

Abstract
Inflammation is a fundamental response of the immune system whose successful termination involves the elimination of the invading pathogens, the resolution of inflammation and the repair of the local damaged tissue. In this context, the interleukin 10 (IL-10)-mediated anti-inflammatory response (AIR) represents an essential homeostatic mechanism that controls the degree and duration of inflammation. Here, we review recent work on the mechanistic characterization of the IL-10-mediated AIR on multiple levels: from the cataloguing of the in vivo genomic targets of STAT3 (the transcription factor downstream of IL-10) to the identification of specific co-factors that endow STAT3 with genomic-binding specificity, and how genomic and computational methods are being used to elucidate the regulatory mechanisms of this essential physiological response in macrophages.
Keywords: IL-10, JAK1, STAT3, anti-inflammatory response, macrophages, transcriptional regulatory modules, bioinformatics
THE IL-10-MEDIATED ANTI-INFLAMMATORY RESPONSE
Inflammation is a crucial response to threats to homeostasis, and the complex molecular events triggering inflammation are best understood in the context of infection and tissue injury in mammals [1–3]. The anti-inflammatory response (AIR) is to a large extent controlled by interleukin 10 (IL-10), a multifunctional cytokine whose vast range of effects in cells of the immune system we are only beginning to understand. A general consensus, however, is that IL-10 is a major immunosuppressive cytokine with such potent effects that its activity must be tightly regulated. Otherwise the outcomes typically include serious diseases related to IL-10 over-production leading to unwanted immunosuppressive effects (e.g. Epstein–Barr virus associated lymphomas, tumour growth and lupus erythematosus), or to IL-10 deficiency leading to unrelenting immune activation (e.g. psoriasis, rheumatoid arthritis and chronic inflammatory bowel diseases) (reviewed in [4]).
IL-10 is the founding member of the so-called IL-10 family (including IL-10, IL-19, IL-20, IL-22, IL-24 and IL-26) [5] as well as one of the best-studied anti-inflammatory cytokines, both in the context of acute and chronic inflammation [4]. IL-10 is produced by nearly all leukocytes, including macrophages, dendritic cells (DC), natural killer (NK) cells, neutrophils, eosinophils, mast cells, B cells and a number of CD4+ T-cell subsets (including Th1, Th2, Th17, Th22, Treg and Tr1) (reviewed in [4]). All these cell types secrete IL-10 in response to different stimuli, thus providing the immune system with an exquisitely regulated mechanism to control the effects of IL-10 in a tissue-specific and temporal manner. For instance, in some of the best-understood examples monocytes and macrophages respond to IL-10 upon TLR4 stimulation with bacterial lipopolysaccharide (LPS) [4], whereas T cells secrete IL-10 upon stimulation of their T-cell receptor (TCR) [6,7]. Whereas IL-10 produced by T cells is required to control chronic inflammation, it seems to be dispensable during acute inflammation [8].
IL-10 exerts its effects by binding to its cognate receptor (IL-10R), a tetramer composed of two distinct chains (IL-10R1 and IL-10R2). IL-10R1 specifically binds IL-10, allowing a conformational change that enables the oligomerization with IL-10R2 [9]. IL-10R2 is relatively widely expressed, whereas IL-10R1 is restricted to leukocytes and lymphoid organs with particularly high levels in monocytes and macrophages (both in human and mouse), although it is also expressed in NK cells, CD4+ and CD8+ T cells, DCs, B cells and mast cells (BioGPS [10]). Therefore, the effects of IL-10 appear to be restricted to differentiated cells of the immune system. In the context of the AIR, IL-10 binding to IL-10R activates the IL-10/JAK1/STAT3 cascade, where phosphorylated STAT3 homodimers translocate to the nucleus within seconds to activate the expression of target genes (Figure 1). Extensive efforts to dissect the IL-10/JAK1/STAT3 anti-inflammatory pathway have led to some clear conclusions, previously summarized by Murray [11]. Most importantly, both IL-10 and STAT3 are essential for the AIR and cannot be replaced by any other cytokine or transcription factor (TF). STAT3 activity by itself does not result in an AIR; instead STAT3 activates a number of effector genes (the AIR factors), which subsequently repress the pro-inflammatory genes primarily at the level of transcription (Figure 1) [12]. The IL-10/STAT3-mediated AIR is known to function in cell types other than macrophages such as DCs, where it works in opposition to the pro-inflammatory IL-6, which also signals via STAT3 [13]. In addition, IL-10 also suppresses cytokine production in neutrophils [14]. On a systemic level, the IL-10 knockout mouse gives rise to chronic intestinal inflammation in a model of Crohn’s disease [15], but the wide expression of the IL-10 receptor throughout the immune system suggests the existence of additional roles. For example, IL-10 regulates apoptosis in B cells [16], and IL-10 produced by Th2 cells directly suppresses Th1 cells and can indirectly suppress Th2 cell activity [17,18]. Indeed, IL-10 was originally discovered (as ‘cytokine synthesis inhibitory factor’) in the context of Th1/Th2 cross-regulation [19]. Still, the mechanisms whereby all of these cell type-specific IL-10 functions combine to orchestrate a systemic IL-10-mediated AIR remain to be elucidated.
