

Abstract
In certain Ras mutant cell lines, the inhibition of extracellular signal-regulated kinase (ERK) signaling increases RhoA activity and inhibits cell motility, which was attributed to a decrease in Fra-1 levels. Here we report a Fra-1-independent augmentation of RhoA signaling during short-term inhibition of ERK signaling. Using mass spectrometry-based proteomics, we identified guanine exchange factor H1 (GEF-H1) as mediating this effect. ERK binds to the Rho exchange factor GEF-H1 and phosphorylates it on S959, causing inhibition of GEF-H1 activity and a consequent decrease in RhoA activity. Knockdown experiments and expression of a nonphosphorylatable S959A GEF-H1 mutant showed that this site is crucial in regulating cell motility and invasiveness. Thus, we identified GEF-H1 as a critical ERK effector that regulates motility, cell morphology, and invasiveness.
INTRODUCTION
Locomotion, and thus invasion and metastasis of tumor cells, is controlled by cytoskeletal reorganizations, which are coordinated by the tightly regulated and localized activation of the Rho family GTPases, namely, RhoA, Rac, and CDC42. Rac and CDC42 are activated mainly at the leading edge, whereas RhoA activity is localized at the rear and front of the moving cell (1–4). In cells randomly migrating on two-dimensional surfaces, RhoA activity precedes the formation of a protrusion, whereas Rac1 and CDC42 activities peak shortly afterwards during the retraction phase (1). Further, RhoA and Rac1 activities are inversely related due to mutual negative feedback connections (5, 6).
In three-dimensional (3D) matrices, cell motility has different characteristics than on two-dimensional surfaces and involves two distinct modes of invasion. Either cells are elongated and move in a matrix metalloprotease (MMP)-dependent mesenchymal fashion or the cells appear rounded and invade in a RhoA-dependent, amoeboid way (7) requiring high Rho kinase (ROCK) activity. RhoA and ROCK control cellular contractility, thus enabling the invading cell to squeeze through the extracellular matrix without the need to degrade it by secreting MMPs. Cells can switch between amoeboid and mesenchymal invasion (5, 8, 9).
The rapid, spatially restricted and controlled activation/deactivation cycle of Rho family GTPases is regulated by a balance of guanine exchange factors (GEFs) and GTPase-activating proteins (GAPs). GEFs binding to RhoA release bound GDP, which is replaced by abundant cellular GTP. GTP binding induces a conformational switch that unmasks binding sites for downstream effectors. Termination of Rho signaling is achieved through the binding of GAPs. These proteins associate with small GTPases and, by creating an active site, dramatically increase their intrinsic GTP hydrolysis activity, thus reverting Rho family member to the inactive GDP-bound state. GEF and GAP activity, as well as their subcellular localization, is controlled by a multitude of external signaling pathways, including Rho/Rac/CDC42-dependent signaling. This high level of regulation, cross talk, and complexity at the GEF/GAP level and the fact that constitutively active GEFs have been identified as oncogenes are driving extensive research interest in these regulatory proteins (for a review, see reference 10).
Recently, guanine nucleotide exchange factor H1 (GEF-H1) (ARHGEF2) was identified as an upstream regulator of leading-edge RhoA activity in migrating cells (11). Depletion of GEF-H1 by small interfering RNA (siRNA) decreased RhoA activity at the leading edge as well as random migration and focal adhesion turnover. As with many GEFs, the regulation of GEF-H1 is complex, involving a multitude of phosphorylations on activating and inactivating sites. Different kinases, including PAK, Aurora A, Cdk1, and PAR1b (12–15), were shown to inactivate GEF-H1 by phosphorylating inhibitory sites, whereas extracellular signal-regulated kinase (ERK) (16, 17) was reported to phosphorylate Thr678, an activating site. Interestingly, regulation of GEF-H1 activity downstream of ERK appears to be more complex, since inhibition of the mitogen-activated protein kinase (MAPK) pathway in unstimulated cells not only enhances RhoA activity but, controversially, increases the phosphorylation of the reported ERK phosphorylation site, Thr678 (18). Further, GEF-H1 is held in an inactive conformation when bound to microtubules. Conversely, microtubule disassembly results in a robust activation of RhoA via GEF-H1 (19).
Here we have shown that under growing conditions, ERK phosphorylates GEF-H1 on an inhibitory site. Inhibition of ERK signaling with chemical MEK inhibitors induces RhoA activation in a GEF-H1-dependent manner. Overexpression of an unphosphorylatable GEF-H1 mutant enhances RhoA activity and blocks cell migration and invasiveness. In addition, preventing ERK inhibition of GEF-H1 induces cells to adopt a rounded shape, and GEF-H1 downregulation interferes with amoeboid invasion.
MATERIALS AND METHODS
Cells and reagents.
Cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 2 mM glutamine and 10% fetal calf serum. Plasmids and siRNA oligonucleotides were transfected with Lipofectamine 2000 using the manufacturer's instructions (Invitrogen, United Kingdom). Enhanced green fluorescent protein-tagged GEF-H1 (eGFP–GEF-H1) was kindly provided by Gary Bokoch (Scripps Institute, La Jolla, CA) and glutathione S-transferase–rhotekin–Rho binding domain (GST-rhotekin-RBD) by Mike Olson (Beatson Institute, Glasgow, United Kingdom). Rat GEF-H1 was cloned from PC12 cDNA by PCR and subsequently cloned into pcDNA3.1 using the NotI and XbaI cloning sites. Flag–GEF-H1-S959A and Flag–GEF-H1-S959D mutants were made using the QuikChange kit (Stratagene, The Netherlands). Antibodies for ERK1, RhoA, and Fra-1 were from Santa Cruz (Clane, United Kingdom) the GEF-H1 and ERK substrate motif (pTP, PXpST) were from Cell Signaling (Hitchin, United Kingdom), ERK1/2 and phospho-ERK1/2 were from Sigma (Gillingham, United Kingdom), the 3DA-luciferase reporter vector and TAT-C3 were a kind gift from Mike Olson (Beatson Institute, Glasgow, United Kingdom), U0126 was from Promega (United Kingdom), and PD0325901 was from Sigma (United Kingdom).
siRNA knockdown.
Forty picomoles of siRNA oligonucleotides were introduced into MDA-MB-231 cells by transfection using the HiPerFect reagent (Qiagen) according to the manufacturer's instructions. SMARTpool siRNAs or single siRNAs (Dharmacon, USA) were used to knock down GEF-H1; a nontargeting siRNA pool (Dharmacon, USA) was used as a control. Oligonucleotides used were no. 1 (GAAUUAAGAUGGAGUUGCA) and no. 2 (GUGCGGAGCAGAUGUGUAA).
