

Abstract
Flock House virus (FHV) is a positive-sense RNA insect virus with a bipartite genome. RNA1 encodes the RNA-dependent RNA polymerase, and RNA2 encodes the capsid protein. A third protein, B2, is translated from a subgenomic RNA3 derived from the 3′ end of RNA1. B2 is a double-stranded RNA (dsRNA) binding protein that inhibits RNA silencing, a major antiviral defense pathway in insects. FHV is conveniently propagated in Drosophila melanogaster cells but can also be grown in mammalian cells. It was previously reported that B2 is dispensable for FHV RNA replication in BHK21 cells; therefore, we chose this cell line to generate a viral mutant that lacked the ability to produce B2. Consistent with published results, we found that RNA replication was indeed vigorous but the yield of progeny virus was negligible. Closer inspection revealed that infected cells contained very small amounts of coat protein despite an abundance of RNA2. B2 mutants that had reduced affinity for dsRNA produced analogous results, suggesting that the dsRNA binding capacity of B2 somehow played a role in coat protein synthesis. Using fluorescence in situ hybridization of FHV RNAs, we discovered that RNA2 is recruited into large cytoplasmic granules in the absence of B2, whereas the distribution of RNA1 remains largely unaffected. We conclude that B2, by binding to double-stranded regions in progeny RNA2, prevents recruitment of RNA2 into cellular structures, where it is translationally silenced. This represents a novel function of B2 that further contributes to successful completion of the nodaviral life cycle.
INTRODUCTION
The nodaviruses are a family of positive-strand RNA viruses that naturally infect insects and fish. They have a bipartite genome that contains approximately 4,500 bases and encodes three proteins. The most thoroughly studied nodavirus is the insect virus Flock House virus (FHV), which has served as an important model system to investigate the structural and molecular basis of RNA replication, virus assembly, and inhibition of antiviral host responses (1). The larger of the two genomic RNAs, RNA1 (3.1 kb), encodes the RNA-dependent RNA polymerase (RdRp; 112 kDa), which is located on the outer membrane of mitochondria in infected cells (2, 3). Viral RNA synthesis induces so-called spherules (4), i.e., membrane invaginations, which are thought to sequester the replication complexes and double-stranded RNA (dsRNA) intermediates to protect them from RNA silencing, a major antiviral pathway activated in insects upon infection with RNA viruses (5). Further protection from RNA silencing is afforded by FHV protein B2 (11.6 kDa), a dsRNA binding protein that is translated from RNA3 (387 nucleotides), a subgenomic RNA derived from the 3′ end of RNA1 (6–8). The capsid protein, protein alpha (43 kDa), is translated from RNA2 (1.4 kb), the second genomic RNA segment.
Although a seemingly simple virus, FHV uses a sophisticated regulatory system to control its gene expression. This regulation occurs at several levels and is currently incompletely understood. For example, replication of RNA2 is dependent on transactivation by the subgenomic RNA3, but RNA2 synthesis subsequently leads to shutdown of RNA3 replication (9–12). How this is accomplished mechanistically is unknown. Replication of RNA1 and RNA2 leads to synthesis of roughly equimolar amounts of the two RNAs throughout infection, yet their respective proteins are generated in widely different ratios, with RNA2 producing massive amounts of capsid protein whereas RNA1 gives rise to comparatively small quantities of RdRp (13, 14). Previous investigations have shown that this is due in part to RNA2 competing more effectively for the translational apparatus, but it is likely that additional aspects underlie this differential regulation (14). Not surprisingly, the synthesis of RdRp and coat protein is also controlled temporally, with RdRp being generated early in infection and coat protein much later (13, 15, 16). This leads to a situation in which progeny RNA is packaged into particles with a significant delay following synthesis, raising the question of where in the cell this RNA is located and how it is stabilized during that time.
Investigations of protein B2 have focused on its function as an inhibitor of RNA silencing. The crystal structure of this protein has shown that it is a homodimer which forms a four-helix bundle by antiparallel association of two monomers (7). It binds dsRNA independent of sequence and length and can interact with both long dsRNAs, preventing their cleavage by Dicer-2, and short interfering RNAs (siRNAs), inhibiting their loading into the RISC complex. Immunoprecipitation experiments of FHV-infected Drosophila melanogaster cell lysates indicated that B2 interacts with the RdRp, and it is thought that this interaction positions B2 next to dsRNA replicative intermediates for their protection from the RNA interference (RNAi) machinery (17). Supporting this data, confocal immunofluorescence analysis showed that B2 is present in close proximity to RdRp in infected cells (18). However, the amount of B2 synthesized in infected Drosophila cells vastly exceeds that of RdRp, and it is clear that not all of it can be physically associated with the polymerase. Indeed, a significant portion of B2 is located away from the replication complexes in the periphery of the cell, where it shows a reticular distribution reminiscent of the capsid protein (18). This points to the possibility that B2 has additional functions associated with this cellular location, but they have remained unexplored.