Figure 1:
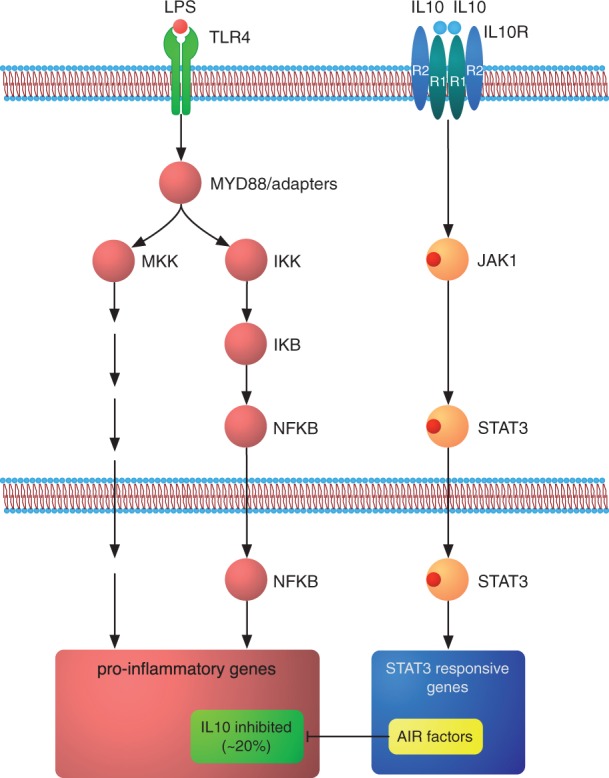
The IL-10 AIR in the context of inflammation. The TLR-initiated inflammatory response activates a set of well-understood signalling pathways, which ultimately lead to the expression of thousands of pro-inflammatory genes. IL-10 initiates the AIR upon binding to its cognate receptor, where JAK1 phosphorylates STAT3. Neither IL-10, the IL-10R, the Janus kinase or STAT3 can be functionally replaced in this pathway. Upon entering the nucleus, STAT3 activates specific target genes among which the ultimate effectors of the AIR (the ‘AIR factors’) are found. The AIR factors repress the expression of pro-inflammatory genes mostly at the transcriptional level, with only ∼20% of the genes activated by LPS in macrophages eventually being repressed by IL-10. Adapted from Murray [11].
The anti-inflammatory effect of IL-10 in macrophages manifests as the transcriptional inhibition of ∼20% of the LPS-induced genes [20], including pro-inflammatory mediators such as cell surface receptors, chemokines and cytokines. As mentioned above, STAT3’s role is to stimulate the expression of specific genes (AIR factors), which in turn suppress the expression of pro-inflammatory genes. The search for the AIR factors has been particular intense, and several factors have been identified. These include Bcl3, which works by impairing NF-κB’s ability to bind DNA, recruits HDAC1 and suppresses TNF-α production [21–23]; Etv3, a transcriptional co-repressor that inhibits NF-κB’s activity [24]; SHIP-1, which acts via a STAT3-independent mechanism to inhibit TNF-α translation [25]; miR-155, which targets SHIP-1 [26]; ABIN-3, which inhibits NF-κB activation but is dispensable in human [27]; Nfil3, which suppresses IL12b expression [28,29] and Zfp36, an RNA-binding protein shown to target AU-rich elements in the 3′ untranslated region (UTR) of TNF-α mRNA [23,30,31] (Table 1). However, this list of factors with anti-inflammatory properties cannot account for the full IL-10-mediated AIR for three reasons: (i) formal proof that their absence partly impairs the IL-10 AIR has only been shown for Bcl3 [21] and Zfp36 [31]; (ii) they cannot account for the inhibition of all pro-inflammatory mediators that are induced by TLR4 stimulation and (iii) not all of them have been shown to be directly regulated by STAT3; indeed, SHIP-1 is completely independent of STAT3 activity [25].
