells (expressing HVEM) transfected with gB, gH, and gL in conjunction with various gD constructs. gD A12W had increased activity, while V231W had activity similar to that of the WT. (D) B78H1 cells (no HSV receptor) were transfected with gB, gH, and gL and various gD constructs. gD A12W was found to have activity similar to that of the WT, while gD V231W had a 3.5-fold increased activity. gD W294A and Δ290-99 had negligible activity, which was lower than the residual activity of the WT in the cell-cell fusion assay.
Cell-cell fusion with and without HSV receptor.
To examine the ability of gD to activate fusion, we chose a system that was previously used to quantitate the cell-cell fusion activity of glycoprotein mutants and that can be used on cells expressing either HVEM, nectin-1, or no receptor (3, 22). We used B78H1 cells (25) and derivative cell lines A10 and C10 that stably express HVEM or nectin-1, respectively. These cells facilitate virus entry only when expressing HSV receptor and produce syncytia in the transfection-based cell-cell fusion assay. To assay fusion, gB, gH, and gL were transfected along with various constructs of gD to directly compare fusion activity. Fixed cells were Giemsa stained, and then the nuclei in the syncytia were counted.
To indicate if either of the mutations broadly disrupted either the structure or function of gD, we asked if the A12W and V231W mutations function in the presence of receptor. Using A10 cells or C10 cells, we quantified the ability of gD constructs to cause cell-cell fusion. On nectin-1-expressing cells, gD A12W and V231W had activity equivalent to that of WT gD, while gD W294A and Δ290-299 had slightly reduced activity (Fig. 2B). On HVEM-expressing cells, gD V231W had activity equivalent to that of WT GD, while gD A12W showed a 40% increase in activity (Fig. 2C). The increased activity by gD A12W on A10 cells fit with the design being based upon the HVEM-bound structure. Mutants W294A and Δ290-299 had a larger decrease in activity on HVEM-expressing cells than on nectin-1-expressing cells, and fusion activity ranged from 50 to 60% of that of the WT for these constructs. Since neither the W294A nor Δ290-299 mutant is involved in the receptor binding interface (16, 17), their reduced cell-cell fusion activity suggested that the C terminus may play another role in activation of fusion, such as interacting with core fusion proteins gH/gL and gB.
To test whether gD containing mutations A12W and V231W would activate fusion in the absence of receptor, we used B78H1 cells, which do not support HSV entry (25). Repeating the fusion assay with WT gD in the absence of receptor, a small number of cells fused to form small syncytia, as others have noted in B78H1 cells (21). Fusion assays on B78H1 cells were extended to 36 h from 24 h, in order to resolve sufficient fusion events for counting. gD V231W was consistently more productive in forming syncytia than WT gD, producing 3.5-fold more fusion events than WT. In contrast, the mutation A12W did not enhance fusion, with the A12W mutant having activity identical to that of WT (Fig. 2D). gD W294A and gD Δ290-299 are both known to dissociate the C terminus from the gD core in the absence of receptor binding (18, 38), yet neither of these constructs increased fusion activity in the absence of receptor. The level of fusion by gD W294A and gD Δ290-299 was decreased to the level of fusion for the vector alone. This reduction in cell-cell fusion activity implied that sequence including W294 is important for efficient activation of fusion by gD, consistent with previous results using virus complementation (18, 19).
Quantitative luciferase assay shows kinetics of fusion when gD V231W is used in place of WT gD.
As further evidence that gD V231W could trigger fusion in the absence of receptor, we employed a kinetic assay of fusion (27, 28). We first compared the ability of full-length gD V231W to that of gD WT to initiate and sustain cell-cell fusion in the presence of gB, gH/gL, and nectin-1 (Fig. 3A). In both cases, gD fusion was initiated within 1 h, and the rate of fusion was also the same over the 4 h of observation. When the target cells contained receptor (Fig. 3A), both gD V231W and WT gD (along with plasmids for gB, gH, and gL) initiated fusion at the same time and fusion occurred at comparable rates. When the same experiment was done using B78H1 cells (which lack a gD receptor), very little fusion occurred when WT gD was employed, even when the rate of fusion was measured over 8 h (Fig. 3B). In contrast, robust fusion occurred with gD V231W in place of WT gD following a lag longer than that seen when receptor was present. The rate of fusion triggered by gD V231W in the absence of receptor was 5-fold greater than that for WT gD, and the total amount of fusion after 8 h was also 5-fold larger. These total numbers agreed well with those obtained by counting of syncytia (Fig. 2). We conclude that gD V231W can cause a significant amount of fusion in cells lacking a gD receptor. The data support the hypothesis that gD V231W loosens the ectodomain C terminus, leading to a partially activated form.
Fig 3.