In addition to insect cells, FHV can also replicate in plants (19), nematodes (20), mammalian cells (21), and yeast (22), indicating that potential host factors required for completion of its infection cycle are highly conserved. Replication in these heterologous systems is typically restricted to a single round, as progeny virus is unable to exit the cell or reinfect fresh cells due to lack of a suitable receptor. Infection of mammalian cells, which requires incubation at a reduced temperature (≤33°C), is usually initiated by transfection of the viral RNAs or plasmids that can launch replication by synthesis of the viral transcripts. Interestingly, it was reported several years ago that FHV RNA replication in BHK21 cells is vigorous in the absence of B2, suggesting that these cells do not use RNA interference as an antiviral pathway (23). Based on this observation, we chose this cell line to generate a mutant of FHV that lacked the gene for B2. Here, we report that while the absence of B2 in BHK21 cells does not interfere with viral RNA synthesis, it leads to defects in the viral life cycle which manifest themselves in extremely low yields of progeny virus. Low viral yield was due to inefficient production of coat protein, which was also observed in the presence of B2 mutants that had reduced dsRNA binding affinity. Additional investigations revealed that in the vast majority of infected cells, the absence of B2 led to recruitment of RNA2, but not RNA1, into cytoplasmic granules, where it appeared to be translationally silenced. These observations suggest that in addition to functioning as an inhibitor of RNA silencing, B2 regulates the specific activities of progeny RNA, controlling the outcome of nodaviral infection at two different levels.
MATERIALS AND METHODS
Cells.
Drosophila melanogaster line 1 (DL-1) cells were maintained at 27°C in Schneider's insect medium as described previously (13). Baby hamster kidney (BHK21) cells were propagated at 37°C (5% CO2) in Dulbecco's modified Eagle medium (DMEM; Invitrogen) containing 5% heat-inactivated fetal bovine serum (FBS) as well as penicillin and streptomycin as described previously (21). All transfection experiments involving these cells and FHV RNAs were carried out at 28°C.
Antibodies.
Rabbit polyclonal antiserum against FHV RdRp was a generous gift from David J. Miller, University of Michigan, Ann Arbor, MI. To generate rabbit polyclonal antiserum against FHV B2, maltose binding protein (MBP)-tagged B2 was expressed and purified by amylose affinity chromatography as described previously (7), followed by removal of MBP by enterokinase cleavage and purification of the cleaved B2 by anion-exchange chromatography using a Mono Q 5/50 GL column and an AKTA purifier chromatography system (GE Healthcare). B2 antiserum was raised in rabbits by Quality Controlled Biochemicals (Hopkinton, MA). Monoclonal mouse antibodies against B2 were a generous gift from Roland R. Rueckert, University of Wisconsin, Madison, WI. Monoclonal mouse antibodies to α-tubulin were obtained from Sigma-Aldrich.
Plasmids.
To generate a plasmid for in vitro transcription of capped RNA1 transcripts, a synthetic DNA fragment containing the restriction sites NsiI, SacII, SnaBI, and XbaI was cloned into the insertion site of pGEM-T vector (Promega), followed by introduction of an EcoRI site directly upstream of the T7 promoter sequence by QuikChange mutagenesis (Agilent Technologies). A DNA fragment comprising the T7 promoter and full-length RNA1 cDNA next was PCR amplified from a previously described plasmid, called FHV(1,0) (23), using as a forward primer an oligonucleotide containing the sequence of the T7 promoter. EcoRI and XbaI sites located proximal to the 5′ and 3′ termini of the PCR product then allowed for insertion of the T7-RNA1 fusion construct into corresponding restriction sites in the modified pGEM-T vector, yielding a transcription plasmid for wt FHV RNA1 (designated pF2100). QuikChange mutagenesis was used to introduce mutations into this plasmid for the generation of constructs R1ΔB2, R1B21-74, and R1KOR3 and for construction of all B2 RNA binding domain mutants (Table 1).
Table 1.
Summary of B2 mutants and corresponding amino acid changes in FHV RdRp as a result of nucleotide changes introduced into RNA1
| B2 mutant | Nucleotide change(s) in RNA1 | Amino acid change(s) in RdRp |
|---|---|---|
| RNA1ΔB2 | T2739C/C2910Aa | None |
| R1KOR3 | G2633T | W865L |
| B2-R36A | A2843G/G2844C | Q935R |
| B2-R36D | A2843G/G2844C/G2845C | Q935R/G936R |
| B2-K47A | A2876G/A2877C | Q946R |
| B2-K47D | A2876G/A2877C/G2878C | Q946R/G947R |
| B2-K62A | A2921G/A2922C | E961G |
| B2-K62D | A2921G/A2922C/A2923C | E961G/T962P |
| B21-74 | G2960T | R974L |
Previously described by Ball (23).
Liposome-mediated RNA transfection.
Total BHK21 cell RNA containing RNA1 served as a source of RNA1 for liposome-mediated transfection. This RNA was prepared in two steps. First, an mMessage machine T7 kit (Ambion) was used to synthesize capped wild-type (wt) or mutant RNA1 transcripts from XbaI-linearized pF2100-based plasmid DNA. Second, the resultant transcripts were transfected into BHK21 cells using Lipofectamine 2000 (Invitrogen), followed by extraction of total cellular RNA at 24 h posttransfection using the RNeasy kit (Qiagen). For transfection experiments, mixtures comprising 500 ng RNA1-containing total RNA and 200 ng capped RNA2 transcripts were added to cells grown in 35-mm cell culture dishes in the presence of a liposome transfection reagent. Transfectin (Bio-Rad) was used for the transfection of DL-1 cells and Lipofectamine 2000 for BHK21 cells. The capped RNA2 transcripts used in these experiments were synthesized from an XbaI-linearized plasmid called p2BS (+)-wt as described previously (24).
