

Abstract
Oncogenic transformation in Ewing sarcoma is caused by EWS/FLI, an aberrant transcription factor fusion oncogene. Glioma-associated oncogene homolog 1 (GLI1) is a critical target gene activated by EWS/FLI, but the mechanism by which GLI1 contributes to the transformed phenotype of Ewing sarcoma was unknown. In this work, we identify keratin 17 (KRT17) as a direct downstream target gene upregulated by GLI1. We demonstrate that KRT17 regulates cellular adhesion by activating AKT/PKB (protein kinase B) signaling. In addition, KRT17 is necessary for oncogenic transformation in Ewing sarcoma and accounts for much of the GLI1-mediated transformation function but via a mechanism independent of AKT signaling. Taken together, our data reveal previously unknown molecular functions for a cytoplasmic intermediate filament protein, KRT17, in coordinating EWS/FLI- and GLI1-mediated oncogenic transformation and cellular adhesion in Ewing sarcoma.
INTRODUCTION
Ewing sarcoma is a highly aggressive bone- and soft tissue-associated malignancy that affects children and young adults (1). The vast majority of these tumors are characterized by a t(11;22)(q24;q12) chromosomal translocation, which generates a fusion oncogene, EWS/FLI (2). Persistent expression of EWS/FLI is necessary for maintenance of the transformed phenotype in Ewing sarcoma (3–5). Previous studies demonstrated that Ewing sarcoma tumors have a relatively low frequency of mutations in known oncogenes and tumor suppressors, supporting the concept that EWS/FLI is largely responsible for oncogenic transformation (6, 7). EWS/FLI functions as an aberrant transcription factor and dysregulates the expression of a myriad of target genes (8–10). Over the years, several critical EWS/FLI target genes have been identified, which are all necessary for maintenance of oncogenic transformation in Ewing sarcoma; however, no target gene alone has proven to be sufficient for EWS/FLI-mediated oncogenic transformation (3, 4). These findings highlight the unique biology of Ewing sarcoma and its sole reliance on a single oncogenic transcription factor, EWS/FLI, as the central regulator of a hierarchy of transcriptional networks.
Hedgehog signaling is of critical importance during development in regulating tissue patterning and stem cell maintenance (11, 12). This signaling pathway is inappropriately activated in a diversity of cancers (13–22). GLI1 is a zinc finger transcription factor and is the principal effector of the Hedgehog signaling pathway (11). Previous microarray studies and a recent RNA sequencing (RNA-seq) experiment have identified GLI1 as an EWS/FLI-upregulated target gene in Ewing sarcoma (3, 10, 23). EWS/FLI has been shown to bind and directly activate transcription from the GLI1 promoter (24). Furthermore, loss-of-function approaches and pharmacological inhibition have demonstrated that GLI1 is necessary for EWS/FLI-mediated oncogenic transformation (24–26). These studies highlight the importance of GLI1 in Ewing sarcoma development.
However, the mechanism underlying GLI1-mediated oncogenesis in Ewing sarcoma and the critical transcriptional network of genes regulated by GLI1 to achieve this function were unknown. Here we sought to define the mechanistic role of GLI1 in Ewing sarcoma, and in doing so, we identified a unique target gene, KRT17, that has novel functions in coordinating parallel functions of cellular adhesion and oncogenic transformation.
MATERIALS AND METHODS
Ethics statement.
Patient tumor specimens were used in a deidentified way and were therefore deemed “nonhuman subject research” by the University of Utah Institutional Review Board via protocol IRB_00035414. Animal experiments were performed following approval from the University of Utah Institutional Animal Care and Use Committee.
Constructs and retroviruses.
The luciferase-RNA interference (Luc-RNAi), EWS/FLI-RNAi (EF-2–RNAi), 3×FLAG-tagged EWS/FLI (3×FLAG EWS/FLI), and 3×FLAG NKX2.2 cDNAs were described previously (3, 10, 27). The GLI1 and KRT17 short hairpin RNAs (shRNAs) were designed to target the cDNA and 3′ untranslated region (UTR), respectively, and were cloned into the pMKO.1 retroviral vector. The GLI1 and KRT17 shRNA sequences are provided in Table S1 in the supplemental material. N-terminally 3×FLAG-tagged constructs for GLI1, KRT17, and KRT17 S44A cDNAs were generated and subcloned into the murine stem cell virus (MSCV) retroviral vector (Clontech). A 1-kb KRT17 promoter including the 5′ UTR was cloned into the pGL3 basic vector (Promega), immediately upstream of the luciferase reporter gene. The constitutively active (myristoylated) AKT in the MSCV retroviral vector was described previously (28).
Cell culture.
Ewing sarcoma cell lines (A673, TC-71, TC-32, SK-N-MC, and EWS502) and HEK293 EBNA cells were infected with retrovirus, and polyclonal populations were grown in the appropriate selection media, as previously described (4, 29). 3T5 growth assays were performed by plating 1 × 105 cells per 10-cm tissue culture plates and counting and replating them at the same density every 3 days as previously described (29).
Soft agar and methylcellulose assays.
Soft agar assays were performed as described previously (29). Methylcellulose assays were performed by plating 1 × 105 cells in 2% methylcellulose mixed with an equal volume of appropriate growth medium, as described previously (30).
Quantitative reverse transcriptase PCR.
Total RNA was extracted by using an RNeasy kit (Qiagen). Total RNA from cells was then amplified and detected by using SYBR green fluorescence for quantitative analysis. Normalized fold enrichment was calculated by determining the fold change under each condition relative to the value for the control (either Luc-RNAi or Luc-RNAi reexpressing an empty vector). The data under each condition were then normalized to values for the internal housekeeping control genes GAPDH (the gene for glyceraldehyde-3-phosphate dehydrogenase) and RPL19. Primer sequences used to amplify target genes by quantitative reverse transcriptase PCR (qRT-PCR) are provided in Table S1 in the supplemental material.
Luciferase reporter assays.
A 1-kb promoter region including the 5′ UTR of KRT17 was cloned into the pGL3 basic vector (Promega) immediately upstream of the luciferase reporter gene. Luciferase reporter assays were performed with HEK293 EBNA cells as previously described (31).
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) was performed as previously described (32), by using anti-FLAG M2 magnetic beads (catalog number M8823; Sigma). Briefly, A673 cells expressing a GLI1-RNAi construct and reexpressing either an empty vector or a 3×FLAG GLI1 cDNA were used for the ChIP experiment. Quantitative PCR was performed with KRT17 gene primers that amplify a region ∼150 bp upstream of the transcription start site (TSS) that includes three putative GLI1 binding sites. Primers that amplify regions 5 kb upstream and 5 kb downstream of the TSS were used as negative controls. ALB (31) and gene desert (33) primers were used as normalization controls. Primer sequences used to amplify the KRT17 promoter regions are provided in the Table S1 in the supplemental material.
Xenograft and intratibial injection assays.
A673 or SK-N-MC Ewing sarcoma cells were infected, selected with a control ERG-RNAi or KRT17-RNAi, and injected into the flanks of nude mice at 1 × 106 cells per flank or 2.5 × 105 cells into the tibias of NOD/SCID mice. For the xenograft tumor assay, five mice each were injected subcutaneously with control knockdown cells or KRT17 knockdown cells. Both flanks of each mouse was injected subcutaneously. Under the control conditions, one mouse died due to the anesthesia and was censored from the analysis. Therefore, 8 control knockdown and 10 KRT17 knockdown tumors were measured. For the intratibial tumor assay, five mice were each injected into the right tibia with infected and selected A673 cells; therefore, 5 tumors were measured per group. Twelve mice were injected in the right tibia with infected and selected SK-N-MC cells; therefore, 12 tumors were measured per group. Tumors were measured by using digital calipers, and three-dimensional (3D) tumor volumes were calculated by using the equation (length × width × depth)/2. The mice in each group were sacrificed once their tumors reached size limits of 2 cm for the subcutaneous injection model and 1.5 cm for the intratibial injection model. The data from both assays were plotted as Kaplan-Meier survival curves by using GraphPad Prism.
Antibodies and reagents.
