

Summary
With our capabilities to culture and sequence the commensal bacteria that dwell on and within a host, we can now study the host in its entirety, as a supraorganism that must be navigated by the pathogen invader. At present, the majority of studies have focused on the interaction between the host’s microbiota and bacterial pathogens. This is not unwarranted, given that bacterial pathogens must compete with commensal organisms for the limited territory afforded by the host. However, viral pathogens also enter the host through surfaces coated with microbial life and encounter an immune system shaped by this symbiotic community. Therefore, we believe the microbiota cannot be ignored when examining the interplay between the host and viral pathogens. Here we review work that details mechanisms by which the microbiota either promotes or inhibits viral replication and virally-induced pathogenesis. The impact of the microbitota on viral infection promises to be a new and exciting avenue of investigation, which will ultimately lead to better treatments and preventions of virally-induced diseases.
Keywords: Microbiota, viruses, immune response, germ-free mice
Introduction
With the current explosion in publications that examine the interactions between higher organisms and their microbiota, one might think these associations only began a decade ago. However, in reality this relationship has been fine tuned over the last 1.2 billion years, and we have only now developed tools powerful enough to dissect its contribution to host physiology (1). Of the vast beneficial contributions that have been uncovered, protection from invading pathogens is one of the most critical to the health of the host.
Commensal organisms protect the host from pathogenic assault by competing for sites of attachment and nutrients and by producing antimicrobial substances, a phenomenon known as colonization resistance (2). The presence of a dense microbial community on exposed surfaces hinders the ability of pathogenic bacteria to take root and establish a niche.
Microbial colonization not only affords protection from pathogens by limiting real estate, but also confers protection by mediating the maturation and regulation of the immune system. To date, the microbiota have been implicated in the development of secondary lymphoid tissues; such as the gut associated lymphoid tissue, isolated lymphoid follicles, Peyer’s patches, mesenteric lymph nodes, and splenic white pulp (3). Furthermore, they promote immune homeostasis by driving both pro- and anti-inflammatory T cell responses (4). In addition, commensal bacteria contribute to production of IgA and antimicrobial peptides (5). Importantly, unlike colonization resistance, the microbiota’s multifaceted influence on the immune system has far-reaching implications for all pathogenic infections: bacterial, fungal, parasitic, and viral.
Viruses are predominantly transmitted through mucosal surfaces: be it fecal-oral transmission, where the virus transits through the gastrointestinal tract; sexual transmission, where the virus contacts the genital mucosa; or aerosol transmission, where the virus encounters commensals occupying the upper airways. In addition, blood-borne viruses that require arthropod vectors to disseminate encounter diverse commensal communities, not only within the mammalian host but also within their insect distributors. Given that nearly all viral pathogens encounter the host’s microbiota, it is likely that these communities modulate viral infection.
The host’s microbiota can potentially influence viral infections in three ways: their presence could be neutral, their presence could hinder viral infection, or their presence could promote viral infection. In our eyes, the latter two scenarios are of primary interest. Therefore, we have chosen to examine foundational work that describes the means by which the commensal population aids or abets viral replication, transmission, and pathogenesis. Given that this field is in its infancy, a consensus has not been reached on the majority mechanisms that we will discuss, but this makes a review of the field ever more valuable.
To study the role of microbiota in viral infections, one needs to compare virus replication/transmission/pathogenesis in organisms with and without commensal bacteria. There are two commonly used approaches that allow ablation of commensals: treatment with the broad-spectrum antibiotics and the production and maintenance of animals under sterile conditions (germ free, GF). Ideally, both approaches should be used in parallel, as each has its unique strengths and weaknesses. Antibiotics can have adverse effects on the host independent of microbial ablation and promote the selection of resistant bacterial species, making it difficult to achieve a truly aseptic system. However, GF animals have immature secondary lymphoid tissues, which can complicate the interpretation of immunological studies. Unfortunately, even with these two approaches in hand, we are restricted to studying the role of the microbiota in viral infections with established animal models.
The microbiota hinder viral replication and virally-induced disease
Influenza virus
In 2009, the World Health Organization made the identification of host factors that influence the severity of influenza infection a priority of its research agenda. As of now, the vast majority of studies have concentrated on the genetic determinants of susceptibility (6–8). However, disease outcome is not solely dictated by the host’s genetic makeup, but also by the commensal community it harbors. Do these commensal constituents govern the host’s susceptibility to influenza infection? Indeed, there is compelling evidence that disruption of the host’s microbiota increases its vulnerability to influenza (9, 10).