Rates of cell fusion for gD V231W in the presence and absence of HSV receptor nectin-1. Rluc8 plasmids were used to measure fusion rates. (A) Target cells consisted of C10 cells transfected with Rluc81-7. (B) Target cells consisted of B78H1 cells (no receptor) plus Rluc81-7. In both experiments, effector cells contained plasmids for Rluc88-11 plus plasmids for gB, gH/gL, and either WT gD, gD V231W, or empty vector. The experiments whose results are presented in panels A and B were done at different times. Target cells were overlaid onto receptor cells that had been preincubated with the EnduRen substrate for 1 h. Luminescence readings were taken at the times indicated on the x axis. The experiments whose results are presented in panels A and B were done independently.
Recombinant protein expression.
Since gD V231W differed from WT in cell-cell fusion without receptor, we aimed to associate this difference in activity with a molecular mechanism by studying a recombinant protein. We did not pursue gD A12W, because its activity did not differ from that of WT on B78H1 cells. To convert gD V231W into a soluble protein, we truncated the construct at position 306 (gD 306t), as shown previously with WT gD (30). gD 306t includes all the residues resolved in the unbound gD crystal structure. As a positive control for displacement of the C terminus, we used a truncation beginning at position 285 (gD 285t), which deletes residues that cover the receptor binding region and is known to bind HSV receptor with increased affinity (40).
Reactivity with MAbs.
Conformational changes in the gD structure both inside and outside the receptor binding pocket were probed using conformationally sensitive MAbs. Native Western blotting was used to directly compare soluble gD V231W and gD 306t (Fig. 4A). DL6 binds the linear epitope from residues 272 to 279, and as expected, DL6 reacted with both V231W and WT equally well by native Western blotting. MAbs with epitopes near the receptor binding region of gD include DL11, MC23, and AP7. DL11 is conformational and neutralizes virus by blocking binding to either HVEM or nectin-1 (41). MC23 is also conformational and neutralizes virus by blocking binding of gD to nectin-1 (5). The reactivity of both MC23 and DL11 was reduced for gD V231W, suggesting that the V231W point mutation was disrupting the receptor binding site of gD. AP7 binds near the receptor binding region, but its epitope spans the N and C termini of gD, as they cover the receptor binding surface (37). The reactivity of AP7 was also reduced, indicating that either the N or C terminus of gD V231W underwent a change in conformation compared to that of gD 306t. MAb epitopes that map to the back side of gD include MC5 and A18. MC5 has a conformational epitope that localizes near amino acid 77, and it neutralizes but does not block binding to receptor (5). A18 is conformationally sensitive and neutralizes viruses but does not compete with receptor (42). The reactivity of MC5 was reduced for gD V231W, while the reactivity of A18 was unchanged. The disparity between the reactivities of these two antibodies that react with the back side of gD suggests that structural changes may be limited to subtle differences in this region of the protein. Overall, gD V231W exhibited changes in MAb epitopes proximal to the receptor binding region.
Fig 4.

Reactivity of gD V231W with conformationally sensitive antibodies. (A) Native Western blotting of gD 306t or V231W with MAb epitopes that are near the receptor binding face of gD (DL11, MC23, AP7) or that map to the opposite side of gD proximal to where the C terminus attaches to the Ig-like core of gD (MC5, A18). Asterisk, full-length gD. (B) gD was adhered to an ELISA plate for MAb binding. Mapping of epitopes to the receptor binding region of gD indicated structural changes associated with V231W, while subtle changes were observed in epitopes not associated with the receptor binding face of gD.
To quantitate these effects, ELISA was used to compare MAb binding to gD 306t, V231W, and 285t (Fig. 4B). gD V231W, 306t, or 285t protein was adhered to an ELISA plate, and reactivity with MAbs was measured. Equal binding was observed for DL6. DL11 reactivity was the highest for gD 285t, indicating that it bound the receptor binding region of gD that is covered by the C terminus in gD 306t. gD V231W had reduced reactivity with DL11, likely because its epitope on the receptor binding face is disrupted by V231W. MC23 had similar reactivity with gD 306t and gD 285t, while gD V231W reactivity was reduced, again likely because of disruption of its epitope. AP7 reacted best with gD 306t. AP7 reactivity was also reduced for gD V231W, indicating that the conformation of either the N or C terminus was altered in gD V231W. Reactivity with MC5 was similar between gD 285t and gD 306t, while gD V231W had fractionally lower binding. The observed reactivity between gD V231W and MC5 differed between the ELISA and native Western blotting assay, leading us to suspect that the epitope on gD V231W was susceptible to differences in experimental setup, such as the exposure to SDS during electrophoresis in the native Western blotting. A18 binding to gD V231W and gD 285t was slightly increased compared to that to gD 306t. Unlike MC5, the native Western blotting results for A18 were qualitatively in agreement with the ELISA results, affirming that changes at the back side of gD were limited to subtle differences, while large changes were evident at the receptor binding site.
HSV receptor binding.
