

Abstract
We have re-examined the utility of native chemical ligation at −Gln/Glu-Cys− [Glx-Cys] and −Asn/Asp-Cys− [Asx-Cys] sites. Using the improved thioaryl catalyst 4-mercaptophenylacetic acid (MPAA), native chemical ligation could be performed at −Gln-Cys− and Asn-Cys− sites without side reactions. After optimization, ligation at a −Glu-Cys− could also be used as a ligation site, with minimal levels of byproduct formation. However, −Asp-Cys− is not appropriate for use as a site for native chemical ligation because of formation of significant amounts of β-linked byproduct. The feasibility of native chemical ligation at −Gln-Cys− enabled a convergent total chemical synthesis of the enantiomeric forms of the ShK toxin protein molecule. The D-ShK protein molecule was ~50,000-fold less active in blocking the Kv1.3 channel than the L-ShK protein molecule. Racemic protein crystallography was used to obtain high resolution X-ray diffraction data for ShK toxin. The structure was solved by direct methods and showed significant differences from the previously reported NMR structures in some regions of the ShK protein molecule.
1. Introduction
The ShK toxin is a cysteine-rich 35-residue protein ion channel ligand that was isolated from the sea anemone Stichodactyla helianthus.1 Its action as a toxin arise from its potent inhibition of the KV1.3 ion channel, which inhibition blocks nerve signal transduction and in combination with other venom components paralyses the prey.1 KV1.3 is a potential therapeutic target for autoimmune diseases including multiple sclerosis, type 1 diabetes mellitus and rheumatoid arthritis.2,3 Although there are several NMR structures of the ShK protein molecule,4 no X-ray crystal structure has been reported. It is important to get the crystal structure in order to better understand binding and interaction of this protein to its target, leading to more informative SAR studies of this ion channel ligand.
Recently, we have reported the facilitated crystallization of proteins from racemic mixtures.5–6 For that reason, we set out to prepare the D-protein and L-protein enantiomers of ShK, and to determine the X-ray crystal structure of this protein toxin by racemic protein crystallography.6 Our first goal was to establish efficient total chemical synthesis using modern chemical ligation methods.7,8 Chemical synthesis of proteins enables the versatile construction of protein molecules of high purity with defined covalent structure containing any of a wide variety of chemical modifications such as the incorporation of non-coded amino acids and the preparation of unnatural mirror image protein molecules made up entirely of D-amino acids and the achiral amino acid glycine.8
A preferred approach to the total chemical synthesis of ShK toxin would involve native chemical ligation at a −Gln16-Cys17− site in the middle of the 35 residue polypeptide chain. First, we carried out model studies of the scope and limitations of native chemical ligation at Glx-Cys, and Asx-Cys sites. Using the improved thioaryl catalyst 4-mercaptophenylacetic acid (MPAA), native chemical ligation could be performed at −Gln-Cys− and Asn-Cys− sites without side reactions. After optimization, ligation at a −Glu-Cys− could also be used as a ligation site, with minimal levels of byproduct formation. However, −Asp-Cys− is not appropriate for use as a site for native chemical ligation because of formation of significant amounts of β-linked byproduct. The feasibility of native chemical ligation at −Gln-Cys− enabled the fully convergent synthesis of L-ShK toxin protein and its mirror image D-ShK. Biological assays of L-ShK toxin and the mirror image D-protein ShK toxin were performed, and it was found that D-ShK had much reduced channel-blocking activity with an IC50 of ~12 microM, i.e. ~50,000-fold less active than the L-ShK protein molecule. The X-ray structure of crystalline ShK toxin was determined by direct methods at atomic resolution (0.97 Angstrom) by racemic protein crystallography. Subsequently, we also determined the X-ray structure of the L-protein ShK toxin at a resolution of 1.06 Angstrom by molecular replacement. Comparison of the X-ray and the previously reported NMR structures of ShK toxin showed significant differences at the 310 helix, the C-terminus of the polypeptide chain, and in the chirality of all three disulfide bonds.
2. Results and Discussion
Amino acid sequence of the ShK polypeptide chain
The amino acid sequence of the ShK toxin protein is shown in Scheme 1. Synthesis of ShK toxin by Fmoc chemistry SPPS has previously been reported.3,9[ In our hands, stepwise synthesis of the 35 residue ShK polypeptide using highly optimized Boc chemistry SPPS10 gave an HPLC yield of only ~5% of the desired polypeptide accompanied by significant amounts of byproducts. For that reason, we decided to establish a more effective total chemical synthesis of the ShK polypeptide chain using modern chemical ligation methods.8 The most effective ligation chemistry is thioester-mediated amideforming native chemical ligation at −Xaa-Cys− sites. There are six Cys residues (3 disulfides) in the native ShK toxin protein.
Scheme 1.