Table 1:
List of factors previously implicated in the AIR, detailing their relationship to STAT3, mechanisms of action and end targets
| AIR factor | Regulated by STAT3? | Nearest STAT3 peak from the gene’s TSS | mRNA differentially regulated after 4 h of IL-10 treatment in peritoneal macrophages? (Hutchins et al. [32]) | End target/mechanism of action? | References |
|---|---|---|---|---|---|
| Bcl3 | Yes, and a common STAT3 target gene in multiple cell types. | −447 bp | Yes, upregulated | Impairs NF-κB’s DNA binding ability; recruits HDAC1 and suppresses TNF-α production. | Kuwata et al. [21]; Wessells et al. [22]; Hutchins et al. [23]. |
| Etv3 | Reported as upregulated in bone marrow-derived macrophages (El Kasmi et al. 2007), but its expression is unchanged in peritoneal macrophages (Hutchins et al.2012). | −187 kb | Noa | Transcriptional co-repressor, inhibits NF-κB transcriptional activity. | El Kasmi et al. [24]; Hutchins et al. [32]. |
| Hmox1 (HO-1) | Thought to be responsible for the entire IL-10/STAT3 AIR, but remains controversial. | >200 kb | Nob | Unknown downstream mechanism. | Lee and Chau [65]. |
| Inpp5d (SHIP-1) | No, it acts via a STAT3-independent pathway. | >200 kb | No | SHIP-1 mediated inhibition of TNF-α translation. | Chan et al. [25]. |
| miR-155 | Unknown if directly targeted by STAT3. | 12 267 bp | Data not availablec | miR-155 targets SHIP-1. | McCoy et al. [26]. |
| Nfil3 | Yes, Nfil3 expression is lost in STAT3 conditional knockouts [28]. | 66 kb | Yes, upregulated | Suppresses Il12b expression | Smith et al. [28]; Kobayashi et al. [29]. |
| Nfkbid (IkBNS) | Unknown if directly targeted by STAT3. | >200 kb | No | NF-κB transcriptional co-repressor. | Kuwata et al.[66]. |
| Sbno2 | Yes | −104 bp | Yes, upregulated | Transcriptional co-repressor, inhibits NF-κB transcriptional activity. | El Kasmi et al. [24]. |
| Tnip3 (ABIN-3) | ABIN-3 can suppress NF-κB activity in human, but is dispensable in mice. | >200 kb | Nod | Inhibits NF-κB activation. | Weaver et al. [27]. |
| Zfp36 | Yes, and a common STAT3 target gene in multiple cell types. | −19 bp | Yes, upregulated | RNA-binding protein against ATTTA elements. Targets TNF-α mRNA. | Schaljo et al. [30]; Gaba et al. [31]; Hutchins et al. [23]. |
aReported as upregulated in bone marrow-derived macrophages (El Kasmi et al. 2007).
bProtein levels are reported as upregulated in peritoneal macrophages (Lee and Chau 2002).
cReported as suppressed by IL-10 (McCoy et al. 2010).
dReported as upregulated in mouse and human mononuclear phagocytes (Weaver et al. 2007).
HIGH-THROUGHPUT GENOMICS AND THE QUEST FOR THE ELUSIVE AIR FACTORS
STAT3 does not ultimately inhibit the transcription of all LPS-induced genes, but only ∼20% [20], although the inhibition of this fraction is sufficient and essential for a functional IL-10-mediated AIR. Therefore, the next logical step is to characterize the genomic-binding sites of STAT3 during the AIR. In the first systematic approach to study the IL-10-mediated AIR using next-generation sequencing, we integrated STAT3 ChIP-seq and RNA-seq with computational analysis to obtain a global understanding of the effect of IL-10 on macrophages [32]. The most important conclusions of this study are: (i) the genome-wide binding patterns of STAT3 are specific to the AIR as the overlap between the STAT3 peaks in unstimulated (n = 602) and IL-10 treated macrophages (n = 1302) is <1%. Moreover, the canonical STAT3-binding motif (the GAS motif) can be retrieved from the vast majority of IL-10-specific peaks, which also appear to be under moderate evolutionary selection and are broadly associated with immune functions. (ii) The major effect of STAT3 on its target genes (by proximity) is to promote their transcriptional activation; therefore, STAT3 functions as a positive transcriptional regulator during the AIR. (iii) As new protein production is essential for the AIR, we reasoned that the ultimate effectors of the AIR might be found among the STAT3 targets that become activated upon IL-10 stimulation [32]. Of those genes with a STAT3-binding site within 20 kb of their transcription start sites (TSS), 351 were found to be up-regulated upon IL-10 stimulation. Among these, previously identified STAT3 targets such as Bcl3 and Socs3 were found. Although we cannot rule out the possibility that the AIR is mediated by certain factors released in a form of paracrine signalling, or by STAT3-regulated factors that compromise the stability of certain mRNA messages or their translation, a likely possibility is that STAT3 up-regulates a number of transcriptional repressors, which in turn suppress the expression of pro-inflammatory genes. Focusing on sequence-specific DNA-binding factors, a filtered list of 42 distinct genes emerged which contains a number of relevant candidates with sustained patterns of gene expression over an 8 h period (thus supporting their putative involvement in the AIR). These candidates may be grouped into distinct categories: (i) repressive TFs previously linked to inflammation (e.g. Ikaros, Ppard, Nfkbiz); (ii) repressive TFs not linked to inflammation before (e.g. Cic, whose Drosophila homologue, Capicua, is a well-known repressor involved in dorsoventral patterning) and (iii) a bunch of TFs about which very little is known (e.g. Vav1, Rlf, Fblim1, Brd2) [32].