Binding of MAbs to gD V231W suggested that significant structural changes in the receptor binding region resulted from the V231W mutation. To understand these changes, we examined the binding of gD V231W to HSV receptors HVEM and nectin-1. Soluble HVEM 200t or nectin-1 245t was adhered to an ELISA plate and then assayed for binding to gD at various concentrations. gD V231W bound HVEM at levels comparable to those for gD 306t, while binding of gD V231W to nectin-1 was essentially undetected (Fig. 5). One possible explanation for the weak binding between gD V231W and nectin-1 may be found in the gD–nectin-1 cocrystal structure. gD V231 is located proximal to nectin-1 residues 130 to 132 (17); therefore, the tryptophan mutation at gD V231 may clash with these nectin-1 residues and disrupt binding. We note that gD mutations proximal to V231 at residues 215, 222, and 223 rendered engineered HSV-2 unable to use nectin-1 for entry (43). The gD-HVEM interface does not contact V231, as seen in the structure of the complex (16), and ELISA binding of gD V231W to HVEM supported this observation.
Fig 5.

ELISA of gD binding to immobilized receptor. (A) HVEM 200t was bound equivalently by gD 306t or gD V231W, while gD 285t bound with much higher affinity. (B) Nectin-1 245t was bound the strongest by gD 285t, while binding by gD 306t was reduced from that by gD 285t. Binding of gD V231W to nectin-1 245t was even weaker than that of gD 306t, with binding barely being observed at 10 μM. OD405, optical density at 405 nm.
Dynamics of the C terminus by biosensor.
The crystal structures of gD bound to either HVEM or nectin-1 depict a large conformational change where receptor displaces the gD C terminus from the receptor binding interface. A comparison of the association rates for gD 306t and gD 285t with HVEM found that gD 285t has a 50-fold higher association rate, which is due to the C terminus actively blocking the receptor binding surface of gD 306t (40). To test if the C terminus was displaced in gD V231W, we measured the binding of both gD V231W and gD 306t to HVEM and nectin-1. The association rate in complex formation reports on the extent that the C terminus blocks HVEM binding. Because gD binding to nectin-1 245t was not seen by ELISA, we examined the binding of gD to both HVEM and nectin-1 in case the complex was too short-lived to survive the ELISA wash protocol, as was the case for a shorter gD truncation (40).
Soluble HVEM 200t (32) was captured with MAb CW10 (34) that was immobilized on a CM5 Biacore chip, facilitating uniform presentation of the ligand on the chip surface. gD 306t or gD V231W was then injected into the flow cell, and binding was measured (Fig. 6A and B). For gD V231W, a global fit to a 1:1 binding model yielded an equilibrium dissociation constant of 13.8 μM, while gD 306t yielded an equilibrium dissociation constant of 12.6 μM (Table 1). We examined the association rates, which imply the extent to which the C terminus blocks the receptor binding region. The association rates for gD 306t and V231W with HVEM were 3.6 M−1 s−1 and 8.9 M−1 s−1, respectively, the latter of which is 2.5-fold higher for gD V231W than for WT. The dissociation rate of gD V231W with HVEM was also higher than that of gD WT 306t, giving rise to the similar equilibrium binding constants. We focused on the association rate for HVEM binding, which indicated that the C terminus of gD V231W has reduced blocking of the receptor binding site in an amount that correlated with the increase in activity of gD V231W on B78H1 cells.
Fig 6.

Biosensor binding of gD to HSV receptor HVEM or nectin-1. HVEM 200t was captured and then bound to gD 306t (A) or gD V231W (B). Nectin-1 245t was captured and bound to gD 306t (C) or gD V231W (D). gD concentrations of 0 μM, 1 μM, 2 μM, 4 μM, 6 μM, 8 μM, and 10 μM were injected in sequential cycles, with the receptor surface being regenerated between cycles. Curves were fit using a 1:1 Langmuir binding model, yielding the binding constants described in Table 1. RU, resonance units.
Table 1.
Binding constants measured by biosensor for gD V231W and gD 306t with HSV receptor captured on a biosensor chipa
| HSV receptor | gD construct | kon (103 M−1 s−1) | koff (10−2 s−1) | KD (μM) | Relative kon |
|---|---|---|---|---|---|
| HVEM 200t | 306t | 3.6 ± 0.2 | 4.5 ± 0.04 | 12.6 ± 0.6 | 1.0 |
| V231W | 8.9 ± 0.3 | 12.2 ± 0.2 | 13.8 ± 0.6 | 2.5 | |
| Nectin-1 245t | 306t | 1.3 ± 0.1 | 0.67 ± 0.01 | 5.2 ± 0.1 | 1.0 |
| V231W | 3.4 ± 0.1 | 8.56 ± 0.03 | 25 ± 0.7 | 2.6 |
kon, rate of association; koff, rate of dissociation; KD, dissociation constant.