Motility assays.
Inverted invasion assays (20) were performed as described previously (21). Cells were allowed to invade toward a gradient of epidermal growth factor (EGF) (30 nM) and 10% serum for 3 days. In the case of A375M2 cells, the Transwell plugs were coated with a 0.001% solution of fibronectin to facilitate migration through the membrane.
Cell treatment, lysis, and immunoprecipitation.
Cells were incubated with U0126 (10 μM) or PD0325901 (2 μM) or serum deprived for 18 h and treated with 20 ng/ml EGF as indicated. Cells were lysed in ice-cold lysis buffer (20 mM HEPES [pH 7.5], 150 mM NaCl, 1% NP-40, and 2 mM EDTA) supplemented with protease (1 mM phenylmlethylsulfonyl fluoride [PMSF], 5 μg/ml leupeptin, 2.2 μg/ml aprotinin, and 2 mM sodium fluoride) and phosphatase (1 mM sodium vanadate, 1 mM sodium pyrophosphate, and 20 mM β-glycerophosphate) inhibitors. Lysates were cleared of debris by centrifugation at 20,000 × g for 10 min in a benchtop centrifuge. For immunoprecipitation antibodies-protein A-agarose beads (GE Healthcare), anti-Flag-M2 beads (Sigma, United Kingdom) or anti-green fluorescent protein (anti-GFP) beads (Chromotek, Germany) were added to the cleared lysates and incubated at 4°C under end-to-end rotation for 2 h. Beads were washed three times with lysis buffer and either eluted with a Flag peptide, boiled off in Laemmli buffer or, if the samples were destined for mass spectrometry, washed twice with lysis buffer devoid of detergents.
Phosphopeptide mapping.
Flag–GEF-H1 was expressed in HEK293 cells and immunoprecipitated with Flag antibody. The immunoprecipitate was extensively washed, equilibrated with ERK kinase buffer (150 mM NaCl, 10 mM MgCl2, 20 mM Tris-HCl, 1 mM dithiothreitol [DTT], 1 mM sodium vanadate, 1 mM sodium pyrophosphate, and 20 mM β-glycerophosphate [pH 7.5]), and incubated with active ERK (Calbiochem, United Kingdom) and 300 μM ATP spiked with 1 μl [γ-32P]ATP (Invitrogen, United Kingdom) for 30 min. As control, ERK was incubated with the widely used substrate myelin basic protein (MBP). The kinase reactions were separated by SDS-PAGE and in gel digested with trypsin (22). The peptide mixture was separated by reverse-phase high-performance liquid chromatography (RP-HPLC). The radioactive fractions were split, with one fraction subjected to Edman degradation and the other to matrix-assisted laser desorption ionization (MALDI)–mass spectrometry (MS). Edman degradation indicated that the major phosphorylation site is located at position 6. MS analysis of the radioactive peptide fraction was performed using a Bruker Ultraflex II time of flight (TOF) instrument in positive ion mode using dihydroxybenzoic acid as a matrix. Resulting spectra were manually searched for tryptic peptides with a modification of +80 and either S/T or Y at position 6.
Alternatively, HEK293 or HCT116 cells were transfected with GFP-GEF-H1 or vector and incubated for 30 min with 10 μM U0126 or DMSO. Immunoprecipitated GEF-H1 was digested on the beads. After washing twice with 300 μl ice-cold phosphate-buffered saline (PBS), beads with bound proteins were eluted in two steps: first by using 60 μl of eluting buffer I (50 mM Tris-HCl [pH 7.5], 2 M urea, and 50 μg/ml trypsin [modified sequencing-grade trypsin; Promega]) and incubating while shaking at 27°C for 30 min and second by adding 25 μl of elution buffer II (50 mM Tris-HCl [pH 7.5], 2 M urea, and 1 mM DTT) twice. Both supernatants were combined and incubated overnight at room temperature.
Samples were alkylated (20 μl iodoacetamide, 5 mg/ml, 30 min in the dark). Then, the reaction was stopped with 1 μl 100% trifluoroacetic acid (TFA), and 100 μl of the sample was immediately loaded into equilibrated handmade C18 StageTips containing octadecyl C18 disks (Supelco, Sigma, United Kingdom). C18 StageTips, spin adaptors, and solvents were prepared as described previously (23). Samples were desalted by washing the StageTips twice with 50 μl of 0.1% TFA and eluted with two lots of 25 μl of 50% acetonitrile (AcN) and 0.1% TFA solution. Final eluates were combined and concentrated until the volume was reduced to 5 μl using a CentriVap concentrator (Labconco, USA). Samples were diluted to obtain a final volume of 12 μl by adding 0.1% TFA and analyzed by MS. The tryptic peptides were analyzed on a Thermo Scientific Q-Exactive mass spectrometer connected to an Ultimate Ultra3000 chromatography system incorporating an autosampler. Five microliters of the resuspended tryptic peptides was loaded onto a homemade column (100-mm length, 75-mm inside diameter [i.d.]) packed with 1.8 μm RepreosilAQ C18 (Dr Maisch, Germany) and separated by an increasing acetonitrile gradient, using a 40-min reverse-phase gradient at a flow rate of 200 nl/min. The mass spectrometer was operated in positive ion mode with a capillary temperature of 220°C, with a potential of 2,000 V applied to the column. Data were acquired with the mass spectrometer operating in automatic data-dependent switching mode, selecting the 12 most intense ions prior to tandem MS (MS/MS) analysis. Mass spectra were analyzed by using the MaxQuant software program. Label-free quantitation was performed using MaxQuant.
RhoA-GTP pulldown assays.
Cells were lysed in ice-cold lysis buffer (20 mM HEPES [pH 7.5], 150 mM NaCl, 1% NP-40, and 2 mM EDTA) supplemented with protease inhibitors (1 mM PMSF, 5 μg/ml leupeptin, 2.2 μg/ml aprotinin, and 2 mM sodium fluoride) and 10 mM MgCl2. Cleared lysates were incubated with 5 μl GST-rhotekin-beads for 30 min at 4°C under end-to-end rotation. The beads were washed, boiled in Laemmli buffer, and Western blotted. The Western blot bands were quantified using the software program ImageJ. Bar graphs represent RhoA-GTP/input RhoA.
Luciferase assay.