RNA analysis.
Total RNA was prepared from transfected DL-1 and BHK21 cells using the RNeasy kit, while a previously described phenol-chloroform extraction method was used to extract RNA from wt and ΔB2 mutant FHV particles (25). Electrophoretic analysis of this RNA was carried out by running it under nondenaturing conditions through 1% agarose gels at 300 V in sodium borate buffer (26), followed by visualization of ethidium bromide (EtBr)-stained RNA with a FluorChem SP gel documentation system (Alpha Innotech). For pulse-chase labeling of FHV RNA in BHK21 cells, complete medium was replaced at 14 h posttransfection with 1 ml serum-free, phosphate-free DMEM (Invitrogen) containing actinomycin D (20 μg/ml). After 1 h of incubation at 28°C, the RNAs were metabolically labeled for 1 h by addition of 32P-labeled UTP (450 μCi per 35-mm dish). Subsequent to removal of the labeling medium, the cells were washed with complete medium and then incubated in this medium for various time intervals. Total RNA was extracted from the cells, electrophoresed through denaturing agarose-formaldehyde gels, and transferred to nylon membranes as described previously (27). Radiolabeled RNA on the resultant blots was visualized using a Storm 820 Phosphorimager (GE Healthcare).
Protein analysis.
To prepare cell lysates for quantitative immunoblot analysis, the cells were washed twice in phosphate-buffered saline (PBS), resuspended in SDS-PAGE loading buffer, and heated to 100°C for 3 min. The samples were then subjected to denaturing SDS-PAGE in NuPage 4 to 12% gels (Invitrogen) and transferred to nitrocellulose membranes by electroblotting. The blots were processed for two-color immunodetection of multiple antigens using an Odyssey infrared imaging system (LI-COR Biosciences) as described previously (28) and analyzed using its associated application software (V3.0). Intracellular tubulin served as a loading control for normalization of levels of RdRp and coat protein.
Purification of virus particles.
To estimate the relative viral yield from transfected BHK21 cells, FHV particles were partially purified using a previously described protocol (29) and subjected to quantitative immunoblot analysis using rabbit anti-FHV serum. In brief, the cells were lysed in 0.5% Nonidet P-40, and cell debris was pelleted at maximum speed for 10 min at 4°C in a microcentrifuge. The resultant supernatant was sedimented through a 30% (wt/wt) sucrose cushion in 50 mM HEPES (pH 7) at 192,000 × g (45,000 rpm in an SW55 rotor) for 45 min at 11°C. The pelleted material was resuspended in SDS-PAGE loading buffer and analyzed by immunoblotting using rabbit anti-FHV serum. A previously described method involving sucrose gradient centrifugation was utilized to purify FHV particles to homogeneity from BHK21 cells (30).
Polyribosome analysis.
BHK21 cells were treated for 15 min with cycloheximide (100 μg/ml) at 12 h posttransfection, washed with ice-cold PBS containing cycloheximide, and resuspended in this buffer with a cell scraper. After this, the cells were pelleted by centrifugation at 250 × g for 5 min at 4°C, washed in 50 mM HEPES (pH 7.2), 25 mM MgCl2, 150 mM KCl (wash buffer), and lysed for 10 min on ice in the same buffer containing 0.5% Nonidet P-40, 15 mM dithiothreitol, 100 μg/ml cycloheximide, 500 μg/ml heparin, and 200 U/ml SUPERase-In (Ambion). The lysates were then centrifuged at 14,000 × g for 5 min at 4°C, and the resultant supernatants were layered on top of 10 to 50% (wt/wt) sucrose gradients in wash buffer, followed by centrifugation at 151,000 × g (35,000 rpm) for 2 h at 4°C in a Beckman SW41 rotor. Gradients were fractionated and polyribosome profiles recorded at a wavelength of 254 nm using an ISCO gradient fractionator. To determine if FHV RNA2 was present within the fractions, RNA was extracted from each fraction using TRIzol reagent (Invitrogen) and then subjected to reverse transcription-PCR (RT-PCR) using the OneStep RT-PCR kit (Qiagen). The forward and reverse primers corresponded to nucleotides 600 to 617 and 880 to 868 of RNA2, respectively; thus, they were specific for the amplification of a 280-bp product. Subsequent to thermal cycling, RT-PCR mixtures were subjected to electrophoretic analysis on 1% agarose gels containing 0.5 μg/ml EtBr in Tris-acetate-EDTA buffer.
Drosophila infection and RNA analysis.
