

Abstract
Ebola virus (EBOV) is the causative agent of a severe hemorrhagic fever in humans with reported case fatality rates as high as 90%. There are currently no licensed vaccines or antiviral therapeutics to combat EBOV infections. Heme oxygenase-1 (HO-1), an enzyme that catalyzes the rate-limiting step in heme degradation, has antioxidative properties and protects cells from various stresses. Activated HO-1 was recently shown to have antiviral activity, potently inhibiting the replication of viruses such as hepatitis C virus and human immunodeficiency virus. However, the effect of HO-1 activation on EBOV replication remains unknown. To determine whether the upregulation of HO-1 attenuates EBOV replication, we treated cells with cobalt protoporphyrin (CoPP), a selective HO-1 inducer, and assessed its effects on EBOV replication. We found that CoPP treatment, pre- and postinfection, significantly suppressed EBOV replication in a manner dependent upon HO-1 upregulation and activity. In addition, stable overexpression of HO-1 significantly attenuated EBOV growth. Although the exact mechanism behind the antiviral properties of HO-1 remains to be elucidated, our data show that HO-1 upregulation does not attenuate EBOV entry or budding but specifically targets EBOV transcription/replication. Therefore, modulation of the cellular enzyme HO-1 may represent a novel therapeutic strategy against EBOV infection.
INTRODUCTION
Ebola virus (EBOV) is an enveloped, nonsegmented, negative-strand RNA virus that, together with Marburg virus (MARV), makes up the family Filoviridae (filovirus) (1). There are five antigenically distinct species of EBOV: Zaire ebolavirus, Sudan ebolavirus, Taï Forest ebolavirus (previously Côte d'Ivoire ebolavirus), Reston ebolavirus, and Bundibugyo ebolavirus (2). Sudan ebolavirus and Zaire ebolavirus are associated with severe outbreaks of hemorrhagic fever in humans, with case fatality rates ranging from 55% to 90% (3). Budibugyo ebolavirus, a recently identified species associated with a 2007 outbreak, produced a case fatality rate of about 25% (4).
The EBOV genome includes seven structural genes. The single surface glycoprotein (GP) mediates virus entry into a variety of different cell types (5–7). Four structural proteins, nucleoprotein (NP), RNA-dependent RNA polymerase (L), VP30, and VP35, are essential for amplification of the viral genome (8). The primary membrane-associated viral protein, VP40, is critical for viral budding (9). The secondary membrane-associated protein, VP24, and VP35 effectively antagonize interferon (IFN) pathways by inhibiting the Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling cascade and by suppressing IFN regulatory factor 3 (IRF3) phosphorylation, respectively (10–13). This efficient suppression of IFN pathways may contribute to EBOV pathogenesis and renders IFN therapeutic treatments ineffective (14, 15).
Currently, there are no approved vaccines or antiviral therapeutics to combat EBOV infection. Given the limited number of biosafety level 4 (BSL4) containment facilities, which are required for EBOV experiments, we developed a biologically contained EBOV, EbolaΔVP30 virus, which lacks the essential VP30 gene, grows only in cells stably expressing the VP30 gene product, and can be studied in BSL3 containment (16, 17). The growth kinetics and morphology of EbolaΔVP30 virus are similar to those of wild-type EBOV (16, 17); therefore, EbolaΔVP30 virus is an ideal surrogate for authentic EBOV to uncover potential therapeutic interventions.
Heme oxygenase-1 (HO-1) is an enzyme that catalyzes the rate-limiting step in heme degradation to carbon monoxide, biliverdin, and free iron (18). HO-1 expression is upregulated not only by its substrate, heme, but also by various nonheme inducers, such as heat shock, inflammatory cytokines, endotoxin, and oxidative stress, suggesting that HO-1 may play a vital role in maintaining cellular homeostasis (19, 20). Increases in HO-1 protein expression and activity have clear anti-inflammatory and antioxidant effects and can protect tissues, organs, and entire animal models from septic shock, oxidative injury, and hypoxia (for recent reviews, see references 21, 22, 23, and 24). In addition, recent studies have demonstrated significant antiviral properties of HO-1. Specifically, upregulation of HO-1 was shown to suppress infection by enterovirus, hepatitis C virus (HCV), hepatitis B virus (HBV), and human immunodeficiency virus (HIV) while protecting infected tissues, such as the liver and lungs, from virus-induced oxidative injury (25–30). However, the effect of HO-1 induction on EBOV infection or EBOV-induced disease is unknown.
Therefore, we used the potent HO-1 inducer cobalt protoporphyrin (CoPP) to examine the effect of HO-1 on EBOV infection in vitro. Here, we provide evidence that HO-1 expression and activation inhibit EBOV replication and may represent a novel and effective strategy against EBOV.
MATERIALS AND METHODS
Cell culture.
Vero VP30 cells (African green monkey kidney cells stably expressing EBOV VP30) were established as previously described (16). Wild-type Vero cells and Vero VP30 cells were grown in Eagle's minimal essential medium (MEM) supplemented with 10% fetal calf serum (FCS), l-glutamine, vitamins, nonessential amino acid solution, and antibiotics. Huh 7.0 VP30 cells (human hepatocyte cells stably expressing EBOV VP30), HEK 293 VP30 cells (human embryonic kidney cells stably expressing EBOV VP30), and HEK 293 VP30/HO-1 cells (HEK 293 cells stably expressing EBOV VP30 and HO-1) were established in a manner similar to that of Vero VP30 cells. Huh 7.0, HEK 293, and 293T (human embryonic kidney cells stably expressing the simian virus 40 [SV40] T antigen) cell lines were grown in high-glucose Dulbecco's modified Eagle's medium (DMEM) containing 10% FCS, l-glutamine, and antibiotics. All cells were maintained at 37°C and 5% CO2.
Chemicals.
CoPP and tin protoporphyrin (SnPP) (Frontier Scientific) were dissolved in dimethyl sulfoxide (DMSO) to a stock concentration of 50 mM and then further diluted in growth medium to a working concentration before each use.
Virus.
EbolaΔVP30 virus, which expresses green fluorescent protein (GFP) instead of the viral gene, VP30, was generated as previously described (16) and was propagated in Vero VP30 cells using propagation medium (MEM supplemented with 2% FCS, l-glutamine, vitamins, nonessential amino acid solution, and antibiotics). EbolaΔVP30 virus is approved for use under BSL3 containment at the University of Wisconsin by the Institutional Internal Biosafety Committee, NIH, and CDC.