The following antibodies were used for immunodetection: M2 anti-FLAG (horseradish peroxidase [HRP]) (catalog number A8592; Sigma), anti-FLI-1 (catalog number sc-356×; Santa-Cruz), anti-α-tubulin (catalog number CP06; Calbiochem), anti-KRT17 (catalog number ab-53707; Abcam), anti-phospho-AKT (S473) (catalog number 9271S; Cell Signaling), anti-AKT (pan) (catalog number 4691S; Cell Signaling), and anti-GLI1 (catalog number 2643S; Cell Signaling). The isozyme-selective AKT inhibitor (Akti1/2) was obtained from Millipore (catalog number 124017). The inhibitor was used at a final concentration of 2 μM. At this concentration, it inhibits all three forms of AKT (AKT1, AKT2, and AKT3). Cells were treated with the inhibitor for 24 h before they were used for experiments.
Adhesion assay.
Ewing sarcoma cells, infected and selected with different constructs, were seeded at 5 ×105 cells per well in a non-extracellular matrix-coated 24-well plate. Cells were allowed to adhere for 2 h at 37°C and were then processed as previously described (34). Cells that adhered were stained with toluidine blue, and the optical density (OD) was measured at 620 nm, as previously described (34).
Immunofluorescence assays.
Sterile coverslips were coated with 10 μg/ml fibronectin in 12-well plates overnight at 4°C. A total of 75 × 103 cells/well were seeded, allowed to adhere for 24 h, and fixed with 3.7% formaldehyde as described previously (34). Cells were stained with antipaxillin antibody (1:100) for 1 h at 37°C and then with Alexa Fluor-secondary antibody (1:200) and Alexa Fluor-phalloidin and were imaged by using a Zeiss Axioskop2 Mot Plus microscope with a 40× objective, as previously described (34).
RNA sequencing analysis, GSEA, and Venn overlaps.
RNA from A673 cells stably infected and selected for expression of a control Luc-RNAi or the KRT17-RNAi was extracted by using the RNeasy kit (Qiagen) with an on-column DNase digestion protocol. Libraries for deep sequencing were prepared according to the manufacturer's instructions (Illumina) and sequenced on an Illumina Hi-Seq instrument with 50 cycles of single-end reads. Sequences were aligned to human genome build hg19. The USeq analysis package was used to identify differentially expressed genes (http://useq.sourceforge.net/). Significance parameters were set to a 2-fold or 4-fold change in expression and a false discovery rate (FDR) of 0.1 (10%) or 1.0 × 10−10.
Overlaps between the different gene sets were generated by using the VennMaster program (http://www.informatik.uni-ulm.de/ni/mitarbeiter/HKestler/vennm/doc.html). Statistical significance of the overlaps was determined by using chi-square analysis. Gene set enrichment analysis (GSEA) was performed by using the GSEA v2.0.10 program (http://www.broad.mit.edu/gsea/). Functional annotation analysis was performed by Database for Annotation, Visualization, and Integrated Discovery (DAVID) analysis (http://david.abcc.ncifcrf.gov/). Identification of potential direct GLI1 target genes was performed by using Find Individual Motif Occurrences (FIMO) (http://meme.nbcr.net/meme/doc/fimo.html).
RNA sequencing accession number.
Raw data from the GLI1 RNA sequencing experiment can be found under NCBI Short Reads Archive number 121863.
RESULTS
GLI1 is a downstream target of EWS/FLI and is necessary for oncogenic transformation.
Previous studies using loss-of-function approaches identified GLI1 as an upregulated target of EWS/FLI (25, 26). To further demonstrate that GLI1 is specifically regulated by EWS/FLI, we used a retroviral-based stable knockdown/rescue approach in A673 cells (a patient-derived Ewing sarcoma cell line). We found that reduction of EWS/FLI levels resulted in a significant reduction in the GLI1 expression level, which was restored by reexpression of an RNAi-resistant EWS/FLI cDNA (Fig. 1A; see also Fig. S1A in the supplemental material). This result demonstrated that GLI1 is specifically upregulated by EWS/FLI and is not an off-target or other nonspecific RNAi effect. EWS/FLI did not regulate GLI2 or GLI3 (see Fig. S1B). GLI1 is not the sole downstream effector of EWS/FLI-mediated oncogenic transformation because GLI1 expression (Fig. 1B) failed to rescue oncogenic transformation following knockdown of EWS/FLI (Fig. 1C; see also Fig. S1C in the supplemental material).
Fig 1.

GLI1 is upregulated by EWS/FLI and is necessary for oncogenic transformation in Ewing sarcoma cells. (A) Western blot analysis to demonstrate EWS/FLI-mediated activation of GLI1. GLI1 and EWS/FLI levels were assessed in A673 cells infected with a control shRNA (Luc) or an shRNA targeting EWS/FLI, followed by rescue with an empty vector or an RNAi-resistant EWS/FLI cDNA using anti-GLI1 and anti-FLI antibodies. Tubulin was used as a loading control. The asterisk indicates the 3×FLAG-tagged EWS/FLI cDNA that runs slightly higher than endogenous EWS/FLI. (B) Western blot analysis to demonstrate expression of the RNAi-resistant 3×FLAG-tagged EWS/FLI cDNA or 3×FLAG-tagged GLI1 cDNA constructs using an anti-FLAG antibody in A673 cells expressing a control shRNA (Luc) or an EWS/FLI shRNA. Tubulin was used as the loading control. (C) Quantification of colonies formed by A673 cells described above for panel B. Error bars indicate standard deviations of duplicate assays. P values were determined by using Student's t test, comparing all conditions to the control knockdown/empty vector condition (∗∗∗, P ≤ 0.001). (D) Western blot analysis of GLI1 levels in A673 cells infected with a control shRNA (Luc) or an shRNA targeting GLI1, followed by rescue with an empty vector or an RNAi-resistant GLI1 cDNA. Tubulin was used as a loading control. The asterisk indicates the 3×FLAG-tagged GLI1 cDNA that runs slightly higher than endogenous GLI1. (E) Growth assays (3T5) for the A673 cells described above for panel D. Student's t test showed no significant difference in growth curves. (F) Quantification of colonies formed in soft agar by A673 cells expressing a control shRNA (Luc) or a GLI1 shRNA, reexpressing an empty vector or RNAi-resistant GLI1 or NKX2.2 cDNA constructs. Error bars indicate standard deviations of duplicate assays. P values were determined by using Student's t test, comparing all conditions to the control knockdown/empty vector condition (∗∗, P ≤ 0.01).
To test the necessity of GLI1 for Ewing sarcoma oncogenesis, we performed GLI1 knockdown/rescue experiments (Fig. 1D; see also Fig. S1D in the supplemental material). In comparison to a control knockdown (Luc-RNAi), GLI1 knockdown did not affect monolayer growth of cells in tissue culture but significantly reduced colony growth in soft agar (Fig. 1E and F). This is not an “off-target” effect because reexpression of GLI1 rescued the loss of transformation induced by GLI1 knockdown (Fig. 1D to F). Importantly, knockdown or reexpression of GLI1 did not affect EWS/FLI expression (see Fig. S1E and F). These results demonstrated that GLI1 is necessary for maintenance of oncogenic transformation in Ewing sarcoma cells.
GLI1 has been shown to transcriptionally activate NKX2.2 (26). NKX2.2 is a critical target of EWS/FLI that is necessary for oncogenic transformation in Ewing sarcoma (3). We therefore asked if NKX2.2 could rescue GLI1 knockdown-mediated loss of transformation. Interestingly, we found that NKX2.2 (see Fig. S1G in the supplemental material) was unable to rescue the loss of transformation mediated by GLI1 knockdown (Fig. 1F), indicating that other GLI1 target genes are necessary for full oncogenic transformation in Ewing sarcoma.
Determining the transcriptional signature of GLI1 in Ewing sarcoma.
