

Abstract
The deubiquitinating enzyme heterodimeric complex USP1-UAF1 regulates the Fanconi anemia (FA) DNA repair pathway. Absence of this complex leads to increased cellular levels of ubiquitinated FANCD2 (FANCD2-Ub) and ubiquitinated PCNA (PCNA-Ub). Mice deficient in the catalytic subunit of the complex, USP1, exhibit an FA-like phenotype and have a cellular deficiency in homologous-recombination (HR) repair. Here, we have characterized mice deficient in the UAF1 subunit. Uaf1+/− mice were small at birth and exhibited reduced fertility, thus resembling Usp1−/− mice. Unexpectedly, homozygous Uaf1−/− embryos died at embryonic day 7.5 (E7.5). These mutant embryos were small and developmentally retarded. As expected, Uaf1 deficiency in mice led to increased levels of cellular Fancd2-Ub and Pcna-Ub. Uaf1+/− murine embryonic fibroblasts (MEFs) exhibited profound chromosome instability, genotoxin hypersensitivity, and a significant defect in homologous-recombination repair. Moreover, Uaf1−/− mouse embryonic stem cells (mESCs) showed chromosome instability, genotoxin hypersensitivity, and impaired Fancd2 focus assembly. Similar to USP1 knockdown, UAF1 knockdown in tumor cells caused suppression of tumor growth in vivo. Taken together, our data demonstrate the important regulatory role of the USP1-UAF1 complex in HR repair through its regulation of the FANCD2-Ub and PCNA-Ub cellular pools.
INTRODUCTION
The integrity of the human genome is essential for the suppression of oncogenesis, and it is maintained by several intricate DNA repair mechanisms. One such mechanism, homologous recombination (HR), is essential for the error-free repair of DNA double-strand breaks (DSBs) (1). Germ line defects in genes regulating homologous recombination result in several human genetic diseases, including ataxia telangiectasia (AT), Fanconi anemia (FA), Bloom's syndrome (BS), and Nijmegen breakage syndrome (NBS). Somatic mutations in genes regulating HR repair, such as the breast and ovarian cancer susceptibility genes BRCA1 and BRCA2, underlie the genomic instability, drug and radiation sensitivity, and clinical progression of cancer.
The core proteins involved in HR repair, such as RAD51 and its paralogs, RAD52, and various recombinases and antirecombinases, have been extensively studied (2). Disruption of the genes encoding these proteins in mouse models can result in embryonic lethality or animals with significant developmental defects and cancer prevalence. More recently, a wide range of HR-regulatory genes have been identified. For instance, functional screening with short hairpin RNA (shRNA) libraries has identified HR-regulatory genes which, when silenced, cause cellular hypersensitivity to radiation or other genotoxic stress (3, 4). Additional HR proteins were identified as substrates of the ATM kinase (5). Among the many HR-regulatory proteins, enzymes that regulate protein ubiquitination and sumoylation have emerged as a prominent class (6).
The deubiquitinating enzyme (DUB) USP1 (ubiquitin-specific protease 1) is a critical regulator of HR repair (7). USP1 is required for the deubiquitination of ubiquitinated FANCD2 (FANCD2-Ub) and ubiquitinated PCNA (PCNA-Ub) (8) and is therefore a regulator of interstrand cross-link (ICL) repair and translesion synthesis (TLS), respectively. Knockdown of USP1 results in elevated levels of FANCD2-Ub and PCNA-Ub and in increased cellular sensitivity to interstrand cross-linking agents, such as mitomycin C (MMC). Knockout (KO) of USP1 in chicken DT40 cells (9) or in a mouse model (10) results in an increase in cross-linker sensitivity and in chromosome radial formation, the hallmark of the FA phenotype.
USP1 binds constitutively to an 80-kDa binding partner, referred to as UAF1 (USP1-associated factor 1) (11). UAF1 has an N-terminal WD40 domain, with eight WD propeller sequences, and a C-terminal coiled-coil domain that targets the DUB complex to its substrates via Sumo-like domain (SLD)-Sumo interaction motif (SIM) interactions (12). Interestingly, the WD40 domain of UAF1 binds and stimulates the ubiquitin protease activity of USP1 (11). UAF1 has also been shown to bind the DUB enzymes USP12 and USP46 (13), although the substrates of the USP12-UAF1 and USP46-UAF1 complexes are unknown. The molecular mechanisms of the USP1-UAF1 complex in DNA repair also remain unknown. Since UAF1 is an abundant cellular protein with many binding partners, one might expect that a murine knockout of Uaf1 would have a more severe phenotype than the Usp1 knockout (10). Recent studies indicate that the USP1-UAF1 complex may play a direct role in regulatory HR repair (14). Accordingly, DT40 clones that are USP1−/−, UAF1−/−, or double knockouts are defective in gene conversion, induced by the endonuclease I-SceI, and hypersensitive to poly(ADP-ribose) polymerase (PARP) inhibitors.
In the current study, we disrupted the Uaf1 gene in a murine model. Uaf1+/− mice, heterozygous for the insertion of a gene trap constraint, were viable but were small and exhibited reduced fertility. Uaf1+/− cells derived from the mice were hypersensitive to genotoxic agents and had a defect in HR repair. Homozygous inactivation of the Uaf1 gene resulted in embryonic lethality at embryonic day 7.5 (E7.5). The identification of Uaf1 as a bona fide HR-regulatory gene has important implications for human cancer genome screening.
MATERIALS AND METHODS
Mice.
Uaf1-deficient mice were generated by homologous insertion of a gene trap cassette. The Uaf1 gene-targeted E14 embryonic stem cells (ESCs) were obtained from the International Gene Trap Consortium (IGTC). Mouse ESC (mESC) clones with a normal karyotype were injected into the blastocysts from the C56BL/6J mice and implanted into the SV-129 mice. The chimeric mice were then bred with C57 BL/6J mice in order to obtain the Uaf1+/− founder mice. The Uaf1+/− mice were then crossed with C57BL/6J mice for nine generations in order to obtain the mutants in a pure genetic background. Uaf1+/− mice in a C57BL/6J background were intercrossed in order to identify Uaf1−/− mice. Genotypes were confirmed by PCR using wild-type (WT) primers (5′-TCA TAA GAA TAT GGA GTG TCA ATC-3′ and 5′-CTC CCT CCC TCC CGT TTG ACT GCA-3′) and KO primers (5′-AGC TGG GCT GTC AGT AAT GCT ATT-3′ and 5′-TTC CGG AGC GGA TCT CAA ACT CTC-3′). All animal experiments were approved by the institutional animal care and use committee of the Dana-Farber Cancer Institute.