Drosophila strains y1w1, y1w1; Dcr-2L811fsX and y1w1; AGO2414 have been described previously (31). These fly stocks were raised on standard cornmeal-agar medium at 25°C. Adult flies 4 to 6 days of age were used in infection experiments. Infections were done by intrathoracic injection (Nanoject II apparatus; Drummond Scientific) with 4.6 nl of a viral suspension in 10 mM Tris-HCl, pH 7.5. For comparative analysis of wt FHV and FHVΔB2, both viral stocks were prepared in BHK21 cells. Injection of the same volume of 10 mM Tris-HCl (pH 7.5) was used as a control. All survival experiments were performed at 22°C. Quantification of viral RNA was based on either RNA blots analyzed with a Bioimager (Fujix BAS 2000) or real-time quantitative RT-PCR (qRT-PCR) as previously described (31). For qRT-PCR, the sequences of the primers for amplification of FHV RNA1 were the following: FHV RNA1 forward, 5′-TTTAGAAGCACATGCGTCCAG-3′; FHV RNA1 reverse, 5′-CGCTCACTTTCTTCGGGTTA-3′. For RNA blotting, the primers used to generate the DNA probes were the following: RpL32 forward, 5′-GTGTATTCCG ACCACGTTACA-3′; RpL32 reverse, 5′-ATACAGGCCCAAGATCGTGA-3′; FHV RNA2 forward, 5′-CGGAACAACTTCAACATCTAGG-3′; and FHV RNA2 reverse, 5′-AACCAAATTGGGCTTATTGTGG-3′.
Indirect immunofluorescence and FISH.
BHK21 cells were transfected with wt FHV RNA2 transcripts and either wt or mutant FHV RNA1 as described above. Transfected cells were incubated at 28°C for 15 h before being fixed in 0.3% glutaraldehyde and 3% paraformaldehyde (in PBS, 1 mM EGTA, 1 mM MgCl2, pH 6.8) for 10 min at room temperature. All indirect immunofluorescence and MitoTracker staining was done as previously reported (32). Monoclonal mouse anti-B2 antibodies were used at a 1:6,000 dilution. Fluorescence in situ hybridization (FISH) was carried out using a QuantiGene ViewRNA in situ hybridization cell kit (Affymetrix) with custom-designed probes directed against positive-sense RNA1 (VF4-14080-01) and RNA2 (VF1-13994-01) according to the manufacturer's instructions. Briefly, prior to in situ hybridization of probes, cells were permeabilized and treated with a 1:4,000 dilution of the provided protease to allow better accessibility of viral RNA to the probes. Thereafter, cells were rinsed and incubated sequentially with the probe, preamplifier, amplifier, and label probe solutions at 40°C, with two 2-min washes between each step. All incubations were for 30 min, except for the probes, which were incubated for 3 h. Cells were then stained with DAPI, washed, and mounted for imaging.
Confocal microscopy and image analysis.
Fluorescently labeled FHV proteins and viral RNA were imaged using a Zeiss LSM 710 confocal microscope at the Scripps Research Institute Core Light Microscopy Facility using standard laser lines for the detection of fluorescein isothiocyanate (FITC)/Alexa Fluor 488 and Cy3/Alexa Fluor 555. Deconvolution of z series images was performed using the constrained iterative algorithm contained in the Zeiss Zen Blue analysis software.
RESULTS
Differential effect of protein B2 on viral RNA and protein synthesis in insect and mammalian cells.
Transfection of cultured Drosophila cells (Schneider's line 1; DL-1) with wild-type (wt) FHV RNA1 and RNA2 gave rise to progeny RNA that was easily detectable in total cellular RNA 24 h later (Fig. 1A). Moreover, the three viral proteins, RdRp, B2, and capsid protein, were sufficiently abundant to be visible in a mixture of total cellular proteins on stained polyacrylamide gels (Fig. 1B). In contrast, neither viral RNA nor protein were detectable by these methods when wt RNA1 was replaced with a mutant version called RNA1ΔB2, in which the B2 open reading frame (ORF) in the subgenomic RNA3 (shifted +1 relative to the ORF of RdRp) was closed due to two nucleotide changes engineered into RNA1 (Table 1) (23). Only traces of RdRp and coat protein were present under these conditions, as revealed by the much more sensitive immunoblot analysis (Fig. 1C). Given the critical function of B2 as an inhibitor of RNA interference in Drosophila cells, these results were unsurprising.
Fig 1.
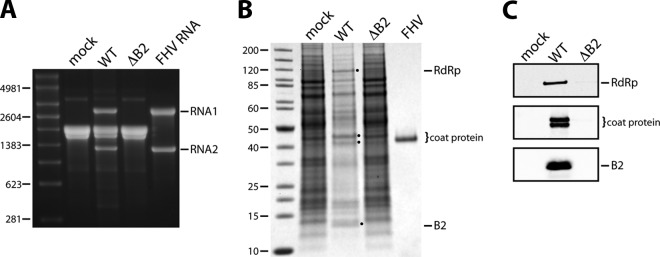
Expression analysis of FHV ΔB2 mutant in Drosophila DL-1 cells. DL-1 cells were transfected with RNA1, wt or ΔB2 mutant, and RNA2 and harvested for analysis at 24 h posttransfection. (A) Electrophoretic analysis of total cellular RNA on a nondenaturing agarose gel. RNA isolated from FHV particles generated by infection of DL-1 cells (FHV RNA) was included as a control. The molecular sizes of RNA markers (in nucleotides) are indicated on the left. (B) Electrophoretic analysis of total cell lysates on an SDS-polyacrylamide gel stained with Coomassie brilliant blue. Purified FHV was included as a marker for coat protein. Molecular size markers are indicated in kDa. Note that FHV infection of DL-1 cells is accompanied by a significant decline in host protein synthesis, explaining the reduced background of cellular proteins in the wt sample. (C) Immunoblot analysis of the lysates shown in panel B for the detection of RdRp, coat protein, and B2. Coat protein and B2 were visualized on the same blot, while RdRp was visualized on a separate blot. Coat protein undergoes an autocatalytic maturation cleavage following assembly into particles, explaining the presence of two bands representing the precursor protein and the mature viral capsid protein.