HCT116 cells were transfected using Lipofectamine 2000 (Invitrogen, United Kingdom) according to the manufacturer's instructions with GEF-H1 plasmids, 3DA-luciferase, and a Renilla control vector. Forty-eight hours later, the cells were lysed and luciferase activity was measured using a dual-luciferase kit (Promega) according to the manufacturer's instructions. Firefly luciferase activity was normalized by the Renilla output.
Statistical analysis.
All experiments were performed in triplicate. Comparisons of RhoA-GTP levels, phosphopeptide concentrations, invasion, and morphological changes were assessed using nonparametric Mann-Whitney U tests. P values of less than 0.05 were considered significant.
RESULTS
MEK inhibition induces RhoA-GTP independent of Fra-1 expression.
Prolonged inhibition of MEK by pharmacological inhibitors was previously shown to increase RhoA activity in several cell lines harboring mutations that result in a sustained activation of the ERK pathway. This increase in RhoA-GTP was attributed to a decrease in Fra-1 expression caused by inhibition of ERK signaling (24, 25). Reduction of Fra-1 levels increased integrin-mediated RhoA activation and permitted the coupling of RhoA activity to stress fiber formation.
In order to explore if short-term inhibition of the ERK pathway regulates RhoA activity, we used HCT116 cells. HCT116 is a human colorectal adenocarcinoma cell line containing a mutated KRASG13D allele, which encodes a constitutively activated protein leading to the chronic stimulation of downstream signaling. We showed previously that HCT116 cells respond to prolonged MEK inhibition with a decrease in Fra-1 and an increase in RhoA activity (24). Since these observations were obtained after overnight inhibition of MEK, we repeated the experiment at shorter time points in order to assess if acute inhibition of the ERK pathway suffices to augment RhoA-GTP. RhoA-GTP levels increased within half an hour of MEK inhibition as measured by a rhotekin pulldown assay (Fig. 1A). Further RhoA activity peaked after 1 h, which coincided with a decrease in general ERK substrate phosphorylation observed using an antibody that recognizes the phosphorylated ERK consensus sequence PXpSP (Fig. 1B). Fra-1 levels did not decrease within this time period but only after prolonged MEK inhibition (Fig. 1C). These results show that short-term inhibition of the ERK pathway is sufficient to increase RhoA activity without downregulation of Fra-1 protein expression. Hence, we concluded that aside from ERK-dependent RhoA regulation via Fra-1, another acute mechanism must exist. Since ERK substrate dephosphorylation and RhoA activation peak at the same time, we hypothesized that an upstream activator of RhoA may be inhibited by ERK phosphorylation.
Fig 1.

MEK inhibition elevates RhoA-GTP levels and reduces ERK association with GEF-H1. (A) RhoA-GTP was precipitated with rhotekin-GST beads from HCT116 cells treated with 10 μM U0126 for the indicated times. Values are means ± SD. (B) Representative Western blot of RhoA-GTP pulled down with rhotekin-GST beads. Cell lysates and rhotekin pulldown were blotted with indicated antibodies. (C) Western blot analysis of the effects of long-term MEK inhibition on pERK and Fra-1 expression in HCT116 cells. (D) HCT116 cells growing in 10% FCS were incubated with DMSO or U0126 (10 μM) for 1 h. Endogenous ERK1 and ERK2 were immunoprecipitated and Western blotted with the indicated antibodies. A mock immunoprecipitation with protein A beads was used as a control.
ERK associates with GEF-H1.
We previously mapped ERK1 interacting proteins by quantitative mass spectrometry (MS) in PC12 cells (22). Proteins that interacted with ERK1 in an EGF-dependent manner included the Rho exchange factor GEF-H1. To confirm the MS data, we immunoprecipitated endogenous ERK1 from serum-starved and EGF-stimulated PC12 cells and examined GEF-H1 association (data are available upon request). GEF-H1 bound to ERK1 in an EGF-dependent manner. The association increased 5 min after EGF addition and started decreasing at 15 min. Expressing exogenous Flag-tagged GEF-H1, we verified the dynamics of the interaction, with ERK1/2 association peaking at 5 min and subsiding to lower levels within 15 min (data are available upon request), correlating with EGF-induced ERK activation dynamics.
Similarly, ERK and GEF-H1 interacted in HCT116 cells in an activation-dependent manner (Fig. 1D). Both proteins could be coimmunoprecipitated in growing cells, and the interaction was disrupted if MEK and consequently ERK activity was inhibited by U0126, thus confirming the results from PC12 cells.
ERK phosphorylates GEF-H1 on S959.
ERK was recently reported to phosphorylate GEF-H1 on T678 and cause its activation (16). In contrast, our results suggested that ERK signaling restricts GEF-H1 activation. Therefore, we determined whether ERK was phosphorylating additional sites on GEF-H1. We introduced eGFP-tagged wild type (wt) GEF-H1 into HEK293 cells and enriched the protein by immunoprecipitation. GEF-H1 can be expressed to high levels in this cell line. Further, expression in a mammalian expression system ensures that the protein is folded correctly. The immunoprecipitated GEF-H1 was phosphorylated with activated recombinant ERK in the presence of [γ-32P]ATP (data are available upon request). 32P-labeled GEF-H1 was digested with trypsin, and the phosphopeptides were separated by HPLC. The radioactive peptides eluted in two peaks (data are available upon request). Edman degradation of both peptide peaks indicated a phosphorylated amino acid at position 6 (data are available upon request). Using MALDI-MS on the radioactive fractions, we identified the peptide in the major first peak as LSPPHpSPR (data are available upon request), which corresponds to S959 in GEF-H1.
We did not identify T678, previously reported as an ERK site (16), in our in vitro assay. This may have been due to low phosphorylation stoichiometry of T678, since the method used may fail to identify low-abundance radiolabeled peptides. Attempts to detect changes in the phosphorylation status of this site using a generic pTP antibody were ambiguous. Therefore, we decided to quantify the phosphorylation sites by using MS. Similar to Western blotting, MS quantification by itself is not able to determine occupancy rates, but thanks to new analysis tools, it is feasible to determine ratio changes by comparing ion intensities across samples without the need to isotopically label them. Thus, we used a label-free quantitative MS method to monitor intensity changes of GEF-H1 phosphorylation in response to MEK inhibition, both in HEK293 (data are available upon request) and HCT116 cells (Fig. 2A). We readily identified multiple GEF-H1 phosphorylation sites in both cell lines. S959 phosphorylation decreased in either cell line upon treatment with U0126. Conversely, T678 phosphorylation was cell type specific. In HEK293 cells, the peptide phosphorylated on T678 was readily identifiable and represented the most intense ion of all the phosphopeptides detected. In accordance with previous reports, U0126 was able to reduce its phosphorylation. Additionally, we identified that phosphorylation of S695 was inhibited by U0126. Surprisingly, both sites were below the detection limit in HCT116 cells, despite this cell line harboring a hyperactivated MAPK pathway. Thus, we concluded that in HCT116 cells, the last two sites appear not to be phosphorylated or are phosphorylated to a level below the detection limit, implying that S959 is the major MEK-dependent phosphorylation site in HCT116 cells. Based on these data, phosphorylation of T678 and S695 is cell type dependent. It has to be noted that despite complete inhibition of ERK phosphorylation for the duration of 1 h, a substantial amount of GEF-H1 was still phosphorylated on Ser959 in HCT116 and HEK293 cells. The same holds true for Thr678 and Ser695 in HEK293 cells. This suggests that ERK is not the sole S959, T678, and S695 kinase and that contributions from other kinases maintain GEF-H1 phosphorylation levels despite the absence of ERK activity.