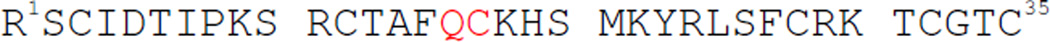
The preferred native chemical ligation site is near the middle of the polypeptide chain i.e. – Gln16-Cys17−. It had been reported that −Gln-Cys− ligations are problematic,11 so we decided to first explore the scope and limitations of native chemical ligation at Glx-Cys, Asx-Cys sites before proceeding to the total synthesis of ShK toxin.
Native Chemical Ligation at -Glx-Cys, -Asx-Cys Sites
In native chemical ligation an unprotected peptide1Xaa-thioester segment is covalently condensed with a Cys-peptide2 via the rearrangement of the initial thioester-linked intermediate to form the full length peptide1Xaa-Cys-peptide2 product with a native peptide bond at the −Xaa-Cys− ligation site7. The C-terminal amino acid in the peptide1-Xaa-thioester segment can in principle be any amino acid, although rates of reaction vary considerably depending on the identity of the C-terminal residue.12 Side reactions have been reported for ligation at some -Xaa-Cys- sites. Camarero et al. mentioned in their review that Xaa can be any residue except Glu, Gln, Asp or Asn.11 Botti et al. observed significant levels of β− and γ−linked byproducts during ligation at -Glu-Cys- and -Asp-Cys- sites (Scheme 2).13
Scheme 2.

Proposed reaction pathway for formation of α- and β-linked peptide products during native chemical ligation at an -Asp-Cys- ligation site.13 R` = CH2CH2CO-Phe-COOH
Since those studies were reported, 4-mercaptophenylacetic acid (MPAA) has been shown to be an improved catalyst for native chemical ligation, speeding up the reaction by as much as an order-of-magnitude.14 In order to evaluate the impact on byproduct formation of using MPAA as a catalyst, we undertook a systematic study of native chemical ligation at −Gln/Glu-Cys− and – Asn/Asp-Cys− sites. The design of our model ligation study was similar to that used by Botti and coworkers.13 We synthesized a Cys-peptide CFYANF and the peptide-thioesters LYRAX-αCOSR`, where X = Q, E, N, D and R` = CH2CH2CO-Phe-COOH. Each peptide-thioester was reacted with the Cys-peptide under standard native chemical ligation conditions (2 mM each peptide-thioester, 2.4 mM Cys-peptide, pH 7.0, 6 M guanidine hydrochloride, 20 mM MPAA, 10 mM TCEP·HCl). The authentic α-linked peptides LYRAXαCFYANF (X = Q, E, N, D), β-linked peptides LYRAXβCFYANF (X = N and D), and γ-linked peptides LYRAXγCFYANF (X = Q and E) were also made as reference standards and used to calibrate the HPLC analyses in order to verify the identity of the ligation products obtained in the test reactions (see Supporting Information).
Initially, we explored native chemical ligation at −Asn-Cys− and −Asp-Cys− sites using the aryl thiol MPAA as catalyst (Figure 1). Next we explored native chemical ligation at -Gln-Cys-and −Glu-Cys- sites using the same MPAA catalyst; representative LC-MS data are shown in Figure 2. The reactions were monitored by LC-MS, and were allowed to run at room temperature (RT) for 18–24 hours. Under these conditions, only trace amounts of β- or γ- linked byproducts were observed during ligation at -Gln-Cys- and -Asn-Cys- sites (0.3–0.4%). Higher levels of byproducts were observed during ligations at -Glu-Cys-, and -Asp-Cys- sites. Under the standard native chemical ligation conditions used, about 25% of β-linked byproduct was formed during the -Asp-Cys- ligation. The amount of β-linked byproduct that was formed was independent of pH over the range pH 6.1–7.2. Results for these model reactions are summarized in Table 1.
Figure 1.

Native chemical ligations between peptide-Asx-αCOSR` and Cys-peptide: * correct product. (Left) −Asn-αCOSR` reaction products [Inset: close-up of ligation products – the vertical bar shows the elution position of the β-linked product]; (Right) −Asp-αCOSR` reaction products – the vertical bar shows the elution position of β-linked product. Reaction conditions: pH 7.0, 20 mM MPAA, 6 M Gu ·HCl, 200 mM Na2HPO4, 10 mM TCEP·HCl, room temperature (RT).
Figure 2.

Native chemical ligations between peptide-Glx-αCOSR` and Cys-peptide: * correct product. (Left) −Gln- COSR reaction products; [Inset: close-up of ligation products – the vertical bar shows the elution position of the γ-linked product] (Right) −Glu- COSR reaction products – the vertical bar shows the elution position of the γ-linked product. Reaction conditions: (Left) pH 7.0, 20 mM MPAA, 6 M Gu·HCl, 200 mM Na2HPO4, 10 mM TCEP·HCl, RT; (Right) pH 6.7, 20 mM MPAA, 6 M Gu·HCl, 200 mM Na2HPO4, 10 mM TCEP·HCl, RT.
Table 1.