We next sought to identify the full complement of genes regulated by GLI1 in Ewing sarcoma. We performed an RNA-seq experiment in A673 cells, comparing genome-wide transcripts from cells expressing control and GLI1-RNAi constructs (Fig. 2A; see also Table S2 in the supplemental material). VennMaster analysis was used to generate overlaps of the upregulated and downregulated gene sets obtained from the GLI1 RNA-seq and the EWS/FLI RNA-seq experiments (23). Of the 1,796 genes upregulated by EWS/FLI, 327 genes were also upregulated by GLI1 (P = 3.19 × 10−162) (Fig. 2B), and of the 2,227 genes repressed by EWS/FLI, 319 genes were also repressed by GLI1 (P = 1.01 × 10−170) (Fig. 2B), demonstrating that GLI1 contributes significantly to the EWS/FLI transcriptional profile in Ewing sarcoma cells. Using very stringent cutoffs of a 4-fold change and an FDR of 1.0 × 10−10, we limited the list to 86 genes upregulated and 55 genes downregulated by GLI1 (see Table S2). We used this stringent set of genes to perform gene set enrichment analysis (GSEA) against EWS/FLI-regulated genes to better determine the relationship between the EWS/FLI and GLI1 transcriptional profiles. We found that the GLI1-upregulated genes clustered strongly with the most highly upregulated EWS/FLI genes (normalized enrichment score [NES] = 2.0; P < 0.001) and vice versa (NES = −1.8; P < 0.001) (Fig. 2C; see also Fig. S2A in the supplemental material), indicating that GLI1-regulated genes make up a significant portion of the EWS/FLI transcriptional signature. We next performed GSEA and VennMaster analysis of the RNA-seq-based GLI1-regulated genes identified in A673 cells against previously identified microarray-based EWS/FLI-regulated genes in TC71 and EWS502 Ewing sarcoma cells (4) and again found significant correlations and overlaps between GLI1 and EWS/FLI in gene regulation (see Fig. S2B to E). We also validated a subset of GLI1-regulated genes identified in the RNA-seq data by quantitative reverse transcriptase PCR (qRT-PCR) (see Fig. S2F).
Fig 2.

GLI1 regulates a significant portion of the EWS/FLI transcription profile in Ewing sarcoma cells. (A) Heat map representation of the rank-ordered expression profiling data from GLI1 RNA-seq analysis. Genes were ranked by mean deviations of the log-transformed fragments per kilobase per million mapped reads. The columns for each shRNA represent three independent biological replicates. Each row represents a different gene. The top 15 upregulated (left) and downregulated (right) genes from the GLI1 RNA-seq analysis are shown. (B) Venn diagram representations of the overlap between the EWS/FLI and the GLI1 transcription profiles, both generated by RNA-seq analysis of A673 cells. The chi-square-determined P values are indicated. (C) Gene set enrichment analysis (GSEA) using the EWS/FLI-regulated genes in A673 cells (RNA-seq) as the rank-ordered data set and the 86 Gli1-upregulated and 55 GLI1-downregulated gene sets (RNA-seq). The normalized enrichment scores (NES) and P values are shown. (D) Top 10 categories identified by DAVID functional analysis of the GLI1-upregulated and -downregulated gene sets. The log-transformed enrichment scores for each category are indicated on the x axis.
To gain further insight into the functional significance of the differentially expressed genes from the GLI1 RNA-seq analysis, we used the functional annotation tools from the Database for Annotation, Visualization, and Integrated Discovery (DAVID). We found that the most significant classes among the GLI1-upregulated genes corresponded to neuronal development and cell cycle regulation (Fig. 2D), which is consistent with the well-studied role of GLI1 in neuronal development (35) and its ability to transcriptionally regulate cell cycle proteins (36). Interestingly, neuronal features have previously been noted for Ewing sarcoma (37, 38), and thus, the RNA-seq data suggest that GLI1 and its downstream target genes may contribute to the neuronal phenotype of Ewing sarcoma. Among the downregulated gene set, the most significant classes were related to signaling and membrane activity (Fig. 2D).
Identification of KRT17 as a direct downstream target of GLI1.
To further investigate the role of GLI1 target genes identified from the RNA-seq analysis, we focused on KRT17, which is the second most upregulated GLI1 target gene (Fig. 2A) and is also regulated by EWS/FLI (Fig. 2B and C; see also Fig. S2A to E in the supplemental material). qRT-PCR analysis demonstrated that both GLI1 and EWS/FLI upregulate KRT17 in multiple Ewing sarcoma cell lines (Fig. 3A to C). These results demonstrate that KRT17 is an upregulated target of GLI1 in Ewing sarcoma.
Fig 3.

KRT17 is regulated by GLI1 in multiple Ewing sarcoma cell lines. (A) Validation of KRT17 as an EWS/FLI and GLI1 target gene. Shown are data for qRT-PCR analysis of KRT17 in A673 cells infected with a control shRNA (Luc), an EWS/FLI shRNA, or a GLI1 shRNA, followed by rescue with an empty vector, an RNAi-resistant EWS/FLI cDNA, or a GLI1 cDNA construct. Error bars indicate standard deviations. P values were determined by using Student's t test, comparing all conditions to the control knockdown/empty vector condition (∗∗, P ≤ 0.01; ∗∗∗, P ≤ 0.001). (B) Western blot analysis of cells described above for panel A, using KRT17, EWS/FLI, and GLI1 antibodies. Tubulin was used as the loading control. The red asterisks indicate the 3×FLAG-tagged EWS/FLI and GLI1 cDNAs. (C) qRT-PCR validation of KRT17 as a GLI1 target gene in multiple patient-derived Ewing sarcoma cell lines (TC71, TC32, SK-N-MC, and EWS502). Cells were infected with a control shRNA (Luc) or a GLI1 shRNA. GLI1 and KRT17 mRNA levels were analyzed. Error bars indicate standard deviations. P values were determined by using Student's t test, comparing all conditions to the control knockdown (Luc-shRNA) (∗∗, P ≤ 0.01; ∗∗∗, P ≤ 0.001). (D) ChIP of 3×FLAG GLI1 at the KRT17 promoter in A673 cells expressing a GLI1-RNAi and reexpressing an empty vector or 3×FLAG GLI1 cDNA. An anti-FLAG antibody was used to chromatin immunoprecipitate 3×FLAG GLI1. The transcriptional start site (TSS) and a region ∼150 bp upstream of the TSS with significant GLI1 binding are indicated. The level of enrichment for 3×FLAG GLI1 at the KRT17 promoter in cells reexpressing the 3×FLAG GLI1 cDNA is plotted as normalized fold enrichment compared to the enrichment in the empty-vector-reexpressing cells, with the fold enrichment for each sample being compared to the average enrichment at two negative-control genes, ALB and a gene desert region in the genome. Enrichment of 3×FLAG GLI1 at regions 5 kb upstream and 5 kb downstream of the FIMO-identified binding sites were used as negative controls to further demonstrate binding specificity for GLI1 at the KRT17 promoter. The error bars indicate standard deviations of a representative experiment. (E) Luciferase reporter assay with HEK293 EBNA cells cotransfected with a 1-kb KRT17 promoter region upstream of luciferase or a control vector (that does not contain the KRT17 promoter) and an empty vector or increasing concentrations of the GLI1 cDNA. Relative luciferase activity is the ratio of firefly luciferase activity to Renilla luciferase activity (to control for transfection efficiency). The red asterisks indicate potential GLI1 binding sites in the KRT17 promoter. The error bars indicate standard deviations. P values were determined by using Student's t test, comparing all GLI1 cDNA-transfected conditions to the vector-transfected conditions (∗∗, P ≤ 0.01).
The GLI1 RNA-seq analysis does not distinguish direct from indirect targets. GLI1 is a well-studied transcription factor, and previous work identified and characterized a conserved 10-bp motif as the preferred binding site (GACCACCCAC/A) for GLI1 on-target gene promoters (39, 40). To predict potential direct targets of GLI1, we used Find Individual Motif Occurrences (FIMO) (41), by combining a previously reported weighted matrix for binding affinity and a weighted matrix for the activation potential of GLI1 at the 10-bp motif, to search for genes in our RNA-seq data set that had a significant match (P value cutoff of 1.0 × 10−5) to the known GLI1 binding motif. We identified 23 potential direct upregulated and 12 direct downregulated targets of GLI1 (see Fig. S3 in the supplemental material). Interestingly, KRT17 was one of the potential direct targets of GLI1 (see Fig. S3). Directed chromatin immunoprecipitation assays demonstrated significant GLI1 binding to the KRT17 promoter-proximal region (Fig. 3D). Luciferase reporter assays demonstrated a dose-dependent increase in luciferase activity from a 1-kb KRT17 promoter region with increasing concentrations of GLI1 cDNA (Fig. 3E). These data indicate that KRT17 is likely a direct upregulated target of GLI1.
KRT17 is expressed in Ewing sarcoma cell lines and primary tumor samples.