Next, we examined gD binding to nectin-1 245t (44). The most striking difference between gD 306t and gD V231W binding was the increased dissociation rate of gD V231W, which was readily visible from the sensograms (Fig. 6C and D). The affinity of gD V231W for nectin-1 245t was 25 μM, as opposed to 5 μM for gD 306t, which corresponded to our expectations of a decreased affinity from ELISA binding. Directly comparing the association rates for nectin-1, the rate for gD V231W was 2.6-fold higher than that for gD 306t. Since this is similar to the 2.5-fold increase in association for gD V231W with HVEM, we were further convinced that the association rate reflected the change in conformation of the gD C terminus and not the dynamics of HVEM or nectin-1.
DISCUSSION
In this study, we tested two mutants of HSV-1 gD that were rationally designed to activate fusion on the basis of our hypothesis that displacement of the gD C terminus permits gD to interact with gH/gL and gB. We found that gD V231W increased fusion without receptor, and we linked the mechanism of this activity to its displacement of the C terminus from the gD core. Displacement of the gD C terminus is proposed to reveal interfaces that are linked to fusion activation by the MC2- and MC5-neutralizing epitopes, which are on the opposite side of gD from the receptor binding site (5). Using cell-cell fusion, we found that residues near W294 in the C terminus itself play a role in activation of fusion, and according to the crystal structure (18), these residues are accessible only when the C terminus is displaced. The single event of C terminus displacement can explain how a number of interfaces on gD that play a role in fusion activation are exposed, leading to the predicted interaction with gH/gL in the fusion pathway.
Effects on cell-cell fusion with and without HSV receptor.
gD V231W triggered fusion on B78H1 cells at a level 3.5-fold increased from that for WT according to Giemsa staining of syncytia, indicating that gD V231W is in an activated state which was capable of inducing fusion. The activity of gD V231W on receptor-negative B78H1 cells was confirmed using a split luciferase assay, which permitted measurement of luminescence in place of counting of syncytia. The luciferase assay measured similar activity for WT gD and gD V231W in the presence of nectin-1 and a 5-fold increase in gD V231W activity over that for WT in the absence of receptor (Fig. 3). Though the result was qualitatively similar to the Giemsa staining result, the luciferase assay observed even more activity than that found by manual counting of syncytia. With the luciferase assay, fusion was also detectable at earlier time points. The experimental setup of the luciferase assay overlaying two cell populations may have permitted this increase in sensitivity, perhaps accounting for the increased difference between WT and V231W.
When the B78H1 cells were transfected with vector alone or if any one of the four HSV glycoproteins was omitted, an average of zero to one syncytium per field was observed. These counts are likely to be false positives, representing the error in manual counting due to cell overlap. The same very low background of the fusion assay was also seen in the split luciferase assay (Fig. 3). On C10 cells, gD V231W triggered cell-cell fusion at levels equivalent to those for WT (Fig. 2B), despite the weak binding between gD V231W and nectin-1 determined by ELISA (Fig. 5B). C10 cells express nectin-1 at a level about 7-fold greater than the level at which A10 cells express HVEM (10), which may have compensated for the high dissociation rate of gD V231W with nectin-1 that was later observed by biosensor (Fig. 6D). gD V231W triggered fusion on A10 cells at levels equivalent to those for WT as well, despite biosensor observations that gD V231W associated with HVEM faster than the WT did. Two factors may best explain this: first, the dissociation rate of gD V231W with HVEM was also higher, resulting in equivalent net binding to WT gD, and second, the kinetics of the interaction required to activate gH/gL and gB at the cell surface are unknown, which leads to difficulty comparing direct binding assays with the cell-based fusion assay.
gD A12W had a 40% increase in activity on HVEM cells, which can be explained by gD A12W stabilizing the N-terminal hairpin of gD that binds HVEM (Fig. 2C), but the fusion activity of A12W was equivalent to that of WT on C10 and B78H1 cells; therefore, gD A12W stabilized the N-terminal hairpin when HVEM was bound but did not augment the conformation of unbound gD or its ability to trigger fusion.
Mutants with the C-terminal mutations W294A and Δ290-299, which had not previously been characterized by cell-cell fusion, had reduced activity on A10 cells and little activity on B78H1 cells. The activity of WT was 4.5-fold greater than that of gD W294A on B78H1 cells, while the activity of gD V231W was 16-fold greater than that of gD W294A on B78H1 cells. The activity of gD Δ290-299 on B78H1 cells was equivalent to that of vector alone. The activity of gD W294A and Δ290-299 on C10 cells was likely aided by high levels of nectin-1 expression, which masked the defects in gD function with these mutants. We interpret the absence in activity on B78H1 cells by mutants with C-terminal gD mutations to mean that residue W294, absent in both gD W294A and Δ290-299, is part of an interface that enables gD to activate fusion efficiently, perhaps by interacting with gH/gL. Our results studying C-terminal mutations in a cell-cell fusion assay are in agreement with previous results with viral entry and complementation (18, 19).
Comparison with other gD mutations that activate fusion.