Native chemical ligation at -Asx-Cys- and -Glx-Cys- sites (reaction conditions: pH 7.0, 20mM MPAA, 6 M Gu·HCl, 200 mM Na2HPO4, 10 mM TCEP·HCl, RT)
| Ligaton site | Percent of α-CONH | Percent of γ/β -CONH |
|---|---|---|
| -Gln-Cys- | >99.6 | <0.4 |
| -Glu-Cys- | 97 | 3 |
| -Asn-Cys- | 99.6 | 0.4 |
| -Asp-Cys- | 75 | 25 |
Detection limit: 0.2%
In contrast to Botti’s results,13 no hydrolysis of the peptide-thioester LYRAD-αCOSR` was observed after HF deprotection and cleavage, nor was significant hydrolysis of LYRAD- αCOSR` observed during the ligation reaction. In a control experiment carried out in the absence of the Cys-peptide and without added MPAA, about 30% hydrolyzed product was formed from the peptide-thioester LYRAD-αCOSR` after 19 h at pH 6.8. Formation of ~30% of a product with mass 18 Da lower than expected for simple hydrolysis of the thioester was also observed under these conditions; this presumably corresponds to formation of a carboxylic anhydride at the C-terminal of the peptide upon loss of thioester. Formation of the anydride would also explain the large amount of hydrolysis observed for the C-terminal Asp-thioester.
For ligation at the −Glu-Cys− site, pH was found to be an important factor in the formation of γ-linked byproduct (Table 2). As the pH increased from pH 6.1 to pH 7.2 the ligation reaction rate was observed to increase, with consequently reduced amounts of γ-linked byproduct formation which fell from ~10% to ~2%. These amounts of γ-linked byproduct formation are significantly lower than the ~20% γ-linked byproduct formation reported by Botti and colleagues during ligation at a -Glu-Cys- site at pH 7.0.13 In a control experiment carried out in the absence of the Cys-peptide and without added MPAA, no significant hydrolysis of the −Glu-α-thioester was observed. The absence of peptide-thioester hydrolysis and the much lower levels of γ-linked byproduct formation observed in the work reported here suggests that peptides with a C-terminal Glu-α-thioester can be used for native chemical ligation with MPAA as catalyst.
Table 2.
Influence of pH on γ-linked byproduct formation during native chemical ligation at -Glu-Cys- site
| pH | 6.1 | 6.7 | 7.0 | 7.2 |
| % α-CONH | 89 | 95 | 97 | 98 |
| % γ-CONH | 11 | 5 | 3 | 2 |
Detection limit: 0.2%
The results reported above are consistent with the formation of β- or γ-linked byproducts via the anhydride formed by attack of the carboxylate side chain on the C-terminal thioester moiety. 13 The anhydride intermediate is formed slowly under ligation conditions,13 and thus speeding up the ligation reaction (but not the rate of anhydride formation) by the use of the improved thiol catalyst MPAA would lead to the observed reduction in the levels of byproduct formation.
Thus, based on these results, a −Glu-Cys− site could be used as a ligation site with only minimal levels of byproduct formation. However, −Asp-Cys− is not appropriate for use as a site for native chemical ligation because of formation of significant amounts of β-linked byproduct. We found that native chemical ligation using MPAA as catalyst could be performed with impunity at −Gln-Cys− and −Asn-Cys− sites without significant side reactions. We were now in a position to use this knowledge in the total chemical synthesis of ShK toxin.
Synthesis of ShK Toxin
The ShK toxin synthetic design is shown in Scheme 3. Native chemical ligation between the unprotected peptide segments [Arg1-Gln16]-α-thioester and [Cys17-Cys35] was conducted under standard conditions (6M Gu HCl, 0.2M Na2HPO4, 10 mM TCEP hydrochloride, 20 mM MPAA, pH=7.0). Data are shown in Figures 3 & 4. The full-length polypeptide ligation products were purified by reverse phase HPLC (Figures 3(b) & 4(b)).
Scheme 3.

Convergent synthesis of ShK toxin by native chemical ligation of two unprotected peptide segments, followed by folding and formation of disulfides.
Figure 3.


(a). (L)-ShK polypeptide ligation T=0, Peak 1 = [Cys17-Cys35]; Peak 2 =[Arg1-Gln16]-α-thioester.
T=19h, *Ligation product.
Early eluting broad peaks are non-peptidic.
(b). Purified L-ShK Polypeptide Mobsd.= 4060.3±0.4Da Mcalc.(av. isotope composition)=4060.8 Da
Figure 4.


(a). (D)-ShK polypeptide ligation T=0, Peak 1’ =D-[Cys17-Cys35]; Peak 2’ = D- [ Arg1 -Gln16]-α-thioester.
T=19h, * Ligation product.
Early eluting broad peaks are non-peptidic.