Western blot analysis revealed that the KRT17 protein is expressed at detectable levels in all Ewing sarcoma cell lines tested albeit at various levels (Fig. 4A). qRT-PCR analysis revealed a significant positive correlation between GLI1 and KRT17 expression in a panel of Ewing sarcoma cell lines (R2 = 0.9) (see Fig. S4A in the supplemental material). To validate this finding in primary Ewing sarcoma tumor samples, we performed maximum threshold cycle RT-PCR with five independent Ewing sarcoma primary tumors, which revealed that KRT17 RNA is expressed in all tumor samples tested (Fig. 4B). We next performed qRT-PCR analysis to correlate expression levels of EWS/FLI and GLI1, and GLI1 and KRT17, in this limited set of primary tumor samples (n = 5). Interestingly, we identified a significant correlation between EWS/FLI and GLI1 expression (R2 = 0.984) (Fig. 4C) and between GLI1 and KRT17 expression (R2 = 0.975) (Fig. 4C), highlighting the significance of the hierarchy of transcriptional regulation between EWS/FLI and GLI1 and between GLI1 and KRT17. To extend our findings further, we next analyzed the expression levels of GLI1 and KRT17 in a publically available microarray data set (42) containing 20 EWS/FLI-positive Ewing sarcoma tumor samples. Again, we identified a moderate correlation (R2 = 0.577) between GLI1 and KRT17 expression in a larger number of Ewing sarcoma tumors (Fig. 4D). These data indicate that GLI1 positively regulates KRT17 expression in Ewing sarcoma cell lines and primary tumors.
Fig 4.

KRT17 is expressed in Ewing sarcoma cell lines and primary tumors. (A) Western blot analysis of KRT17 expression in multiple patient-derived Ewing sarcoma cell lines (A673, TC71, TC32, SKNMC, SKES1, and EWS502). Tubulin was used as the loading control. (B) Maximum threshold cycle RT-PCR analysis of KRT17 transcript levels in five independent Ewing sarcoma patient tumor samples compared to KRT17 transcript levels in A673 cells infected with a control shRNA (Luc) or a KRT17 shRNA as well as a water negative control. (C) Linear regression analysis to determine the correlation between EWS/FLI and GLI1 expression and between GLI1 and KRT17 expression in the patient tumor samples described above for panel B. The expression levels were determined by qRT-PCR and plotted as a percentage of the expression level of the housekeeping gene (GAPDH). The R2 value is indicated. (D) Linear regression analysis to determine the correlation between GLI1 and KRT17 expression in a publically available microarray data set (Ohali sarcoma data set) consisting of 20 Ewing sarcoma tumor samples (42). The mRNA expression values for GLI1 and KRT17 in each tumor sample were obtained through Oncomine (https://www.oncomine.org/) and plotted as a percentage of the value for the housekeeping gene (GAPDH). The R2 value is indicated.
KRT17 is necessary for oncogenic transformation in vitro and in vivo.
KRT17 is a cytoplasmic intermediate filament protein (43) that is overexpressed in several cancers (44–50). High KRT17 expression levels correlate with poor prognosis in breast, pancreatic, and gastric adenocarcinomas (51–53). Basal cell carcinomas, which are associated with aberrant hedgehog signaling and, in turn, high GLI levels, express high levels of KRT17, which promotes tumor growth by modulating the immune response (44). However, it is unknown whether KRT17 plays a more direct role in oncogenic transformation.
To determine if KRT17 is involved in oncogenic transformation in Ewing sarcoma, we performed knockdown/rescue of KRT17 in A673, EWS502, and SK-N-MC Ewing sarcoma cells. We found that knockdown of KRT17 had no effect on cell growth in tissue culture but significantly reduced colony formation in soft agar (Fig. 5A to C; see also Fig. S5A to C in the supplemental material). Furthermore, reexpression of KRT17 in knockdown cells (see Fig. S5E) restored their ability to form colonies in soft agar, demonstrating a specific function of KRT17 in maintaining the transformed phenotype of Ewing sarcoma cells (Fig. 5C). Importantly, qRT-PCR analysis of endogenous KRT17 transcript levels demonstrated that the KRT17 knockdown was maintained even in the KRT17 cDNA rescue samples (see Fig. S5F), suggesting that the rescue of oncogenic transformation was not merely due to a loss of KRT17-RNAi. KRT17 knockdown had no effect on oncogenic transformation in a non-Ewing sarcoma cell line, HEK293 EBNA (human embryonic kidney cells) (see Fig. S5D), suggesting that KRT17 is specifically required for oncogenic transformation in Ewing sarcoma.
Fig 5.

KRT17 is necessary for GLI1-mediated oncogenesis in Ewing sarcoma. (A) Western blot analysis of KRT17 in A673 cells infected with a control shRNA (Luc) or two different shRNA constructs targeting KRT17. Tubulin was used as the loading control. (B) Growth assays (3T5) for the A673 cells described above for panel A. Student's t test showed no significant difference in growth curves. (C) Quantification of colonies formed in methylcellulose by A673 cells expressing a control shRNA (Luc) or two different KRT17 shRNAs, reexpressing an empty vector or an RNAi-resistant KRT17 cDNA construct. Error bars indicate standard deviations of duplicate assays. P values were determined by using Student's t test, comparing all conditions to the control knockdown/empty vector condition (∗∗, P ≤ 0.01). (D to F) Survival curves for immunodeficient mice subjected to subcutaneous or intratibial injections with A673 cells or SK-N-MC cells expressing a control shRNA (ERG) or a KRT17 shRNA. Five mice and 12 mice were used per condition for the A673 cells and the SK-N-MC cells, respectively. For the subcutaneous model, both flanks of each mouse were injected subcutaneously. Under the control conditions, one mouse died due to the anesthesia and was censored from the analysis. Therefore, 8 and 10 tumors were measured for the control knockdown and KRT17 knockdown groups, respectively. For the intratibial model, the right tibia of each mouse was injected, and therefore, 5 tumors for the A673 group and 12 tumors for the SK-N-MC group were measured for both the control (ERG) knockdown and the KRT17 knockdown groups. The mice in each group in the subcutaneous model were sacrificed once their tumors reached a size limit of 2 cm3. The mice in each group in the intratibial model for both A673 and SK-N-MC cells were sacrificed once their tumors reached a size limit of 1.5 cm3. Percent survival was plotted for both models as Kaplan-Meier survival curves by using GraphPad Prism. The P values determined by log-rank test (Mantel-Cox test) using GraphPad Prism are indicated. (G) Western blot analysis of control (ERG) shRNA- or KRT17 shRNA-expressing tumors from the subcutaneous injection model described above for panel D. KRT17 levels in the tumors were compared to levels in the parental A673 cells expressing either the control shRNA or KRT17 shRNA, used to inject mice. Tubulin was used as the loading control. (H) Quantification of colonies formed in methylcellulose by A673 cells expressing a control shRNA (Luc) or a GLI1 shRNA and reexpressing the empty vector, 3×FLAG-tagged GLI1, or 3×FLAG-tagged KRT17 cDNA constructs. Error bars indicate standard deviations of duplicate assays. The P value was determined by using Student's t test, comparing the GLI1 knockdown/empty vector conditions to the control knockdown/empty vector conditions (∗∗∗, P ≤ 0.001).
We next used two in vivo tumor models, a subcutaneous model and an orthotopic intratibial model, to evaluate the role of KRT17 in tumor growth in vivo. We noted a significant improvement in overall survival of immunocompromised mice injected with KRT17 knockdown A673 cells compared to those with control (ERG) knockdown cells in both in vivo models (Fig. 5D and E). We extended these findings using SK-N-MC Ewing sarcoma cells in the orthotopic intratibial model and again noted a significant improvement in overall survival in the KRT17 knockdown group compared to the control (ERG) knockdown group (Fig. 5F). In the tumors that did form in mice injected with KRT17 knockdown cells, we noted that the knockdown effect was lost in tumors that grew actively, while the slow-growing (indolent) tumors from the opposite flanks of a few mice still maintained the KRT17 knockdown (Fig. 5G; see also Fig. S5G in the supplemental material), indicating that KRT17 is necessary for more aggressive tumor growth in vivo.
To evaluate if KRT17 was a critical target gene downstream of GLI1, we performed anchorage-independent colony-forming assays with A673 cells following control or GLI1 knockdown and reexpression of an empty vector or the GLI1 or KRT17 cDNA construct (see Fig. S5H in the supplemental material). Surprisingly, expression of the KRT17 cDNA rescued the GLI1 knockdown-mediated loss of transformation (Fig. 5H). In cells harboring GLI1-RNAi and reexpressing the KRT17 cDNA, maintenance of GLI1 knockdown and a lack of rescue of GLI1 target genes were demonstrated by qRT-PCR analysis (see Fig. S5I and J), indicating that the rescue of oncogenic transformation was not due to reexpression of GLI1 when KRT17 was expressed. Importantly, knockdown or reexpression of KRT17 did not have any effect on EWS/FLI or GLI1 expression (see Fig. S5K to N in the supplemental material). Taken together, these results demonstrate that KRT17 is necessary for maintaining the transformed phenotype of Ewing sarcoma cells both in vitro and in vivo and that KRT17 is a critical target gene downstream of GLI1 that contributes significantly to GLI1-mediated maintenance of oncogenic transformation in Ewing sarcoma.