Fig 2.

ERK phosphorylation of S959 inhibits GEF-H1 activity. (A) GEF-H1 phosphopeptides and corresponding peptides were identified by MS/MS from HCT116 cells transfected with eGFP–GEF-H1. Forty-eight hours posttransfection, the cells were treated with 10 μM U0126 for 1 h, and GEF-H1 was enriched with anti-eGFP–agarose. Bar graphs represent the normalized intensities of phosphorylated peptides derived from the ion intensities determined by the MaxQuant software program. The bar graphs represent means for three independent experiments ± SEM. ∗, P < 0.05. (B) MCF7 cells were transfected with Flag–GEF-H1 wild type (WT) or mutant Flag–GEF-H1 S959A (S-A), serum starved overnight, pretreated with 10 μM U0126 or DMSO for 30 min, and stimulated with 20 ng/ml EGF for 5 min. Immunoprecipitated Flag–GEF-H1 was Western blotted with antibodies against Flag and the ERK consensus phosphorylation site PXpSP. (C) MCF7 cells were transfected with Flag–GEF-H1 WT, serum starved overnight, pretreated with 10 μM U0126, 2 μM PD0325901, or DMSO for 30 min, stimulated with 20 ng/ml EGF for 5 min, and Western blotted as for panel C. (D) Mammalian GEF-H1 isoforms were aligned by Clustal-Ω. Amino acids which form the D domain (red) and the ERK phosphorylation motif (blue) are conserved across the species. CANFA, Canis familiaris. (E) HCT116 cells were transfected with empty vector (EV), Flag–GEF-H1 wt, and the S959A (S-A) mutant. RhoA-GTP was precipitated with rhotekin-GST beads 48 h posttransfection. The bar graph represents the average of RhoA-GTP levels observed in three independent experiments. Error bars represent SD. (F) Representative Western blots of RhoA-GTP precipitated with rhotekin-GST beads and of total lysates from one of the experiments shown in panel D. (G) HCT116 cells were transfected with Flag–GEF-H1 wt and the S959A (S-A) mutant and incubated with DMSO or 10 μM U0126 for 1 h, 48 h posttransfection. RhoA-GTP was precipitated with rhotekin-GST beads. Cell lysates and rhotekin pulldown were blotted with indicated antibodies. Intensities were quantified by densitometry.
Due to the absence of T678 and S695 phosphorylation, we focused on the characterization of S959, which is the major ERK-regulated GEF-H1 phosphorylation site in HCT116 cells. S959 has been previously shown to be a direct substrate of CDK1 and Aurora B, as well as being required for PAR1b regulation of GEF-H1 activity (12, 14). First, we established that S959 is the major target phosphorylation site for ERK in GEF-H1. GEF-H1 was constitutively phosphorylated under growing conditions in HCT116 cells (Fig. 2A). Phosphorylation was detected using an antibody that selectively recognizes a phosphorylated serine on a perfect ERK consensus motif (PXpSP). This motif is unique to S959 within the GEF-H1 sequence, and the phosphorylation signal could be reduced by about 50% by the U0126 and PD0325901 MEK inhibitors (data are available upon request). These results suggest that GEF-H1 is phosphorylated by ERK on S959 in a MEK-dependent manner. To independently confirm the ERK dependency of S959 phosphorylation in another cell line, we transfected MCF7 cells with wt and S-to-A mutant (S-A) Flag–GEF-H1 (Fig. 2B). In the latter construct, S959 was mutated to an alanine to prevent phosphorylation. The cells were serum starved and stimulated with EGF in the presence or absence of a MEK inhibitor. The phosphorylation of S959 increased within 5 min of EGF treatment. The augmentation was blocked by U0126, confirming the MEK dependency that we observed in HCT116 cells. No signal was detectable if S959 was mutated to alanine, confirming the specificity of the PXpSP antibody and that S959 is phosphorylated in response to acute growth factor stimulation. Further, to demonstrate that the reduction of S959 phosphorylation was not due to an off-target effect of U0126, we repeated the experiment with an alternative MEK inhibitor, PD0325901. Conversely, PD0325901 reduced EGF-induced S959 phosphorylation (Fig. 2C). Having established that ERK binds in an activation dependent manner and phosphorylates GEF-H1 on S959, we wanted to ascertain if GEF-H1 contains any putative ERK/MAPK binding motifs. ERKs can specifically bind to a DEF motif. and MAPKs can bind to D domains (26). We used the software program Scansite 2.0 (27) to predict putative interaction domains and phosphorylation sites of human GEF-H1. We detected both a DEF domain and a D domain in the C terminus of the protein. In addition, only S959 and S955 were predicted to be MAPK substrate sites (data are available upon request). The DEF domain was not conserved across mammals, whereas both the predicted phosphorylation sites and the D domain are conserved (Fig. 2D). Thus, we found a conserved MAPK-binding and phosphorylation site within the vicinity of each other, suggesting that this is the ERK interaction domain which targets the C-terminal phosphorylation of GEF-H1.
Phosphorylation on S959 inhibits GEF-H1 activity.