STAT3 IS A PLEIOTROPIC TF WITH CELL TYPE-SPECIFIC AND UNIVERSAL FUNCTIONS
STAT3 has a crucial role in the IL-10 mediated AIR but it is also involved in a great diversity of cellular functions, which naturally leads one to questioning the underlying mechanisms that make this possible. For instance, what properties determine how STAT3 can be anti-inflammatory in one cell type and pro-inflammatory in another? Indeed, even within the same cell type, STAT3 can be both pro- and anti-inflammatory: when DCs are stimulated by IL-6, STAT3 is pro-inflammatory, but when stimulated by IL-10, STAT3 is anti-inflammatory [13]. By seeking to understand how STAT3 can function in a diversity of cellular contexts, we might be able to unveil the mechanisms whereby STAT3 specifically activates an anti-inflammatory programme in macrophages and DCs. STAT3 is constitutively expressed, in contrast with other TFs whose expression is restricted to a single cell type [e.g. OCT4/Pou5f1 is virtually only expressed in embryonic stem cells (ESC)], or to single germ lineages, such as STAT4 (mesodermal) or SoxB1 subfamily members (ectodermal) [33]. Therefore, STAT3 is the quintessential pleiotropic TF as it is involved in the regulation of a myriad processes, including metabolism (with specific effects on obesity and glucose tolerance [34–38]), development (e.g. cardiomyocytes [39], liver regeneration [40], haematopoiesis [41–44]) and in orchestrating responses to environmental stimuli, particularly the previously described anti-inflammatory effect in IL-10 stimulated myeloid cells [20,32,45–47]). Not surprisingly, mutations in the coding region of STAT3, which can have both activating and inactivating effects, are responsible for many diseases, including the Hyper-IgE syndrome and cancers [48,49]. Given the plethora of functions that STAT3 regulates, it is intuitive to think that STAT3 regulates distinct genes in different cell types. This was first shown for a small selection of genes (summarized in [50]), and more recently genome wide by combining ChIP-seq, microarrays and RNA-seq in ESCs [51], CD4+ T cells [52,53], macrophages [32], AtT-20 (pituitary) cells [54] and conventional DCs [44].
Our analysis of the genome-wide binding patterns of STAT3 in ESCs, CD4+ T cells, macrophages and AtT-20 cells unveiled two important regulatory mechanisms encoded in distinct sets of STAT3-binding sites [23]: (i) the vast majority of STAT3-binding events are cell type-specific and likely reflect the different functions that STAT3 performs in these cells and (ii) a small core of 35 STAT3-binding events is found in all four cell types. We call these the ‘cell type-specific’ and ‘universal’ binding modes of STAT3. Whereas the former seems to encapsulate the specific functions that STAT3 executes in distinct cells, the universal core of 35 STAT3-binding sites encodes a crucial self-regulatory mechanism of STAT3 signalling, which: (i) perpetuates Stat3’s transcription by binding to its own promoter; (ii) is a master regulator of important TFs downstream of STAT3; (iii) transcribes specific signalling modulators and cytoplasmic enzymes that fine-tune STAT3 signalling and (iv) ensures a robust cell division programme and the maintenance of a stable phenotype [23]. Some of the target genes of STAT3 in all cell types include Socs3 (which blocks the IL-6 mediated pro-inflammatory response by binding to the IL-6 receptor [55]), Bcl3 (which suppresses TNF-α expression in IL-10-stimulated macrophages [21]) and the protein tyrosine phosphatase Ptpn1, a negative regulator of JAK-STAT signalling that works by dephosphorylating phospho-STAT3 [56]).
In summary, STAT3 is capable of binding to widely divergent sites in distinct cell types, as well as to a small core of evolutionarily conserved sites in all cell types that regulates its own signalling. Collectively, the STAT3-binding sites as determined by ChIP-seq amount to a few thousands at most in any of the cell types investigated [23]. However, in the mouse genome there are potentially over 1.3 million STAT3-binding sites [57], yet ChIP-seq experiments conclusively demonstrate that STAT3 is only ever recruited to a few thousand binding sites at most [23]. An outstanding question here is: How does STAT3 select its ∼1700 genomic targets during the AIR [23,32]?
SPECIFIC CO-FACTORS DETERMINE STAT3’S FUNCTIONS DURING THE AIR
As the functions of STAT3 are directly linked to its ability to bind specific genomic sites and thus regulate the expression of specific genes, working out the underlying mechanism is essential for understanding how the AIR is regulated at the genomic binding level. The DNA-binding preferences of STAT3 are indistinguishable from those of most other STAT family members, a ‘DNA binding degeneracy’ trait common to virtually all TF families as shown by protein-binding microarray and high-throughput SELEX technologies [58–60]. Moreover, the analysis of the STAT3-binding sites identified by ChIP-seq in macrophages, CD4+ T cells, ESCs and AtT-20 cells showed that, although small variations of the GAS motif were identified, these alone cannot account for specific STAT3 functions in any one cell type, and they probably represent alternate methods for STAT3 recruitment [23].