Uaf1−/− mESC generation.
In order to generate Uaf1−/− mESCs, Uaf1+/− mESCs were plated (at 2 × 105/10 cm cell density) in gelatin-coated dishes, along with wild-type feeder murine embryonic fibroblasts (MEFs) (15). G418 was added at various concentrations (1 to 3 mg/ml). After 5 days in culture, G418-resistant ESCs were cultured with fresh medium, and colonies were grown in 2.5 mg/ml of G418. Twenty ESC clones were chosen and plated into 24-well plates, along with feeder cells in the absence of G418. The Uaf1−/− ESCs were expanded and identified by PCR.
Cell culture.
Primary MEFs were generated from E13.5 to E15 embryos and cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 15% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin (Invitrogen). mESCs were cultured in DMEM (high glucose; Invitrogen) containing 15% fetal bovine serum, 2% penicillin-streptomycin (Invitrogen), 1% nucleoside mix, 1% l-glutamine, 1% nonessential amino acids (NEAA), 10−4 M beta mercaptoethanol, and 103 U/ml of leukemia-inhibitory factor (LIF) in 0.1% gelatin-coated plates. U2OS cells were grown in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin (Invitrogen). All of the cell types were incubated at 37°C with 5% CO2.
Immunohistochemistry and skeletal staining.
Mouse tissues (ovaries, testes, and skin) were fixed in formalin and prepared in paraffin blocks as described previously (10). The samples were stained with hematoxylin and eosin (H&E). For skeletal staining, samples were fixed in 100% ethanol for 48 h and stained with alcian blue staining solution (0.03% alcian blue, 80% ethanol, 20% acetic acid) for 2 days, followed by staining with alizarin red solution (0.03% alizarin red, 1% KOH, and H2O) for 2 days. The skeletons were cleared with 20% glycerol-ethanol and stored in 80% glycerol-ethanol solution.
Western blot analysis.
Cell lysates were prepared as described previously (10, 16), and Western blotting was performed using the following antibodies: anti-murine Usp1, Fancd2, Uaf1, and PCNA (Santa Cruz); Fanci (Bethyl Laboratories); gamma-H2AX (Cell Signaling Technology); and BRCA2(EMD) and antitubulin (Santa Cruz). The small interfering RNAs (siRNAs) against human USP1, UAF1, USP12, and USP46, as well as shRNA against human UAF1, were as described previously (11, 13).
Immunofluorescence.
The Fancd2 and 53BP1 foci were detected by immunofluoresecnce as described previously (10) using anti-Fancd2 (10) or anti-53BP1 (Bethyl Laboratories) primary and Alexa Fluor 488-conjugated secondary antibodies. Briefly, the MEFs and mESCs were prepared with 0.5% Triton–phosphate-buffered saline (PBS) preextraction and fixed with 4% formaldehyde, followed by staining with the antibodies. The quantification of cells with foci was performed by counting the cells with foci using a Nikon Zenesis microscope, and images were acquired using Axiovision 4.8.2 software.
Cell survival analysis.
MEFs and mESCs were prepared for colony assays. After the treatments with MMC, etoposide, UV, infrared (IR), or acetaldehyde (Sigma), the cells were plated in 6-well plates for clonogenic assays in triplicate. After 2 weeks in culture, colonies were fixed with methanol and stained with crystal violet.
MMC-induced chromosome breakage analysis.
Chromosome breakage analysis was performed as described previously (10). MEFs or mESCs were exposed to MMC for 48 h. Colcemid (0.1 μg/ml) was added to the medium 2 h before harvesting the cells, and 50 metaphases were analyzed for chromosomal abnormalities for each sample.
HR analysis.
HR activity was analyzed by direct-repeat green fluorescent protein (DR-GFP) reporter assay as previously described (10, 17, 18). U2OS–DR-GFP cells carrying a chromosomally integrated single copy of HR repair substrate or MEFs engineered to express DR-GFP were used. DSB-induced HR after I-SceI expression in these cells results in restoration and expression of GFP. Briefly, the DR-GFP cells were infected with retrovirus encoding I-SceI or transfected with an I-SceI plasmid. Forty-eight to 96 h after induction of chromosomal DSBs through the expression of I-SceI, DR-GFP cells were subjected to fluorescence-activated cell sorter (FACS) analysis to quantify the percentage of viable GFP-positive cells.
qPCR analysis.
The mESCs were cultured without LIF to induce differentiation, and the cell pellets were harvested at different time points (days 0 to 8) for RNA extraction. Also, U2OS cells with siRNA against control, USP12, and USP46 were cultured and harvested for RNA extraction using a TRIzol reagent. For quantitative PCR (qPCR) analysis, the following primers were used: Id1 primers (5′-TCT ACG ACA TGA ACG GCT GCT A-3′ and 5′-ACT CCG AGT TCA GCT CCA GCA-3′), Uaf1 primers (5′-AAG AAG AGA ATG AAG TGA ACC ATG-3′ and 5′-ATT TTT ATC CAC AGT GAT GTC AAT-3′), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers (5′-CAT,GTT CCA GTA TGA CTC CAC TCA CG-3′ and 5′-CCA GTA GAC TCC ACG ACA TAC TCA GCA-3′), USP12 primers (5′-CTT ACC GGG TAG TTT TTC CTT TAG-3′ and 5′-CCC GTA GAA TTC TTC AAT AGC TTG-3′), and USP46 primers (5′-ATG GAG CAG CTG CAC AGA TAC ACC-3′ and 5′-ATT TTC TCT ACA ATG TCA TCA TCA-3′).
TPA treatment in mice.
The cell proliferation assay in mouse skin was employed as described previously (16). Briefly, the dorsal skin of 6-week-old Uaf1+/+ and Uaf+/− female mice was shaved. The next day, the mice were treated with 2.5 μg 12-O-tetradecanoylphorbol-13-acetate (TPA) (LC Laboratories) or vehicle (acetone) 5 times a week for 4 weeks. The skin was fixed with formalin and stained with H&E for histological analysis.