Focus-forming assay.
A focus-forming assay was performed to determine viral titers. Briefly, 10-fold dilutions of virus were adsorbed onto confluent Vero VP30 cells for 1 h at 37°C, after which any unbound virus was removed by washing the cells with propagation medium. The cells were then overlaid with propagation medium containing 1.5% methyl cellulose (Sigma). Six days after infection, the cells were fixed with 10% buffered formaldehyde, permeabilized with 0.25% Triton X-100 in phosphate-buffered saline solution (PBS) for 10 min, and blocked with 5% goat serum and 1% bovine serum albumin (BSA) in PBS for 1 h. The cells were then incubated for 1 h with a 1:10,000 dilution of a mouse monoclonal antibody against EBOV VP40 (16), washed with PBS, and incubated for 1 h with a 1:1,000 dilution of an anti-mouse IgG peroxidase-conjugated secondary antibody. After being washed with PBS, the cells were incubated with 3,3′-diaminobenzidine tetrahydrochloride (Sigma) in PBS. The reaction was stopped by rinsing the cells with water. Foci that stained positive for EBOV VP40 were counted.
siRNA treatment.
Small interfering RNAs (siRNAs) targeting the human HO-1-coding sequences (Hs_HMOX1_1 and Hs_HMOX_10) were purchased from Qiagen. AllStars Negative Control siRNA (Qiagen) was used as the nontargeted siRNA control. The siRNAs were transfected into Vero VP30 or Huh 7.0 VP30 cells at a final concentration of 60 nM by using the HiPerfect transfection reagent (Qiagen) and incubated for 24 h. The cells were then treated with the indicated concentrations of CoPP or SnPP for 24 h prior to infection with EbolaΔVP30 virus at a multiplicity of infection (MOI) of 0.001. The efficiency of HO-1 knockdown was assessed by Western blot analysis using a specific HO-1 antibody (Stressgen). EBOV titers in the siRNA-treated cells were determined as described for the focus-forming assay.
Luciferase activity-based EBOV minireplicon assay.
To examine viral transcription/replication, a luciferase activity-based EBOV minireplicon assay was performed as described previously (31). Briefly, 293T cells or HEK 293 VP30/HO-1 cells were seeded into 12-well tissue culture plates and transfected with the following EBOV protein expression plasmids: pCAGGS EBOV NP (0.75 μg), pCAGGS EBOV VP35 (0. 5 μg), pCAGGS EBOV VP30 (0.5 μg), and pCAGGS EBOV L (2 μg). To generate virus-like RNA, an EBOV minireplicon plasmid (EBOV pol-luc; 0.5 μg) encoding firefly luciferase under the polymerase I promoter was also transfected by using TransIt-LT1 (Mirus) (32). To test the effect of transient HO-1 overexpression on EBOV transcription/replication in the minireplicon assay, 293T cells were cotransfected with a HO-1 protein expression plasmid under a cytomegalovirus (CMV) promoter (Origene). To test the effect of CoPP treatment on EBOV transcription/replication in the minireplicon assay, 1 day following transfection, 293T cells were washed and treated with the indicated concentrations of CoPP or SnPP for 24 h. As a control for luciferase activity and cell viability, cells were transfected with a reporter plasmid containing firefly luciferase under the control of the CMV promoter, pGL2 (1 μg [Promega]). At completion of the experiments, the cells were lysed in 300 μl of Glo-lysis buffer (Promega) and analyzed for firefly luciferase activity using the Steady-Glo luciferase assay system (Promega) according to the manufacturer's instructions.
VSV pseudovirus infectivity assay.
VSVΔG virus possessing luciferase instead of the vesicular stomatitis virus (VSV) G gene and pseudotyped with EBOV GP (VSVΔG-EbGP-luc) was prepared as previously described (33). Vero VP30 and Huh 7.0 VP30 cells were treated with 10 μM or 15 μM CoPP, respectively, for 16 h prior to infection with VSVΔG-EbGP-luc. One hour postinfection, unbound virus was removed by washing the cells three times, and growth medium containing the indicated concentrations of CoPP was added to the cells. Twenty-four hours later, the cells were lysed with 300 μl of Glo-lysis buffer (Promega) and analyzed for firefly luciferase activity by using the Steady-Glo luciferase assay system (Promega) according to the manufacturer's instructions.
EBOV budding assay.
In 35-mm tissue culture dishes, 293T cells were treated with 10 μM CoPP for 16 h, washed, and transfected with the protein expression plasmid pCAGGS EBOV VP40 (2 μg), using TransIt-LT1. The cells were washed 6 h after transfection, and the medium containing 10 μM CoPP was replaced. Two days after transfection, cell supernatants containing virus-like particles (VLPs) were harvested and centrifuged through 20% sucrose for 1.5 h at 27,000 rpm. Cell lysates were also harvested and, together with the VLP pellets, were resuspended in 1× sample buffer (20 mM Tris-HCl, 2 mM EDTA, 1 mM Na3VO4, 2 mM dithiothreitol [DTT], 2% SDS, 20% glycerol, and protease inhibitors [Roche]) and analyzed by Western blotting using a mouse EBOV VP40 antibody.
Western blot analysis.
Cell lysates were mixed with a 4× SDS loading buffer (40% glycerol, 240 mM Tris, 8% SDS, 0.04% bromophenol blue, and 5% β-mercaptoethanol) and then incubated at 95°C for 5 min. Samples were then centrifuged (8,000 rpm for 2 min) and loaded onto a 10% SDS-polyacrylamide gel (Bio-Rad). After electrophoresis, proteins were transferred onto a nitrocellulose membrane (Invitrogen) and blocked for 1 h at room temperature with 5% nonfat dry milk-PBS solution containing 0.05% Tween 20 (PBS-T) (Sigma). The membranes were incubated overnight at 4°C with an anti-HO-1 antibody (Cell Signaling Technologies) or an anti-EBOV VP40 antibody in PBS-T containing 5% bovine serum albumin (Sigma). The membranes were then washed three times with PBS-T for 5 min, incubated with a secondary antibody coupled to horseradish peroxidase in 5% skim milk–PBS-T solution for 1 h, and washed three times with PBS-T for 5 min each time. Bound antibody was detected with Dura chemiluminescence reagent (Thermo Scientific) and a FluorChem HD2 imager (Alpha Innotech).
Cell viability assay.