GEF-H1 has been shown to be regulated by phosphorylation on multiple sites (12–14, 16, 28). Interestingly, phosphorylation on both S885 and S959 was recently reported to inhibit GEF-H1 activity (12, 14). Therefore, we examined the effects of wt GEF-H1 and the S959A (S-A) mutant on RhoA activity in growing HCT116 cells using rhotekin pulldown assays (Fig. 2E and F). While expression of wt GEF H1 elevated RhoA-GTP levels only marginally and insignificantly, the S-A mutation induced significant increases, confirming previous reports that S959 is an inhibitory site (12, 14). Surprisingly, the phosphomimetic S-D mutation also increased RhoA-GTP levels to an extent similar to that with the S-A mutation. The similar effects of the nonphosphorylatable alanine and aspartate mutations indicate that size and negative charge of the carboxyl group are insufficient to mimic the acidity and size of the phosphoric acid residue. In these cases, substitutions by alanine are functionally equivalent to substitutions by phosphomimetic amino acids. Therefore, we conducted subsequent experiments using the S-A mutant. In order to establish if another phosphorylation site on GEF-H1 mediates the MEK-dependent regulation of its activity, we transfected HCT116 cells with the wt and S-A mutant and treated the cells with U0126. As expected, both the S-A mutation and U0126 treatment increased RhoA activity in comparison to that for wt-transfected cells, whereas MEK inhibition had no effect on cells transfected with the S-A mutant (Fig. 2G). This result suggests that S959 is the main regulator of GEF-H1 activity downstream of the MAPK pathway in growing HCT116 cells.
GEF-H1 has been reported to bind to microtubules when inactive. Therefore, we examined whether the inhibitory effect of S959 phosphorylation by ERK may be due to induction of microtubule binding of GEF-H1. We transfected COS-1 cells with the eGFP-tagged wt and S-A mutant and treated the cells with 10 μM U0126 or DMSO. In accordance with previous reports, we detected wt GEF-H1 localized at microtubules and at the plasma membrane. Treatment with U0126 or the S-A mutant changed the localization only marginally to a more diffuse cytoplasmatic localization. The change in localization was only slight (data are available upon request). Therefore, we cannot conclude that the inhibitory effect of S959 phosphorylation is due to induction of microtubule binding.
S959 phosphorylation is crucial for invasiveness.
Efficient cell migration depends on the close spatial and temporal coordination of RhoA, Rac, and CDC42 activities. GEF-H1 can promote directional migration and regulate RhoA activity at the leading edge in moving HeLa cells (11). Additionally, MEK inhibition can impair cell migration, which led us to investigate whether the GEF-H1-mediated cross talk between ERK and RhoA signaling may regulate cell motility. Tumor cells can move into a three-dimensional environment in two basic modes: they can invade in a Rac-dependent mesenchymal or a RhoA-dependent amoeboid fashion (7, 29). Cells can switch between these modes of invasion (29), and high levels of RhoA activity can induce a more rounded amoeboid shape. Since HCT116 cells poorly invade Matrigel, which is used for three-dimensional invasion assays, we used MDA-MB-231 cells to further investigate the roles of GEF-H1 and S959 phosphorylation in three-dimensional motility. MDA-MB-231 cells invade in a mesenchymal and collective fashion. Similar to the HCT116 cell line, MDA-MB-231 cells harbor a mutant K-RASG13D allele, which should reduce the endogenous GEF activity through stimulation of S959 phosphorylation.
In order to test this hypothesis, we knocked down GEF-H1 by siRNAs or inhibited the ERK pathway using a chemical MEK inhibitor and monitored RhoA activity by using rhotekin pulldown experiments (Fig. 3A). Knocking down GEF-H1 approximately halved the basal RhoA activity. Moreover, RhoA-GTP levels increased upon administering the U0126 MEK inhibitor in a time dependent manner. Intriguingly, this increase was completely blocked by GEF-H1 downregulation. Thus, at least half of the RhoA activity in growing MDA-MB-231 cells is due to GEF-H1, while the increase in RhoA-GTP induced by short-term MEK inhibition is completely dependent on GEF-H1. In order to show that the observed increase of RhoA upon MEK inhibition is not due to an off-target effect, we treated MDA-MB-231 cells with an alternative MEK inhibitor and were able to show that both U0126 and PD0325901 increased RhoA-GTP in MDA-MB-231 cells (Fig. 3B). Additionally, we could show that both inhibitors reduce GEF-H1 S959 phosphorylation and ERK binding in MDA-MB-231 cells (Fig. 3C).
Fig 3.

ERK signaling regulates motility and cell morphology via GEF-H1. (A) GEF-H1 was knocked down by siRNA in MDA-MB-231 cells. Forty-eight hours later, cells were treated with a 10 μM concentration of the MEK inhibitor U0126 for the indicated times. RhoA-GTP was precipitated with rhotekin-GST beads. The bar graphs represent means of data from three independent experiments ± SD. Western blots show a representative example. (B) RhoA-GTP was precipitated with rhotekin-GST beads from MDA-MB-231 cells treated with DMSO, 10 μM U0126 or 2 μM PD0325901 for 1 h. Cell lysates and rhotekin pulldown were blotted with indicated antibodies. Intensities were quantified by densitometry. (C) MDA-MB-231 cells were transfected with Flag–GEF-H1 and at 24 h posttransfection were incubated with 10 μM U0126, 2 μM PD0325901, or DMSO in 10% serum for 1 h. Immunoprecipitated Flag–GEF-H1 and lysates were Western blotted with antibodies as indicated. (D) MDA-MB-231 cells were transfected with wt GEF-H1 (WT), the S-A mutant, or empty vector (EV) and subjected to an invasion assay 48 h posttransfection. The expression of the GEF-H1 constructs was monitored by Western blotting. Invasion was quantified by measuring the fluorescence intensity of cells penetrating the Matrigel at ≥45 μm. ∗, P < 0.05. (E) MDA-MB-231 cells were transfected with eGFP, eGFP–GEF-H1 wt, or the S959A (S-A) mutant. RhoA-GTP was precipitated with rhotekin-GST beads 48 h posttransfection. Lysate and rhotekin pulldown were blotted with indicated antibodies. Intensities were quantified by densitometry. (F) Graph representing invasion, normalized to the control, of MDA-MB-231 cells incubated with DMSO, 10 μM U0126, or 2 μM PD0325901. Invasion was quantified by measuring the fluorescence intensity of cells penetrating the Matrigel at ≥45 μm. ∗, P < 0.05 (G) Cells transfected and treated as indicated were stained with calcein acetoxymethyl ester (calcein-AM) and photographed using confocal microscopy at the 30-μm plane of penetration into Matrigel. Morphological changes were quantified using the ImageJ program. Error bars in all panels represent SEM. ∗, P < 0.05.
To test the role GEF-H1 plays in invasion, we overexpressed wt and S-A mutant GEF-H1 in MDA-MB-231 cells by transient transfection. Overexpression of wt GEF-H1 did not significantly affect three-dimensional invasion into Matrigel or RhoA activity, whereas the S-A mutant inhibited invasiveness and increased Rho-GTP levels (Fig. 3D and E; tiled images of sections are available upon request). Likewise, U0126 and PD0325901also severely reduced invasion (Fig. 3F).