Mouse xenograft experiments.
A mouse xenograft model was established as described previously (16). Human A549 cells (2 × 106) with shRNA against UAF1 (shUAF1) or shRNA against Scramble (shScramble) were prepared with 200 μl of PBS-Matrigel gel (BD Bioscience; 1:1 ratio) and injected subcutaneously into the flanks of athymic nude mice (two-site injections). Tumor growth was monitored by measuring the size, and tumor volume (V) was calculated using the following equation: V = lw2/2, where l is the length and w is the width of the tumor.
RESULTS
Homozygous inactivation of Uaf1 causes embryonic lethality.
Initially, we identified a murine ESC line containing a gene trap insertion in intron 2 of the Uaf1 gene (Fig. 1A) and used the cells to generate Uaf1-deficient mice. The ESCs were injected into host blastocysts to generate chimeric mice. The heterozygote founders, containing one copy of the mutant Uaf1 allele, were mated with C57BL/6J wild-type mice in order to generate Uaf1+/− animals. The mutant animals with the Uaf1 gene trap insertion (Uaf1+/−) were then backcrossed with C57BL/6J mice in order to generate Uaf1 heterozygous animals in a pure genetic background.
Fig 1.

Homozygous inactivation of murine Uaf1 causes embryonic lethality. (A) Map of the genomic Uaf1 gene, showing disruption by a gene trap vector. The gene trap vector was designed in three parts: SA (the splice acceptor sequence), a Trap cassette (the selection and reporter construct, a neo selection), and pA (the polyadenylation signal). The gene trap vector was inserted in intron 2 (between exon 2 and exon 3) of the Uaf1 gene. The locations of WT and KO primers for PCR genotyping are shown. (B) Viability of Uaf1-deficieint mice at various stages of development. (C) Representative images of Uaf1+/+ and Uaf1−/− embryos at E7.5. (D) Appearance of Uaf1+/− and Uaf1+/+ mice at birth. (E) Skeletal staining of 3.5-week-old Uaf1+/− and Uaf1+/+ mice. (F) Growth curve of Uaf1+/− and Uaf1+/+ mice (n = 30) from the newborn stage to 49 days after birth. *, P < 0.001. The error bars indicate standard deviations of 30 mice. (G) Representative photographs of testes from 12-week-old Uaf1+/− and Uaf1+/+ male mice. (H) Comparison of the testis size and oocyte numbers in Uaf1+/− and Uaf1+/+ mice. Testes were obtained from 12-week-old male Uaf1+/− and Uaf1+/+ mice (n = 20). For the quantitative analysis of oocyte numbers, visible oocytes were counted per section, and the numbers of oocytes in 12-week-old female Uaf1+/− and Uaf1+/+ mice (n = 20) were determined for each section; 7 sections per female were analyzed. *, P < 0.001. (I and J) Histology of the testes and ovaries from two representative 12-week-old Uaf1+/− and Uaf1+/+ mice. H&E staining of tissue sections of ovaries and testes is shown (scale bar, 200 μm). The arrows indicate smaller seminiferous tubules in the testes and smaller numbers of oocytes in the ovaries. (C to J) Representative analyses; n = 4 to 20 for each genotype.
Uaf1+/− animals were intercrossed to generate Uaf1−/− embryos. No viable Uaf1−/− mice were identified among the 204 pups genotyped at the newborn stage from Uaf1+/− matings (Fig. 1B). Homozygous inactivation of Uaf1 therefore results in embryonic lethality. To determine the consequences of Uaf1 loss for embryonic development, embryos from heterozygous Uaf1 intercrosses were analyzed at different stages of gestation. At E7.5, only four Uaf1−/− embryos (4 of 29 pups) were detected, which is lower than the expected Mendelian ratio (Fig. 1B). The Uaf1−/− embryos showed abnormal morphology and less growth than the Uaf1+/+ embryos (Fig. 1C). No Uaf1−/− embryos (0/82) were detected at the E10.5 stage. The Uaf1+/− mice were born at the expected Mendelian frequencies. However, at birth, the Uaf1+/− mice were smaller than their wild-type littermates (0.5 ± 0.28 and 1.2 ± 0.3 g body weight for Uaf1+/− and Uaf1+/+ newborn mice) (Fig. 1D and F). In addition, Uaf1+/− mice exhibited a defect in skeletal development (Fig. 1E) similar to the skeletal deformity observed in Usp1−/− mice (19). Collectively, these data suggest that, unlike Usp1, Uaf1 deficiency results in early embryonic lethality.
Gonadal dysfunction in Uaf1+/− mice.
Usp1−/− mice exhibit gonadal dysfunction, and specifically, the Usp1−/− male mice are infertile (10). We next tested if Uaf1 deficiency also causes gonadal dysfunction. Uaf1+/− male and female mice were able to produce offspring, but at reduced frequencies compared to the wild-type sibling controls (data not shown). Severe abnormalities were observed in the gonads of Uaf1+/− mice. The testes from adult Uaf1+/− male mice were strikingly smaller than those of wild-type littermates (Fig. 1G and H). Histological analysis of testes from adult Uaf1+/− mice revealed significant atrophy with distorted seminiferous tubules, fewer germ cells, and limited spermatogenesis compared to the wild-type littermate controls (Fig. 1I). The ovaries of 20 Uaf1+/− female mice had fewer oocytes than the wild-type sibling controls (Fig. 1H and J).
Mitomycin C hypersensitivity and enhanced Fancd2 monoubiquitination in Uaf1+/− MEFs.
Initially, we generated Uaf1+/− and Uaf1+/+ primary MEFs from E13.5 embryos and immortalized these cells with simian virus 40 (SV40) T antigen. The MEFs were analyzed for expression of DNA repair proteins and for sensitivity to various genotoxic agents (Fig. 2). Previous studies have shown that UAF1 binds, stabilizes, and promotes the activity of the USP1 deubiquitinating enzyme (11). Moreover, disruption of the USP1-UAF1 complex in eukaryotic cells, such as DT40 cells, results in elevated levels of FANCD2-Ub (9). As predicted from these studies, Uaf1+/− MEFs had decreased Uaf1 and Usp1 protein levels and an increase in the level of monoubiquitinated Fancd2 (Fig. 2A). Interestingly, the Uaf1+/− MEFs also exhibited an increased level of phosphorylated H2AX, indicating an increased baseline level of DNA damage and suggesting that the cells have an underlying defect in double-strand break repair.