To explain how STAT3 discriminates and selects specific-binding sites during the IL-10-mediated AIR, we developed a novel method for identifying the co-factors that endow STAT3 with DNA-binding selectivity and thus constitute what are called ‘transcriptional regulatory modules’ (TRMs) [23]. The assembly of TFs and co-factors around master regulatory TFs has been shown to operate in ESCs (OCT4, SOX2, NANOG, KLF4, SMAD1, ESRRB and STAT3 [51]), B lymphocytes (PU.1 centres around E2A, EBF1 and FOXO1 [61,62]) and macrophages (PU.1, CEBPA, CEBPB and AP-1 [61]). Our method, called rTRM, systematically reconstructs TRMs by integrating motif enrichment analysis on sets of experimentally characterized binding regions (by ChIP-seq), gene expression data and protein–protein interactions (PPIs) (Figure 2A). By applying rTRM we predicted that STAT3 assembles around two distinct TRMs to enact the AIR and control its cell type-independent (universal) self-regulatory network (Figure 2B) [23]. The macrophage-specific TRM suggests that STAT3 is being recruited to a network containing AP-1 TF family members as well as ETS family members. Intriguingly, CEBPA is a member of the TRM: CEBPA and AP-1 family members together are cell type-specific determinants for macrophages [61], suggesting that STAT3 is co-operating with cell type-specific factors to enact the AIR. This situation is strongly reminiscent of that observed by Natoli and co-workers in their studies of inflammation in macrophages. The authors found that challenging the macrophages with an inflammatory signal leads to NF-κB recruitment at genomic-binding sites already ‘primed’ by the presence of cell type-specific TFs [63,64]. In this way it appears that cell type-determining TFs, besides specifying the macrophage cell type also encode the repertoire of possible signalling responses. Indeed, the macrophage cell type-determining factors PU.1, CEBPA and CEBPB are pre-bound at sites of future STAT3 recruitment: of 1724 STAT3-binding sites, 498 sites are also bound by PU.1, 434 by CEBPA, 474 by CEBPB and 181 by JUNB [23,33,61,63]. Remarkably for PU.1, CEBPA and CEBPB, all three factors are bound to a core of 339 binding sites of future STAT3 recruitment. This finding suggests that the macrophage determining factors PU.1, CEBPA and CEBPB function to specify the macrophage cellular type as well as the genomic locations of future STAT3 binding during the AIR. The experimental exploration of these TRMs will be of great value in understanding macrophage responses and the IL-10/STAT3 AIR.
Figure 2:

Characterization of TRMs. (A) The goal of the rTRM method is to reconstruct TRMs by integrating multiple data sources. Starting with a set of experimentally characterized genomic-binding sites (e.g. from ChIP-seq) (1), these sites are investigated for the presence of over-represented TF binding motifs using computational approaches (2). The over-represented motifs are matched to their corresponding TFs, and these are further filtered by expression data from the same cell type (3). Finally, rTRM harnesses the power of PPIs by investigating the entire PPI space from the BioGRID database to build TRMs from the TFs that have survived the previous filtering steps. (B) The genomic-binding patterns of STAT3 encode two distinct modes of regulation: a ‘macrophage-specific’ regulatory mode characterized by a specific set of co-TFs and co-factors (left panel) and a ‘universal’ regulatory mode characterized by a distinct TRM. Whereas the universal mode of STAT3 regulation serves to perpetuate STAT3 signalling and fine-tune the JAK-STAT pathway, the macrophage-specific mode is responsible for the AIR.
CONCLUDING REMARKS AND PERSPECTIVES
Despite the unquestionable importance of the IL-10/JAK1/STAT3 AIR in the negative regulation of both acute and chronic inflammation, a number of essential aspects still elude our understanding. These include the identities of the AIR factors and the multiple levels of regulation of the AIR. Many putative AIR factors have so far been described (Table 1), but no single factor has yet been able to account for the complete AIR. Emerging evidence suggests that the AIR is a multi-factorial response and that we may never identify a single, universal AIR factor downstream of STAT3. Therefore, systematic experimental and computational approaches will be invaluable in teasing apart the many combinations of factors required to enact a full AIR. The putative AIR factors described so far will need to be explored further using a combination of functional genomics screens, AIR factor-target gene relationships (e.g. by ChIP-seq), as well as in the context of other AIR factors to gain a global understanding of the regulation of the AIR by STAT3-regulated genes. The ultimate aim of research into the AIR should be to achieve a network-based mechanistic understanding with a view to developing tailored and tissue-specific anti-inflammatory therapies. We should be able to cancel specific pro-inflammatory molecules without suppressing the entire inflammatory response, which would leave us defenseless against pathogens (Figure 3).
Figure 3:
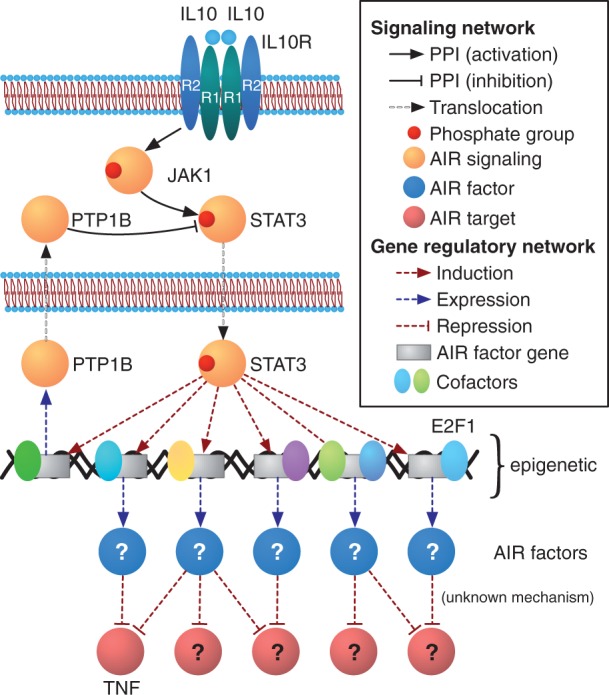
The IL-10/JAK1/STAT3 anti-inflammatory pathway: current state of affairs. The ChIP-seq of STAT3 in IL-10-stimulated macrophages, coupled with RNA-seq, has unveiled the repertoire of genes that are activated by STAT3 during the AIR. Among these the AIR factors shall be found, followed by a detailed characterization of the AIR factor-target gene relationships, and their mechanisms. Another fundamental aspect of the AIR that needs addressing is the role that the epigenetic landscape plays in regulating the AIR, as well as the potential regulation of the AIR by STAT3-induced enzymes, such as PTP1B, a tyrosine phosphatase previously shown to target phospho-STAT3 [56]. An integrated understanding of the AIR factors and their mechanisms of action on their targets, the genomic regulation of the AIR by specific TRMs and by the epigenetic landscape, as well as the cytoplasmic-level regulation of the AIR will provide us with an unprecedented degree of detail on this essential physiological response that we may harness to develop tailored anti-inflammatory therapies.