KRT17-mediated activation of AKT signaling is necessary and sufficient to regulate cellular adhesion in Ewing sarcoma.
KRT17 is known to regulate protein synthesis and epithelial cell growth by inducing phosphorylation and activation of the AKT protein (54). We therefore asked whether KRT17 regulated AKT phosphorylation downstream of GLI1 and if this genetic interaction was necessary for KRT17 function in Ewing sarcoma. GLI1 knockdown significantly reduced AKT phosphorylation levels in A673 cells, and this effect was rescued by GLI1 or KRT17 reexpression (Fig. 6A) but not by expression of the KRT17 S44A mutant (Fig. 6B), a previously described mutant that fails to induce phosphorylation of AKT (54, 55), demonstrating that KRT17 is the critical mediator of AKT phosphorylation downstream of GLI1.
Fig 6.

KRT17 is necessary and sufficient for AKT phosphorylation-mediated cellular adhesion in Ewing sarcoma cells. (A) Western blot analysis of A673 cells infected with a control shRNA (Luc) or the GLI1 shRNA and reexpressing the empty vector, GLI1, or KRT17 cDNA constructs. The protein lysates from these cells were probed with phosphorylated AKT (S473), total AKT, and FLAG antibodies. Tubulin was used as a loading control. (B) Western blot analysis of A673 cells infected with a control shRNA (Luc) or a KRT17 shRNA and reexpressing an empty vector, the KRT17 wild-type, or an S44A mutant KRT17 cDNA construct. The protein lysates from these cells were probed with phosphorylated AKT (S473), total AKT, and KRT17 antibodies. The asterisks indicate 3×FLAG-tagged KRT17 and 3×FLAG-tagged S44A KRT17 cDNA constructs, which run slightly higher than endogenous KRT17. Tubulin was used as a loading control. (C) Immunofluorescence images of A673 cells infected with control shRNA (Luc), KRT17 shRNA, or EWS/FLI shRNA stained for focal adhesions (paxillin antibody) and for actin filaments (phalloidin). Arrowheads indicate paxillin-rich focal adhesions. (D) Adhesion assay with A673 cells infected with a control shRNA (Luc) or a KRT17 shRNA and reexpressing the empty vector, KRT17 wild-type, or S44A mutant KRT17 cDNA constructs. Error bars indicate standard deviations. P values were determined by using Student's t test, comparing all conditions to the control knockdown/empty vector conditions (∗∗∗, P ≤ 0.001). (E) Western blot analysis of A673 cells infected with a control shRNA (Luc) or a KRT17 shRNA and reexpressing an empty vector, KRT17 cDNA, or a constitutively active (myristoylated) form of AKT. The protein lysates from these cells were probed with KRT17, phosphorylated AKT (S473), and total AKT antibodies. Tubulin was used as a loading control. (F) Adhesion assay with the A673 cells described above for panel E. Error bars indicate standard deviations. The P value was determined by using Student's t test, comparing the KRT17 knockdown/empty vector conditions to the control knockdown/empty vector conditions (∗∗∗, P ≤ 0.001). (G) Adhesion assay with A673 cells treated with the selective AKT inhibitor or the vehicle control for 24 h. Error bars indicate standard deviations. The P value was determined by using Student's t test, comparing the inhibitor-treated conditions to the vehicle-treated conditions (∗∗∗, P ≤ 0.001).
To characterize the functional significance of KRT17-mediated activation of AKT signaling, we performed immunofluorescence studies on A673 cells expressing reduced levels of KRT17. The paxillin protein is a well-characterized marker of focal adhesions in cells (34). We therefore used paxillin staining as a measure of the adhesive capability of Ewing sarcoma cells. Interestingly, we noted a significant decrease in staining for paxillin protein in cells expressing reduced KRT17 levels in comparison to control cells (Fig. 6C). As a control, EWS/FLI knockdown cells expressed higher levels of paxillin (Fig. 6C), as noted previously (34). To test if KRT17 is directly involved in regulating cellular adhesion in Ewing sarcoma cells, we performed cellular adhesion assays with KRT17 knockdown cells reexpressing wild-type KRT17 or the S44A mutant. Interestingly, wild-type KRT17, but not the S44A mutant, rescued basal levels of cellular adhesion in Ewing sarcoma cells (Fig. 6D).
To directly test the contribution of active AKT signaling to cellular adhesion mediated by KRT17, we took two complementary approaches: (i) a genetic approach by expressing a constitutively active form of AKT (myristoylated AKT) (28) and (ii) a pharmacological approach using a selective AKT inhibitor (Akti1/2; Millipore). We found that expression of the constitutively active AKT following knockdown of endogenous KRT17 (Fig. 6E) phenocopied KRT17-mediated cellular adhesion (Fig. 6F). We also found that selective inhibition of AKT by the pharmacological inhibitor significantly decreased the basal levels of cellular adhesion in Ewing sarcoma cells, similar to levels achieved with KRT17 knockdown (Fig. 6G). These data clearly define the genetic and functional interaction between GLI1, KRT17, and active AKT signaling in regulating cellular adhesion in Ewing sarcoma cells.
KRT17-mediated oncogenic transformation is independent of the AKT pathway.
AKT signaling is frequently activated in cancer (56). We therefore asked whether AKT phosphorylation was necessary for KRT17-mediated oncogenic transformation in Ewing sarcoma. Interestingly, the KRT17 S44A mutant, which failed to phosphorylate AKT (Fig. 6B), retained the ability to rescue KRT17 knockdown-mediated loss of transformation to an extent comparable to that of wild-type KRT17 (Fig. 7A), suggesting that oncogenic transformation by KRT17 is independent of the AKT pathway.
Fig 7.
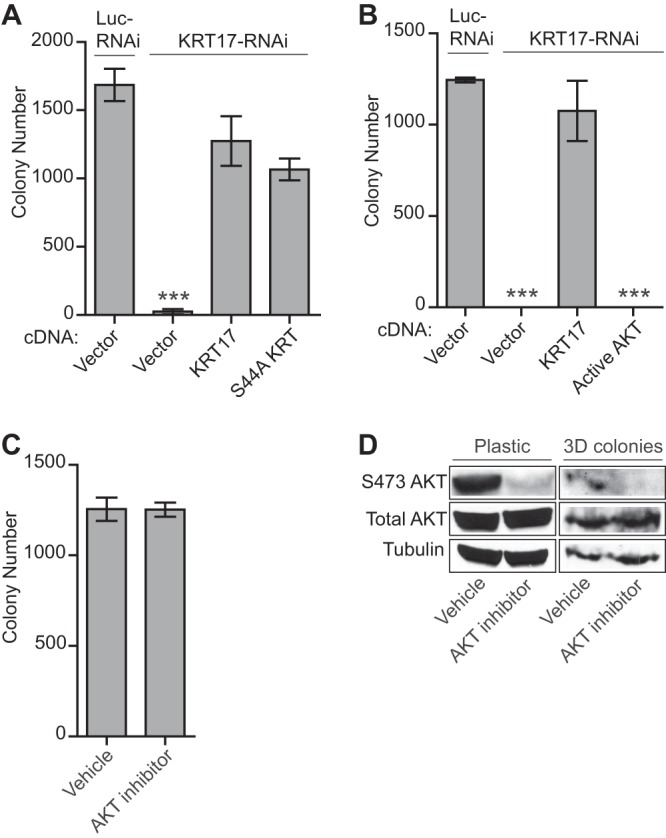
KRT17-mediated oncogenic transformation is independent of AKT signaling. (A) Quantification of colonies formed in methylcellulose by A673 cells infected with a control shRNA (Luc) or a KRT17 shRNA and reexpressing the empty vector, the KRT17 wild-type, or the S44A mutant KRT17 cDNA construct. Error bars indicate standard deviations of duplicate assays. The P value was determined by using Student's t test, comparing the conditions of a KRT17 knockdown rescued with an empty vector to those of the control knockdown rescued with an empty vector (∗∗∗, P ≤ 0.001). (B) Quantification of colonies formed in methylcellulose by A673 cells infected with a control shRNA (Luc) or a KRT17 shRNA and reexpressing an empty vector, KRT17 cDNA, or a constitutively active (myristoylated) form of AKT. Error bars indicate standard deviations of duplicate assays. P values were determined by using Student's t test, comparing all conditions to the control knockdown/empty vector conditions (∗∗∗, P ≤ 0.001). (C) Quantification of colonies formed in methylcellulose by A673 cells treated with a selective AKT inhibitor or a vehicle control for 24 h. Error bars indicate standard deviations of duplicate assays. (D) Western blot analysis of the A673 cells described above for panel C. Protein lysates from treated cells and from 3D colonies at the end of the anchorage-independent colony-forming assay were probed with phosphorylated AKT (S473) and total AKT antibodies. Tubulin was used as the loading control. The total amount of protein obtained from the 3D colonies was much smaller than that obtained from cells grown and treated on plastic.