We selected residue V231W by design, but other mutations have arisen naturally through selection of virus and have been found to partially uncouple receptor binding from gD activity. S140N was first described as an adaptation to selective pressure by MAb DL11, which binds gD and blocks receptor binding (45). Independently, the S140N mutation arose when virus was selected on a lineage of BHK (TK−) cells that do not express HSV receptor (46). S140 is immediately next to V231 in the structure (asterisk in upper right of Fig. 1B). We selected V231W for study because the mutation is centrally located on the receptor binding interface, in a region that straddles the HVEM and nectin-1 binding sites. V231 also forms one side of a hydrophobic pocket which binds either W294 of the C terminus or receptor residue Y23 from HVEM or F99 from nectin-1. While the tryptophan mutation of V231W is a large amino acid, it is nonetheless a single amino acid, and its position in the structure affords plenty of solvent-exposed area to accommodate the larger amino acid. Mutation S140N introduces an N-linked glycan consensus sequence, and this glycan has been confirmed biochemically (45). The glycan would likely occlude the entire receptor binding face of gD, a dramatic change to the surface of gD that may have unpredicted effects on the structure. While S140N and the attached glycan have a much bigger footprint than V231W, it is interesting to consider that both mutations may operate by a common mechanism, given their proximity to each other. S140K, which does not introduce an N-linked glycan, was also found to reduce gD dependence on receptor, although it was characterized in the context of hyperfusogenic gH and gB mutations that already greatly enhanced fusion activity without HSV receptor (21).
Another mutation that arose through viral selection for receptor-independent entry is A185T (46). Position 185 is near residues 276 to 278 of the C terminus, where the C terminus sits atop the Ig-like gD core in the unbound crystal structure. The epitope for MAb MC14, residues 264 to 275, is nearby, and MC14 is known to alter the C terminus to increase the accessibility of the MC2-neutralizing epitope (5). Mutations V231W and S140N are linked to A185T by their proximity to the C terminus, which runs between these mutations. Therefore, all receptor-independent mutations for gD are notable for their potential effect on the conformation of the C terminus, suggesting that the C terminus is the physical link between receptor binding and exposure of interfaces on gD that activate fusion.
Structural differences highlighted by MAbs.
Several antigenic differences between gD V231W and WT indicated that structural changes resulted from the mutation. gD V231W had reduced binding to both DL11 and MC23, which suggests that V231W partially disrupted these epitopes and indicates that the receptor binding site was altered. Indeed, the inability of gD V231W to bind to nectin-1 by ELISA corroborated the finding that the receptor binding interface was disrupted (Fig. 5). gD V231W also had reduced binding to AP7, an epitope that spans the N and C termini (37, 47). gD 285t has a truncated C terminus, which directly disrupted the AP7 epitope but revealed the DL11 epitope, which is obscured by the C terminus (45). MAbs that bound outside the receptor binding region of gD illustrated only subtle changes in affinity for gD V231W. While MC5 bound weakly to gD V231W compared to the binding of WT by native Western blotting, this result differed from the ELISA results, which found only a slight reduction in binding to MC5. This discrepancy is best explained by the difference in the conditions between the two assays; in particular, the SDS that is present in the native Western blotting may disrupt an epitope that may already be more labile in gD V231W. A18 type specificity for HSV-1 has localized its epitope to the region of amino acid 246, which is near where the C terminus connects to the Ig-like core of gD, opposite the receptor binding region. Both gD 285t and gD V231W had modest increases in A18 reactivity, hinting at exposure of the epitope by the deleted or displaced C terminus.
V231W effect on receptor binding.
Using recombinant protein, we measured the direct binding of gD V231W to HSV receptors HVEM 200t and nectin-1 245t by ELISA. We found that gD V231W had dramatically reduced binding to nectin-1 compared to that of gD 306t, but binding to HVEM was similar to that of WT (Fig. 5). The molecular interfaces observed in the cocrystal structure support these observations, as V231 is adjacent to nectin-1 residues 130 to 132 in the cocrystal structure, and the added size of tryptophan at gD position 231 may clash with nectin-1 and prevent it from docking into its binding site (17). The cocrystal structure of gD and HVEM illustrates that the N-terminal hairpin of gD separates V231 from HVEM, permitting plenty of room to accommodate the V231W mutation (16).
Conformation of the C terminus measured by biosensor.
We used a biosensor to measure the rates of association between gD and soluble receptor constructs, which indicated the extent to which the C terminus of gD blocks the receptor binding site. We measured binding under identical conditions for gD 306t and gD V231W, which was important, since this study used antibody to capture receptor to facilitate uniform presentation on the biosensor chip rather than directly coupling of receptor to the chip surface (18, 38, 40, 44). Antibody capture also permitted fresh HSV receptor to be captured after chip regeneration. We measured a 2.5-fold relative increase in the association rate for gD V231W binding to HVEM and a 2.6-fold increase in the association rate for gD V231W binding to nectin-1 (Fig. 6; Table 1). gD V231W also had a higher dissociation rate for both HVEM and nectin-1, which indicated that the gD V231W mutation had a destabilizing effect on the receptor-bound complex. The increased rate of association of gD V231W with both HVEM and nectin-1 was evidence that the association rate was a function of the conformation of gD and not that of the receptor and that the extent that the C terminus blocked receptor binding was reduced for gD V231W.