Vero VP30 and Huh 7.0 VP30 cells were seeded in 96-well plates and treated with various concentrations of CoPP for 3, 5, or 7 days at 37°C. 293T cells were seeded in 96-well plates and treated with various concentrations of CoPP for 1, 2, or 3 days at 37°C. Following these incubations, cell viability was assessed by using the Non-Radioactive Cell Proliferation Assay (Promega) according to the manufacturer's protocol. Absorbance was measured at 490 nm on a Tecan M1000 plate reader.
Statistical analysis.
Student's two-tailed paired and unpaired t test was used to assess statistical differences between samples. Significance levels were set at a P value of ≤0.05.
RESULTS
CoPP upregulates HO-1 expression and attenuates EBOV replication in vitro without altering cellular viability.
Upregulation of HO-1 suppresses infection by certain viruses, including enterovirus, HCV, and HIV (25–30). However, there are currently no data concerning the effect of HO-1 induction on EBOV infection. To determine whether upregulation of HO-1 attenuates EBOV infection, we first examined whether CoPP, a potent HO-1 inducer, increases HO-1 expression in cell lines relevant to this study. Vero VP30, Huh 7.0 VP30, and 293T cells were treated with 10 μM CoPP, cell lysates were harvested at the indicated time points, and HO-1 expression was assessed by Western blot analysis. We found that CoPP induced HO-1 expression in all three cell lines (Fig. 1A). To ensure that CoPP treatment was not cytotoxic to any of our cell lines, we performed viability assays. Treatment of Vero VP30 cells with 10 μM CoPP and treatment of Huh VP30 cells with 10 μM and 15 μM CoPP for 3, 5, and 7 days had no significant effect on cell viability (Fig. 1B and C). Similarly, treatment of 293T cells with 10 μM CoPP for 1, 2, and 3 days did not attenuate cell viability (Fig. 1D).
Fig 1.

CoPP induces HO-1 expression without affecting cell viability. (A) Representative Western blot demonstrating HO-1 induction in Vero VP30, Huh 7.0 VP30, and 293T cells after treatment with 10 μM CoPP from 0 to 24 h. IB, immunoblot. (B to D) Percent viability (compared to vehicle-treated cells) of Vero VP30 or Huh 7.0 VP30 cells on days 3, 5, and 7 or 293T cells on days 1, 2, and 3 posttreatment with the indicated concentrations of CoPP. The data are presented as means ± standard deviations (SD) and are representative of at least three independent experiments.
Next, we evaluated whether CoPP treatment attenuates EBOV growth kinetics. Vero VP30 and Huh 7.0 VP30 cells were pretreated with the indicated concentrations of CoPP for 8 h (Fig. 2A and B). The medium was removed, and the cells were washed once with medium and then infected with EbolaΔVP30 virus at an MOI of 0.001 for 1 h. After infection, the cells were washed three times with medium to remove unbound virus, and fresh medium containing CoPP was added to the cells. For postinfection treatment, the cells were infected as described above and medium containing CoPP was added to the cells only after infection. Supernatants were harvested 3, 5, and 7 days postinfection, and virus titers were determined in Vero VP30 cells by a focus-forming assay. Treatment of Vero VP30 cells pre- and postinfection with 10 μM CoPP reduced virus titers by 2.5 log units (Fig. 2A) compared to cells treated with vehicle. CoPP was even more effective against virus growth in Huh 7.0 VP30 cells (Fig. 2B), since pretreatment with 15 μM CoPP resulted in a 3-log-unit reduction in the viral titer compared with cells treated with vehicle. Virus titers from Huh 7.0 VP30 cells pretreated with a lower concentration of CoPP (10 μM) or treated postinfection with 15 μM CoPP were similarly attenuated after infection. Conversely, similar concentrations of CoPP had no effect on VSV titers when tested pre- or postinfection at multiple MOIs (data not shown). Together, these data demonstrate that CoPP upregulates HO-1 and inhibits EBOV growth without affecting cell viability.
Fig 2.
CoPP attenuates EBOV growth in Vero VP30 and Huh 7.0 VP30 cells. (A and B) Growth kinetics (FFU/ml) of EbolaΔVP30 virus in Vero VP30 cells (A) and Huh 7.0 VP30 cells (B) after treatment with the indicated concentrations of CoPP 8 h prior to infection (pretreatment) or immediately following infection (posttreatment) with EbolaΔVP30 virus at an MOI of 0.001. All values for CoPP-treated cells (pre- and postinfection) are statistically significant (P ≤ 0.05) compared to the values for vehicle-treated cells on the same day, except the 10 μM posttreatment in Vero VP30 cells on day 3 at an MOI of 0.001. (C) Growth kinetics of EbolaΔVP30 in Vero VP30 cells following direct incubation with vehicle or 15 μM CoPP for 1 h. The data are presented as means ± SD (for small SD, the error bars may not be clearly visible) and are representative of at least three independent experiments.
CoPP is not directly virucidal to EBOV.
Several porphyrins and porphyrin derivatives are virucidal, that is, able to reduce virus infectivity after direct contact with the virus (34–36). To determine whether CoPP affects EBOV virulence through this mechanism, 100 μl of EbolaΔVP30 virus (at 1 × 106 focus-forming units [FFU]/ml) was diluted in medium containing CoPP at a final concentration of 15 μM and incubated for 1 h at 37°C. Virus titers were determined in Vero VP30 cells. There was no significant difference in the number of viral foci formed between virus treated with vehicle and virus treated with CoPP (Fig. 2C), indicating that CoPP is not virucidal at the concentrations used in this study. These results indicate that CoPP does not inhibit EBOV growth via cellular toxicity (see above) or direct virucidal properties.
CoPP-mediated attenuation of EBOV growth is HO-1 dependent.
To confirm that attenuation of EBOV growth by CoPP is dependent upon HO-1 induction, siRNAs specific to HO-1 were transfected into Huh 7.0 VP30 or VeroVP30 cells. The cells were then treated with the indicated concentrations of CoPP for 24 h and infected with EbolaΔVP30 virus at an MOI of 0.001 for 4 days. HO-1 siRNA resulted in 74% and 99% decreases in CoPP-induced HO-1 protein in Huh 7.0 VP30 and Vero VP30 cells, respectively (Fig. 3A). Compared with cells treated with CoPP and transfected with a nontargeting siRNA, HO-1 siRNA partially rescued virus growth from CoPP-dependent attenuation, significantly increasing virus titers by 0.7 and 0.8 log units in Vero VP30 (P = 0.001) and Huh 7.0 VP30 (P = 0.0003) cells, respectively (Fig. 3B). These data suggest that CoPP-dependent inhibition of EBOV growth is partially dependent upon HO-1 upregulation.