As demonstrated by Abt et al, influenza pathogenesis was greatly exaggerated in both antibiotic-treated and GF mice (Figure 1A). Specifically, these mice exhibited an increase in viral titer, which correlated with a precipitous decrease in survival. Furthermore, their lung epithelium displayed morphological features of degeneration and necrosis, a hallmark of disease severity that was not evident in specific pathogen free (SPF) animals. The administration of a purified innate immune agonist (poly I:C) only partially rescued the mortality of antibiotic treated mice (10), suggesting that full protection against influenza likely requires activation of more than one innate immune pathway (Figure 1A).
Figure 1. A) The host’s microbiota protect against viral infection.

Influenza [based on (10)]. Influenza virus was administered intranasally to conventionally raised, antibiotic-treated (+Abx), and Germ-free C57BL/6 mice. Compared to conventionally raised mice, antibiotic-treated (sterilized conditions) and GF animals exhibited an increase in viral titers within the lung, which correlated with augmented mortality. This phenotype was partially rescued by the addition of the innate immune agonist, poly I:C (+Abx, pIC).
HIV. Top panel [based on (19)]. The diffusion rate of HIV-1 was measured in cervicovaginal mucus (CVM) isolated from healthy subjects. HIV was trapped in lactobacilli acidified CVM compared to neutralized CMV, as the virus diffusion was slowed by 1,000-fold. Bottom panel [based on (20)]. Infectivity of HIV-1 incubated with L-lactic acid at acidic or neutral pH was assessed in an in vitro assay. When compared to neutralized L-lactic acid, acidified L-lactic acid reduced HIV-1 infectivity by approximately 10,000 times.
Dengue. Top and middle panels [based on (25)]. Aedes aegypti were treated with an antibiotic cocktail that ablated all cultivable bacteria (+Abx). After which, they were fed dengue-infected blood meal. Mosquitos that had been treated with antibiotics exhibited a 3-fold increase in viral titers within their midgut. Bottom [based on (24)]. Mosquitos treated with antibiotics were colonized with a single commensal isolate (Proteus) (+Abx, Proteus). The addition of Proteus to aseptic mosquitos returned the virus titer to levels observed in those that were untreated.
B) The host’s microbiota enhance susceptibility to viral infection.
Poliovirus/Reovirus [based on (26)]. Mice expressing the human poliovirus receptor were treated with an antibiotic cocktail that ablated cultivable bacteria (+Abx). After which, either poliovirus or reovirus was administered via the oral route. Elimination of microbiota resulted in a decrease in virus titers and -improved survival. Colonization of antibiotic-treated mice with an antibiotic resistant bacterium (+Abx, Abx resistant bacteria) rescued polivirus replication and virus-induced pathogenesis.
MuLV. Top and middle panels [based on (31) and (32)]. MuLV-infected GF mice demonstrated higher viremia and resisted leukemia compared to infected conventionally raised mice. Bottom (31). Immunization of germ free mice with sheep red blood cells (SRBCs) increased the mortality of these mice upon MuLV infection. However, the cause of death was unclear, as leukemia was not documented in these animals. MMTV [based on (36)]. Transmission of MMTV was compared in conventionally raised, antibiotic-treated (+Abx), and GF mice. While the virus persisted indefinitely in conventionally raised animals, it was eliminated from both antibiotic-treated and GF mouse families. Notably, ablation of the MyD88 immune adaptor (MyD88−/− mice) rescued the virus persistence in GF mice. MyD88 functions within the TLR7 innate immune pathway, which senses retroviruses (46).
Like antibiotic-treated mice, GF mice infected with influenza also demonstrated exacerbated lung pathology and increased mortality (10), which indicates the microbiota play a role in protection against the disease. However, the disease exacerbation in influenza-infected GF and antibiotic-treated SPF mice may be due to the loss of different protective mechanisms in these two groups of mice. Whereas antibiotic treatment caused a significant decrease in virus-specific IgG antibodies (Abs) in SPF mice, virus-specific IgG responses were not affected in GF animals (10). It is not clear whether such a disparity was reflected in the cell-mediated immune response; as virus-specific cytotoxic T lymphocyte (CTL) responses, which were significantly decreased in antibiotic-treated mice, were not reported for infected GF mice (10).
Surprisingly, the aggravated disease phenotype observed in mice treated with sterilizing antibiotic cocktail (10) was identical to the disease phenotype seen in SPF mice treated with a non-sterilizing concentration of antibiotics (9). In the latter study, the exacerbated disease phenotype was attributed to a significant decrease in virus-specific IgG Abs and CTLs, which was associated with a reduction in neomycin sensitive bacteria and a bloom in enterobacteria (9). Thus, the increased virus pathogenesis could be due to either a drastic depletion of the total microbial population or a dysregulation of the microbiota. Consequently, a more detailed comparison of antibiotic-treated SPF and GF infected animals is required to uncover how the microbiota modulate the host’s susceptibility to this pathogen.