Cumulative model of gD activation of fusion.
Epitopes for neutralizing antibodies MC2 and MC5 that block the postulated association with gH/gL (5) are on the opposite side of gD from the receptor binding site. These epitopes represent regions on the Ig-like core of gD that are critical for fusion activation. Soluble constructs of gD, which can also activate fusion, implicated residues 260 to 285, which are located in the C terminus where it lies across the top of the Ig-like core of gD (19). This region was independently described using gD chimeras constructed from HSV and PRV (48). A third region of gD, which consists of the C terminus where it covers the receptor binding site and which is inclusive of W294, plays a dual role by both activating fusion (19) and blocking receptor binding (18). Here we describe how displacement of the C terminus, using a point mutation, V231W, that is outside all of these fusion-activating interfaces on gD, is sufficient to trigger fusion in the absence of receptor. Using biosensor measurements of gD associating with receptor, we observe that gD V231W promotes the open conformation of the C terminus. We associate this change in conformation with its activity on receptor-negative cells and exposure of interfaces that activate fusion. For WT gD, this means that receptor binding acts by displacing the C terminus, leading to the exposure of gD interfaces that are postulated to bind gH/gL (Fig. 7) and initiating the cascade of events leading to fusion.
Fig 7.
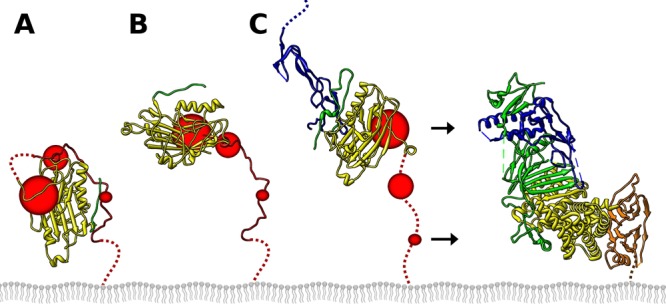
Model of gD activation. (A) On the viral envelope, gD is autoinhibited by the C terminus of the gD ectodomain, inhibiting activation of fusion. The C terminus transiently dissociates (B), allowing HSV receptor (blue) to capture gD while the C terminus is dissociated (C). The extended C terminus of gD exposes neutralizing epitopes near residues 242 and 70 (large red circle); residues 260 to 285, known to be important by soluble gD C-terminal truncations (medium red circle); and residues in the C terminus that have a dual role in blocking receptor and activating fusion, such as W294 (small red circle). The extended conformation of gD can then interact with gH/gL, leading to fusion catalyzed by gB. The point mutation V231W acts on gD by promoting the initial opening of the C terminus shown in panel B. At the concentrations of gD used in a cell-cell fusion assay, this was sufficient to interact with gH/gL to trigger fusion. The gH/gL color scheme is as published previously (49).
In summary, we designed a mutation at the receptor binding face of gD to displace the C terminus, hypothesizing that this would activate fusion even in the absence of bound receptor. We found that by introducing a tryptophan mutation in place of valine at HSV-1 gD position 231, cell-cell fusion activity in the absence of HSV receptor was increased 3.5-fold. A second mutation at position 12 of the N terminus that was also designed to displace the C terminus did not induce cell-cell fusion on receptor-negative cells, although it did increase activity on HVEM-expressing cells. MAb epitopes on gD V231W were disrupted at the receptor binding face proximal to the mutation, but at changes associated with the C terminus elsewhere in the molecule were also hinted. Finally, we measured the rate of association of gD V231W with HVEM and nectin-1 to determine if the V231W mutation affected the conformation of the C terminus. We found the C terminus to have reduced blocking of receptor binding to gD V231W, validating our prediction that V231W would displace the C terminus of gD. Therefore, gD V231W is in an activated state which can directly activate gH/gL and gB, and displacement of the gD C terminus is the mechanism by which the front of gD communicates receptor binding to the opposite side of gD. We conclude that the gD C terminus acts as a switch that plays a dual role by directly inhibiting fusion when covering the receptor binding interface, as well as promoting fusion by exposing interfaces on gD that activate the fusion pathway when the C terminus is displaced.
ACKNOWLEDGMENTS
The research reported in this publication was supported by NIH grant R37-AI-18289 to G.H.C. and NIH grants R01-AI-076231 and R01-AI-056045 to R.J.E., and J.R.G. was supported by NIH training grant T32-AI-07234.
We thank all members of our laboratories for their helpful advice.