(b). Purified D-ShK Polypeptide Mobsd.= 4060.1±0.4Da
ShK toxin folding
A number of different folding conditions were explored.3 In our hands, optimal folding conditions were: air oxidation (gentle stirring open to the air), 50 mM NH4Ac, pH=8.0.
Folded L-ShK and D-ShK protein products were purified by reverse phase HPLC and characterized by analytical LCMS (Figures 5(b), 6(b)). The observed masses were consistent with the formation of three disulfide bonds in each synthetic protein.
Figure 5.


(a). (L)-ShK polypeptide folding. (Upper) Reduced polypeptide; (Lower) After air oxidation for 63h. *Folded product
(b). HPLC purified L-ShK toxin Mobs.= 4054.2±0.4Da Mcalc. (av. isotope composition)=4054.8
Figure 6.


(a). (D)-ShK polypeptide folding (Upper) Reduced polypeptide; (Lower) After air oxidation for 42h. *Folded product
(b). HPLC purified D-ShK toxin Mobs.= 4054.3±0.4Da
Kv1.3 channel blocking activities
To examine channel-blocking abilities of L- and D- ShK toxins, we used the cut-open oocyte voltage clamp method15 to measure potassium ionic currents from Xenopus laevis oocytes that contained expressed human Kv1.3 (hKv1.3) channels, before and after addition of toxin. The L-protein form of ShK toxin blocked potassium currents in a concentration dependent manner, with an IC50 value of 250 ± 20 picoM for L-ShK (Figure 7). This result indicates that the synthetic L-ShK toxin is a potent blocker of the hKv1.3 channel at picomolar concentrations, which is consistent with previous reports.3,9
Figure 7.

Functional blockade of hKv1.3 currents by L- and D- ShK synthetic toxins using the cut-open oocyte voltage clamp method15 to measure potassium ionic currents from Xenopus laevis oocytes that contained expressed human Kv1.3 (hKv1.3) channels. Peak currents were recorded during a 150ms step to + 30 mV from a holding voltage of −90 mV (upper inset). The fractions of current blockade are plotted for test concentration of L-ShK (filled squares, n = 3–5 cells) and D-ShK (open squares, n = 3–6 cells). Dotted lines indicate dose-response curves fitted by the Hill equation. Error bars in figure and uncertainties in table indicate standard error of the mean.
In initial experiments, the D-protein form of ShK also showed channel-blocking activity with an IC50 of 450 ± 110 nanoM, ~2,000-fold less active than L-ShK in the same assay. We suspected that this apparent activity of the D-protein form of ShK might have arisen from contamination by the L-protein ShK that had been purified on the same HPLC column. D-ShK protein was resynthesized, purified on a virgin HPLC column, and re-assayed. This new preparation of D-ShK had much reduced channel-blocking activity with an IC50 of ~12 microM, i.e. ~50,000-fold less active than the L-ShK protein molecule. Based on these results, it is likely that the D-protein form of ShK toxin has no intrinsic affinity for the human Kv1.3 ion channel.
ShK toxin crystallization and X-ray structure determinations
Crystallization of L-ShK toxin was attempted using a screen of 96 conditions from the Hampton Index (HR2-144). Purified L-ShK toxin was dissolved in water to a concentration of 25 mg/mL. Each condition used 1 mL of well solution with hanging drops consisting of 1 µL well solution and 1 µL protein solution. The plates were kept in an incubator at 19 °C. Only one condition (2.1 M DL-Malic acid, pH=7.0) gave crystals after one week. Using the same 96 conditions, a racemic mixture of {D- ShK toxin & L- ShK toxin} at 25 mg/mL (12.5 mg/mL of L- ShK toxin & 12.5 mg/mL of D- ShK toxin) produced crystals from seven conditions within 3 days. We optimized three of these conditions by varying precipitant concentrations. The crystal that gave the best diffraction results was grown from 0.2 M Lithium sulfate monohydrate, 0.1 M Tris, pH 8.5, 25 % Polyethylene glycol 3,350, 20 mM NaCl at 19 °C.
Diffraction data were collected at an energy level of 18 keV using the NE-CAT beamline 24-ID-C at the Advanced Photon Source at Argonne National Laboratory. Diffraction was observed to a resolution of 1.06 Å for L-ShK toxin crystal and 0.97 Å for the racemic DL-protein crystal. Diffraction intensity statistics revealed that the L-ShK toxin crystallized in space group C2221, with one molecule in the asymmetric unit. The racemic DL-protein mixture crystallized in space group P21/c with one enantiomer in the asymmetric unit.
Initially, we attempted to solve the structure by molecular replacement using the previously reported NMR structure as a search model.4 Neither the racemic protein nor L-ShK protein gave a solution by molecular replacement. Previously, we have had success using direct methods to solve racemic protein structures.16–19 With atomic-resolution diffraction data from the DL-protein racemate in hand, we explored the use of direct methods20–21 to obtain phasing information for solving the structure. The program SHELXS22 gave a clear solution (see Figure 8(b)) from a 2-h run on a 2.7 GHz Intel Core i7 processor. After preliminary refinement with SHELXL22, a well-defined map of continuous electron density was obtained. The protein model was built manually using COOT23 and refined in space group P21/c using Phenix24 to a crystallographic R-factor of 0.153 (R-free 0.165), corresponding to an R-factor of 0.10 for a non-centrosymmetric space group.25
Figure 8.