Fig 3.
CoPP-induced attenuation of EBOV is HO-1 dependent. siRNAs specific to HO-1 were transfected into Vero VP30 or Huh 7.0 VP30 cells, which were then treated with vehicle or CoPP (10 μM and 15 μM, respectively) for 24 h. Then, the cells were infected with EbolaΔVP30 virus at an MOI of 0.001 for 4 days. (A) Western blots demonstrating the downregulation of CoPP-induced HO-1 by siRNAs on day 4 postinfection. (B) Growth of EbolaΔVP30 virus in Vero VP30 or Huh 7.0 VP30 cells on day 4 postinfection. The data are presented as means ± SD and are representative of at least three independent experiments.
To further support the hypothesis that HO-1 is a mediator of CoPP-dependent inhibition of EBOV infection, we tested whether direct HO-1 expression could inhibit EBOV growth kinetics. HEK 293 cell lines stably expressing VP30 (HEK 293 VP30) or both VP30 and HO-1 (HEK 293 VP30/HO-1) were infected with EbolaΔVP30 virus at an MOI of 0.001. Supernatants were harvested 3, 5, and 7 days postinfection, and virus titers were determined in Vero VP30 cells by a focus-forming assay. We found that stable HO-1 expression significantly inhibited EBOV growth at all three time points (all values from the HEK 293 VP30/HO-1 cell line were statistically significant compared to the HEK 293 VP30 cell line) (Fig. 4). These data indicate that HO-1 expression is able to directly inhibit EBOV infection.
Fig 4.
Stable expression of HO-1 attenuates EBOV growth kinetics. Shown are the growth kinetics of EbolaΔVP30 virus in HEK 293 VP30 and HEK 293 VP30/HO-1 cells infected at an MOI of 0.001. The numbers of focus-forming units on days 3, 5, and 7 postinfection are presented as means ± SD and are representative of two independent experiments. The asterisks indicate statistical significance compared to the control (P ≤ 0.05).
To determine whether HO-1 enzymatic activity is necessary for CoPP-mediated attenuation of EBOV, we tested if SnPP, a porphyrin that upregulates HO-1 protein levels but inhibits HO-1 activity (25, 37–39), attenuates EBOV growth. First, cells were treated with SnPP (10 μM for Vero VP30 and 293T cells and 15 μM for Huh 7.0 VP30 cells) for 24 h. Cell lysates were harvested at the indicated time points, and HO-1 expression was assessed by Western blot analysis. We found that the inhibitor SnPP does induce HO-1 expression in all three cell lines (Fig. 5A). Next, Huh 7.0 VP30 and Vero VP30 cells were pretreated with the indicated concentrations of CoPP or SnPP for 8 h. The medium was removed, and the cells were washed and infected with EbolaΔVP30 virus at an MOI of 0.01. After infection, fresh medium containing CoPP or SnPP was added. Cell supernatants from EbolaΔVP30 virus-infected cells were harvested 5 days postinfection, and virus titers were determined in Vero VP30 cells by a focus-forming assay. SnPP treatment had no significant effect on EBOV titers (Fig. 5B). These data support our hypothesis that inhibition of EBOV growth by CoPP is dependent upon HO-1 activity.
Fig 5.
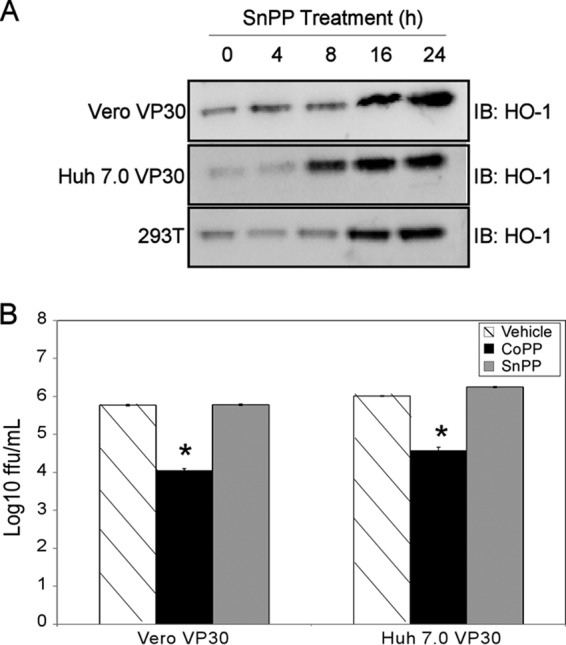
The competitive HO-1 inhibitor SnPP does not attenuate EBOV growth. (A) Western blot demonstrating the induction of HO-1 in Vero VP30, Huh 7.0 VP30, and 293T cells by the selective HO-1 enzymatic inhibitor SnPP. (B) Growth of EbolaΔVP30 virus in Vero VP30 or Huh 7.0 VP30 cells 5 days posttreatment with CoPP or SnPP (10 μM in Vero VP30 and 15 μM in Huh 7.0 VP30 cells) and infection at an MOI of 0.01. All data are presented as means and SD and are representative of at least three independent experiments. The asterisks indicate statistical significance compared to the control (P ≤ 0.05).
CoPP-induced attenuation of EBOV growth is independent of EBOV entry and budding.
To determine the stage(s) of the EBOV life cycle that is targeted by HO-1, we examined the effect of CoPP on EBOV entry and budding. First, Vero VP30 and Huh 7.0 VP30 cells were treated with 10 or 15 μM CoPP, respectively, for 16 h. Then, the cells were infected with a luciferase-expressing VSV lacking VSV G and pseudotyped with EBOV GP (VSVΔG-EbGP-luc) at MOIs of 0.1, 0.01, and 0.001. One hour after infection, the cells were washed and the medium containing CoPP was replaced. Twenty-four hours later, the cells were lysed and luciferase activity was assessed. When CoPP-treated cells were compared to cells treated with vehicle, we found no statistically significant attenuation of luciferase activity (Fig. 6A and B), suggesting that CoPP had no effect on EBOV GP-dependent cell entry. To examine the effect of CoPP on EBOV budding, 293T cells were treated with 10 μM CoPP for 16 h and then transfected with EBOV VP40, the viral protein responsible for the formation and budding of VLPs (40). Two days posttransfection, VP40 expression in cell lysate and VLPs was assessed via Western blotting. The expression levels of VP40 in cell lysates and VLPs (Fig. 6C), as well as endogenous calnexin levels in cell lysates (data not shown) harvested from CoPP-treated cells and cells treated with vehicle, were similar, indicating that CoPP had no effect on EBOV VP40-dependent budding (Fig. 6C).