Inflammation is not the principal cause of many diseases, such as asthma, obesity, cancer atherosclerosis and rheumatoid arthritis, but it is a very important aggravating factor [67]. Current cytokine-based anti-inflammatory therapies include the use of anti-TNF-α humanized monoclonal antibodies and soluble TNF-α receptors, which only target specific factors. The identification of the AIR factors that are specifically activated by STAT3 upon IL-10 stimulation, and the mechanisms whereby these interfere with pro-inflammatory genes alone or in combination with other AIR factors, is an obvious outstanding question. However, the IL-10/JAK1/STAT3 AIR, like all signalling pathways, has multiple levels of regulation that should also be taken into account as these may illuminate additional points for therapeutic intervention.
Key Points.
The IL-10/JAK1/STAT3 AIR is an essential negative regulator that controls the degree and duration of inflammation.
Combined ChIP-seq/RNA-seq studies in IL-10 stimulated macrophages have generated an important list of putative effectors of the AIR (the AIR factors) that are activated by STAT3 and which potentially suppress the expression of pro-inflammatory genes.
Genomic studies across various cell types have unveiled two modes of STAT3 binding: a macrophage-specific mode (responsible for the AIR), and a universal (cell type-independent) mode that regulates STAT3’s own signalling and the fine-tuning of the JAK-STAT pathway.
The reconstruction of TRMs using a combination of experimental and computational techniques, have shown that STAT3 combines with specific factors to perform the AIR.
Additional levels of regulation of the AIR include the combination of AIR factors with other co-factors to target and suppress the expression of pro-inflammatory genes, the regulation of specific AIR factors in the context of the local epigenetic environment and the regulation of the JAK-STAT pathway by cytoplasmic enzymes.
The ultimate aim of research into the AIR is achieving a network-based understanding of this essential physiological response with a view to developing tailored and tissue-specific anti-inflammatory therapies.
Acknowledgements
We thank Mr Shaq Liu for excellent systems support and Ms Mineko Tanimoto for secretarial support.
Biographies
Andrew P. Hutchins read for a PhD at the University of East Anglia (UK) and the John Innes Centre under the supervision of Profs. John Doonan and Clive Lloyd (2004). He later joined the Genome Institute of Singapore as a post-doctoral research fellow in Prof. Paul Robson’s laboratory (2004–10). He has been a member of the Bioinformatics and Genomics laboratory at IFReC since 2010.
Diego Diez obtained a PhD in biochemistry from the Universidad Autónoma de Madrid working under Prof. Juan Bernal (2006). This was followed by a post-doctoral stay in the Bioinformatics Center of Kyoto University under the supervision of Profs. Minoru Kanehisha and Susumu Goto (2006–12). He joined the Bioinformatics and Genomics laboratory in January 2012.
Diego Miranda-Saavedra did his PhD in bioinformatics with Prof. Geoff Barton at the Wellcome Trust Biocentre of the University of Dundee (2007). This was followed by a postdoc in Prof. Bertie Göttgens’ laboratory of molecular hematology at the Cambridge Institute for Medical Research (2007–09). He is currently an associate professor at Osaka University and PI of the Bioinformatics and Genomics laboratory at IFReC since January 2010.
FUNDING
Work in the DMS laboratory is funded by the Japan Society for the Promotion of Science (JSPS) through the WPI-IFReC Research Program and a Kakenhi grant; the Kishimoto Foundation and the ETHZ-JST Japanese-Swiss Cooperative Program.