Crystal structure of ShK DL-protein racemate. (a), Cartoon representation of the packing of D-ShK (Cyan) and L-ShK (Green) toxin in unit cell in P21/c, unit cell composed of 4 molecules, 2 L-ShK and 2 D-ShK. (b), SigmaA-weighted 2Fo - Fc electron density map at a σ level of 2, showing Tyr23 that derived from the best SHELXS22 solution (CFOM = 0.14).
The packing of D-ShK toxin and L-ShK toxin in the unit cell is shown in Figure 8(a). There are five pairs of reciprocal interactions at the interface between the D-protein and L-protein enantiomers as shown in Figure 9. They are: (a), Hydrogen bonding between (D)-Thr6 and (L)-Arg11 and (L)-Arg11, (L)-, (L)-Thr6 and (D)-Arg11 (one face); (b), Hydrogen bonding between side chain of (D)-His19 and main chain of (L)-Cys32, side chain of (L)-His19 and main chain of (D)-Cys32, also hydrogen bonding connected by water molecules at multiple sites; interface between side chain of (L)-His19 and main chain of (D)-Cys32 is not shown here. Water molecules are shown as small red spheres (two faces); (c), Hydrogen bonding between side chain of (L)-Ser2 and main chain of (D)-Lys9, side chain (D)-Ser2 and main chain of (L)-Lys9, also hydrogen bonding connected by water molecules at multiple sites (one face). (d). Hydrophobic interaction between Leu25 and Met21(one face).
Figure 9.

Close up view of the interfaces between the enantiomeric L-ShK (Green) and D-ShK (Cyan) molecules in the DL-protein racemate crystal. (a), Hydrogen bonding between (D)-Thr6 and (L)-Arg11, (L)-Thr6 and (D)-Arg11; (b), Hydrogen bonding between side chain of (D)-His19 and backbone of (L)-Cys32, also hydrogen bonding connected by water molecules at multiple sites, Water molecules are shown as small red spheres; (c), Hydrogen bonding between side chain of (L)-Ser2 and main chain of (D)-Lys9, side chain (D)-Ser2 and main chain of (L)-Lys9, also hydrogen bonding connected by water molecules at multiple sites; (d), Weak hydrophobic interaction between Leu25 and Met21.
The X-ray structure of L-ShK toxin was also solved with diffraction data from crystals of the L-protein, by molecular replacement26 using as a search model the L-protein structure obtained by racemic crystallography. The final model was refined to a crystallographic R-factor of 0.133 (R-free 0.157) using Phenix.24
A cartoon representation of the X-ray structure of ShK toxin is shown in Figure 10(a). The alignment of a previously reported NMR model4 with the X-ray structure of ShK toxin is shown in Figure 10(b). The structure of L-ShK toxin obtained from the DL-protein racemate and the structure of L-ShK crystallized on its own were in good agreement with a Cα rms difference of 0.47 Å. The structures are compared in Figure 10c. The major secondary structures in the crystal structure of ShK toxin are Lys9-Arg11 310 helix, Ala14-His19 α helix, Met21-Leu25 α helix, Ser26-Phe27 hydrogen bond turn (α turn) and Arg29-Gly33 hydrogen bond turn (α turn). These structural elements are similar to the secondary structures seen in the NMR structure. However, close comparison of the X-ray and NMR structures of ShK toxin shows significant differences at the 310 helix, the C-terminus, and in the orientation of all three disulfide bonds. In the NMR structure, there is ambiguity as to whether the sequence Lys9-Ser10-Arg11 forms a 310 helix or forms a turn, while in the crystal structure it is clear that the Lys9-Ser10-Arg11 region forms a short 310 helix. The disulfide bonds between cysteines12–28 and 17–32 in the crystal structure are well defined and adopt a left handed conformation whereas in those 20 NMR models reported,4 the majority adopted a right handed conformation. The 3–35 disulfide bond is also well defined in the crystal structure and adopts a right handed conformation whereas in the NMR structure the C terminus of the polypeptide chain is not well structured, and this disulfide is not well defined.
Figure 10.

(a) Cartoon representation of the L-ShK toxin. The structure was solved as the racemate at 0.97 Å X-ray resolution by direct methods. The protein fold is shown as ribbons with three S-S bonds shown in stick mode. (b) Superposition of the NMR structure4 (PDB ID: 1ROO) of ShK toxin (in red) and the present L-enantiomer from the X-ray racemic crystal structure (in green); the selected NMR model (#10) displays the minimal Cα r.m.s. deviation of 1.8 Å.27 (c) Superposition of the L-ShK crystal structure (in blue) and L-ShK enantiomer structure from the DL-protein racemate (Green).