It must be noted that the most severe and fatal cases of influenza are associated with pneumonia induced by bacterial co-infection (11–13). Patients that arrive at the hospital with symptoms indicative of pneumonia receive antibiotic treatment independent of their influenza status (13). Given that either the dysregulation or ablation of commensal organisms enhances disease severity, it is worth evaluating the consequences and benefits of administering antibiotic therapy in the context of influenza virus infection. To date, there has not been a comprehensive investigation comparing the affects of such therapy on disease outcome in co-infected individuals.
Human Immunodeficiency Virus
Per exposure event, the probability of women becoming infected with HIV-1 via vaginal intercourse is significantly lower than that of rectal or parenteral transmission (14). However, imbalance in the vaginal flora due to bacterial vaginosis (BV) increases the risk of acquiring and transmitting HIV-1(15) (16). This suggests the balanced microbial consortium associated with a healthy vagina plays a protective role in HIV-1 acquisition.
A healthy vaginal flora is dominated by lactobacilli that acidify the vagina with lactic acid, a known antimicrobial agent (17, 18). Lactobacilii generate L-lactic acid as a metabolic byproduct while hydrolyzing glycogen stored by mucosal epithelial cells. Two studies provide strong evidence that L-lactic acid has broad-spectrum HIV virucidal activity at low pH. Lai et al established that lactic acid-acidified cervicovaginal mucus (CVM) trapped HIV-1 within the vaginal mucus layer (Figure 1A), where the virus diffused approximately 100 times more slowly than in neutralized mucus (19). This phenomenon required the presence of the lactic acid molecule, as acidified conditions alone did not alter the rate of HIV-1 diffusion. Recent studies published by Aldunate et al. reported that L-lactic acid was also capable of inactivating both HIV-1 and HIV-2 in vitro (20). The HIV virucidal activity of lactic acid was pH dependent, as neutralized L-lactic acid lost its ability to inhibit HIV-1 infectivity (Figure 1A). Thus, lactic acid produced by lactobacilli can directly suppress HIV infectivity. In addition, lactic acid kills BV-associated bacteria under physiological conditions (21). Therefore, lactic acid can also have an indirect impact on HIV transmission by preventing BV, which is associated with an increased risk of HIV acquisition (15, 16, 22).
Dengue virus
Dengue viruses, the causative agents of dengue fever in humans, are predominantly transmitted by the Aedes aegypti mosquito (23). The virus, taken up with dengue laden blood, first infects the mosquito gut tissue and then migrates to the salivary glands, where it can be transmitted to another host during feeding (23). The fact that both the virus and microbiota inhabit the arthropod midgut prompted the investigation of the microbiota’s role in dengue virus replication/transmission.
It was observed that the commensal microbiota of Aedes aegypti is capable of controlling Dengue virus replication, as virus titers were significantly increased in mosquitoes whose commensals were ablated by antibiotics (Figure 1A) (24). In addition, the virus resistance phenotype in aseptic mosquitos was rescued by colonization with a single commensal isolate, Proteus sp. (Figure 1A) (24), indicating that a single bacterial species can mitigate virus infection.
To gain insights into the microbiota-mediated virus restriction, the authors compared the transcript abundance in the midgut of mosquitos fed on dengue-infected or naïve blood. This analysis revealed an elevation in expression of Toll pathway-regulated antimicrobial peptide genes in septic but not aseptic mosquitoes, suggesting that the endogenous bacterial flora were stimulating immune gene expression in response to the virus. Repression of the Toll pathway through MyD88 gene silencing resulted in elevated dengue titers, which mirrored those of the aseptic MyD88-sufficient mosquitos (Figure 1A) (24).
The fact that the virus alone was a poor activator of Toll and that the bacterial flora did not influence virus infectivity in vitro (25) indicates that the microbiota have an indirect role in modulating dengue virus infection, possibly through basal level stimulation of the Toll immune pathway. However, how this bacterial community mediates the anti-viral immune response remains to be elucidated.
The host’s microbiota facilitate viral replication and pathogenesis
Picornavirus/reovirus
Throughout its transmission, poliovirus is intimately associated with the microbiota of the human host, as it is spread via the fecal-oral route. Remarkably, this association is required for the success of virus transmission and also influences subsequent pathogenesis (26).