References
- 1.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–35. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 3.Simonatto M, Natoli G. Functional genomics of the inflammatory response: Where are we now? Brief Funct Genomics. 2013 doi: 10.1093/bfgp/elt023. [Epub ahead of print] PMID: 23814131. [DOI] [PubMed] [Google Scholar]
- 4.Sabat R, Grutz G, Warszawska K, et al. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010;21:331–44. doi: 10.1016/j.cytogfr.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Volk H, Asadullah K, Gallagher G, et al. IL-10 and its homologs: important immune mediators and emerging immunotherapeutic targets. Trends Immunol. 2001;22:414–7. doi: 10.1016/s1471-4906(01)01985-8. [DOI] [PubMed] [Google Scholar]
- 6.Saraiva M, Christensen JR, Veldhoen M, et al. Interleukin-10 production by Th1 cells requires interleukin-12-induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose. Immunity. 2009;31:209–19. doi: 10.1016/j.immuni.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–81. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 8.Roers A, Siewe L, Strittmatter E, et al. T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J Exp Med. 2004;200:1289–97. doi: 10.1084/jem.20041789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoon SI, Logsdon NJ, Sheikh F, et al. Conformational changes mediate interleukin-10 receptor 2 (IL-10R2) binding to IL-10 and assembly of the signaling complex. J Biol Chem. 2006;281:35088–96. doi: 10.1074/jbc.M606791200. [DOI] [PubMed] [Google Scholar]
- 10.Wu C, Orozco C, Boyer J, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr Opin Pharmacol. 2006;6:379–86. doi: 10.1016/j.coph.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci U S A. 2005;102:8686–91. doi: 10.1073/pnas.0500419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braun DA, Fribourg M, Sealfon SC. Cytokine response is determined by duration of receptor and signal transducers and activators of transcription 3 (STAT3) activation. J Biol Chem. 2013;288:2986–93. doi: 10.1074/jbc.M112.386573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cassatella MA, Tamassia N, Crepaldi L, et al. Lipopolysaccharide primes neutrophils for a rapid response to IL-10. Eur J Immunol. 2005;35:1877–85. doi: 10.1002/eji.200526088. [DOI] [PubMed] [Google Scholar]
- 15.Kuhn R, Lohler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 16.Itoh K, Hirohata S. The role of IL-10 in human B cell activation, proliferation, and differentiation. J Immunol. 1995;154:4341–50. [PubMed] [Google Scholar]
- 17.Grunig G, Corry DB, Leach MW, et al. Interleukin-10 is a natural suppressor of cytokine production and inflammation in a murine model of allergic bronchopulmonary aspergillosis. J Exp Med. 1997;185:1089–99. doi: 10.1084/jem.185.6.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hawrylowicz CM, O'Garra A. Potential role of interleukin-10-secreting regulatory T cells in allergy and asthma. Nat Rev Immunol. 2005;5:271–83. doi: 10.1038/nri1589. [DOI] [PubMed] [Google Scholar]
- 19.Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170:2081–95. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lang R, Patel D, Morris JJ, et al. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–63. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- 21.Kuwata H, Watanabe Y, Miyoshi H, et al. IL-10-inducible Bcl-3 negatively regulates LPS-induced TNF-alpha production in macrophages. Blood. 2003;102:4123–9. doi: 10.1182/blood-2003-04-1228. [DOI] [PubMed] [Google Scholar]
- 22.Wessells J, Baer M, Young HA, et al. BCL-3 and NF-kappaB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J Biol Chem. 2004;279:49995–50003. doi: 10.1074/jbc.M404246200. [DOI] [PubMed] [Google Scholar]
- 23.Hutchins AP, Diez D, Takahashi Y, et al. Distinct transcriptional regulatory modules underlie STAT3's cell type-independent and cell type-specific functions. Nucleic Acids Res. 2013;41:2155–70. doi: 10.1093/nar/gks1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El Kasmi KC, Smith AM, Williams L, et al. Cutting edge: a transcriptional repressor and corepressor induced by the STAT3-regulated anti-inflammatory signaling pathway. J Immunol. 2007;179:7215–9. doi: 10.4049/jimmunol.179.11.7215. [DOI] [PubMed] [Google Scholar]
- 25.Chan CS, Ming-Lum A, Golds GB, et al. Interleukin-10 inhibits lipopolysaccharide-induced tumor necrosis factor-alpha translation through a SHIP1-dependent pathway. J Biol Chem. 2012;287:38020–7. doi: 10.1074/jbc.M112.348599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCoy CE, Sheedy FJ, Qualls JE, et al. IL-10 inhibits miR-155 induction by toll-like receptors. J Biol Chem. 2010;285:20492–8. doi: 10.1074/jbc.M110.102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weaver BK, Bohn E, Judd BA, et al. ABIN-3: a molecular basis for species divergence in interleukin-10-induced anti-inflammatory actions. Mol Cell Biol. 2007;27:4603–16. doi: 10.1128/MCB.00223-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith AM, Qualls JE, O'Brien K, et al. A distal enhancer in Il12b is the target of transcriptional repression by the STAT3 pathway and requires the basic leucine zipper (B-ZIP) protein NFIL3. J Biol Chem. 2011;286:23582–90. doi: 10.1074/jbc.M111.249235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi T, Matsuoka K, Sheikh SZ, et al. NFIL3 is a regulator of IL-12 p40 in macrophages and mucosal immunity. J Immunol. 2011;186:4649–55. doi: 10.4049/jimmunol.1003888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schaljo B, Kratochvill F, Gratz N, et al. Tristetraprolin is required for full anti-inflammatory response of murine macrophages to IL-10. J Immunol. 2009;183:1197–206. doi: 10.4049/jimmunol.0803883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaba A, Grivennikov SI, Do MV, et al. Cutting edge: IL-10-mediated tristetraprolin induction is part of a feedback loop that controls macrophage STAT3 activation and cytokine production. J Immunol. 2012;189:2089–93. doi: 10.4049/jimmunol.1201126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hutchins AP, Poulain S, Miranda-Saavedra D. Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood. 2012;119:e110–9. doi: 10.1182/blood-2011-09-381483. [DOI] [PubMed] [Google Scholar]