3. Summary and Conclusions
In conclusion, native chemical ligation using MPAA as catalyst could be performed at −Gln-Cys− and Asn-Cys− sites without side reactions. After optimization, ligation at a −Glu-Cys− could also be used as a ligation site, with minimal levels of byproduct formation. However, − Asp-Cys− is not appropriate for use as a site for native chemical ligation because of formation of significant amounts of β-linked byproduct. The feasibility of native chemical ligation at −Gln-Cys− enabled a convergent total chemical synthesis of the enantiomeric forms of the ShK toxin protein molecule. The D-ShK protein molecule was ~50,000-fold less active in blocking the Kv1.3 channel than the L-ShK protein molecule. Racemic protein crystallography was used to obtain high resolution X-ray diffraction data for ShK toxin. The structure was solved by direct methods and showed significant differences from the previously reported NMR structures in some regions of the ShK protein molecule.
4. Experimental
Peptide synthesis
Peptide segments were synthesized by stepwise SPPS using manual in situ neutralization Boc chemistry protocols10 on Boc-Phe-OCH2-Pam-resin at a 0.3 mmol scale. The peptide-thioesters were synthesized on trityl-SCH2CH2CO-Phe-OCH2-Pam-resin12 at a 0.3 mmol scale. Details of all peptide syntheses and characterization data for the synthetic products are given in the Supporting Information.
Model native chemical ligations of LYRAX-αCOSR` and CFYANF were performed by dissolving LYRAX-αCOSR`(~0.35 mg, ~2mM) and CFYANF (~0.37 mg, ~2.4 mM) in ligation buffer (0.2 mL) containing 6 M guanidine hydrochloride, 200 mM Na2HPO4, 10 mM TCEP hydrochloride and 20 mM MPAA. The pH was adjusted to 7.0 unless otherwise indicated. The ligation buffer was purged with helium for 15 min before use. All the reactions were monitored by LC-MS, and were allowed to run at room temperature (RT) for 18–24 hours.
HPLC and LC-MS analysis
Analytical reverse phase HPLC reported in this work was performed on an Agilent C-8 (3.5 µm, 300 Å) 4.6 × 150 mm silica column at a flow rate of 1 mL/min or an in-house packed C-18 (Microsorb, 3 µm, 300 Å), 2.1 × 50 mm silica column at a flow rate of 0.5 mL/min using a linear gradient of 5–65% of buffer B in buffer A over 15 min, 5–45% of buffer B in buffer A over 40 min or 5–45% of buffer B in buffer A over 80 min at 40 °C (buffer A= 0.1% TFA in H2O; buffer B = 0.08% TFA in acetonitrile). The absorbance of the column elute was monitored at 210 nm.
Peptide masses were measured by on-line LC-MS using an Agilent 1100 LC/MSD ion trap. Calculated masses were based on average isotope composition, unless otherwise stated. Preparative reverse phase HPLC of crude peptides was performed with an Agilent 1100 prep system on in-house packed C-18 (10 µm, 300 Å), 10 × 250 mm columns at 40 °C using an appropriate shallow gradient of increasing concentration of buffer B in buffer A at a flow rate of 5 mL/min. Fractions containing the purified target peptide were identified by LC-MS. Selected fractions were then combined and lyophilized.
Chemical synthesis of L-ShK toxin and D-ShK toxin
L-ShK toxin was prepared by native chemical ligation of two unprotected L-peptide segments, then purification of the product 35 residue polypeptide, followed by folding/disulfide bond formation. L-[Arg1-Gln16]-α-thioester (30 mg, 14.6 µmole) and L-[Cys17-Cys35] (35 mg, 15.5 µmole) were dissolved in 5 mL ligation buffer containing 6 M Gu HCl, 0.2 M Na2HPO4, 10 mM TCEP hydrochloride, 20 mM MPAA, that had been degassed for 15 minutes before use. The pH was adjusted to 7.0 and the reaction kept for 19 hours and purified by reverse phase semi-preparative HPLC. The purified linear (1–35) peptide product was then dissolved in 42 mL 50 mM NH4·OAc, and the pH was adjusted to 8.04. The resulting solution was stirred under air at RT for 63h, then purified and lyophilized to afford L-ShK toxin (26 mg, 6,4 µmole, 44% based on limiting peptide).
D-ShK toxin was synthesized using D-[ Arg1-Gln16]-α-thioester (25 mg, 12.2 µmole) and D-[ Arg1-Gln16]-α-thioester (25 mg, 12.2 µmole) and D-[ Cys17-Cys35] (31 mg, 13.7 µmole) following the same procedures described for L-ShK. Note that for D-Thr and D-Ile, both chiral centers in each amino acid are inverted from the corresponding L-Thr and L-Ile.28 The overall yield of D-ShK was 21mg (5.2 µmole, 43% based on limiting peptide).