To directly test the contribution of active AKT signaling to Ewing sarcoma oncogenesis, we used the constitutively active form of AKT (myristoylated AKT) (28) and the selective AKT inhibitor (Akti1/2; Millipore). The constitutively active form of AKT failed to rescue the loss of oncogenic transformation following KRT17 knockdown, even though high levels of AKT phosphorylation were achieved (Fig. 6E and 7B). Pharmacological inhibition of AKT phosphorylation also had no effect on oncogenic transformation of Ewing sarcoma cells (Fig. 7C). Maintenance of AKT inhibition in the anchorage-independent environment was ensured by assessing the phosphorylation status of AKT in the colonies that did form (Fig. 7D). These results suggest that AKT signaling is completely dispensable for the anchorage-independent colony-forming phenotype of Ewing sarcoma cells. Taken together, our data highlight a central role for KRT17 downstream of GLI1 in coordinating two important, but independent, phenotypes of cancer cells, oncogenic transformation and cellular adhesion.
DISCUSSION
In this work, we identified KRT17 as an upregulated target of EWS/FLI and GLI1 in Ewing sarcoma. Furthermore, we unraveled novel functional roles for KRT17 in regulating oncogenic transformation and cellular adhesion in Ewing sarcoma: KRT17 induces AKT signaling to mediate cellular adhesion, while KRT17 modulates oncogenic transformation (as measured by colony formation under anchorage-independent conditions and by xenograft tumor formation) independent of AKT signaling. To our knowledge, this is the first demonstration of such a coordinating function for an intermediate-filament protein for these cancer-relevant phenotypes.
Hyperactive AKT signaling is characteristic of several cancers (56). Interestingly, oncogenic transformation mediated by KRT17 is independent of the AKT signaling pathway in Ewing sarcoma. Consequently, inhibition of the AKT signaling pathway had no impact on growth or oncogenic transformation of Ewing sarcoma cells. These observations suggest that cooperating molecules or pathways necessary for AKT to mediate oncogenic transformation in other cancers may be absent in Ewing sarcoma cells. Indeed, polymerization of KRT17 with KRT5/6α/6β is required to form stable cytoskeletal structures (43), and mutations in KRT17 or its partner KRT5, -6α, or -6β result in human genetic diseases. We inspected our global transcriptional profiling data sets and found very low, if any, expression for KRT5, -6α, or -6β in Ewing sarcoma cells, suggesting that KRT17 functions in a novel capacity to regulate oncogenic transformation. This also indicates that in addition to regulating AKT signaling, KRT17 might impinge on multiple critical growth factor signaling pathways in the context of Ewing sarcoma cells, all of which together contribute to the transformed phenotype. Further studies are ongoing to identify the precise mechanism by which KRT17 regulates oncogenic transformation in Ewing sarcoma.
Importantly, we demonstrate in this report that KRT17-mediated AKT phosphorylation is necessary and sufficient for regulating cellular adhesion. There is a growing body of evidence indicating that alterations in the adhesion properties of cells play a pivotal role in the development and progression of cancer (57). Expression of EWS/FLI has profound effects on adhesion and cytoskeletal architecture of Ewing sarcoma cells (34). In support of this are transcriptional profiling data for EWS/FLI in Ewing sarcoma cells that reveal a significant downregulation of adhesion and cytoskeletal proteins, suggesting that Ewing sarcoma cells have low basal levels of cellular adhesion (10). Therefore, EWS/FLI is necessary for oncogenic transformation in Ewing sarcoma, and EWS/FLI globally represses adhesion of Ewing sarcoma cells. In fact, cellular adhesion is dramatically increased upon EWS/FLI knockdown in Ewing sarcoma cells (34). GLI1 is a direct activated target of EWS/FLI that is necessary for oncogenic transformation. Transcription profiling data suggest that GLI1-regulated genes in Ewing sarcoma function to both activate and repress cellular adhesion. Therefore, as we move lower in the hierarchy of transcriptional regulation away from EWS/FLI, we start segregating genes that differentially regulate oncogenic transformation versus cellular adhesion. Our data suggest that the direct GLI1 target gene KRT17 functions to positively regulate oncogenic transformation in the same way as EWS/FLI and GLI1. However, contrary to the role of EWS/FLI in significantly repressing cellular adhesion or the role of GLI1 in activating as well as repressing genes that regulate cellular adhesion, KRT17 functions to positively regulate cellular adhesion in Ewing sarcoma.
Ewing sarcoma is a highly metastatic tumor, and in the absence of chemotherapy, the vast majority of patients die from metastatic disease, suggesting that most patients have micrometastases at presentation (58, 59). In support of this, circulating tumor cells can be identified in Ewing sarcoma patients (60). These observations suggest that, in contrast to epithelial cell cancers, which are thought to follow a multistep process for metastasis, a mesenchymal tumor such as Ewing sarcoma may display metastatic dissemination of tumor cells early in the disease process (34). The ability of Ewing sarcoma tumor cells to readily disseminate clearly highlights the importance of regulating adhesion levels in these tumors. Although EWS/FLI largely inhibits cellular adhesion proteins likely to promote metastatic dissemination in Ewing sarcoma, these tumor cells still need to maintain low basal levels of adhesion to be able to form tumors and to adhere to and colonize secondary sites of metastasis. Our data suggest that KRT17 is one of the critical cytoskeletal proteins downstream of EWS/FLI and GLI1 that is necessary to maintain basal levels of cellular adhesion in Ewing sarcoma by activating the AKT signaling pathway. Interestingly, the AKT signaling pathway was previously shown to activate focal adhesion kinase (FAK)-dependent adhesion in cancer (61), further supporting our finding that AKT signaling regulates cellular adhesion in Ewing sarcoma.
Our data in this study suggest that AKT signaling uncouples KRT17-mediated cellular adhesion and oncogenic transformation in Ewing sarcoma. A similar uncoupling of cellular adhesion and oncogenic transformation was previously noted for activated Src kinase signaling. Src kinase expression/activity is frequently increased in various cancers, where it affects oncogenic transformation by activating RAS, phosphatidylinositol 3-kinase (PI3K), and STAT signaling pathways (62). Activated mutants of Src play a role in oncogenic transformation and affect morphological changes, including cellular adhesion (63). Interestingly, it has been shown that integrin α5β3 signaling regulates Src kinase-mediated oncogenic transformation, but this interaction does not affect Src-mediated cellular adhesion (64). Our data suggest that signaling downstream of KRT17 may occur through multiple independent pathways, one of which is AKT signaling, which is necessary for cellular adhesion but dispensable for oncogenic transformation.
Based on our findings, we hypothesize that inhibition of the AKT signaling pathway alone would be an ineffective therapy for Ewing sarcoma patients. In support of this are the findings that insulin-like growth factor 1 receptor (IGF1R) antagonists that have shown efficacy in phase I/II clinical trials for the treatment of Ewing sarcoma patients (65, 66) inhibit not only PI3K-AKT signaling but also the RAS–mitogen-activated protein kinase (MAPK) and JAK-STAT pathways. Therefore, inhibition of multiple crucial signaling pathways may be necessary to inhibit growth and transformation of Ewing sarcoma cells. Also, targeting of pathways downstream of IGF1R with MEK/MAPK inhibitors (PD98059 and U0126) and a PI3K inhibitor (LY294002) decreases Ewing sarcoma cell survival and increases sensitivity to doxorubicin (67). Interestingly, blocking of AKT activation alone did not have any effect on survival or proliferation of Ewing sarcoma cells (S. Sankar, unpublished observations). Our results demonstrate that active AKT signaling is not required for proliferation or oncogenic transformation in Ewing sarcoma.