- 33.Hutchins AP, Diez D, Miranda-Saavedra D. Genomic and computational approaches to dissect the mechanisms of STAT3's universal and cell type-specific functions. JAK-STAT. 2013;2:e25097. doi: 10.4161/jkst.25097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cernkovich ER, Deng J, Bond MC, et al. Adipose-specific disruption of signal transducer and activator of transcription 3 increases body weight and adiposity. Endocrinology. 2008;149:1581–90. doi: 10.1210/en.2007-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui Y, Huang L, Elefteriou F, et al. Essential role of STAT3 in body weight and glucose homeostasis. Mol Cell Biol. 2004;24:258–69. doi: 10.1128/MCB.24.1.258-269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao Q, Wolfgang MJ, Neschen S, et al. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A. 2004;101:4661–6. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gorogawa S, Fujitani Y, Kaneto H, et al. Insulin secretory defects and impaired islet architecture in pancreatic beta-cell-specific STAT3 knockout mice. Biochem Biophys Res Commun. 2004;319:1159–70. doi: 10.1016/j.bbrc.2004.05.095. [DOI] [PubMed] [Google Scholar]
- 38.Buettner C, Pocai A, Muse ED, et al. Critical role of STAT3 in leptin's metabolic actions. Cell Metab. 2006;4:49–60. doi: 10.1016/j.cmet.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacoby JJ, Kalinowski A, Liu MG, et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A. 2003;100:12929–34. doi: 10.1073/pnas.2134694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haga S, Ogawa W, Inoue H, et al. Compensatory recovery of liver mass by Akt-mediated hepatocellular hypertrophy in liver-specific STAT3-deficient mice. J Hepatol. 2005;43:799–807. doi: 10.1016/j.jhep.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 41.Welte T, Zhang SS, Wang T, et al. STAT3 deletion during hematopoiesis causes Crohn's disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci U S A. 2003;100:1879–84. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee CK, Raz R, Gimeno R, et al. STAT3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation. Immunity. 2002;17:63–72. doi: 10.1016/s1074-7613(02)00336-9. [DOI] [PubMed] [Google Scholar]
- 43.Laouar Y, Welte T, Fu XY, et al. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity. 2003;19:903–12. doi: 10.1016/s1074-7613(03)00332-7. [DOI] [PubMed] [Google Scholar]
- 44.Wan CK, Oh J, Li P, et al. The cytokines IL-21 and GM-CSF have opposing regulatory roles in the apoptosis of conventional dendritic cells. Immunity. 2013;38:514–27. doi: 10.1016/j.immuni.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murray PJ, Smale ST. Restraint of inflammatory signaling by interdependent strata of negative regulatory pathways. Nat Immunol. 2012;13:916–24. doi: 10.1038/ni.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 47.Williams L, Bradley L, Smith A, et al. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. 2004;172:567–76. doi: 10.4049/jimmunol.172.1.567. [DOI] [PubMed] [Google Scholar]
- 48.Pilati C, Amessou M, Bihl MP, et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J Exp Med. 2011;208:1359–66. doi: 10.1084/jem.20110283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 50.Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2002;109:1143–8. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen X, Xu H, Yuan P, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–17. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 52.Durant L, Watford WT, Ramos HL, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–15. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kwon H, Thierry-Mieg D, Thierry-Mieg J, et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity. 2009;31:941–52. doi: 10.1016/j.immuni.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langlais D, Couture C, Balsalobre A, et al. The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol Cell. 2012;47:38–49. doi: 10.1016/j.molcel.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 55.Yasukawa H, Ohishi M, Mori H, et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4:551–6. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- 56.Gu F, Dube N, Kim JW, et al. Protein tyrosine phosphatase 1B attenuates growth hormone-mediated JAK2-STAT signaling. Mol Cell Biol. 2003;23:3753–62. doi: 10.1128/MCB.23.11.3753-3762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vallania F, Schiavone D, Dewilde S, et al. Genome-wide discovery of functional transcription factor binding sites by comparative genomics: the case of Stat3. Proc Natl Acad Sci U S A. 2009;106:5117–22. doi: 10.1073/pnas.0900473106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Badis G, Berger MF, Philippakis AA, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–3. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berger MF, Badis G, Gehrke AR, et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell. 2008;133:1266–76. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jolma A, Yan J, Whitington T, et al. DNA-binding specificities of human transcription factors. Cell. 2013;152:327–39. doi: 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 61.Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin YC, Jhunjhunwala S, Benner C, et al. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol. 2010;11:635–43. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ostuni R, Piccolo V, Barozzi I, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–71. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 64.Ghisletti S, Barozzi I, Mietton F, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–28. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 65.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–6. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 66.Kuwata H, Matsumoto M, Atarashi K, et al. IkappaBNS inhibits induction of a subset of Toll-like receptor-dependent genes and limits inflammation. Immunity. 2006;24:41–51. doi: 10.1016/j.immuni.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 67.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–82. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]