Before we delve into the results of this study, it is imperative to address the species tropism of poliovirus. Poliovirus is tropic for the human species; only humans express the receptor required for virus attachment, poliovirus receptor (PVR). However, using transgenic mice expressing the human PVR (PVRtg) can circumvent this tropism, which allows investigators to examine the virus in a tractable system. Consequently, the authors utilized PVRtg mice in conjunction with antibiotic therapy to address whether the intestinal microbiota effect poliovirus infection.
Antibiotic treated mice exposed to poliovirus via the oral route exhibited a significant decrease in mortality relative to their untreated counterparts (Figure 1B). Notably, this effect was not unique to poliovirus; as antibiotic treatment decreased the pathogenesis of reovirus, an enteric pathogen from a divergent viral family. In addition, colonization of antibiotic treated mice with a single antibiotic resistant bacterium made the animals susceptible to poliovirus infection, recapitulating the phenotype observed in mice colonized with their natural flora.
The enhanced susceptibility of microbially replete animals was a result of increased poliovirus replication. To gain insight into the microbiota-enhanced virus replication, the authors utilized an in vitro assay where susceptible cells were exposed to poliovirus pretreated with fecal material with or without microbiota. The virus that came in contact with commensal organisms was significantly more infectious than that exposed to fecal material lacking a microbial population (derived from GF mice), indicating that microbiota directly impact virus infectivity. Furthermore, exposure of poliovirus to a broad spectrum of individual bacteria recapitulated the enhanced infectivity phenotype. Further experiments showed that N-acetylglucosamine (GlcNAc)-containing surface polysaccharides such as peptidoglycan and Lipopolysaccharid (LPS) increased virus infectivity. These bacterial products, bound to virus particles, enhanced virus attachment to the target cells (26) and increased virus stability (Dr. Pfeiffer, personal communication). Additionally, GlcNAc containing compounds of non-bacterial origin could also promote virus infectivity (Dr. Pfeiffer, personal communication). The exact mechanism by which GlcNAc containing molecules abet viral infectivity however, is yet to be determined.
Murine Leukemia Virus
Murine Leukemia Virus (MuLV) is a gammaretrovirus, which causes leukemia/lymphomas in mice from susceptible backgrounds. Exogenous virus can be transmitted either through the blood (birth, bites, and scratches), or through the oral route via the milk of an infected mother. Interestingly, the oral route appears to be the most efficient for virus transmission. This is best exemplified by a recent study that compared post-partum and intra-partum transmission rates of MuLV (27). The key message to take away from this work is when pups born to an uninfected mother were transferred to an infected lactating female, the rate of transmission was 97% compared to 78% observed in mice born to an infected female and transferred to an uninfected lactating female (27).
Like many retroviruses, which induce malignancies, MuLV does not encode an appreciated oncogene in its genome. Tumor induction by these viruses is explained by proviral integration in the proximity of a cellular proto-oncogene, which activates its expression. Activation of an oncogene leads to immortalization of the target cell. However, the malignant transformation of immortalized cells requires additional steps, often referred to as ‘second hits’.
MuLV-induced leukemia requires a series of events beginning with early infection of hematopoietic stem cells (HSCs) in the bone marrow and development of spleen hyperplasia. The latter is the consequence of a compensatory hematopoiesis resulting from diminished normal hematopoiesis in the infected bone marrow (28, 29). The preleukemic HSC that has undergone an initial transformation event (up-regulation of an oncogene, for example) will have acquired additional mutations necessary to progress to leukemia (30).
Given the most successful route of infection houses an abundant microbial population, is there evidence that the microbiota plays a role in MuLV infection and subsequent pathogenesis? Two papers (31, 32), and our own unpublished study, examined the contribution of the microbiota to MuLV-induced leukemia in mice susceptible to infection. Accordingly, conventionally raised and germ-free BALB/c mice were infected with MuLV via intraperitoneal injection, and the disease process was monitored. In all studies, GF mice were significantly more resistant to MuLV-induced leukemia and exhibited a lower mortality rate than their conventionally raised counterparts (31, 32) (Figure 1B). Importantly, the level of viremia was as much as 10-fold lower, relative to SPF mice, during the period preceding the development of leukemia (32).
MuLV requires dividing cells for successful replication. Therefore, Kouttab et al proposed that the relative resistance of GF mice to leukemogenic effects of MuLV could be due to either decreased frequency or low permissiveness of target cells. To test this possibility, they stimulated the immune system of MuLV-infected GF mice via injection of sheep erythrocytes (SRBC) and monitored their survival. Although infected SRBC-injected GF mice died at the same rate as their conventionally raised infected counterparts, none of them developed symptoms of leukemia (31) (Figure 1B). Whether the microbiota modulates MuLV-induced disease directly, by influencing virus replication, or indirectly, by contributing to the ‘second hit’ in the transformation pathway, is yet to be determined.