In conclusion, we have defined a new pathway downstream of GLI1 in Ewing sarcoma that highlights the central role of KRT17 in coordinating both oncogenic transformation and cellular adhesion in Ewing sarcoma. Future work will be required to identify the critical factors and pathways downstream of KRT17 that affect oncogenic transformation. These studies will be key to a better understanding of the biology of Ewing sarcoma and may lead to more effective targeted therapies for patients with this devastating disease.
Supplementary Material
ACKNOWLEDGMENTS
S.S. acknowledges support from the HHMI Med into Grad program at the University of Utah (U2M2G). This work was supported by NIH/NCI grants R01 CA140394 (to S.L.L.) and P30 CA042014 (to the Huntsman Cancer Institute).
We thank Alana Welm for discussions and critical reading of the manuscript and for generously providing us with immunodeficient mice, Michael Monument for providing the Ewing sarcoma patient tumor RNA, and Wen Luo and members of the Lessnick and Beckerle laboratories for helpful discussions and reagents.
Footnotes
Published ahead of print 16 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00241-13.
REFERENCES
- 1.Arndt CA, Crist WM. 1999. Common musculoskeletal tumors of childhood and adolescence. N. Engl. J. Med. 341:342–352 [DOI] [PubMed] [Google Scholar]
- 2.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, Aurias A, Thomas G. 1992. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 359:162–165 [DOI] [PubMed] [Google Scholar]
- 3.Smith R, Owen LA, Trem DJ, Wong JS, Whangbo JS, Golub TR, Lessnick SL. 2006. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing's sarcoma. Cancer Cell 9:405–416 [DOI] [PubMed] [Google Scholar]
- 4.Kinsey M, Smith R, Lessnick SL. 2006. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing's sarcoma. Mol. Cancer Res. 4:851–859 [DOI] [PubMed] [Google Scholar]
- 5.Prieur A, Tirode F, Cohen P, Delattre O. 2004. EWS/FLI-1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3. Mol. Cell. Biol. 24:7275–7283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shukla N, Ameur N, Yilmaz I, Nafa K, Lau CY, Marchetti A, Borsu L, Barr FG, Ladanyi M. 2012. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin. Cancer Res. 18:748–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang HY, Illei PB, Zhao Z, Mazumdar M, Huvos AG, Healey JH, Wexler LH, Gorlick R, Meyers P, Ladanyi M. 2005. Ewing sarcomas with p53 mutation or p16/p14ARF homozygous deletion: a highly lethal subset associated with poor chemoresponse. J. Clin. Oncol. 23:548–558 [DOI] [PubMed] [Google Scholar]
- 8.May WA, Lessnick SL, Braun BS, Klemsz M, Lewis BC, Lunsford LB, Hromas R, Denny CT. 1993. The Ewing's sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol. Cell. Biol. 13:7393–7398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.May WA, Gishizky ML, Lessnick SL, Lunsford LB, Lewis BC, Delattre O, Zucman J, Thomas G, Denny CT. 1993. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc. Natl. Acad. Sci. U. S. A. 90:5752–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Owen LA, Kowalewski AA, Lessnick SL. 2008. EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing's sarcoma. PLoS One 3:e1965. 10.1371/journal.pone.0001965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kasper M, Regl G, Frischauf AM, Aberger F. 2006. GLI transcription factors: mediators of oncogenic Hedgehog signalling. Eur. J. Cancer 42:437–445 [DOI] [PubMed] [Google Scholar]
- 12.Ingham PW, McMahon AP. 2001. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15:3059–3087 [DOI] [PubMed] [Google Scholar]
- 13.Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, Sorensen N, Berthold F, Henk B, Schmandt N, Wolf HK, von Deimling A, Wainwright B, Chenevix-Trench G, Wiestler OD, Wicking C. 1997. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 57:2085–2088 [PubMed] [Google Scholar]
- 14.Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgard R, Unden AB. 2006. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J. Pathol. 208:17–25 [DOI] [PubMed] [Google Scholar]
- 15.Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, Negus K, Smyth I, Pressman C, Leffell DJ, Gerrard B, Goldstein AM, Dean M, Toftgard R, Chenevix-Trench G, Wainwright B, Bale AE. 1996. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85:841–851 [DOI] [PubMed] [Google Scholar]
- 16.Sheng T, Li C, Zhang X, Chi S, He N, Chen K, McCormick F, Gatalica Z, Xie J. 2004. Activation of the hedgehog pathway in advanced prostate cancer. Mol. Cancer 3:29. 10.1186/1476-4598-3-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernandez-del Castillo C, Yajnik V, Antoniu B, McMahon M, Warshaw AL, Hebrok M. 2003. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425:851–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bian YH, Huang SH, Yang L, Ma XL, Xie JW, Zhang HW. 2007. Sonic hedgehog-Gli1 pathway in colorectal adenocarcinomas. World J. Gastroenterol. 13:1659–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chi S, Huang S, Li C, Zhang X, He N, Bhutani MS, Jones D, Castro CY, Logrono R, Haque A, Zwischenberger J, Tyring SK, Zhang H, Xie J. 2006. Activation of the hedgehog pathway in a subset of lung cancers. Cancer Lett. 244:53–60 [DOI] [PubMed] [Google Scholar]
- 20.Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, Beachy PA. 2003. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 425:846–851 [DOI] [PubMed] [Google Scholar]
- 21.Ruiz i Altaba A, Sanchez P, Dahmane N. 2002. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat. Rev. Cancer 2:361–372 [DOI] [PubMed] [Google Scholar]
- 22.Riobo NA, Lu K, Emerson CP., Jr 2006. Hedgehog signal transduction: signal integration and cross talk in development and cancer. Cell Cycle 5:1612–1615 [DOI] [PubMed] [Google Scholar]
- 23.Sankar S, Bell R, Patel M, Davis IJ, Lessnick SL, Luo W. 17 May 2013. EWS and RE1-silencing transcription factor inhibit neuronal phenotype development and oncogenic transformation in Ewing sarcoma. Genes Cancer [Epub ahead of print.] 10.1177/1947601913489569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beauchamp E, Bulut G, Abaan O, Chen K, Merchant A, Matsui W, Endo Y, Rubin JS, Toretsky J, Uren A. 2009. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 284:9074–9082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zwerner JP, Joo J, Warner KL, Christensen L, Hu-Lieskovan S, Triche TJ, May WA. 2008. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 27:3282–3291 [DOI] [PubMed] [Google Scholar]
- 26.Joo J, Christensen L, Warner K, States L, Kang HG, Vo K, Lawlor ER, May WA. 2009. GLI1 is a central mediator of EWS/FLI1 signaling in Ewing tumors. PLoS One 4:e7608. 10.1371/journal.pone.0007608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braunreiter CL, Hancock JD, Coffin CM, Boucher KM, Lessnick SL. 2006. Expression of EWS-ETS fusions in NIH3T3 cells reveals significant differences to Ewing's sarcoma. Cell Cycle 5:2753–2759 [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Lodish HF. 2004. Constitutive activation of the MEK/ERK pathway mediates all effects of oncogenic H-ras expression in primary erythroid progenitors. Blood 104:1679–1687 [DOI] [PubMed] [Google Scholar]
- 29.Lessnick SL, Dacwag CS, Golub TR. 2002. The Ewing's sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell 1:393–401 [DOI] [PubMed] [Google Scholar]
- 30.Sankar S, Bell R, Stephens B, Zhuo R, Sharma S, Bearss DJ, Lessnick SL. 26 November 2012. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene [Epub ahead of print.] 10.1038/onc.2012.525 [DOI] [PubMed] [Google Scholar]