The content of this paper is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 18 September 2013
REFERENCES
- 1.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heldwein EE, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 65:1653–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atanasiu D, Cairns TM, Whitbeck JC, Saw WT, Rao S, Eisenberg RJ, Cohen GH. 2013. Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors. mBio 4(2):e00046-13. 10.1128/mBio.00046-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lazear E, Whitbeck JC, Ponce-de-Leon M, Cairns TM, Willis SH, Zuo Y, Krummenacher C, Cohen GH, Eisenberg RJ. 2012. Antibody-induced conformational changes in herpes simplex virus glycoprotein gD reveal new targets for virus neutralization. J. Virol. 86:1563–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436 [DOI] [PubMed] [Google Scholar]
- 7.Kwon BS, Tan KB, Ni J, Oh KO, Lee ZH, Kim KK, Kim YJ, Wang S, Gentz R, Yu GL, Harrop J, Lyn SD, Silverman C, Porter TG, Truneh A, Young PR. 1997. A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J. Biol. Chem. 272:14272–14276 [DOI] [PubMed] [Google Scholar]
- 8.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620 [DOI] [PubMed] [Google Scholar]
- 9.Takahashi K, Nakanishi H, Miyahara M, Mandai K, Satoh K, Satoh A, Nishioka H, Aoki J, Nomoto A, Mizoguchi A, Takai Y. 1999. Nectin/PRR: an immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J. Cell Biol. 145:539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krummenacher C, Baribaud F, Ponce de Leon M, Baribaud I, Whitbeck JC, Xu R, Cohen GH, Eisenberg RJ. 2004. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology 322:286–299 [DOI] [PubMed] [Google Scholar]
- 11.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22 [DOI] [PubMed] [Google Scholar]
- 12.Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246:179–189 [DOI] [PubMed] [Google Scholar]
- 13.Menotti L, Cerretani A, Campadelli-Fiume G. 2006. A herpes simplex virus recombinant that exhibits a single-chain antibody to HER2/neu enters cells through the mammary tumor receptor, independently of the gD receptors. J. Virol. 80:5531–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menotti L, Cerretani A, Hengel H, Campadelli-Fiume G. 2008. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J. Virol. 82:10153–10161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou G, Roizman B. 2006. Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13alpha2 receptor. Proc. Natl. Acad. Sci. U. S. A. 103:5508–5513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC. 2001. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 8:169–179 [DOI] [PubMed] [Google Scholar]
- 17.Di Giovine P, Settembre EC, Bhargava AK, Luftig MA, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C, Carfi A. 2011. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog. 7:e1002277. 10.1371/journal.ppat.1002277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. 2005. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 24:4144–4153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cocchi F, Fusco D, Menotti L, Gianni T, Eisenberg RJ, Cohen GH, Campadelli-Fiume G. 2004. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc. Natl. Acad. Sci. U. S. A. 101:7445–7450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazear E, Carfi A, Whitbeck JC, Cairns TM, Krummenacher C, Cohen GH, Eisenberg RJ. 2008. Engineered disulfide bonds in herpes simplex virus type 1 gD separate receptor binding from fusion initiation and viral entry. J. Virol. 82:700–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uchida H, Chan J, Shrivastava I, Reinhart B, Grandi P, Glorioso JC, Cohen JB. 2013. Novel mutations in gB and gH circumvent the requirement for known gD receptors in herpes simplex virus 1 entry and cell-to-cell spread. J. Virol. 87:1430–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoon M, Zago A, Shukla D, Spear PG. 2003. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J. Virol. 77:9221–9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324 [DOI] [PubMed] [Google Scholar]
- 24.Turner A, Bruun B, Minson T, Browne H. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72:873–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller CG, Krummenacher C, Eisenberg RJ, Cohen GH, Fraser NW. 2001. Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol. Ther. 3:160–168 [DOI] [PubMed] [Google Scholar]
- 26.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atanasiu D, Saw WT, Gallagher JR, Hannah BP, Matsuda Z, Whitbeck JC, Cohen GH, Eisenberg RJ. 2013. Dual split protein-based fusion assay reveals that mutations to herpes simplex virus (HSV) glycoprotein gB alter the kinetics of cell-cell fusion induced by HSV entry glycoproteins. J. Virol. 87:11332–11345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishikawa H, Meng F, Kondo N, Iwamoto A, Matsuda Z. 2012. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng. Des. Sel. 25:813–820 [DOI] [PubMed] [Google Scholar]