Fig 6.
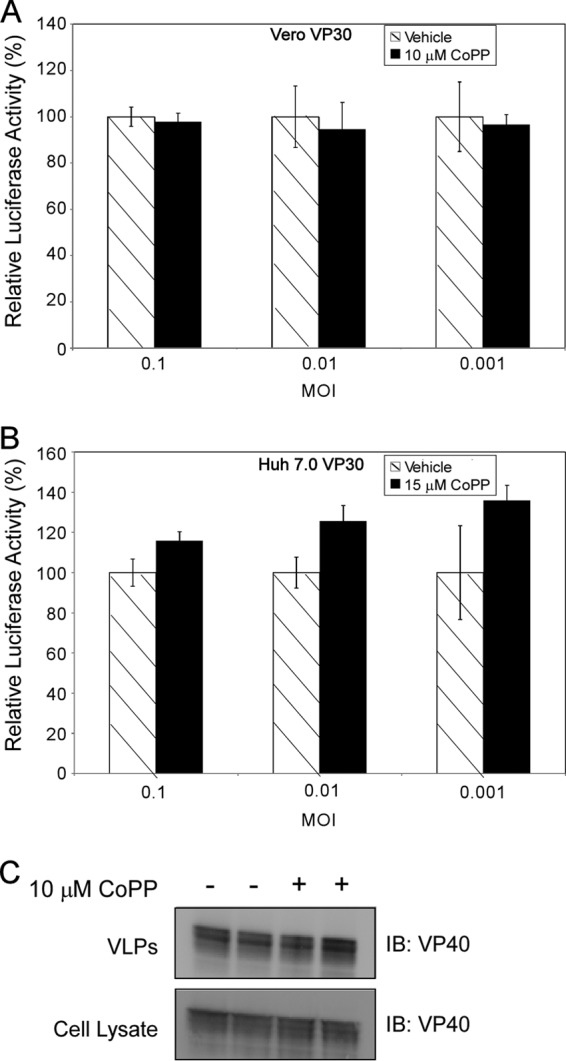
CoPP treatment does not affect EBOV entry or budding. (A and B) Relative luciferase activity in Vero VP30 cells (A) or Huh 7.0 VP30 cells (B) after treatment with 10 μM or 15 μM CoPP for 16 h and infection with VSVΔG EbGP Luc at MOIs of 0.1, 0.01, and 0.001. (C) Representative Western blot demonstrating VP40 expression in VLPs and cell lysate from 293T cells treated with 10 μM CoPP for 16 h and transfected with a VP40 protein expression plasmid for 2 days. The error bars indicate SD.
HO-1 expression attenuates EBOV transcription/replication.
To further examine the mechanism underlying CoPP-dependent inhibition of EBOV growth, we tested the effects of CoPP treatment and HO-1 expression on EBOV transcription and replication in a luciferase-based EBOV minireplicon system. To generate virus-like RNA, 293T cells were transfected with an EBOV minireplicon plasmid encoding firefly luciferase under the control of the RNA polymerase I promoter, along with plasmids encoding EBOV proteins required for transcription and replication, i.e., NP, VP30, VP35, and L (31). To test the effect of CoPP treatment, cells were washed 6 h posttransfection and treated with 10 μM CoPP or 10 μM SnPP for 48 h, after which cell lysates were harvested and assessed for luciferase activity. CoPP treatment, but not treatment with SnPP, significantly attenuated viral replication/transcription in the EBOV minireplicon assay (Fig. 7A). To test the effect of transient HO-1 expression, 293T cells were transfected with EBOV minireplicon plasmids and a HO-1 plasmid under the control of a CMV promoter for 48 h, after which cell lysates were harvested and assessed for luciferase activity. As demonstrated in Fig. 7B, transient expression of HO-1 significantly attenuated viral replication/transcription in the EBOV minireplicon assay. CoPP treatment, SnPP treatment, and the transient expression of HO-1 had no statistically significant effect on the luciferase activity from a control firefly luciferase reporter plasmid (pGL2), indicating that the attenuation was not due to general inhibition of cellular transcription or translation (Fig. 7A and B). Finally, HEK 293 VP30/HO-1 and HEK 293 VP30 cells were transfected with EBOV minireplicon plasmids for 48 h and processed as described above. Stable expression of HO-1 in HEK 293 VP30/HO-1 cells significantly attenuated viral replication/transcription in the EBOV minireplicon assay compared to the HEK 293 VP30 cell line but had no significant effect on the luciferase activity from a control pGL2 plasmid (Fig. 7C). These data demonstrate that HO-1 upregulation interferes with the transcription and replication stages of the EBOV life cycle.
Fig 7.
HO-1 expression attenuates EBOV transcription/replication. (A) Relative polymerase activity (compared to vehicle-treated cells) from an EBOV minireplicon assay in 293T cells after treatment with 10 μM CoPP or SnPP for 2 days. (B) Relative polymerase activity (compared to empty vector) from an EBOV minireplicon assay in 293T cells that were transfected with a HO-1 overexpression plasmid for 2 days. (C) Relative polymerase activity (compared to HEK 293 VP30 cells) 2 days posttransfection with EBOV minireplicon plasmids in HEK 293 VP30/HO-1 cells. The data are presented as means ± SD and are representative of at least three independent experiments. The asterisks indicate statistical significance compared to the control (P ≤ 0.05).
DISCUSSION
Induction of HO-1 has pronounced anti-inflammatory and antioxidant effects, protecting tissues and organs from numerous stresses, including septic shock, virus-induced oxidative injury, and hemorrhage-induced hypoxia (26, 29, 41, 42). Moreover, recent studies have demonstrated that HO-1 induction and activity have antiviral properties. Here, we demonstrate that increased levels of HO-1 suppress EBOV in different cell culture assays. These findings, together with the fact that HO-1 can be induced by several drugs already approved for human use, makes the targeting of HO-1 induction/activity an attractive therapeutic possibility in the battle against EBOV and EBOV-induced disease. To test our hypothesis that upregulation of HO-1 attenuates EBOV growth, we used the potent HO-1 inducer CoPP to induce high HO-1 levels in both Vero cells (an African green monkey kidney cell line devoid of an intact interferon system [43]), and Huh 7.0 VP30 cells (a human hepatocyte cell line). CoPP treatment, both pre- and immediately postinfection, significantly attenuated EBOV growth in both cell lines, indicating that EBOV is sensitive to the antiviral properties of HO-1 (Fig. 2). Additionally, unlike HBV or HCV, which directly target HO-1 for downregulation as a means of optimizing viral replication (28, 44), our study indicates that EBOV infection does not attenuate CoPP-induced HO-1 levels (data not shown).