Mouse Mammary Tumor Virus
Mouse Mammary Tumor Virus (MMTV) is a betaretrovirus that has been circulating in mice for over 20 million years. Exogenous MMTV is passed from mothers to their offspring through milk and infects lymphoid cells before spreading to mammary epithelial cells where is causes mammary tumors (33). In 1985, Outzen et al found that C3H/HeJ mice carrying a mutation in the innate immune Toll-like 4 (TLR4) gene exhibited a delay in mammary tumor development (34). However, the underlying cause for this decrease in pathogenesis remained elusive until our discovery of a TLR4-dependent mechanism used by MMTV to evade the anti-virus immune response (35). In the course of our studies, we found that MMTV interacts with TLR4 and triggers production of the immunosuppressive cytokine, interleukin 10 (IL-10), which blocks the anti-viral response (35). MMTV-induced signaling through TLR4 required bacterially produced LPS, a well-characterized TLR4 ligand. In fact, LPS-free MMTV stocks failed to induce IL-10 production. Furthermore, SPF mice deficient in either TLR4 and IL-10, as well as GF wild type mice, were unable to transmit infectious virus through successive generations (36).
Despite exhibiting overall similarities, the phenotypes of these SPF and GF mice were not identical. Whereas GF mice eliminated the virus in one generation, it took multiple generations of IL-10- and TLR4-deficient SPF mice to eradicate the virus, even though the virus titer was reduced with each passage (36). Since GF mice were infected via peritoneal injection (a single virus dose) and SPF mice ingested the virus with mother’s milk (suckled for 17 days), the differences in the phenotypes between GF and SPF mice could be explained by the different routes of infection or by variation in virus dose. To discriminate between these two possibilities, we limited the exposure of TLR4-deficient or TLR4-sufficient newborn mice to milk-borne virus by only allowing them to suckle for 2 days on infected mothers. They were aged and screened for infection. Whereas 100% of TLR4-sufficient mice maintained the virus, 70% of TLR4-deficient mice were virus-free (Figure 2). Thus, the reduction in the dose of orally-transmitted virus resulted in accelerated virus eradication in resistant SPF mice, indicating the dose rather than the route of infection determines successful virus transmission.
Figure 2. High virus titers can overcome host protective responses.
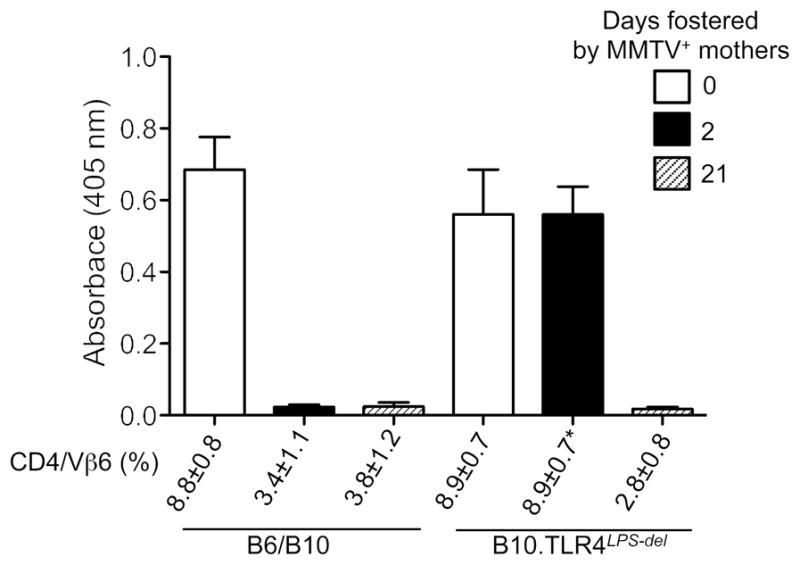
Mice of indicated strains were fostered by MMTV(LA)-infected females (47) for 2 or 21 days. As all productively infected mice show deletion of superantigen (SAg) cognate T cells (48), infection was evaluated by determining percentages of SAg cognate Vβ6+ T cells among CD4+ T cells via FACS analysis at 8 weeks of age. Fostered and control mice were immunized at 9 weeks of age with MMTV(LA) in complete Freund adjuvant as described (49). Production of virus-specific Abs was tested by ELISA (49). Sera from six to twelve mice were used per group. All graphs show mean of OD ± SD. B6 and B10 mice were used interchangeably. Where 100% (7/7) of TLR4-sufficient mice fostered by viremic mothers for 2 days became infected, only 3/11 (30%) of TLR4-deficient mice were infected. Virus-free TLR4-deficient mice (8/11) were exclusively used in immunization studies.