- 29.Krummenacher C, Baribaud I, Ponce de Leon M, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. 2000. Localization of a binding site for herpes simplex virus glycoprotein D on herpesvirus entry mediator C by using antireceptor monoclonal antibodies. J. Virol. 74:10863–10872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sisk WP, Bradley JD, Leipold RJ, Stoltzfus AM, Ponce de Leon M, Hilf M, Peng C, Cohen GH, Eisenberg RJ. 1994. High-level expression and purification of secreted forms of herpes simplex virus type 1 glycoprotein gD synthesized by baculovirus-infected insect cells. J. Virol. 68:766–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen GH, Isola VJ, Kuhns J, Berman PW, Eisenberg RJ. 1986. Localization of discontinuous epitopes of herpes simplex virus glycoprotein D: use of a nondenaturing (“native” gel) system of polyacrylamide gel electrophoresis coupled with Western blotting. J. Virol. 60:157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitbeck JC, Connolly SA, Willis SH, Hou W, Krummenacher C, Ponce de Leon M, Lou H, Baribaud I, Eisenberg RJ, Cohen GH. 2001. Localization of the gD-binding region of the human herpes simplex virus receptor, HveA. J. Virol. 75:171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krummenacher C, Nicola AV, Whitbeck JC, Lou H, Hou W, Lambris JD, Geraghty RJ, Spear PG, Cohen GH, Eisenberg RJ. 1998. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J. Virol. 72:7064–7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stiles KM, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C. 2010. Herpes simplex virus glycoprotein D interferes with binding of herpesvirus entry mediator to its ligands through downregulation and direct competition. J. Virol. 84:11646–11660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Cohen GH, Eisenberg RJ. 2003. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/HVEM binding interface. J. Virol. 77:8127–8140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Eisenberg RJ, Cohen GH. 2002. Structure-based analysis of the herpes simplex virus glycoprotein D binding site present on herpesvirus entry mediator HveA (HVEM). J. Virol. 76:10894–10904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiang HY, Cohen GH, Eisenberg RJ. 1994. Identification of functional regions of herpes simplex virus glycoprotein gD by using linker-insertion mutagenesis. J. Virol. 68:2529–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willis SH, Rux AH, Peng C, Whitbeck JC, Nicola AV, Lou H, Hou W, Salvador L, Eisenberg RJ, Cohen GH. 1998. Examination of the kinetics of herpes simplex virus glycoprotein D binding to the herpesvirus entry mediator, using surface plasmon resonance. J. Virol. 72:5937–5947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huber MT, Wisner TW, Hegde NR, Goldsmith KA, Rauch DA, Roller RJ, Krummenacher C, Eisenberg RJ, Cohen GH, Johnson DC. 2001. Herpes simplex virus with highly reduced gD levels can efficiently enter and spread between human keratinocytes. J. Virol. 75:10309–10318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rux AH, Willis SH, Nicola AV, Hou W, Peng C, Lou H, Cohen GH, Eisenberg RJ. 1998. Functional region IV of glycoprotein D from herpes simplex virus modulates glycoprotein binding to the herpesvirus entry mediator. J. Virol. 72:7091–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muggeridge MI, Isola VJ, Byrn RA, Tucker TJ, Minson AC, Glorioso JC, Cohen GH, Eisenberg RJ. 1988. Antigenic analysis of a major neutralization site of herpes simplex virus glycoprotein D, using deletion mutants and monoclonal antibody-resistant mutants. J. Virol. 62:3274–3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scheffer AJ, Koedijk DG, Abee T, Osterhaus AD. 1984. Monoclonal antibodies against herpes simplex viruses. Dev. Biol. Stand. 57:269–274 [PubMed] [Google Scholar]
- 43.Wang K, Kappel JD, Canders C, Davila WF, Sayre D, Chavez M, Pesnicak L, Cohen JI. 2012. A herpes simplex virus 2 glycoprotein D mutant generated by bacterial artificial chromosome mutagenesis is severely impaired for infecting neuronal cells and infects only Vero cells expressing exogenous HVEM. J. Virol. 86:12891–12902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krummenacher C, Rux AH, Whitbeck JC, Ponce-de-Leon M, Lou H, Baribaud I, Hou W, Zou C, Geraghty RJ, Spear PG, Eisenberg RJ, Cohen GH. 1999. The first immunoglobulin-like domain of HveC is sufficient to bind herpes simplex virus gD with full affinity, while the third domain is involved in oligomerization of HveC. J. Virol. 73:8127–8137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muggeridge MI, Wu TT, Johnson DC, Glorioso JC, Eisenberg RJ, Cohen GH. 1990. Antigenic and functional analysis of a neutralization site of HSV-1 glycoprotein D. Virology 174:375–387 [DOI] [PubMed] [Google Scholar]
- 46.Rauch DA, Rodriguez N, Roller RJ. 2000. Mutations in herpes simplex virus glycoprotein D distinguish entry of free virus from cell-cell spread. J. Virol. 74:11437–11446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Minson AC, Hodgman TC, Digard P, Hancock DC, Bell SE, Buckmaster EA. 1986. An analysis of the biological properties of monoclonal antibodies against glycoprotein D of herpes simplex virus and identification of amino acid substitutions that confer resistance to neutralization. J. Gen. Virol. 67(Pt 6):1001–1013 [DOI] [PubMed] [Google Scholar]
- 48.Zago A, Jogger CR, Spear PG. 2004. Use of herpes simplex virus and pseudorabies virus chimeric glycoprotein D molecules to identify regions critical for membrane fusion. Proc. Natl. Acad. Sci. U. S. A. 101:17498–17503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 17:882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]