Biological activity assays: Kv1.3 block
Preparation of cells and recording procedures
The cDNA encoding hKv1.3 was cloned in pBSTA vectors containing the Kozak sequence of Xenopus laevis beta-globin gene 5’-gccgccatgg.29 Plasmids were transcribed using in vitro transcription kits, mMESSAGE T7, from Ambion (Austin, TX). Freshly isolated oocytes were injected with 20 ng of cRNA and kept in SOS incubation solution (100 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 10 mM HEPES, pH 7.4) for 1–4 days at 18 °C.
Oocytes were mounted in the Cut-open oocyte voltage-clamp configuration (COVC) 1–4 day after injection as described previously.15,30 To record potassium currents, recording solutions were prepared as follows: extracellular solution: 113 mM n-methylglucamine (NMG)-methylsufonate (Mes), 7 mM K-Mes, 2mM Ca(Mes)2, 20 mM HEPES, pH 7.4, and internal solution: 120 mM K-Mes, 2mM EGTA, 20 mM HEPES, pH 7.4, respectively.
L- and D- ShK toxin proteins were separately diluted in the extracellular solution containing 0.1% BSA and added to the top and guard chambers. After applying each toxin, currents were monitored by a 150ms step pulse every 60 s from a holding voltage of −90 mV for at least 10 minutes. After confirming ShK toxin blockade was saturated, currents were recorded by a 150ms step pulse to + 30 mV from a holding voltage of −90 mV.
Protein crystallization
Crystals of the synthetic proteins were grown as described in the text.
X-ray diffraction data collection
For low temperature data collection, selected crystals were briefly transferred to the cryoprotectant (reservoir solution plus 20% (v/v) glycerol) and flash-frozen in liquid nitrogen. The atomic resolution X-ray diffraction data were collected at 100K at the Argonne National Laboratory (Advanced Photon Source, beamline 24ID-C equipped with a PILATUS detector) using 0.688 Å synchrotron radiation. Diffraction images were integrated, scaled, and merged with HKL2000.31 Examination of the systematic absences of the diffraction intensities for the racemic DL-ShK protein crystals revealed that the unit cell automatically chosen by the program HKL2000 corresponded to the non-standard space group setting P21/a. The final data were therefore reindexed in the standared space group setting P21/c and used in the subsequent structure determination steps. The diffraction data for the L-ShK toxin protein were processed in space group C 2221.
X-ray structure determination
The racemic DL-ShK protein crystal structure was solved by direct methods using the program SHELXS22 in a 2-h run on a 2.7 GHz Intel Core i7 processor. The positions for most of the atoms were revealed in the best solution with figure-of-merit of 0.14. The initial model was built manually using COOT23 and the structure was refined using Phenix.24
The L-ShK toxin crystal structure was solved by molecular replacement26 using the software PHASER and using as a search model the L-enantiomer coordinates from the structure obtained by racemic protein crystallography. The final model of the native ShK protein was refined to a crystallographic R-factor of 0.133 (R-free 0.157) using Phenix.24 All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were included in the model and individual hydrogen atom coordinates were refined isotropically for both the racemic and native Shk protein structure. Molecular graphics were generated using MacPymol. The main chain torsion angles for all residues are in the allowed regions and additional allowed regions of the Ramachandran plot.
Crystal structure data of the racemic and the native ShK have been deposited in the Protein Data Bank with PDB codes 4LFS and 4LFQ respectively.
Supplementary Material
Acknowledgements
This research was supported in part by funds from NIH Grants U54 GM087519, and R01-GM030376 to F. Bezanilla and by American Heart Association (13POST14800031) to T. Kubota. We thank Dr. Dirk J. Snyders for providing the hKv1.3 construct, Ms. Ludivine Frezza for her technical support and Dr. Dhakshnamoorthy Balasundaresan for the X-ray data collection support. Use of NE-CAT beamline 24-ID at the Advanced Photon Source is supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Footnotes
Associated Content
Reagents. Methods of peptide synthesis and peptides synthesized. Native chemical ligations of model peptides. Identification of ligation products. X-ray data statistics. Tables of H-bond lengths. This material is available free of charge via the Internet at http://pubs.acs.org
Contributor Information
Bobo Dang, Email: bbdang@uchicago.edu.
Tomoya Kubota, Email: tomoyak@uchicago.edu.
Kalyaneswar Mandal, Email: kmandal@uchicago.edu.
Francisco Bezanilla, Email: fbezanilla@peds.bsd.uchicago.edu.
Stephen B. H. Kent, Email: skent@uchicago.edu.