Virus persistence in naturally infected mice (dependent on the functional TLR4 and microbiota) correlated with their inability to respond to viral antigens upon immunization later in life (Figure 2) (36). These data suggest that the virus may induce tolerance to itself by exploiting the tolerogenic properties of the microbiota. Interestingly, TLR4-deficient mice which maintained infectious virus remained virus tolerant (Figure 2). The ability of orally transmitted MMTV to induce tolerance in TLR4-defficient mice indicates the existence of a TLR4-independent pathway capable of suppressing anti-MMTV immunity. This pathway, however, is less potent than that triggered by bacterial LPS, because TLR4-deficient mice reduced virus titers within each consecutive generation (36).
HIV
One major hallmark of HIV infection is chronic immune activation (37). This phenomenon is a powerful predictor of disease outcome, as immune activation correlates with a precipitous decrease in survival (38). Notably, immune activation is not entirely dependent on viral replication, but on a stimulus that is uniquely present in HIV-1-infected individuals.
Infected individuals have a compromised gastrointestinal epithelium [reviewed in (37)], which is partially attributed to virally encoded gp120Env, as purified envelope protein is capable of disrupting the epithelial barrier (39). It has been proposed that barrier disruption leads to the translocation of microbial ligands, which results in aberrant immune activation. This hypothesis is corroborated by the presence of LPS, a potent immunostimulus, in the sera of HIV-1 infected patients (40).
LPS has been directly visualized in the mesenteric lymph nodes of Simian Immunodeficiency Virus (SIV)-infected rhesus macaques (41). Within the lymph node, LPS was surrounded by a suite of macrophages and dendritic cells, all of which expressed high levels of the proinflammatory cytokine IL-18, a primary indicator of activation of these cell subsets. Thus, disruption of the gastrointestinal epithelial barrier allows LPS to traverse the epithelium and disseminate throughout the infected macaques, promoting systemic immune activation.
While there is strong evidence for LPS translocation, there is scant evidence to support the translocation of Gram-negative commensal microbes, producers of LPS, across the intestinal epithelial barrier. Though LPS was visualized in the aforementioned study, there was no evidence for the dissemination of commensal bacteria in any of the major organs or the mesenteric lymph nodes of infected macaques (41). Moreover, sepsis in HIV-infected individuals is typically reported as a result of co-infection with an enteric pathogen, rather than the translocation of commensal organisms (42). Therefore, it is likely that the term ‘microbial translocation’ primarily refers to the systemic influx of microbial products, and not the microbiota.
Does the translocation of microbial products and the resulting immune activation benefit HIV-1? Even though non-proliferating quiescent T cells can be infected by HIV-1, they harbor the virus in an inactive state, and transition to productive infection is dependent upon mitogenic stimulation of these cells (43, 44). Thus, it is possible that the immune activation coinciding with HIV-1 infection is required for productive infection of the target cells. Furthermore, there may be a direct interaction between the virus and LPS, as HIV-1 gp120Env binds LPS in vitro (45). Therefore, it is possible that HIV and SIV may also take advantage of commensal bacteria to assure successful propagation and spread. However, to test such a hypothesis, HIV-1 must be isolated from infected individuals and the associated LPS must be determined.
Concluding remarks and open questions
As we showed in this review, the microbiota of the host affects the replication and transmission of a diverse array of viral pathogens. Therefore, it is tempting to speculate that the composition of an individual’s microbiota can influence disease outcome during viral infection, making it a potential target for therapeutic intervention. However, the dual role of the microbiota in viral infection necessitates the development of therapies that are uniquely tailored to the infectious agent. In the case of microbially-mediated anti-virus protection the exploration of probiotic therapy might be beneficial. In the case of microbially-promoted viral infection, one would need to develop a molecular scalpel to replace the unwieldy ax of broad-spectrum antibiotics. Creation of both types of therapy would require identification of either the individual bacterial species or bacterial ligands responsible for anti- and pro-viral effects.
Acknowledgments
The authors thank Alexander Chervonsky for helpful discussion and to Patrick Lane for help with Figure 1. This work was supported in part by PHS grant AI090084 to T.V.G. This work was also supported by a grant (P30 CA014599) to the University of Chicago.