Fig 2.

Uaf1+/− MEFs are hypersensitive to DNA-damaging agents. (A) Immunoblots of the lysates from primary Uaf1+/− and Uaf1+/+ MEFs. Usp1 antibody against the C-terminal epitope of Usp1 was used for immunoblots. (B to F) Survival plots of Uaf1+/− and Uaf1+/+ MEFs exposed to various DNA-damaging agents. The MEFs were exposed to MMC, UV, IR, etoposide, or AZD 2281, and survival was determined using a colony assay. P < 0.005 for Uaf1+/− versus Uaf1+/+ MEFs. (G to I) The Uaf1+/− and Uaf1+/+ MEFs were exposed to MMC for 48 h, and metaphase spreads of chromosomes were scored for chromosomal abnormalities. (G and H) Aberrations per cell (G) and percentages of cells with radials (H). (I) Representative images of metaphase spreads of the chromosomes. The arrows indicate radials in Uaf1+/− MEFs upon MMC exposure. The data in panels B to H are representative of two independent experiments in triplicate, and means ± standard errors are shown.
We next evaluated the responses of Uaf1+/− MEFs to various DNA-damaging agents (Fig. 2B to F). The cells were exposed to various genotoxic agents, and survival was determined. As expected, Uaf1+/− MEFs showed elevated sensitivity to a DNA cross-linking agent, MMC (Fig. 2B). Interestingly, Uaf1+/− MEFs were also hypersensitive to IR, UV, etoposide, and a PARP inhibitor (AZD2281) compared to the wild-type MEFs (Fig. 2C to F). The Uaf1+/− MEFs also exhibited increased MMC-induced chromosomal aberrations and radial forms (Fig. 2G to I), the hallmark feature of Fanconi anemia cells (20). Taking the data together, similar to Usp1-deficient cells, Uaf1-deficient mouse cells also exhibit cellular phenotypes of Fanconi anemia. Unlike Usp1 deficiency, however, Uaf1 deficiency causes more severe cellular impairment in DNA repair.
Uaf1+/− MEFs have a defect in homologous-recombination repair.
Usp1−/− MEFs exhibit HR defects (10). The hypersensitivity of Uaf1+/− cells to PARP inhibitor and MMC suggested an impairment in HR-mediated DNA repair. We therefore next determined whether Uaf1-deficient MEFs also have an HR defect. We used a cell-based assay, which measures the efficiency of DR recombination of two GFP alleles in cells (17, 18). We established Uaf1+/+ and Uaf1+/− MEFs with a stably integrated DR-GFP reporter, and HR was induced by transducing cells with retrovirus encoding I-Scel. After induction of I-SceI, Uaf1+/− MEFs showed decreased DR-GFP expression compared to Uaf1+/+ MEFs (1.2% versus 7%) (Fig. 3A). The similar efficiencies of I-Scel expression in Uaf1+/− and Uaf1+/+ MEFs were confirmed by Western blotting (Fig. 3B).
Fig 3.

Uaf1-deficient cells are defective in HR repair. (A) Defective HR repair in Uaf1+/− MEFs. Uaf1+/− and Uaf1+/+ MEFs harboring DR-GFP reporter were infected with retroviruses encoding hemagglutinin (HA)-tagged I-SceI or vector. The GFP expression was then determined after 96 h using flow cytometry, and the GFP expression as a readout of HR activity is shown. The data are from three Uaf1+/+ and four Uaf1+/− MEF lines. P < 0.001 for Uaf1+/− versus Uaf1+/+ MEFs after I-SceI. (B) Immunoblots of lysates from Uaf1+/− and Uaf1+/+ MEFs showing equal expression of HA-tagged I-SceI. (C) Defective HR repair in U2OS cells with knockdown of Uaf1. U2OS cells with DR-GFP reporter were transfected with siRNAs against UAF1, USP1, USP12, USP46, or BRCA2 (as a positive control), followed by transfection with I-SceI reporter plasmid. The GFP expression was then determined after 48 h using flow cytometry. The GFP expression as a readout of HR activity is shown. Two independent experiments were performed in triplicate, and means and standard errors are shown. (D) Survival curves of U2OS cells exposed to ABT-888. U2OS cells were transfected with siRNAs against control (LacZ), UAF1, USP1, USP12, USP46, or FANCD2 (as a positive control) and exposed to ABT-888. Five days after the exposure, the cell viability was determined. Two independent experiments were performed in triplicate, and means ± standard errors are shown. (E) Immunoblots of the lysates from U2OS cells transfected with control siRNA or siRNA against UAF1, USP1, or BRCA2 are shown on the left. qPCR analysis of RNA from U2OS cells transfected with control siRNA or siRNA against USP12 and USP46 is shown on the right.
Previous studies have indicated that besides USP1, UAF1 also regulates other DUBs, including USP12 and USP46 (13). We next examined the HR defect in USP12- and USP46-deficient U2OS cells harboring a DR-GFP reporter. The siRNA-mediated knockdown of UAF1 or USP1 in U2OS cells conferred HR deficiency, as measured by decreased DR-GFP expression in the I-Scel-transfected cells (Fig. 3C and E). Knockdown of USP12 did not alter DR-GFP expression (Fig. 3C and E), and knockdown of USP46 increased the DR-GFP expression. To further confirm the HR defects, we exposed DR-GFP U2OS cells with siRNAs against USP1, USP12, USP46, UAF1, and FANCD2 (as a positive control) to ABT-888, a PARP inhibitor, and performed a survival assay. The knockdown of UAF1, USP1, or FANCD2 in U2OS cells conferred sensitivity to ABT-888 treatment. Also, U2OS cells with siRNA against USP46, but not USP12, showed hypersensitivity to ABT-888 (Fig. 3D and E). We recently identified a PCNA-interacting protein, PARI, which inhibits recombination by interfering with the formation of RAD51-DNA HR structures (21). Knockdown of PARI leads to increased HR and increased genomic instability (data not shown). Similar to PARI, USP46 may also regulate HR and increase DNA damage after ABT-888 exposure.