Induction and activation of HO-1 are stimulated by porphyrins, which are organic compounds that contain sets of four pyrrole rings, such as heme or CoPP. Certain porphyrins and porphyrin derivatives are broadly virucidal against a host of viruses, including EBOV and MARV (34–36). We therefore assessed the potential virucidal effects of CoPP. However, direct incubation of EBOV with CoPP had no effect on focus numbers, indicating that CoPP is not virucidal and attenuates EBOV growth via an intracellular mechanism (Fig. 2C).
To determine the specificity of HO-1 induction in CoPP-mediated attenuation of EBOV growth, we first utilized HO-1 siRNAs to downregulate CoPP-induced HO-1 protein levels in Vero VP30 and Huh 7.0 VP30 cell lines. HO-1 downregulation resulted in a modest but statistically significant rescue of EBOV from CoPP-induced attenuation (Fig. 3B). In addition to its effect on HO-1, CoPP treatment may also modulate the expression of other gene products that, together with HO-1, could influence EBOV infection. Recent studies have suggested that CoPP treatment may also inhibit the activation of inducible nitric oxide synthase (iNOS) and c-Jun N-terminal kinase (JNK) in certain cell types in a HO-1-independent manner (45). Although we have found that specific inhibitors of JNK and nitric oxide (NO) production do not attenuate EBOV growth individually (data not shown), it is possible that, together with HO-1, they contribute to the overall antiviral effect of CoPP treatment on EBOV. To determine whether HO-1 activity was necessary for inhibition of EBOV growth, we utilized SnPP, a molecule that upregulates HO-1 protein expression levels yet inhibits HO-1 activity (25, 37, 38, 39). SnPP treatment did not attenuate EBOV growth (Fig. 5B), indicating that the enzymatic activity of HO-1 is required for EBOV attenuation.
Several studies have shown that activation of HO-1 markedly attenuates the infection of a diverse population of viruses, including enterovirus, HBV, HCV, and HIV. However, there are some viruses, such as influenza A virus, whose replication appears unaffected by inducers of HO-1 (26). Here, we observed differences in the effectiveness of HO-1 inducers against viral infection. Specifically, we found that activation of HO-1 greatly attenuated EBOV infection but had no major effect on the replication of wild-type VSV at several MOIs (data not shown). It is reasonable to assume that these differences are due to the specific mechanisms behind the antiviral properties of HO-1 and what part(s) of the viral replication cycle is disrupted. Previous studies suggest that HO-1 targets viral transcription/replication (25, 29, 46). Our data are consistent with these findings, since both stable and transient expression of HO-1 significantly attenuated EBOV in a minireplicon assay (Fig. 7B and C). Although there is currently no literature indicating that HO-1 may target host ribosomal factors, we cannot rule out the possibility that CoPP treatment/HO-1 overexpression may affect viral translation. In contrast, CoPP-induced attenuation of EBOV infection is independent of viral entry and budding (Fig. 6A to C). How an increase in HO-1 protein/activity leads to the inhibition of viral replication/transcription remains to be elucidated. Recent reports have provided evidence that HO-1 enzymatic products may interfere directly with the viral replication machinery or that increased HO-1 protein expression/enzymatic products may lead to the upregulation of IFN-stimulated genes and the initiation of an antiviral state (27, 47, 48). In the case of EBOV, the latter seems less likely, in that EBOV VP24 and VP35 effectively antagonize IFN pathways and viral inhibition occurs in CoPP-treated Vero cells, which are void of an intact interferon system (43). Another intriguing possibility is that the intrinsic antioxidant properties of HO-1 contribute to its inhibition of viral growth. Besides contributing to the pathogenesis of several viruses, reactive oxygen species can directly affect viral replication through modulation of cell signaling factors, such as kinases and transcription factors (49, 50). Indeed, several reports have demonstrated that antioxidants can greatly attenuate viral infectivity (30, 51–53). However, we found that basic antioxidants, like N-acetylcysteine, and specific inhibitors of NO production could not attenuate EBOV infection independently (data not shown). Interestingly, while the manuscript was in preparation, Panchal et al. reported the identification of an antioxidant compound with potent antiviral activity against EBOV and MARV (54). The authors went on to demonstrate, however, that several other antioxidants could not attenuate filovirus infection in their system. Together, these data suggest that the mechanisms behind the antiviral action of HO-1 are complex and that the antioxidative properties of HO-1 are likely contributory but not sufficient to inhibit EBOV infection.
In summary, stimulation of HO-1 by CoPP attenuates EBOV infection in several cell types. This CoPP-mediated inhibition targets EBOV transcription/replication and is dependent upon HO-1 expression/activity.
ACKNOWLEDGMENTS
We acknowledge membership within and support from the Region X “Pacific Northwest” RCE (NIH award 1-U54-AI-081680).
We thank Susan Watson for scientific editing.