- 31.Gangwal K, Sankar S, Hollenhorst PC, Kinsey M, Haroldsen SC, Shah AA, Boucher KM, Watkins WS, Jorde LB, Graves BJ, Lessnick SL. 2008. Microsatellites as EWS/FLI response elements in Ewing's sarcoma. Proc. Natl. Acad. Sci. U. S. A. 105:10149–10154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hollenhorst PC, Shah AA, Hopkins C, Graves BJ. 2007. Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev. 21:1882–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oler AJ, Alla RK, Roberts DN, Wong A, Hollenhorst PC, Chandler KJ, Cassiday PA, Nelson CA, Hagedorn CH, Graves BJ, Cairns BR. 2010. Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat. Struct. Mol. Biol. 17:620–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaturvedi A, Hoffman LM, Welm AL, Lessnick SL, Beckerle MC. 2012. The EWS/FLI oncogene drives changes in cellular morphology, adhesion, and migration in Ewing sarcoma. Genes Cancer 3:102–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hui CC, Angers S. 2011. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 27:513–537 [DOI] [PubMed] [Google Scholar]
- 36.Yoon JW, Kita Y, Frank DJ, Majewski RR, Konicek BA, Nobrega MA, Jacob H, Walterhouse D, Iannaccone P. 2002. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J. Biol. Chem. 277:5548–5555 [DOI] [PubMed] [Google Scholar]
- 37.Cavazzana AO, Miser JS, Jefferson J, Triche TJ. 1987. Experimental evidence for a neural origin of Ewing's sarcoma of bone. Am. J. Pathol. 127:507–518 [PMC free article] [PubMed] [Google Scholar]
- 38.Lipinski M, Hirsch MR, Deagostini-Bazin H, Yamada O, Tursz T, Goridis C. 1987. Characterization of neural cell adhesion molecules (NCAM) expressed by Ewing and neuroblastoma cell lines. Int. J. Cancer 40:81–86 [DOI] [PubMed] [Google Scholar]
- 39.Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J. 2006. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell 124:47–59 [DOI] [PubMed] [Google Scholar]
- 40.Winklmayr M, Schmid C, Laner-Plamberger S, Kaser A, Aberger F, Eichberger T, Frischauf AM. 2010. Non-consensus GLI binding sites in Hedgehog target gene regulation. BMC Mol. Biol. 11:2. 10.1186/1471-2199-11-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27:1017–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohali A, Avigad S, Zaizov R, Ophir R, Horn-Saban S, Cohen IJ, Meller I, Kollender Y, Issakov J, Yaniv I. 2004. Prediction of high risk Ewing's sarcoma by gene expression profiling. Oncogene 23:8997–9006 [DOI] [PubMed] [Google Scholar]
- 43.Coulombe PA, Tong X, Mazzalupo S, Wang Z, Wong P. 2004. Great promises yet to be fulfilled: defining keratin intermediate filament function in vivo. Eur. J. Cell Biol. 83:735–746 [DOI] [PubMed] [Google Scholar]
- 44.Depianto D, Kerns ML, Dlugosz AA, Coulombe PA. 2010. Keratin 17 promotes epithelial proliferation and tumor growth by polarizing the immune response in skin. Nat. Genet. 42:910–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turashvili G, Bouchal J, Baumforth K, Wei W, Dziechciarkova M, Ehrmann J, Klein J, Fridman E, Skarda J, Srovnal J, Hajduch M, Murray P, Kolar Z. 2007. Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer 7:55. 10.1186/1471-2407-7-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smedts F, Ramaekers F, Troyanovsky S, Pruszczynski M, Link M, Lane B, Leigh I, Schijf C, Vooijs P. 1992. Keratin expression in cervical cancer. Am. J. Pathol. 141:497–511 [PMC free article] [PubMed] [Google Scholar]
- 47.Bournet B, Pointreau A, Souque A, Oumouhou N, Muscari F, Lepage B, Senesse P, Barthet M, Lesavre N, Hammel P, Levy P, Ruszniewski P, Cordelier P, Buscail L. 2012. Gene expression signature of advanced pancreatic ductal adenocarcinoma using low density array on endoscopic ultrasound-guided fine needle aspiration samples. Pancreatology 12:27–34 [DOI] [PubMed] [Google Scholar]
- 48.Zhang J, Wang K, Liu SS, Dai L, Zhang JY. 2011. Using proteomic approach to identify tumor-associated proteins as biomarkers in human esophageal squamous cell carcinoma. J. Proteome Res. 10:2863–2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi I, Hashemi Sadraei N, Duan ZH, Shi T. 2011. Aberrant signaling pathways in squamous cell lung carcinoma. Cancer Inform. 10:273–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ossandon FJ, Villarroel C, Aguayo F, Santibanez E, Oue N, Yasui W, Corvalan AH. 2008. In silico analysis of gastric carcinoma. Serial analysis of gene expression libraries reveals different profiles associated with ethnicity. Mol. Cancer 7:22. 10.1186/1476-4598-7-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van de Rijn M, Perou CM, Tibshirani R, Haas P, Kallioniemi O, Kononen J, Torhorst J, Sauter G, Zuber M, Kochli OR, Mross F, Dieterich H, Seitz R, Ross D, Botstein D, Brown P. 2002. Expression of cytokeratins 17 and 5 identifies a group of breast carcinomas with poor clinical outcome. Am. J. Pathol. 161:1991–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ide M, Kato T, Ogata K, Mochiki E, Kuwano H, Oyama T. 2012. Keratin 17 expression correlates with tumor progression and poor prognosis in gastric adenocarcinoma. Ann. Surg. Oncol. 19:3506–3514 [DOI] [PubMed] [Google Scholar]
- 53.Sarbia M, Fritze F, Geddert H, von Weyhern C, Rosenberg R, Gellert K. 2007. Differentiation between pancreaticobiliary and upper gastrointestinal adenocarcinomas: is analysis of cytokeratin 17 expression helpful? Am. J. Clin. Pathol. 128:255–259 [DOI] [PubMed] [Google Scholar]
- 54.Kim S, Wong P, Coulombe PA. 2006. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature 441:362–365 [DOI] [PubMed] [Google Scholar]
- 55.Pan X, Kane LA, Van Eyk JE, Coulombe PA. 2011. Type I keratin 17 protein is phosphorylated on serine 44 by p90 ribosomal protein S6 kinase 1 (RSK1) in a growth- and stress-dependent fashion. J. Biol. Chem. 286:42403–42413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Altomare DA, Testa JR. 2005. Perturbations of the AKT signaling pathway in human cancer. Oncogene 24:7455–7464 [DOI] [PubMed] [Google Scholar]
- 57.Okegawa T, Pong RC, Li Y, Hsieh JT. 2004. The role of cell adhesion molecule in cancer progression and its application in cancer therapy. Acta Biochim. Pol. 51:445–457 [PubMed] [Google Scholar]
- 58.Dahlin DC, Coventry MB, Scanlon PW. 1961. Ewing's sarcoma. A critical analysis of 165 cases. J. Bone Joint Surg. Am. 43-A:185–192 [PubMed] [Google Scholar]
- 59.Wang CC, Schulz MD. 1953. Ewing's sarcoma; a study of fifty cases treated at the Massachusetts General Hospital, 1930-1952 inclusive. N. Engl. J. Med. 248:571–576 [DOI] [PubMed] [Google Scholar]
- 60.Dubois SG, Epling CL, Teague J, Matthay KK, Sinclair E. 2010. Flow cytometric detection of Ewing sarcoma cells in peripheral blood and bone marrow. Pediatr. Blood Cancer 54:13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang S, Basson MD. 2011. Protein kinase B/AKT and focal adhesion kinase: two close signaling partners in cancer. Anticancer Agents Med. Chem. 11:993–1002 [DOI] [PubMed] [Google Scholar]
- 62.Irby RB, Yeatman TJ. 2000. Role of Src expression and activation in human cancer. Oncogene 19:5636–5642 [DOI] [PubMed] [Google Scholar]
- 63.Yeatman TJ. 2004. A renaissance for SRC. Nat. Rev. Cancer 4:470–480 [DOI] [PubMed] [Google Scholar]
- 64.Huveneers S, Arslan S, van de Water B, Sonnenberg A, Danen EH. 2008. Integrins uncouple Src-induced morphological and oncogenic transformation. J. Biol. Chem. 283:13243–13251 [DOI] [PubMed] [Google Scholar]
- 65.Olmos D, Postel-Vinay S, Molife LR, Okuno SH, Schuetze SM, Paccagnella ML, Batzel GN, Yin D, Pritchard-Jones K, Judson I, Worden FP, Gualberto A, Scurr M, de Bono JS, Haluska P. 2010. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing's sarcoma: a phase 1 expansion cohort study. Lancet Oncol. 11:129–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kelleher FC, Thomas DM. 2012. Molecular pathogenesis and targeted therapeutics in Ewing sarcoma/primitive neuroectodermal tumours. Clin. Sarcoma Res. 2:6. 10.1186/2045-3329-2-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benini S, Manara MC, Cerisano V, Perdichizzi S, Strammiello R, Serra M, Picci P, Scotlandi K. 2004. Contribution of MEK/MAPK and PI3-K signaling pathway to the malignant behavior of Ewing's sarcoma cells: therapeutic prospects. Int. J. Cancer 108:358–366 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.