Analogous experiments in mammalian BHK21 cells yielded significantly different outcomes. Although transfection with wt RNAs produced essentially the same results as those observed in Drosophila cells, the use of RNA1ΔB2 gave rise to abundant amounts of progeny RNA as well. However, despite high levels of FHV RNA2, only trace amounts of coat protein were detectable (Fig. 2). Robust replication of FHV RNAs in the absence of B2 in BHK21 cells has previously been reported, but production of viral protein under these conditions had not been investigated (23). To further probe this phenomenon, we performed a detailed time course experiment, in which the accumulation of viral RNAs and proteins in BHK21 cells was monitored following transfection with wt RNA2 and either wt RNA1 or RNA1ΔB2. As shown in Fig. 2A, in both cases progeny RNA was first detectable on ethidium bromide-stained agarose gels at 8 h posttransfection (hpt). The levels rapidly increased thereafter and closely matched or even exceeded those of ribosomal RNAs at 24 and 48 h. RNA3, the subgenomic RNA derived from RNA1, became faintly visible at these later points.
Fig 2.

Time course of FHV RNA and protein accumulation in BHK21 cells in the presence or absence of B2. BHK21 cells were transfected with RNA2 in vitro transcripts together with either wt or ΔB2 mutant RNA1 and harvested at 0, 4, 8, 12, 24, and 48 h posttransfection (HPT). (A) Electrophoretic analysis of total cellular RNA on a nondenaturing agarose gel. RNA isolated from FHV particles generated in DL-1 cells (FHV RNA) was included as a control. The molecular sizes of RNA markers (in nucleotides) are indicated on the left. (B) Immunoblot analysis of RdRp, coat protein, and B2 in total cell lysates at various times after transfection. Tubulin was included as a loading control. (C) Graphical representation of the time course of RdRp and coat protein accumulation in the presence and absence of B2. Intracellular protein levels were normalized to the level of tubulin and expressed as a percentage of the maximal signal intensity values for the respective proteins.
Accumulation of viral proteins was monitored in parallel by immunoblot analysis (Fig. 2B). As expected, cells transfected with RNA1ΔB2 did not show evidence for the presence of B2, and there was no significant difference in the accumulation of RdRp relative to cells transfected with wt RNA1, except at the 8-h time point, when the latter clearly contained more of this protein. A dramatic difference, however, was detected for the viral coat protein. In cells containing B2, it was first detectable 8 hpt, and its level steadily increased over the next 40 h. In stark contrast, coat protein was undetectable until 24 hpt in cells lacking B2, and even then it was only faintly visible on the blot. Although the signal increased somewhat over the next 24 h, it did not reflect the large amount of RNA2 that was present at this point in the same cells (compare Fig. 2A to B). Quantification of immunoblot signals using fluorescently labeled secondary antibodies further confirmed that throughout the time course, the amount of coat protein was substantially lower in cells lacking B2 than in those containing B2 (Fig. 2C). In addition, the analysis revealed that the level of RdRp was also reduced, although to a much lesser degree.
Not surprisingly, the considerable reduction of coat protein synthesis in the absence of B2 led to a steep decrease in the yield of progeny virus particles. As shown in Fig. 3A, only a minute amount of FHV was produced in the absence of B2, and recovery of particles after sucrose gradient sedimentation was challenging. Nonetheless, we were able to obtain sufficient amounts of purified particles to permit extraction of encapsidated RNA and visualization on ethidium bromide-stained agarose gels (Fig. 3B). This analysis confirmed that the particles contained RNA1 and RNA2, excluding the possibility that B2 plays a significant role in specific packaging of the viral genome.
Fig 3.