References
- 1.Castaneda O, Sotolongo V, Amor AM, Stocklin R, Anderson AJ, Harvey AL, Engstrom A, Wernstedt C, Karlsson E. Toxincon. 1995;33:603. doi: 10.1016/0041-0101(95)00013-c. [DOI] [PubMed] [Google Scholar]
- 2.Wulff H, Castle NA, Pardo LA. Nat. Rev. Drug Discov. 2009;8:982. doi: 10.1038/nrd2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chi V, Pennington MW, Norton RS, Tarcha EJ, Londono LM, SimsFahey B, Upadhyay SK, Lakey JT, Iadonato S, Wulff H, Beeton C, Chandy KG. Toxicon. 2012;59:529. doi: 10.1016/j.toxicon.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tudor JE, Pallaghy PK, Pennington MW, Norton RS. Nat. Struct. Biol. 1996;3:317. doi: 10.1038/nsb0496-317. [DOI] [PubMed] [Google Scholar]
- 5.Pentelute BL, Gates ZP, Tereshko V, Dashnau JL, Vanderkooi JM, Kossiakoff AA, Kent SBH. J. Am. Chem. Soc. 2008;130:9695. doi: 10.1021/ja8013538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeates TO, Kent SBH. Ann Rev Biophys. 2012;41:41. doi: 10.1146/annurev-biophys-050511-102333. [DOI] [PubMed] [Google Scholar]
- 7.Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 8.Kent SBH. Chem. Soc. Rev. 2009;38:338. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- 9.Pennington MW, Byrnes ME, Zaydenberg I, Khaytin I, Dechastonay J, Krafte DS, Hill R, Mahnir VM, Volberg WA, Gorczyca W, Kem WR. Int. J. Pept. Prot. Res. 1995;46:354. doi: 10.1111/j.1399-3011.1995.tb01068.x. [DOI] [PubMed] [Google Scholar]
- 10.Schnölzer M, Alewood P, Jones A, Alewood D, Kent SBH. Int. J. Pept. Protein Res. 1992;40:180. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 11.Camarero JA, Muir TW. Curr. Protoc. Protein Sci. 2001 doi: 10.1002/0471140864.ps1804s15. Chapter 18Unit18 4. [DOI] [PubMed] [Google Scholar]
- 12.Hackeng TM, Griffin JH, Dawson PE. Proc. Natl. Acad. Sci. USA. 1999;96:10068. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villain M, Gaertner H, Botti P. Eur. J. Org. Chem. 2003:3267. [Google Scholar]
- 14.Johnson ECB, Kent SBH. J. Am. Chem. Soc. 2006;128:6640. doi: 10.1021/ja058344i. [DOI] [PubMed] [Google Scholar]
- 15.Stefani E, Bezanilla F. Methods Enzymol. 1998;293:300. doi: 10.1016/s0076-6879(98)93020-8. [DOI] [PubMed] [Google Scholar]
- 16.Mandal K, Pentelute BL, Tereshko V, Kossiakoff AA, Kent SBH. J. Am. Chem. Soc. 2009;131:1362. doi: 10.1021/ja8077973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mandal K, Pentelute BL, Tereshko V, Thammavongsa V, Schneewind O, Kossiakoff AA, Kent SBH. Protein Sci. 2009;18:1146. doi: 10.1002/pro.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banigan JR, Mandal K, Sawaya MR, Thammavongsa V, Hendrickx AP, Schneewind O, Yeates TO, Kent SBH. Protein Sci. 2010;19:1840. doi: 10.1002/pro.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pentelute BL, Mandal K, Gates ZP, Sawaya MR, Yeates TO, Kent SBH. Chem. Commun. 2010;46:8174. doi: 10.1039/c0cc03148h. [DOI] [PubMed] [Google Scholar]
- 20.Hauptman H. Science. 1986;233:178. doi: 10.1126/science.233.4760.178. [DOI] [PubMed] [Google Scholar]
- 21.Karle J. Science. 1986;232:837. doi: 10.1126/science.232.4752.837. [DOI] [PubMed] [Google Scholar]
- 22.Sheldrick GM, Dauter Z, Wilson KS, Hope H, Sieker LC. Acta Crystallogr. D. 1993;49:18. doi: 10.1107/S0907444992007364. [DOI] [PubMed] [Google Scholar]
- 23.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta. Cryst. 2010;D66:486. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr. D. 2010;66:213. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luzzati PV. Acta. Cryst. 1952;5:802. [Google Scholar]
- 26.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J. Appl. Crystallogr. 2007;40:658. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maiti R, Van Domselaar GH, Zhang H, Wishart DS. Nucleic Acids Res. 2004;32:W590. doi: 10.1093/nar/gkh477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pure & Appl. Chem. 1984;56(pNo. 5):595. [Google Scholar]
- 29.Kozak M. J. Biol. Chem. 1991;266:19867. [PubMed] [Google Scholar]
- 30.Chanda B, Bezanilla F. J. Gen. Physiol. 2002;120:629. doi: 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. In: Methods in Enzymology. part A. Carter CW Jr, Sweet RM, editors. Vol. 276. New York: Academic Press; 1997. p. 307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.