References
- 1.Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol. 2008;6:776–788. doi: 10.1038/nrmicro1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiological reviews. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 3.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith PM, Garrett WS. The gut microbiota and mucosal T cells. Frontiers in microbiology. 2011;2:111. doi: 10.3389/fmicb.2011.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sommer F, Backhed F. The gut microbiota--masters of host development and physiology. Nat Rev Microbiol. 2013;11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L, Katz JM, Gwinn M, Dowling NF, Khoury MJ. Systems-based candidate genes for human response to influenza infection. Infection, genetics and evolution : journal of molecular epidemiology and evolutionary genetics in infectious diseases. 2009;9:1148–1157. doi: 10.1016/j.meegid.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horby P, et al. What is the evidence of a role for host genetics in susceptibility to influenza A/H5N1? Epidemiology and infection. 2010;138:1550–1558. doi: 10.1017/S0950268810000518. [DOI] [PubMed] [Google Scholar]
- 8.Horby P, Nguyen NY, Dunstan SJ, Baillie JK. The role of host genetics in susceptibility to influenza: a systematic review. PLoS One. 2012;7:e33180. doi: 10.1371/journal.pone.0033180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ichinohe T, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abt MC, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. 2012;37:158–170. doi: 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beadling C, Slifka MK. How do viral infections predispose patients to bacterial infections? Curr Opin Infect Dis. 2004;17:185–191. doi: 10.1097/00001432-200406000-00003. [DOI] [PubMed] [Google Scholar]
- 12.McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clinical microbiology reviews. 2006;19:571–582. doi: 10.1128/CMR.00058-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chertow DS, Memoli MJ. Bacterial coinfection in influenza: a grand rounds review. Jama. 2013;309:275–282. doi: 10.1001/jama.2012.194139. [DOI] [PubMed] [Google Scholar]
- 14.Hladik F, McElrath MJ. Setting the stage: host invasion by HIV. Nat Rev Immunol. 2008;8:447–457. doi: 10.1038/nri2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taha TE, et al. Bacterial vaginosis and disturbances of vaginal flora: association with increased acquisition of HIV. AIDS. 1998;12:1699–1706. doi: 10.1097/00002030-199813000-00019. [DOI] [PubMed] [Google Scholar]
- 16.Cohen CR, et al. Bacterial vaginosis associated with increased risk of female-to-male HIV-1 transmission: a prospective cohort analysis among African couples. PLoS Med. 2012;9:e1001251. doi: 10.1371/journal.pmed.1001251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ravel J, et al. Vaginal microbiome of reproductive-age women. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(Suppl 1):4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gajer P, et al. Temporal dynamics of the human vaginal microbiota. Science translational medicine. 2012;4:132ra152. doi: 10.1126/scitranslmed.3003605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai SK, et al. Human immunodeficiency virus type 1 is trapped by acidic but not by neutralized human cervicovaginal mucus. Journal of virology. 2009;83:11196–11200. doi: 10.1128/JVI.01899-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aldunate M, et al. Vaginal concentrations of lactic acid potently inactivate HIV. The Journal of antimicrobial chemotherapy. 2013 doi: 10.1093/jac/dkt156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boris S, Barbes C. Role played by lactobacilli in controlling the population of vaginal pathogens. Microbes Infect. 2000;2:543–546. doi: 10.1016/s1286-4579(00)00313-0. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell C, et al. Interaction between lactobacilli, bacterial vaginosis-associated bacteria, and HIV Type 1 RNA and DNA Genital shedding in U.S. and Kenyan women. AIDS Res Hum Retroviruses. 2013;29:13–19. doi: 10.1089/aid.2012.0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gubler DJ. Dengue and dengue hemorrhagic fever. Clinical microbiology reviews. 1998;11:480–496. doi: 10.1128/cmr.11.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramirez JL, et al. Reciprocal tripartite interactions between the Aedes aegypti midgut microbiota, innate immune system and dengue virus influences vector competence. PLoS neglected tropical diseases. 2012;6:e1561. doi: 10.1371/journal.pntd.0001561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xi Z, Ramirez JL, Dimopoulos G. The Aedes aegypti toll pathway controls dengue virus infection. PLoS pathogens. 2008;4:e1000098. doi: 10.1371/journal.ppat.1000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuss SK, et al. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. 2011;334:249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duggan J, Okonta H, Chakraborty J. Transmission of Moloney murine leukemia virus (ts-1) by breast milk. J Gen Virol. 2006;87:2679–2684. doi: 10.1099/vir.0.82015-0. [DOI] [PubMed] [Google Scholar]