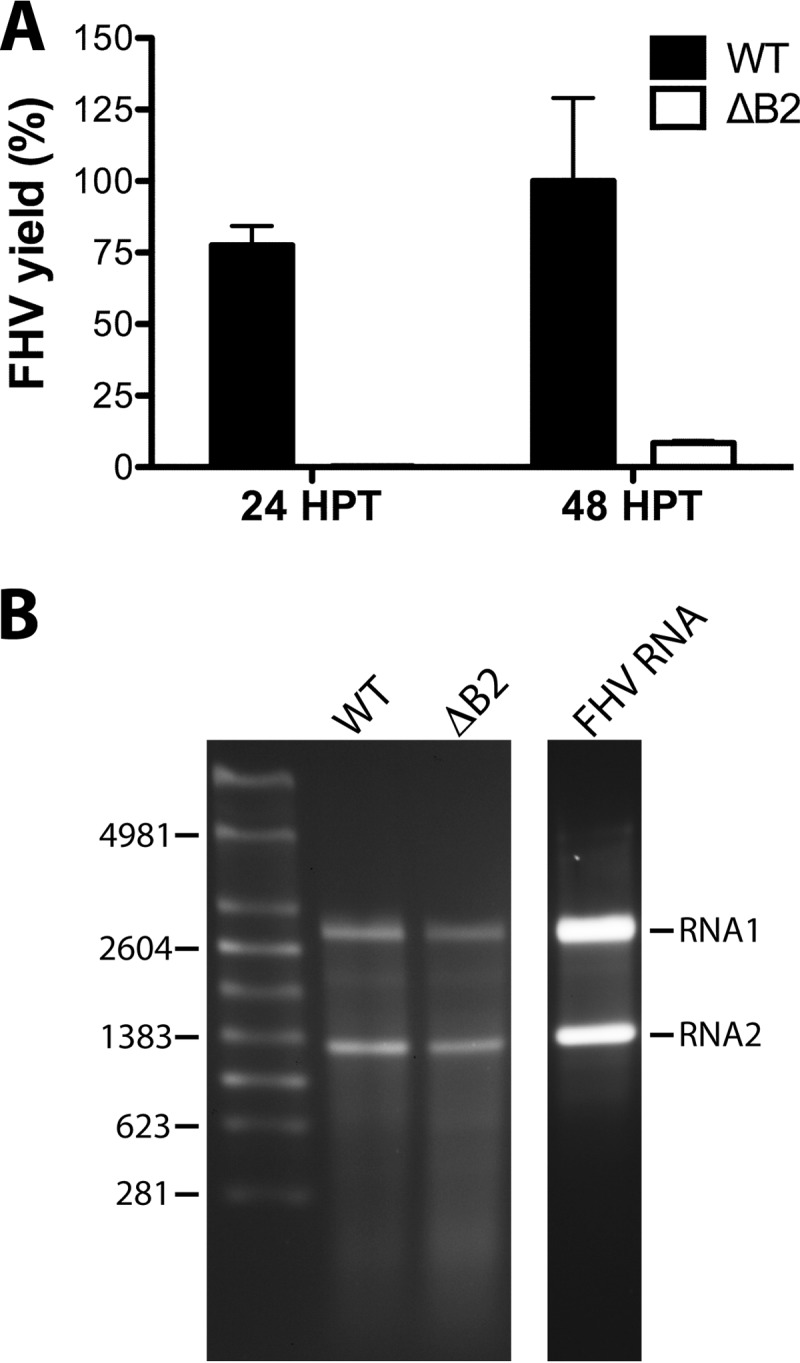
Comparison of yield and RNA packaging phenotypes of FHV particles assembled in the presence or absence of B2 in BHK21 cells. (A) Graphical representation of the relative amounts of wt and ΔB2 FHV particles obtained at 24 and 48 h posttransfection (HPT). FHV yield was estimated by quantitative immunoblot analysis from signal intensity values for FHV coat protein in partially purified particle preparations and were expressed as a percentage of the maximal yield. Error bars represent the standard errors of the means for two independent FHV preparations. (B) Electrophoretic analysis of RNA extracted from wild-type and ΔB2 particles at 48 h posttransfection. RNA isolated from wt FHV particles generated in DL-1 cells (FHV RNA) was included as a control. The molecular sizes of RNA markers (in nucleotides) are indicated on the left.
As expected, FHVΔB2 particles were not infectious to wt Drosophila flies but infected and killed mutant flies lacking a functional Dicer-2 or Argonaute-2 protein (Fig. 4). Similar results have been reported previously (33). More interestingly, when we compared the infectivity of FHVΔB2 particles to that of wt FHV in Drosophila strains defective for Dicer-2 or Argonaute-2, we observed a notable delay in the time to death for flies infected with FHVΔB2 (Fig. 4C and E) as well as a significant reduction in the yield of progeny virus (Fig. 4D and F). Thus, even in the absence of a functional RNAi pathway, FHVΔB2 showed reduced virulence compared to wt FHV, pointing to a potential secondary function of B2 independent of its role as an inhibitor of RNAi.
Fig 4.

FHVΔB2 is less pathogenic than wt FHV in mutant Drosophila flies whose RNAi pathways are inactivated. (A, C, and E) Survival curves of wt and mutant flies (20 per group) after infection with either FHVΔB2 or wt FHV (1,000 PFU per fly). Data represent the means and standard deviations from duplicate experiments. P < 0.005 (**) and P < 0.0001 (****) by log-rank (Mantel-Cox) test. (B, D, and F) Viral titer in wt and mutant flies as determined by quantitative RT-PCR 3 days after infection with FHVΔB2 or wt FHV (1,000 PFU per fly). Total RNA isolated from five flies was used in each qRT-PCR analysis. Bars represent the means and standard deviations from five independent experiments. P < 0.05 (*) and P < 0.0001 (****) by Student's t test.
Investigation of BHK21 cellular responses to FHV infection in the absence of B2.