Collectively, these results suggested that, similar to USP1, UAF1 is also required for efficient HR repair.
DNA repair defects in Uaf1−/− mESCs.
Since we were unable to establish Uaf1−/− MEFs, we next established Uaf1−/− mESCs instead. To accomplish this goal, we cultured Uaf1+/− mESCs in the presence of increasing concentrations of G418 (15). Two Uaf1−/− mESC clones were isolated, and their genotypes were confirmed (data not shown). As predicted, Uaf1−/− mESCs did not express the Uaf1 protein and had a significant decrease in the level of Usp1 protein (Fig. 4A). Also as predicted, these cells had elevated levels of monoubiquitinated Fancd2 compared to the Uaf1+/+ mESCs and a slight elevation of phosphorylated H2AX, indicating the presence of endogenous double-strand DNA breaks.
Fig 4.

Uaf1−/− mESCs are hypersensitive to DNA-damaging agents. (A) Immunoblots of the lysates from Uaf1−/− or Uaf1+/+ mESCs. Usp1 antibody against the C-terminal epitope of Usp1 was used for the immunoblots. (B to E) Survival plots of Uaf1−/−, Uaf1+/−, and Uaf1+/+ mESCs exposed to various DNA-damaging agents. mESCs were plated in gelatin-coated 6-well plates without feeder cells and exposed to MMC, UV, IR, or acetaldehyde. The cells were then cultured for 2 weeks, and the colonies were fixed with methanol, stained with crystal violet, and counted. P < 0.001 for Uaf1−/− versus Uaf1+/+ mESCs. (F to H) The Uaf1−/− and Uaf1+/+ mESCs were exposed to MMC for 48 h, and metaphase spreads of chromosomes were scored for chromosomal abnormalities. (F) Number of aberrations per cell. (G) Percentages of cells with radials. (H) Representative images of metaphase spreads of the chromosomes. The arrow indicates radials in Uaf1−/− mESCs upon MMC exposure. The data in panels B to G are representative of the two independent experiments in triplicate, and means ± standard errors are shown.
We next tested the sensitivity of Uaf1-deficient mESCs to various genotoxic agents. The mESCs were cultured on 0.1% gelatin-coated plates and exposed to MMC, IR, or UV (Fig. 4B, C, and D). Uaf1−/− mESCs were hypersensitive to MMC, IR, and UV, and the Uaf1+/− mESCs had intermediate sensitivity compared to the Uaf1+/+ mESCs. The Uaf1-deficient mESCs also were hypersensitive to acetaldehyde (Fig. 4E), consistent with recent studies indicating that the FA pathway controls acetaldehyde toxicity (22). In addition, following MMC treatment, Uaf1−/− mESCs exhibited increased chromosomal aberrations and radial forms, a hallmark feature of Fanconi anemia (Fig. 4F to H). Taking the data together, Uaf1 deficiency in mESCs causes Fanconi anemia-like cellular phenotypes and DNA repair defects.
Increased Fancd2-Ub but impaired Fancd2 foci in Uaf1−/− mESCs.
As expected, Uaf1−/− mESCs exhibited an increase in Fancd2 and PCNA monoubiquitination levels. The monoubiquitination was further enhanced upon exposure of cells to DNA-damaging agents (Fig. 5A, lanes 6 to 8). Usp1 protein levels were decreased in Uaf1−/− mESCs, consistent with previous studies indicating that UAF1 stabilizes its binding partner, USP1 (13). Monoubiquitinated FANCD2 and FANCI are targeted to chromatin upon DNA damage and form DNA repair foci; however, the focus formation is impaired in Usp1-deficient cells (10). We next determined whether monoubiquitinated Fancd2 still forms foci in Uaf1-deficient mESCs. Notably, Uaf1−/− mESCs were impaired in both spontaneous and DNA damage-inducible Fancd2 focus formation despite the high levels of ubiquitination, as observed in Usp1−/− cells (Fig. 5B) (10). There was no difference in 53BP1 focus formation between Uaf1+/+ and Uaf1−/− mESCs after IR treatment (Fig. 5C). Collectively, Uaf1-deficient ESCs have elevated levels of Fancd2-Ub but impaired focus formation of Fancd2.
Fig 5.

Uaf1-deficient mESCs exhibit increased Fancd2 monoubiquitination and decreased Fancd2 nuclear foci. (A) Immunoblots of the lysates from Uaf1−/− and Uaf1+/+ mESCs exposed to DNA-damaging agents. Usp1 antibody against the C-terminal epitope of Usp1 was used for immunoblots. (B and C) Fancd2 or 53BP1 foci in Uaf1−/− and Uaf1+/+ mESCs 3 h after IR (10 Gy) exposure. Quantification of cells with more than 5 foci/cell representing at least 100 nuclei each in three independent experiments was carried out. *, P < 0.001 for Uaf1−/− and Uaf1+/+ mESCs after IR exposure. Means and standard errors are shown in panels B and C.
Uaf1 expression is required for efficient expression of Id1.
A recent study showed that USP1 is the critical deubiquitinating enzyme that controls the cellular level of ID1 (19). ID1 is an inhibitor of basic helix-loop-helix (bHLH) transcription factors and functions to promote cellular proliferation and to block differentiation. Indeed, Usp1 knockout in a mouse results in decreased Id1 cellular levels and abnormal mesenchymal (bone) differentiation (19). We next determined the possible role of Uaf1 in Id1 regulation. Similar to Usp1 knockout mice, Uaf1+/− mice exhibited abnormal bone growth (Fig. 1E) and reduced cellular levels of Id1 protein in bone (Fig. 6A and B, lanes 2 to 4). Moreover, the Uaf1−/− mESCs failed to regulate Id1 levels upon differentiation following LIF withdrawal (Fig. 6C to E). Taken together, these results further indicate that the USP1-UAF1 complex functions normally to limit ID1 from proteasome-mediated degradation.
Fig 6.