Interestingly, GEF-H1 S-A and MEK inhibition not only decreased the invasiveness of MDA-MB-231 cells but also changed the morphology of the remaining invasive cells from an elongated and mesenchymal to a rounded phenotype (Fig. 3G), which is indicative of high RhoA activity. The cells expressing GEF-H1 S-A or treated with MEK inhibitor appeared rounded, very similar to A375-M2 cells, which exhibit the prototypical amoeboid mode of invasion (30), suggesting that GEF-H1 hyperactivity switches invading cells to the amoeboid morphology.
Since the transition from a mesenchymal to an amoeboid phenotype could be induced by either hyperactive GEF-H1 or MEK inhibition, we hypothesized that knocking down GEF-H1 should rescue some effects of MEK inhibition, i.e., promote invasion and/or inhibit the transition to the round phenotype. Inhibition of RhoA activation by TAT-C3 or knockdown of GEF-H1 partially salvaged cell invasion inhibited by U0126 or PD0325901 (Fig. 4A and B; tiled images of sections are available upon request). Additionally, the change in morphology was indeed RhoA dependent, since we could rescue the phenotype with TAT-C3 or GEF-H1 downregulation (Fig. 4C and D). We thus conclude that the shift from mesenchymal to rounded morphology upon MEK inhibition is transmitted via GEF-H1 being dephosphorylated on S959, which leads to an increase in GEF-H1 and consequently RhoA activity, inducing the cell shape changes in a 3D matrix.
Fig 4.

The effects of MEK inhibition on invasion and morphology are specifically mediated by GEF-H1. (A) Graph representing invasion of MDA-MB-231 cells untreated or treated with 10 μM U0126, 2 μM PD0325901, or the Rho inhibitor TAT-C3 and transfected with nontargeting siRNA (NT) or oligonucleotides against GEF-H1. Invasion was quantified by measuring the fluorescence intensity of cells penetrating the Matrigel at ≥45 μm. ∗, P < 0.05. (B) Western blot showing GEF-H1 knockdown efficiency after 48 h. (C) Cells transfected and treated as indicated were stained with calcein-AM and imaged by confocal microscopy at the 0- and 30-μm Matrigel penetration planes. In all panels, error bars represent SEM. (D) Quantitation of elongated and rounded cells of panel C. ∗, P < 0.05.
GEF-H1 is dispensable in mesenchymal invasion but essential in amoeboid invasion.
The reversal of the U0126-induced morphological transition by GEF-H1 depletion suggested that GEF-H1 may promote amoeboid invasion. Thus, we investigated how GEF-H1 knockdown by siRNA influenced mesenchymal and amoeboid invasion. Notably, the reduction in GEF-H1 had opposite effects on mesenchymal and amoeboid invasion. Reduced expression of GEF-H1 in the mesenchymally and collective invading cell line MDA-MB-231 increased its invasive potential by ∼60% (Fig. 5A; tiled images of sections are available upon request). In contrast, the invasion of A375M2 cells, which predominantly invade amoeboidly, was more than halved (Fig. 5B; tiled images of sections are available upon request). This observation indicates that GEF-H1 is dispensable (and maybe even inhibitory) for mesenchymal invasion but necessary for amoeboid invasion. Therefore, we speculated that if we were able to induce a mesenchymal-to-amoeboid transition (MAT) in MDA-MB-231 cells, their invasion should switch from a GEF-H1-independent to a GEF-H1-dependent mechanism. Mesenchymal invasion requires the degradation of surrounding tissue or matrix by the matrix-metalloproteases (MMPs), and evidence suggests that MAT can be induced by inhibiting MMPs (31). When the broad-band MMP inhibitor GM6001 was applied, the cells became rounded and morphologically underwent MAT (Fig. 5C). When we reduced GEF-H1 levels with siRNA and monitored both MDA-MB-231 morphology and motility, GM6001 not only induced MAT but also inhibited invasion (Fig. 5D; tiled images of sections are available upon request). GEF-H1 knockdown in combination with GM6001 did not further inhibit invasion but induced an amoeboid-to-mesenchymal transition (AMT), characterized by the reappearance of elongated, mesenchymal cells in the invading fraction (Fig. 5C). In contrast to MAT induced by MEK inhibition, the knockdown and subsequent AMT did not rescue the invasiveness of the cells, probably because the presence of MMP inhibitors prevented the degradation of the Matrigel required for efficient mesenchymal invasion. The rounding of the cells is apparently independent of the ERK pathway, since GM6001 treatment on its own only marginally reduced ERK phosphorylation in MDA-MB-231 (Fig. 5E).
Fig 5.

GEF-H1 is necessary for amoeboid transition and invasion. (A) Graph representing mesenchymal invasion of MDA-MB-231 cells transfected with siRNA SMARTpools (SP) or single oligonucleotides (#) against GEF-H1 as indicated. Nontargeting siRNA (NT) was used as a control. Invasion was quantified by measuring the fluorescence intensity of cells penetrating the Matrigel at ≥45 μm. ∗, P < 0.05. (B) Graph representing amoeboid invasion of A375M2 cells transfected with siRNA SMARTpools against GEF-H1 as indicated. Invasion was quantified as for panel A. ∗, P < 0.05. (C) Cells transfected and treated as indicated were stained with calcein-AM and imaged by confocal microscopy at the 30-μm Matrigel penetration plane. Morphological changes were quantified in ImageJ. ∗, P < 0.05. (D) Graph representing invasion of MDA-MB-231 cells treated with the MMP inhibitor GM6001 and transfected with siRNA SMARTpools against GEF-H1 as indicated. Invasion was quantified as for panel A. ∗, P < 0.05. (E) MDA-MB-231 cells were seeded onto a thick layer of polymerized 4% collagen and treated with DMSO, 10 μM U0126. or 10 μM GM6001 for 24 h. Cells were lysed, and collagen was separated by centrifugation. Cleared lysates were Western blotted with indicated antibodies.
Taken together, these data confirm our hypothesis that GEF-H1 can drive amoeboid invasion and is pivotal for MAT induced by MMP inhibitors.