In contrast to insect cells, mammalian cells do not use RNA interference as a major antiviral pathway. Instead, they employ alternative mechanisms that are triggered by cytoplasmic dsRNA, including activation of protein kinase R (PKR) with resulting phosphorylation of eIF2α and global translational shutoff and/or degradation of RNA through activation of 2′-5′ oligoadenylate synthetase with resulting activation of RNaseL. Since FHV replication in the absence of B2 likely led to cytoplasmic exposure of dsRNA, either through the presence of viral replication intermediates or partially folded single-stranded RNA (ssRNA) progeny, we sought to determine whether these pathways were activated to explain the decrease in coat protein synthesis. However, metabolic labeling of cellular and viral proteins with [35S]methionine/cysteine from 13 to 14 hpt in the presence or absence of B2 did not reveal any differences in the amount of radioactivity incorporated into total cellular protein, excluding translational shutdown as the likely explanation (data not shown). Moreover, we were unable to detect phosphorylated eIF2α by immunoblot analysis with antibodies specific to the phosphorylated form of this protein (data not shown). It also seemed unlikely that RNaseL was activated, given the large amounts of viral RNAs present in the absence of B2. However, it was possible that viral RNA was subject to higher turnover in the absence of B2, which, while not manifesting itself in reduced steady-state levels, could have resulted in reduced translational efficiency. To test this, we monitored stability of newly synthesized viral RNAs by pulse-chase analysis. Specifically, at 14 hpt, BHK21 cells transfected with RNA1 (wt or ΔB2) and RNA2 were incubated for 1 h in serum-free, phosphate-free medium containing actinomycin D to inhibit RNA transcription from cellular DNA. [32P]Orthophosphoric acid was added, and the cells were incubated for another hour. The medium was then replaced with standard medium, and total RNA was extracted after various times following radiolabeling. Analysis of the RNA by gel electrophoresis and phosphorimaging revealed similar rates of RNA degradation in the presence and absence of B2 (Fig. 5). Altogether, these data suggested that the reduction in coat protein synthesis was not due to general cellular responses triggered by the presence of dsRNA in mammalian cells but that other mechanisms led to reduced translational efficiency in the absence of B2.
Fig 5.

Pulse-chase labeling of viral RNA synthesized in the presence or absence of B2. BHK21 cells transfected with the indicated RNA species were metabolically labeled with 32P-labeled UTP at 14 h posttransfection. Labeling was carried out for 1 h in the presence of actinomycin D and terminated by replacement of the labeling medium with fresh growth medium. At the indicated times, total RNA was extracted from the cells, electrophoresed through denaturing agarose-formaldehyde gels, and transferred to nylon membranes. Bands representing viral RNAs were visualized on a phosphorimager and quantified using the software ImageQuant.
Differences in translational efficiency may arise from variations in ribosome occupancy of a given mRNA; therefore, we investigated the association of RNA2 with polyribosomes in the presence and absence of B2. To this end, transfected BHK21 cells were lysed at 12 hpt following treatment with the translation elongation inhibitor cycloheximide, and the clarified lysates were centrifuged through a linear sucrose gradient. Fractionation of the gradients with continuous absorbance showed that the presence or absence of B2 had no effect on the polyribosome profile itself (Fig. 6). RNA was then extracted from each fraction and subjected to RT-PCR using RNA2-specific primers. After 20 cycles of PCR, the anticipated 280-bp cDNA product of RNA2 was readily detectable in fractions containing polyribosomes prepared in the presence of B2 (Fig. 6A) but not in its absence (Fig. 6B). In the latter case, 10 additional PCR cycles were required to visualize this product (data not shown). Taken together, these results suggested that reduced translational efficiency of coat protein in the absence of B2 was a consequence of decreased ribosomal occupancy of RNA2.
Fig 6.

Distribution of RNA2 in polyribosome fractions from BHK21 cells transfected with R1WT+R2 (A) or R1ΔB2+R2 (B). (Top) Polyribosome profiles of clarified extracts from these cells after sedimentation through 10 to 50% sucrose gradients. Sedimentation is from left to right. Peaks corresponding to 40S and 60S ribosomal subunits, as well as 80S ribosomes, are indicated. (Middle) Electrophoretic analysis of 18S and 28S rRNA extracted from fractions collected across each gradient. Shown are ethidium bromide-stained agarose gels in inverse color. Note that there is only a rough correspondence between the lanes of the gels and the trace of the gradient profile shown above. (Bottom) RT-PCR analysis for the specific detection of RNA2 in the same fractions. Primers used for this analysis directed amplification of a 280-bp cDNA fragment. Shown are ethidium bromide-stained agarose gels in inverse color.
The C terminus of B2 is not required for efficient coat protein synthesis or inhibition of RNAi.
We next turned our attention to B2 to identify regions that might play a role in the observed effect on translational efficiency of coat protein. The X-ray structure of B2 has provided detailed insights into the domain that is involved in binding to dsRNA (residues 1 to 73), but the structure lacks the C-terminal 33 amino acids, which were deleted to promote crystallization (7). To test the potential function of this C-terminal region, we generated a truncation mutant by engineering a stop codon into the B2 ORF on RNA1 immediately following amino acid 74 (B21-74). This involved the introduction of a single-nucleotide substitution (G2960T), which changed the corresponding amino acid in the RdRp ORF from Arg to Leu (residue 974) (Table 1). Drosophila and BHK21 cells were then transfected with the mutated RNA1 and wt RNA2, and RNA replication and coat protein synthesis were analyzed 24 h later. As controls for this experiment, we included the RNA1ΔB2 mutant as well as an RNA1 mutant that was unable to generate the subgenomic RNA3 because of a single-nucleotide change in its promoter region on RNA1 (Table 1).