Footnotes
Published ahead of print 9 October 2013
REFERENCES
- 1.Feldmann H, Geisbert TW, Jahrling PB, Klenk HD, Netesov SV, Peters CJ, Sanchez A, Swanepoel R, Volchkov VE. 2004. Filoviridae, p 645–653 In Fauquet C, Mayo MA, Maniloff J, Desselberger U, Ball LA. (ed), Virus taxonomy: 8th report of the International Committee on Taxonomy of Viruses. Elsevier, London, United Kingdom [Google Scholar]
- 2.Kuhn JH, Becker S, Ebihara H, Geisbert TW, Johnson KM, Kawaoka Y, Lipkin WI, Negredo AI, Netesov SV, Nichol ST, Palacios G, Peters CJ, Tenorio A, Volchkov VE, Jahrling PB. 2010. Proposal for a revised taxonomy of the family Filoviridae: classification, names of taxa and viruses, and virus abbreviations. Arch. Virol. 155:2083–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanchez A, Geisbert TW, Feldmann HG. 2007. Filoviridae: Marburg and Ebola viruses, p 1409–1448 In Knipe DM, Howley PM, Griffin DE, Martin MA, Lamb RA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4.MacNeil A, Farnon EC, Wamala J, Okware S, Cannon DL, Reed Z, Towner JS, Tappero JW, Lutwama J, Downing R, Nichol ST, Ksiazek TG, Rollin PE. 2010. Proportion of deaths and clinical features in Bundibugyo Ebola virus infection, Uganda. Emerg. Infect. Dis. 16:1969–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan SY, Empig CJ, Welte FJ, Speck RF, Schmaljohn A, Kreisberg JF, Goldsmith MA. 2001. Folate receptor-alpha is a cofactor for cellular entry by Marburg and Ebola viruses. Cell 106:117–126 [DOI] [PubMed] [Google Scholar]
- 6.Manicassamy B, Wang J, Jiang H, Rong L. 2005. Comprehensive analysis of ebola virus GP1 in viral entry. J. Virol. 79:4793–4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimojima M, Takada A, Ebihara H, Neumann G, Fujioka K, Irimura T, Jones S, Feldmann H, Kawaoka Y. 2006. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J. Virol. 80:10109–10116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muhlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. 1999. Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J. Virol. 73:2333–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panchal RG, Ruthel G, Kenny TA, Kallstrom GH, Lane D, Badie SS, Li L, Bavari S, Aman MJ. 2003. In vivo oligomerization and raft localization of Ebola virus protein VP40 during vesicular budding. Proc. Natl. Acad. Sci. U. S. A. 100:15936–15941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, Klenk HD, Palese P, Garcia-Sastre A. 2003. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 77:7945–7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basler CF, Wang X, Muhlberger E, Volchkov V, Paragas J, Klenk HD, Garcia-Sastre A, Palese P. 2000. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc. Natl. Acad. Sci. U. S. A. 97:12289–12294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, Volchkov VE, Nichol ST, Basler CF. 2006. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J. Virol. 80:5156–5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reid SP, Valmas C, Martinez O, Sanchez FM, Basler CF. 2007. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J. Virol. 81:13469–13477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowen ETW, Baskerville A, Cantell K, Mann GF, Simpson DIH, Zuckerman AJ. 1978. The effect of interferon on experimental Ebola virus infection in rhesus monkeys, p 245–253 In Pattyn SR. (ed), Ebola virus haemorrhagic fever. Elsevier, Amsterdam, The Netherlands [Google Scholar]
- 15.Jahrling PB, Geisbert TW, Geisbert JB, Swearengen JR, Bray M, Jaax NK, Huggins JW, LeDuc JW, Peters CJ. 1999. Evaluation of immune globulin and recombinant interferon-alpha2b for treatment of experimental Ebola virus infections. J. Infect. Dis. 179(Suppl 1):S224–S234 [DOI] [PubMed] [Google Scholar]
- 16.Halfmann P, Kim JH, Ebihara H, Noda T, Neumann G, Feldmann H, Kawaoka Y. 2008. Generation of biologically contained Ebola viruses. Proc. Natl. Acad. Sci. U. S. A. 105:1129–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halfmann P, Ebihara H, Marzi A, Hatta Y, Watanabe S, Suresh M, Neumann G, Feldmann H, Kawaoka Y. 2009. Replication-deficient ebolavirus as a vaccine candidate. J. Virol. 83:3810–3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tenhunen R, Marver HS, Schmid R. 1968. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. U. S. A. 61:748–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maines MD. 1988. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 2:2557–2568 [PubMed] [Google Scholar]
- 20.Applegate LA, Luscher P, Tyrrell RM. 1991. Induction of heme oxygenase: a general response to oxidant stress in cultured mammalian cells. Cancer Res. 51:974–978 [PubMed] [Google Scholar]
- 21.Pae HO, Chung HT. 2009. Heme oxygenase-1: its therapeutic roles in inflammatory diseases. Immune Network 9:12–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farombi EO, Surh YJ. 2006. Heme oxygenase-1 as a potential therapeutic target for hepatoprotection. J. Biochem. Mol. Biol. 39:479–491 [DOI] [PubMed] [Google Scholar]
- 23.Chung SW, Hall SR, Perrella MA. 2009. Role of haem oxygenase-1 in microbial host defence. Cell. Microbiol. 11:199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gozzelino R, Jeney V, Soares MP. 2010. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 50:323–354 [DOI] [PubMed] [Google Scholar]
- 25.Devadas K, Dhawan S. 2006. Hemin activation ameliorates HIV-1 infection via heme oxygenase-1 induction. J. Immunol. 176:4252–4257 [DOI] [PubMed] [Google Scholar]
- 26.Hashiba T, Suzuki M, Nagashima Y, Suzuki S, Inoue S, Tsuburai T, Matsuse T, Ishigatubo Y. 2001. Adenovirus-mediated transfer of heme oxygenase-1 cDNA attenuates severe lung injury induced by the influenza virus in mice. Gene Ther. 8:1499–1507 [DOI] [PubMed] [Google Scholar]
- 27.Lehmann E, El-Tantawy WH, Ocker M, Bartenschlager R, Lohmann V, Hashemolhosseini S, Tiegs G, Sass G. 2010. The heme oxygenase 1 product biliverdin interferes with hepatitis C virus replication by increasing antiviral interferon response. Hepatology 51:398–404 [DOI] [PubMed] [Google Scholar]
- 28.Protzer U, Seyfried S, Quasdorff M, Sass G, Svorcova M, Webb D, Bohne F, Hosel M, Schirmacher P, Tiegs G. 2007. Antiviral activity and hepatoprotection by heme oxygenase-1 in hepatitis B virus infection. Gastroenterology 133:1156–1165 [DOI] [PubMed] [Google Scholar]