DISCUSSION
In migrating cells, leading-edge RhoA activity regulates protrusion dynamics. This highly dynamic process requires a fast response to intrinsic and extrinsic stimuli, as well as coordinated cycling between active and inactive states. Previous studies have revealed a mutual antagonism between Rac and Rho signaling, whose coordination is important for the regulation of cell motility, invasiveness, and mode of invasiveness (for a recent review, see reference 31). High Rac activity promotes the mesenchymal mode of invasion, while high Rho activity facilitates amoeboid movement. The chain of events regulating Rac activation in migration was recently elucidated as a cascade of the adaptor protein NEDD9 recruiting the Rac-specific GEF DOCK3, which activates Rac1 (5). However, the GEF which activates Rho is unknown. Our data suggest that this GEF is GEF-H1.
The known properties of GEF-H1 are consistent with a pivotal role upstream of RhoA in regulating RhoA-mediated protrusion dynamics. GEF-H1 activity is tightly regulated by microtubule binding, and microtubule disassembly by nocodazole induces a robust and fast activation of RhoA via GEF-H1 (19, 28). In addition to microtubule binding, GEF-H1 activity is suppressed intrinsically by its C-terminal domain. Cleavage of this regulatory domain is sufficient to transform GEF-H1 into a potent oncogene (32). The C-terminal regulatory domain contains a multitude of phosphorylation sites and functions as a hub for integrating upstream signaling. Aside from PAK (13, 28), Aurora A, Cdk1/cyclin B, and Par1b (12) phosphorylate GEF-H1 on inhibitory sites in the C terminus. Here we have identified ERK as a kinase that can inhibit GEF-H1 activity by phosphorylating S959 in this C-terminal region in response to acute growth factor stimulation or under growing conditions. Our results contrast with previous reports which have shown a MEK-dependent activation of GEF-H1 via a direct ERK phosphorylation. Although at first sight our results are diametrically opposite, ERK-dependent activation of RhoA and GEF-H1 has been observed downstream of stress signaling, such as tumor necrosis factor alpha (TNF-α) and membrane depolarization. It is plausible that upon activation of stress signals, several pathways are activated which result in the formation of an ERK/Scaffold/GEF-H1 complex, which enables ERK to phosphorylate Thr678 efficiently. Additionally, we have observed that in embryo-derived HEK293 cells, T678, S695, and S959 phosphorylations are reduced upon MEK inhibition, whereas in the colon cancer cell line HCT116, only S959 phosphorylation is detectable and responds to U0126 treatment. These data suggest that the pattern of GEF-H1 phosphorylation is cell type dependent.
MEK inhibition or the GEF-H1 S959A mutation has dramatic effects on cells embedded in a three-dimensional matrix. MDA-MB-231 cells invade into collagen or Matrigel as elongated cells in a mesenchymal fashion. Inhibition of the ERK pathway almost completely prevents the cells from invading toward a gradient of serum supplemented with EGF and more interestingly also causes the cells to change their morphology. They become rounded and resemble cells which invade in an amoeboid fashion. This change in shape is also enforced by expressing a GEF-H1 mutant that cannot be phosphorylated on S959. This switch in morphology and to a lesser extent the deficient motility can be rescued by either suppressing RhoA signaling or reducing endogenous levels of GEF-H1. Taken together, these data reveal a mechanism of how ERK can regulate cell motility and invasiveness by cross talking to the RhoA pathway (Fig. 6). They also suggest that the transition from mesenchymal to amoeboid morphology can be induced by GEF-H1 and that amoeboid invasion in A375M2 cells is dependent on GEF-H1.
Fig 6.
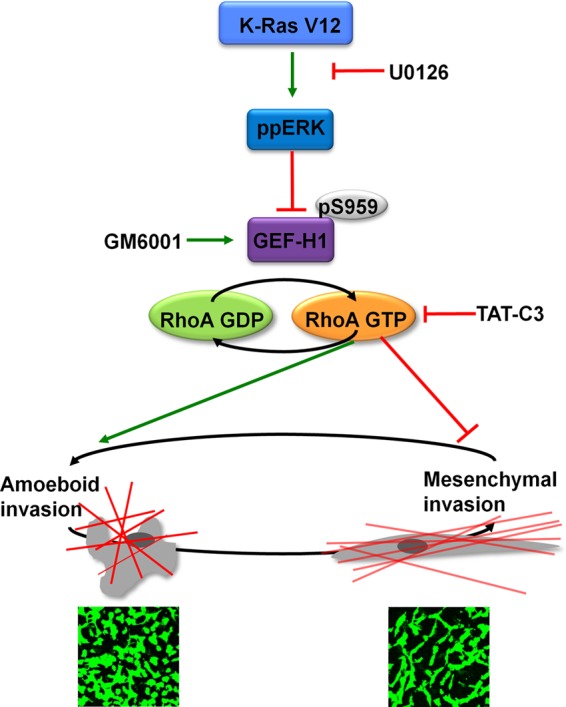
Summary of experimental findings. In K-Ras V12 mutant cell lines, high levels of active ERK inhibit GEF-H1 by phosphorylating the exchange factor at S959. This leads to a decrease in RhoA-GTP levels, which promotes mesenchymal invasion in vitro. In contrast, inhibition of ERK signaling with U0126 stimulates RhoA activity and promotes amoeboid invasion.
GEF-H1 is frequently expressed to high levels in cancers (www.proteinatlas.org), and we were surprised to find that GEF-H1 expression was subduing the invasive potential of MDA-MB-231 cells. A plausible explanation is that the ability of cancer cells to switch between mesenchymal and amoeboid modes of movement may be advantageous for invading surrounding tissue. GEF-H1 can easily be inactivated by high ERK activity when mesenchymal invasion is preferred. On the other hand, high levels of GEF-H1 activity will facilitate transition to an amoeboid type of invasion. This plasticity would enable a cell to maintain high invasive motility regardless of its environment.
ACKNOWLEDGMENTS
We thank Gary Bokoch and Mike Olson for sharing their plasmids and reagents with us, David Matallanas and Dan Croft for helping us with the RhoA-GTP pulldowns, and Natalia Volinsky and Drieke Vandamme for comments.
We thank Cancer Research UK, the European Union FP6 interaction proteome project, and Science Foundation Ireland (grant 06/CE/B1129) for financial support.