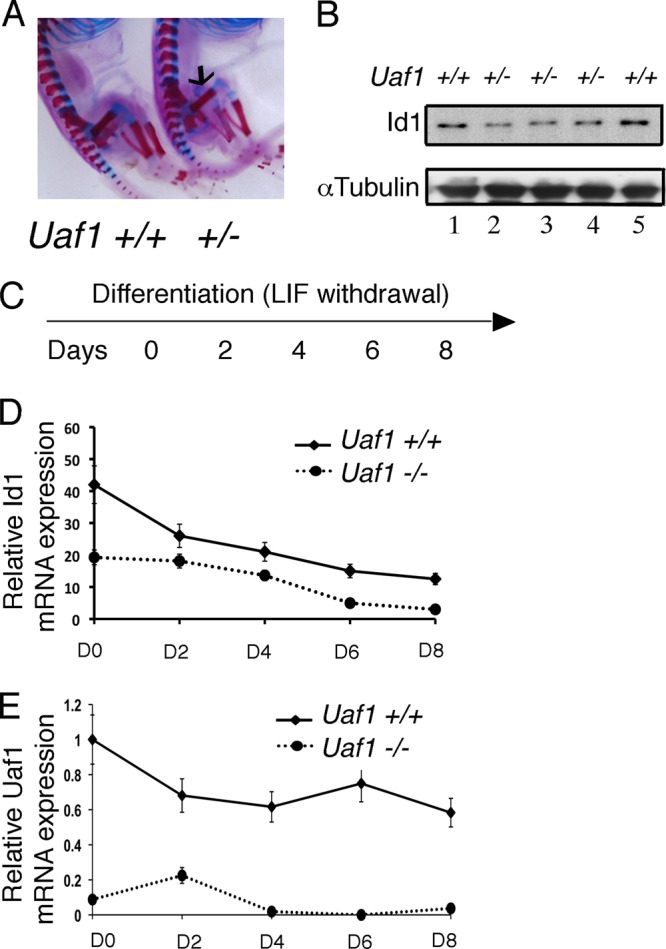
Uaf1 deficiency results in decreased Id1 expression. (A) Skeletal staining of Uaf1+/− and Uaf1+/+ E19 embryos. The arrow indicates the location of the femur. (B) Immunoblots of the lysates from femurs (in panel A) of Uaf1+/− and Uaf1+/+ E19 embryos. (C to E) Uaf1−/− and Uaf1+/+ mESCs were cultured without LIF in order to promote differentiation. RNA was then isolated at days (D) 0, 2, 4, 6, and 8 for quantification of Id1 and Uaf1 by qPCR. Id1 or Uaf1 expression upon differentiation of Uaf1−/− and Uaf1+/+ mESCs is shown. The data are shown as relative expression of Id1 or Uaf1 mRNA in Uaf1−/− mESCs compared to the wild-type controls. The samples were normalized using GAPDH expression.
Uaf1 deficiency inhibits cellular proliferation and suppresses tumor growth.
We have previously shown that knockdown of Usp1 results in elevation of Fancd2-Ub levels and increased transcriptional activation of the epithelial tumor suppressor protein, TAp63 (16). We also showed that Usp1 inhibits Ras-mediated tumorigenesis via Fancd2 deubiquitination (16). We therefore assessed the role of Uaf1 in tumorigenesis. Interestingly, Uaf1+/− mice exhibited elevated TAp63 levels in newborn skin (data not shown), indicating that Usp1 cooperates with Uaf1 in regulating TAp63 expression. Uaf1+/− mice also had reduced epithelial growth (thickening) following topical TPA exposure (Fig. 7A), as observed in Usp1-deficient mice (16). Since Uaf1 deficiency promotes the elevation of Fancd2-Ub and inhibits skin proliferation, we reasoned that it may also inhibit tumorigenesis in a Ras-driven tumor model. To test this hypothesis, we transduced a retrovirus encoding shRNA to UAF1 into a RAS-driven tumor cell line. UAF1 knockdown resulted in decreased cellular levels of USP1 and UAF1 (Fig. 7B) and reduced the growth of tumor cells in a soft-agar assay in vitro (Fig. 7C). Finally, knockdown of UAF1 suppressed the growth of tumor cells in vivo in a mouse xenograft model (Fig. 7D and E). Collectively, these data suggest that UAF1 knockdown, like USP1 knockdown, can inhibit tumorigenesis.
Fig 7.
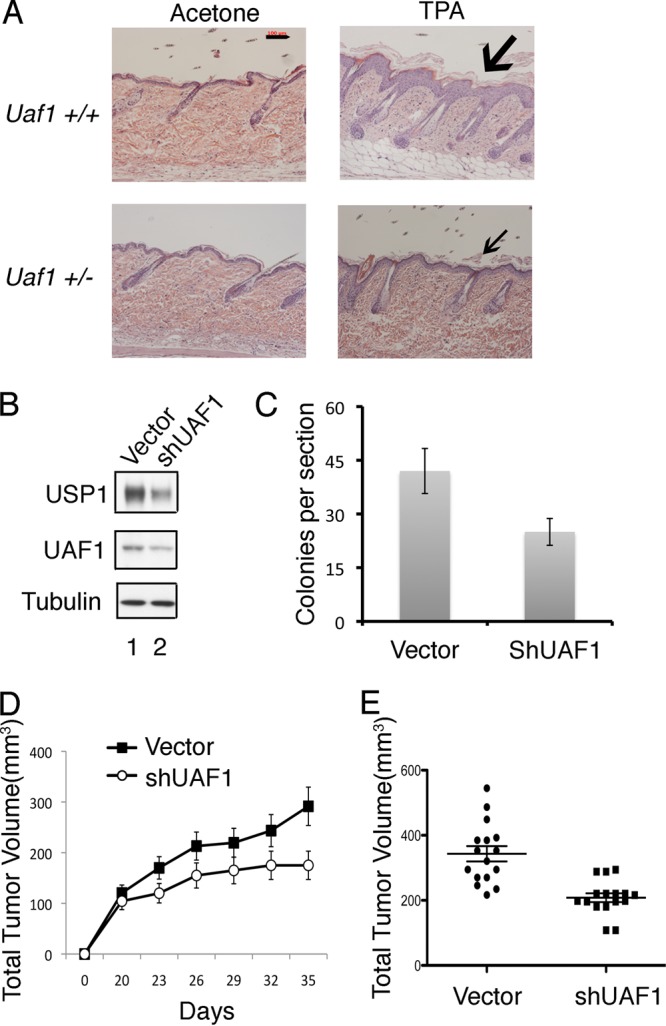
Uaf1 deficiency causes decreased cell proliferation and inhibits tumorigenesis. (A) Uaf1+/− mice have decreased TPA-induced epithelial proliferation. Six-week-old Uaf1+/− and Uaf1+/+ mice were exposed topically to TPA for 4 weeks. The mice were sacrificed, and the skin histology was evaluated by microscopy. H&E staining of tissue sections of skin (representative analysis; n = 4 for each genotype) is shown. The scale bar represents 100 μm, and the arrows indicate epithelial layers. (B to E) UAF1 knockdown in lung cancer cells suppresses growth in soft agar and reduces tumorigenesis in nude mice. (B) The cDNA encoding a UAF1 shRNA or shScramble (Vector) was stably expressed in the RAS-driven human lung adenocarcinoma epithelial cell line A549. The cells were subjected to growth assay or analyzed for tumorigenicity in nude mice. Immunoblots of the A549 cell lysates are shown. (C) Usp1 antibody against the N-terminal epitope of Usp1 was used for immunoblots. A549 cells were plated in soft agar, and transformed foci were counted (at ×100 magnification) after 2 weeks in culture. The data are means ± standard errors of three independent experiments. Each experiment was performed in triplicate plates (P < 0.001; t test). (D and E) Tumorigenicity of A549 cells in xenograft mouse models. UAF1 shRNA- or shScramble-transfected A549 cells were injected subcutaneously as xenografts in nude mice (two-site injections), and every 3 days the tumor volume was measured. The data represent the average tumor size for 10 mice in each group (more than 10 tumors, due to the two-site injections; P < 0.001). Quantification of the tumor volume of the xenografts at day 35 is shown in panel E. The data are means ± standard errors.