- 29.Zhu Z, Wilson AT, Mathahs MM, Wen F, Brown KE, Luxon BA, Schmidt WN. 2008. Heme oxygenase-1 suppresses hepatitis C virus replication and increases resistance of hepatocytes to oxidant injury. Hepatology 48:1430–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tung WH, Hsieh HL, Lee IT, Yang CM. 2011. Enterovirus 71 induces integrin beta1/EGFR-Rac1-dependent oxidative stress in SK-N-SH cells: role of HO-1/CO in viral replication. J. Cell. Physiol. 226:3316–3329 [DOI] [PubMed] [Google Scholar]
- 31.Jasenosky LD, Neumann G, Kawaoka Y. 2010. Minigenome-based reporter system suitable for high-throughput screening of compounds able to inhibit Ebolavirus replication and/or transcription. Antimicrob. Agents Chemother. 54:3007–3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neumann G, Fujii K, Kino Y, Kawaoka Y. 2005. An improved reverse genetics system for influenza A virus generation and its implications for vaccine production. Proc. Natl. Acad. Sci. U. S. A. 102:16825–16829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takada A, Robison C, Goto H, Sanchez A, Murti KG, Whitt MA, Kawaoka Y. 1997. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 94:14764–14769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo H, Pan X, Mao R, Zhang X, Wang L, Lu X, Chang J, Guo JT, Passic S, Krebs FC, Wigdahl B, Warren TK, Retterer CJ, Bavari S, Xu X, Cuconati A, Block TM. 2011. Alkylated porphyrins have broad antiviral activity against hepadnaviruses, flaviviruses, filoviruses, and arenaviruses. Antimicrob. Agents Chemother. 55:478–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vzorov AN, Dixon DW, Trommel JS, Marzilli LG, Compans RW. 2002. Inactivation of human immunodeficiency virus type 1 by porphyrins. Antimicrob. Agents Chemother. 46:3917–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen-Collins AR, Dixon DW, Vzorov AN, Marzilli LG, Compans RW. 2003. Prevention of poxvirus infection by tetrapyrroles. BMC Infect. Dis. 3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sardana MK, Kappas A. 1987. Dual control mechanism for heme oxygenase: tin(IV)-protoporphyrin potently inhibits enzyme activity while markedly increasing content of enzyme protein in liver. Proc. Natl. Acad. Sci. U. S. A. 84:2464–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siner JM, Jiang G, Cohen ZI, Shan P, Zhang X, Lee CG, Elias JA, Lee PJ. 2007. VEGF-induced heme oxygenase-1 confers cytoprotection from lethal hyperoxia in vivo. FASEB J. 21:1422–1432 [DOI] [PubMed] [Google Scholar]
- 39.Marinissen MJ, Tanos T, Bolos M, de Sagarra MR, Coso OA, Cuadrado A. 2006. Inhibition of heme oxygenase-1 interferes with the transforming activity of the Kaposi sarcoma herpesvirus-encoded G protein-coupled receptor. J. Biol. Chem. 281:11332–11346 [DOI] [PubMed] [Google Scholar]
- 40.Noda T, Sagara H, Suzuki E, Takada A, Kida H, Kawaoka Y. 2002. Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. J. Virol. 76:4855–4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takaki S, Takeyama N, Kajita Y, Yabuki T, Noguchi H, Miki Y, Inoue Y, Nakagawa T. 2010. Beneficial effects of the heme oxygenase-1/carbon monoxide system in patients with severe sepsis/septic shock. Intensive Care Med. 36:42–48 [DOI] [PubMed] [Google Scholar]
- 42.Vallabhaneni R, Kaczorowski DJ, Yaakovian MD, Rao J, Zuckerbraun BS. 2010. Heme oxygenase 1 protects against hepatic hypoxia and injury from hemorrhage via regulation of cellular respiration. Shock 33:274–281 [DOI] [PubMed] [Google Scholar]
- 43.Emeny JM, Morgan MJ. 1979. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J. Gen. Virol. 43:247–252 [DOI] [PubMed] [Google Scholar]
- 44.Abdalla MY, Britigan BE, Wen F, Icardi M, McCormick ML, LaBrecque DR, Voigt M, Brown KE, Schmidt WN. 2004. Down-regulation of heme oxygenase-1 by hepatitis C virus infection in vivo and by the in vitro expression of hepatitis C core protein. J. Infect. Dis. 190:1109–1118 [DOI] [PubMed] [Google Scholar]
- 45.Lin HY, Shen SC, Lin CW, Wu MS, Chen YC. 2009. Cobalt protoporphyrin inhibition of lipopolysaccharide or lipoteichoic acid-induced nitric oxide production via blocking c-Jun N-terminal kinase activation and nitric oxide enzyme activity. Chem. Biol. Interact. 180:202–210 [DOI] [PubMed] [Google Scholar]
- 46.Shan Y, Zheng J, Lambrecht RW, Bonkovsky HL. 2007. Reciprocal effects of micro-RNA-122 on expression of heme oxygenase-1 and hepatitis C virus genes in human hepatocytes. Gastroenterology 133:1166–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Z, Wilson AT, Luxon BA, Brown KE, Mathahs MM, Bandyopadhyay S, McCaffrey AP, Schmidt WN. 2010. Biliverdin inhibits hepatitis C virus nonstructural 3/4A protease activity: mechanism for the antiviral effects of heme oxygenase? Hepatology 52:1897–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tzima S, Victoratos P, Kranidioti K, Alexiou M, Kollias G. 2009. Myeloid heme oxygenase-1 regulates innate immunity and autoimmunity by modulating IFN-beta production. J. Exp. Med. 206:1167–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwarz KB. 1996. Oxidative stress during viral infection: a review. Free Radic. Biol. Med. 21:641–649 [DOI] [PubMed] [Google Scholar]
- 50.Peterhans E. 1997. Oxidants and antioxidants in viral diseases: disease mechanisms and metabolic regulation. J. Nutr. 127:962S–965S [DOI] [PubMed] [Google Scholar]
- 51.Kalebic T, Kinter A, Poli G, Anderson ME, Meister A, Fauci AS. 1991. Suppression of human immunodeficiency virus expression in chronically infected monocytic cells by glutathione, glutathione ester, and N-acetylcysteine. Proc. Natl. Acad. Sci. U. S. A. 88:986–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gonzalez O, Fontanes V, Raychaudhuri S, Loo R, Loo J, Arumugaswami V, Sun R, Dasgupta A, French SW. 2009. The heat shock protein inhibitor Quercetin attenuates hepatitis C virus production. Hepatology 50:1756–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garozzo A, Tempera G, Ungheri D, Timpanaro R, Castro A. 2007. N-acetylcysteine synergizes with oseltamivir in protecting mice from lethal influenza infection. Int. J. Immunopathol. Pharmacol. 20:349–354 [DOI] [PubMed] [Google Scholar]
- 54.Panchal RG, Reid SP, Tran JP, Bergeron AA, Wells J, Kota KP, Aman J, Bavari S. 2012. Identification of an antioxidant small-molecule with broad-spectrum antiviral activity. Antiviral Res. 93:23–29 [DOI] [PubMed] [Google Scholar]