Footnotes
Published ahead of print 16 September 2013
REFERENCES
- 1.Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. 2009. Coordination of Rho GTPase activities during cell protrusion. Nature 461:99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurokawa K, Matsuda M. 2005. Localized RhoA activation as a requirement for the induction of membrane ruffling. Mol. Biol. Cell 16:4294–4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pertz O, Hodgson L, Klemke RL, Hahn KM. 2006. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature 440:1069–1072 [DOI] [PubMed] [Google Scholar]
- 4.McGhee EJ, Morton JP, Von Kriegsheim A, Schwarz JP, Karim SA, Carragher NO, Sansom OJ, Anderson KI, Timpson P. 2011. FLIM-FRET imaging in vivo reveals 3D-environment spatially regulates RhoGTPase activity during cancer cell invasion. Small GTPases 2:239–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. 2008. Rac activation and inactivation control plasticity of tumor cell movement. Cell 135:510–523 [DOI] [PubMed] [Google Scholar]
- 6.Rottner K, Hall A, Small JV. 1999. Interplay between Rac and Rho in the control of substrate contact dynamics. Curr. Biol. 9:640–648 [DOI] [PubMed] [Google Scholar]
- 7.Sahai E, Marshall CJ. 2003. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 5:711–719 [DOI] [PubMed] [Google Scholar]
- 8.Pankova K, Rosel D, Novotny M, Brabek J. 2010. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell. Mol. Life Sci. 67:63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS, Friedl P. 2007. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 9:893–904 [DOI] [PubMed] [Google Scholar]
- 10.Rossman KL, Der CJ, Sondek J. 2005. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6:167–180 [DOI] [PubMed] [Google Scholar]
- 11.Nalbant P, Chang YC, Birkenfeld J, Chang ZF, Bokoch GM. 2009. Guanine nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the leading edge. Mol. Biol. Cell 20:4070–4082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Birkenfeld J, Nalbant P, Bohl BP, Pertz O, Hahn KM, Bokoch GM. 2007. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev. Cell 12:699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Callow MG, Zozulya S, Gishizky ML, Jallal B, Smeal T. 2005. PAK4 mediates morphological changes through the regulation of GEF-H1. J. Cell Sci. 118:1861–1872 [DOI] [PubMed] [Google Scholar]
- 14.Yamahashi Y, Saito Y, Murata-Kamiya N, Hatakeyama M. 2011. Polarity-regulating kinase partitioning-defective 1b (PAR1b) phosphorylates guanine nucleotide exchange factor H1 (GEF-H1) to regulate RhoA-dependent actin cytoskeletal reorganization. J. Biol. Chem. 286:44576–44584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshimura Y, Miki H. 2011. Dynamic regulation of GEF-H1 localization at microtubules by Par1b/MARK2. Biochem. Biophys. Res. Commun. 408:322–328 [DOI] [PubMed] [Google Scholar]
- 16.Fujishiro SH, Tanimura S, Mure S, Kashimoto Y, Watanabe K, Kohno M. 2008. ERK1/2 phosphorylate GEF-H1 to enhance its guanine nucleotide exchange activity toward RhoA. Biochem. Biophys. Res. Commun. 368:162–167 [DOI] [PubMed] [Google Scholar]
- 17.Guilluy C, Swaminathan V, Garcia-Mata R, O'Brien ET, Superfine R, Burridge K. 2011. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat. Cell Biol. 13:722–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kakiashvili E, Speight P, Waheed F, Seth R, Lodyga M, Tanimura S, Kohno M, Rotstein OD, Kapus A, Szaszi K. 2009. GEF-H1 mediates tumor necrosis factor-alpha-induced Rho activation and myosin phosphorylation: role in the regulation of tubular paracellular permeability. J. Biol. Chem. 284:11454–11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang YC, Nalbant P, Birkenfeld J, Chang ZF, Bokoch GM. 2008. GEF-H1 couples nocodazole-induced microtubule disassembly to cell contractility via RhoA. Mol. Biol. Cell 19:2147–2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hennigan RF, Hawker KL, Ozanne BW. 1994. Fos-transformation activates genes associated with invasion. Oncogene 9:3591–3600 [PubMed] [Google Scholar]
- 21.von Thun A, Birtwistle M, Kalna G, Grindlay J, Strachan D, Kolch W, von Kriegsheim A, Norman JC. 2012. ERK2 drives tumour cell migration in three-dimensional microenvironments by suppressing expression of Rab17 and liprin-beta2. J. Cell Sci. 125:1465–1477 [DOI] [PubMed] [Google Scholar]
- 22.von Kriegsheim A, Baiocchi D, Birtwistle M, Sumpton D, Bienvenut W, Morrice N, Yamada K, Lamond A, Kalna G, Orton R, Gilbert D, Kolch W. 2009. Cell fate decisions are specified by the dynamic ERK interactome. Nat. Cell Biol. 11:1458–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rappsilber J, Ishihama Y, Mann M. 2003. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75:663–670 [DOI] [PubMed] [Google Scholar]
- 24.Pollock CB, Shirasawa S, Sasazuki T, Kolch W, Dhillon AS. 2005. Oncogenic K-RAS is required to maintain changes in cytoskeletal organization, adhesion, and motility in colon cancer cells. Cancer Res. 65:1244–1250 [DOI] [PubMed] [Google Scholar]
- 25.Vial E, Sahai E, Marshall CJ. 2003. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell 4:67–79 [DOI] [PubMed] [Google Scholar]
- 26.Tanoue T, Nishida E. 2003. Molecular recognitions in the MAP kinase cascades. Cell Signal. 15:455–462 [DOI] [PubMed] [Google Scholar]
- 27.Obenauer JC, Cantley LC, Yaffe MB. 2003. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31:3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zenke FT, Krendel M, DerMardirossian C, King CC, Bohl BP, Bokoch GM. 2004. p21-activated kinase 1 phosphorylates and regulates 14-3-3 binding to GEF-H1, a microtubule-localized Rho exchange factor. J. Biol. Chem. 279:18392–18400 [DOI] [PubMed] [Google Scholar]
- 29.Friedl P. 2004. Prespecification and plasticity: shifting mechanisms of cell migration. Curr. Opin. Cell Biol. 16:14–23 [DOI] [PubMed] [Google Scholar]
- 30.Wilkinson S, Paterson HF, Marshall CJ. 2005. Cdc42-MRCK and Rho-ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat. Cell Biol. 7:255–261 [DOI] [PubMed] [Google Scholar]
- 31.Symons M, Segall JE. 2009. Rac and Rho driving tumor invasion: who's at the wheel? Genome Biol. 10:213. 10.1186/gb-2009-10-3-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitehead I, Kirk H, Tognon C, Trigo-Gonzalez G, Kay R. 1995. Expression cloning of lfc, a novel oncogene with structural similarities to guanine nucleotide exchange factors and to the regulatory region of protein kinase C. J. Biol. Chem. 270:18388–18395 [DOI] [PubMed] [Google Scholar]