DISCUSSION
Increasing evidence indicates that protein ubiquitination is an important regulatory mechanism for DNA repair pathways (6). Protein ubiquitination is regulated by the coordinated activity of E3 ubiquitin ligases and DUBs. The mechanisms by which protein ubiquitination regulates DNA repair are largely unknown. On one hand, protein monoubiquitination results in the recruitment of key effector proteins with monoubiquitin binding domains, such as UBZ or UBM domains (23). For instance, PCNA monoubiquitination recruits UBZ-containing TLS polymerases to sites of DNA lesion bypass. Also, FANCD2 monoubiquitination recruits the UBZ-containing FAN1 nuclease to sites of DNA cross-link repair. On the other hand, protein polyubiquitination promotes recruitment of other DNA repair proteins to sites of damaged chromatin (6).
The mechanisms by which DUB enzymes regulate DNA repair are largely unknown. Multiple DUBs, including USP1 (7), USP3 (24), USP11 (25), and USP28 (26), have been implicated in DNA repair or cell cycle checkpoint regulation. USP1 regulates DNA repair by controlling the monoubiquitination states of its two substrates, FANCD2-Ub and PCNA-Ub, and USP1 is bound and activated by the UAF1 protein (11). Efficient DNA repair requires monoubiquitination of these two substrates by the FA core complex and Rad18, respectively. The deubiquitination of the two substrates is also critical in mediating DNA repair. Disruption of either ubiquitination or deubiquitination, therefore, results in abnormal DNA repair.
Our current study indicates that Uaf1, like its binding partner, Usp1, is critical for HR repair. Previous studies indicated that the DUB complex, USP1-UAF1, is targeted to its ubiquitinated substrates, FANCD2-Ub and PCNA-Ub, through SLD2-SIM interactions (12). Specifically, a Sumo-like domain (SLD2) at the C terminus of UAF1 binds to a SIM on either the FANCI-FANCD2 complex or the ELG1-PCNA complex, thus localizing the active DUB to its substrates.
Similar to the Usp1 knockout mice, Uaf1+/− mice exhibit FA-like phenotypes, including growth retardation and gonadal defects, as well as cellular hypersensitivity to the DNA-cross-linking agent MMC. However, the Uaf1−/− mice exhibit a more severe phenotype, with embryonic lethality at E7.5, than the Usp1 knockout mice (10). UAF1 is a more general targeting subunit, and it also binds to other DUBs, including USP12 and USP46 (13). It is therefore not surprising that the Uaf1−/− mice have a more severe phenotype. The Usp46-deficient mice are viable and show depression-like behavior (27, 28). The role of Usp12 in mouse embryonic development remains unknown, since Usp12-deficient mice have not yet been generated. However, Usp12, but not USP46, regulates early development of Xenopus embryos (29). The embryogenesis defects in Uaf1−/− mice, therefore, may be attributed, at least in part, to the dysfunction of USP12. The substrates of the Usp12-Uaf1 and Usp46-Uaf1 DUB complexes remain unknown.
How USP1-UAF1 regulates HR repair is also unknown. The regulation is derived, at least in part, from its ability to deubiquitinate FANCD2-Ub, a key player in HR repair. However, Uaf1-deficient cells are also hypersensitive to other genotoxic stresses, such as UV light, suggesting that USP1-UAF1 plays a broader role in DNA repair regulation. Other recent studies indicate that UAF1 may interact with other HR substrates, such as RAD51AP1 (30).
Recent studies indicate that the USP1-UAF1 complex regulates other cellular functions beyond DNA repair. Our studies indicate that this DUB complex may control cell fate decisions by stabilizing ID1. The USP1-UAF1 complex can deubiquitinate polyubiquitinated ID1 and thereby rescue this differentiation inhibitor from degradation. Disruption of the USP1-UAF1 complex, with siRNA or a small molecule, may therefore destabilize ID1 and promote cellular differentiation.
Finally, recent studies indicate that FANCD2-Ub, a substrate of USP1-UAF1, is a transcription factor that stimulates expression of TAp63 (16) and other genes. Elevated Fancd2-Ub results in increased RAS-induced senescence and provides a barrier to tumorigenesis (16). Our current study indicates that Uaf1−/− mESCs also have elevated levels of Fancd2-Ub. Moreover, knockdown of UAF1 inhibits Ras-induced proliferation and tumorigenesis. Taken together, the genetic studies described above provide new evidence that Usp1 and its obligate binding partner, Uaf1, control multiple cellular functions, including HR repair, embryonic development, and a barrier to tumorigenesis. The work further suggests that inhibitors of the USP1-UAF1 complex may have anticancer activity by inhibiting DNA repair, promoting ID1 degradation, and promoting RAS-induced cellular senescence.