- 28.Hanna MG, Jr, Walburg HE, Jr, Tyndall RL, Snodgrass MJ. Histoproliferative effect of Rauscher leukemia virus on lymphatic tissue. II. Antigen-stimulated germfree and conventional BALB-c mice. Proc Soc Exp Biol Med. 1970;134:1132–1141. doi: 10.3181/00379727-134-34959. [DOI] [PubMed] [Google Scholar]
- 29.Belli B, Fan H. The leukemogenic potential of an enhancer variant of Moloney murine leukemia virus varies with the route of inoculation. Journal of virology. 1994;68:6883–6889. doi: 10.1128/jvi.68.11.6883-6889.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banerjee P, Crawford L, Samuelson E, Feuer G. Hematopoietic stem cells and retroviral infection. Retrovirology. 2010;7:8. doi: 10.1186/1742-4690-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kouttab NM, Jutila JW. Friend leukemia virus infection in germfree mice following antigen stimulation. Journal of immunology. 1972;108:591–595. [PubMed] [Google Scholar]
- 32.Isaak DD, Bartizal KF, Caulfield MJ. Decreased pathogenicity of murine leukemia virus-Moloney in gnotobiotic mice. Leukemia. 1988;2:540–544. [PubMed] [Google Scholar]
- 33.Nandi S, McGrath CM. Mammary neoplasia in mice. Adv Cancer Res. 1973;17:353–414. [Google Scholar]
- 34.Outzen HC, Corrow D, Shultz LD. Attenuation of exogenous murine mammary tumor virus virulence in the C3H/HeJ mouse substrain bearing the Lps mutation. J Natl Cancer Inst. 1985;75:917–923. doi: 10.1093/jnci/75.5.917. [DOI] [PubMed] [Google Scholar]
- 35.Jude BA, et al. Subversion of the innate immune system by a retrovirus. Nat Immunol. 2003;4:573–578. doi: 10.1038/ni926. [DOI] [PubMed] [Google Scholar]
- 36.Kane M, et al. Successful transmission of a retrovirus depends on the commensal microbiota. Science. 2011;334:245–249. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klatt NR, Funderburg NT, Brenchley JM. Microbial translocation, immune activation, and HIV disease. Trends Microbiol. 2013;21:6–13. doi: 10.1016/j.tim.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salazar-Gonzalez JF, et al. Increased immune activation precedes the inflection point of CD4 T cells and the increased serum virus load in human immunodeficiency virus infection. J Infect Dis. 1998;178:423–430. doi: 10.1086/515629. [DOI] [PubMed] [Google Scholar]
- 39.Nazli A, et al. Exposure to HIV-1 directly impairs mucosal epithelial barrier integrity allowing microbial translocation. PLoS pathogens. 2010;6:e1000852. doi: 10.1371/journal.ppat.1000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brenchley JM, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 41.Estes JD, et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS pathogens. 2010;6:e1001052. doi: 10.1371/journal.ppat.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kankwatira AM, Mwafulirwa GA, Gordon MA. Non-typhoidal salmonella bacteraemia--an under-recognized feature of AIDS in African adults. Tropical doctor. 2004;34:198–200. doi: 10.1177/004947550403400404. [DOI] [PubMed] [Google Scholar]
- 43.Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen IS. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell. 1990;61:213–222. doi: 10.1016/0092-8674(90)90802-l. [DOI] [PubMed] [Google Scholar]
- 44.Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J. 1990;9:1551–1560. doi: 10.1002/j.1460-2075.1990.tb08274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Majerle A, Pristovsek P, Mancek-Keber M, Jerala R. Interaction of the HIV-1 gp120 viral protein V3 loop with bacterial lipopolysaccharide: a pattern recognition inhibition. J Biol Chem. 2011;286:26228–26237. doi: 10.1074/jbc.M111.220434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kane M, Case LK, Wang C, Yurkovetskiy L, Dikiy S, Golovkina TV. Innate Immune Sensing of Retroviral Infection via Toll-like Receptor 7 Occurs upon Viral Entry. Immunity. 2011;35:135–145. doi: 10.1016/j.immuni.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Piazzon I, Goldman A, Torello S, Nepomnaschy I, Deroche A, Dran G. Transmission of an Mls-1a-like superantigen to BALB/c mice by foster-nursing on F1 Mls-1bxa mothers. Sex-influenced onset of clonal deletion. J Immunol. 1994;153:1553–1562. [PubMed] [Google Scholar]
- 48.Marrack P, Kushnir E, Kappler J. A maternally inherited superantigen encoded by mammary tumor virus. Nature. 1991;349:524–526. doi: 10.1038/349524a0. [DOI] [PubMed] [Google Scholar]
- 49.Purdy A, et al. Unique resistance of I/LnJ mice to a retrovirus is due to sustained interferon gamma-dependent production of virus-neutralizing antibodies. The Journal of experimental medicine. 2003;197:233–243. doi: 10.1084/jem.20021499. [DOI] [PMC free article] [PubMed] [Google Scholar]