ACKNOWLEDGMENTS
We thank members of the D'Andrea laboratory for helpful discussions. We also thank Yuko Fujiwara, Frank Godinho, and Stuart H. Orkin for their help in generating murine embryonic stem cells.
E.P. was supported by postdoctoral fellowships from the Susan G. Komen Foundation (KG101186). This study was supported by NIH grants R01DK43889, R01HL52725, and P01HL048546 to A.D.D. This work was also supported in part by a Center of Excellence in Molecular Hematology grant (5P30 DK49216-19).
Footnotes
Published ahead of print 3 September 2013
REFERENCES
- 1.Symington LS, Gautier J. 2011. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45:247–271 [DOI] [PubMed] [Google Scholar]
- 2.San Filippo J, Sung P, Klein H. 2008. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 77:229–257 [DOI] [PubMed] [Google Scholar]
- 3.O'Connell BC, Adamson B, Lydeard JR, Sowa ME, Ciccia A, Bredemeyer AL, Schlabach M, Gygi SP, Elledge SJ, Harper JW. 2010. A genome-wide camptothecin sensitivity screen identifies a mammalian MMS22L-NFKBIL2 complex required for genomic stability. Mol. Cell 40:645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ. 2012. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 14:318–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316:1160–1166 [DOI] [PubMed] [Google Scholar]
- 6.Jackson SP, Durocher D. 2013. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49:795–807 [DOI] [PubMed] [Google Scholar]
- 7.Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R. 2005. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell 17:331–339 [DOI] [PubMed] [Google Scholar]
- 8.Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D'Andrea AD. 2006. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 8:339–347 [DOI] [PubMed] [Google Scholar]
- 9.Oestergaard VH, Langevin F, Kuiken HJ, Pace P, Niedzwiedz W, Simpson LJ, Ohzeki M, Takata M, Sale JE, Patel KJ. 2007. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol. Cell 28:798–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, D'Andrea AD. 2009. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev. Cell 16:314–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, D'Andrea AD. 2007. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol. Cell 28:786–797 [DOI] [PubMed] [Google Scholar]
- 12.Yang K, Moldovan GL, Vinciguerra P, Murai J, Takeda S, D'Andrea AD. 2011. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev. 25:1847–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohn MA, Kee Y, Haas W, Gygi SP, D'Andrea AD. 2009. UAF1 is a subunit of multiple deubiquitinating enzyme complexes. J. Biol. Chem. 284:5343–5351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murai J, Yang K, Dejsuphong D, Hirota K, Takeda S, D'Andrea AD. 2011. The USP1/UAF1 complex promotes double-strand break repair through homologous recombination. Mol. Cell. Biol. 31:2462–2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mortensen RM, Conner DA, Chao S, Geisterfer-Lowrance AA, Seidman JG. 1992. Production of homozygous mutant ES cells with a single targeting construct. Mol. Cell. Biol. 12:2391–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park E, Kim H, Kim JM, Primack B, Vidal-Cardenas S, Xu Y, Price BD, Mills AA, D'Andrea AD. 2013. FANCD2 activates transcription of TAp63 and suppresses tumorigenesis. Mol. Cell 50:908–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pierce AJ, Hu P, Han M, Ellis N, Jasin M. 2001. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 15:3237–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinstock DM, Nakanishi K, Helgadottir HR, Jasin M. 2006. Assaying double-strand break repair pathway choice in mammalian cells using a targeted endonuclease or the RAG recombinase. Methods Enzymol. 409:524–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams SA, Maecker HL, French DM, Liu J, Gregg A, Silverstein LB, Cao TC, Carano RA, Dixit VM. 2011. USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell 146:918–930 [DOI] [PubMed] [Google Scholar]
- 20.Auerbach AD. 2009. Fanconi anemia and its diagnosis. Mutat. Res. 668:4–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moldovan GL, Dejsuphong D, Petalcorin MI, Hofmann K, Takeda S, Boulton SJ, D'Andrea AD. 2012. Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol. Cell 45:75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. 2011. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475:53–58 [DOI] [PubMed] [Google Scholar]
- 23.Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR, Hofmann K, Dikic I. 2005. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 310:1821–1824 [DOI] [PubMed] [Google Scholar]
- 24.Nicassio F, Corrado N, Vissers JH, Areces LB, Bergink S, Marteijn JA, Geverts B, Houtsmuller AB, Vermeulen W, Di Fiore PP, Citterio E. 2007. Human USP3 is a chromatin modifier required for S phase progression and genome stability. Curr. Biol. 17:1972–1997 [DOI] [PubMed] [Google Scholar]
- 25.Wiltshire TD, Lovejoy CA, Wang T, Xia F, O'Connor MJ, Cortez D. 2010. Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. J. Biol. Chem. 285:14565–14571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang D, Zaugg K, Mak TW, Elledge SJ. 2006. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell 126:529–542 [DOI] [PubMed] [Google Scholar]
- 27.Tomida S, Mamiya T, Sakamaki H, Miura M, Aosaki T, Masuda M, Niwa M, Kameyama T, Kobayashi J, Iwaki Y, Imai S, Ishikawa Abe AK, Yoshimura T, Nabeshima T, Ebihara S. 2009. Usp46 is a quantitative trait gene regulating mouse immobile behavior in the tail suspension and forced swimming tests. Nat. Genet. 41:688–695 [DOI] [PubMed] [Google Scholar]
- 28.Imai S, Mamiya T, Tsukada A, Sakai Y, Mouri A, Nabeshima T, Ebihara S. 2012. Ubiquitin-specific peptidase 46 (Usp46) regulates mouse immobile behavior in the tail suspension test through the GABAergic system. PLoS One 7:e39084. 10.1371/journal.pone.0039084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joo HY, Jones A, Yang C, Zhai L, Smith AD, Zhang Z, Chandrasekharan MB, Sun ZW, Renfrow MB, Wang Y, Chang C, Wang H. 2011. Regulation of histone H2A and H2B deubiquitination and Xenopus development by USP12 and USP46. J. Biol. Chem. 286:7190–7201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sowa ME, Bennett EJ, Gygi SP, Harper JW. 2009. Defining the human deubiquitinating enzyme interaction landscape. Cell 138:389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]