As expected, neither of these control constructs, when transfected together with wt RNA2 into Drosophila cells, gave rise to progeny RNA or viral proteins (Fig. 7A). In BHK21 cells, on the other hand, both yielded progeny RNA1, but only RNA1ΔB2 gave rise to RNA2 and RNA3 (Fig. 7B). This was anticipated, as RNA3 is required in trans for the replication of RNA2. When transfected with RNA1 encoding the truncated B21-74 and wt RNA2, both types of cells contained abundant progeny RNAs and coat protein 24 h later, indicating that the C-terminal 32 amino acids of B2 are dispensable for viral RNA replication and efficient translation (Fig. 7A and B, lanes 4). Moreover, there was no change in the yield or specific infectivity of progeny virus compared to that of virus generated in the presence of full-length B2 (data not shown). To further confirm that virus encoding B21-74 was phenotypically indistinguishable from wt virus, we infected wt Drosophila flies with either viral isolate and monitored viral load and survival. Viral load was indistinguishable, as reflected by statistically insignificant differences in the levels of progeny RNA2 (Fig. 7C). However, a small but statistically significant difference in the rate with which the flies succumbed to infection was observed (Fig. 7D). The importance of this minor difference is currently unclear but could reflect altered effects on host gene expression by full-length versus truncated B2 protein (31). Taken together, the results suggested that the C-terminal 32 amino acids of protein B2 are nonessential for virus accumulation in vitro and in vivo. These findings differ from a report by Singh et al. (34), who found that the C terminus is required for the function of B2 as an inhibitor of RNAi. This inconsistency will be discussed below.
Fig 7.

Effect of removal of the C-terminal 32 amino acids of B2 on viral replication in DL-1 and BHK21 cells and in Drosophila flies. (A) Expression analysis of R1B21-74 in DL-1 cells at 24 h posttransfection. Shown is an image of an EtBr-stained gel on which total RNA extracts from cells transfected with the indicated constructs were loaded (top panel), as well as immunoblots for the detection of coat protein and B2 in corresponding cell lysates (lower panels). R1KOR3 refers to the RNA1 construct in which synthesis of RNA3 was knocked out. (B) Analogous analysis for the expression of R1B21-74 in BHK21 cells. (C) Quantification of viral RNA2 in flies 4 days postinfection with wt or B21-74 mutant FHV particles (1,000 PFU/fly). Data are normalized relative to RpL32 and represent the means and standard deviations of results from three independent experiments. The P value from Student's t test is indicated. (D) Survival curves for adult flies (10 per group) injected with wt or B21-74 mutant FHV particles (1,000 PFU/fly). Data represent the means and standard deviations from 3 independent groups. **, P < 0.005 by log-rank (Mantel-Cox) test.
Mutations in the B2 dsRNA binding domain affect translational efficiency of viral RNAs.
We next focused on the dsRNA binding domain of B2. B2 is a dimer that forms a four-helix bundle by antiparallel association of two monomers (7). One face of the helical bundle binds the phosphodiester backbone of dsRNA in a manner that is independent of the sequence and length of the RNA substrate. Many of the contacts established by B2 are electrostatic in nature and involve positively charged amino acid side chains, including those of Arg36, Lys47, and Lys62. We mutated these residues to Ala or Asp to reduce or inhibit the ability of B2 to bind dsRNA and then tested the effect on viral RNA replication and protein synthesis in BHK21 cells. Note that because of the 2-fold symmetry of the B2 dimer, a single mutation occurs twice in the dsRNA binding domain. As shown in Fig. 8, the mutations had little, if any, effect on RNA replication, but introducing the negatively charged Asp at any of the three positions drastically compromised accumulation not only of viral coat protein but also of B2. Even the steady-state levels of RdRp appeared to be affected, although to a much lesser degree. The presence of Ala had a milder effect: it was well tolerated at positions 36 and 47 but significantly reduced viral protein accumulation when located at position 62. The effect of the Asp mutants was enhanced when they were introduced in duplicate or triplicate at all three sites (Fig. 8). These results indicated that the dsRNA binding function of B2 was required for efficient translation of coat protein as well as of B2 itself and, to a lesser extent, of RdRp. As expected, when the Asp mutants were tested in Drosophila cells, progeny RNA was not detectable on ethidium bromide-stained agarose gels. However, trace amounts of coat protein and B2 were detectable by immunoblot analysis in total cell lysates, indicating that RNA replication was not completely inhibited, although it was greatly decreased (data not shown).
Fig 8.

Effect of mutations in the dsRNA binding domain of B2 on FHV RNA and protein synthesis in BHK21 cells. (Top) Electrophoretic analysis of total RNA extracted from cells transfected with wt RNA2 and the indicated mutant RNA1 species, harboring single, double, or triple point mutations in the B2 ORF. Total RNA from mock-transfected cells and cells transfected with wt R1+R2 or R1ΔB2+R2 served as controls. (Lower) Immunoblots comparing cellular levels of coat protein and B2 for all constructs tested in this study. Tubulin served as a loading control.
Distribution of FHV proteins in BHK21 cells.
Mammalian cells subjected to environmental stresses, such as viral infection, may respond by sequestering mRNAs into cytoplasmic bodies called stress granules, in which these mRNAs are translationally silenced. Using antibodies against stress granule markers, such as TIA-1 and G3BP, we probed for the presence of stress granules in BHK21 cells transfected with wt RNA2 and either wt RNA1 or RNA1ΔB2 by confocal fluorescence microscopy. However, there was no evidence for stress granule formation at 15 hpt when large amounts of viral RNA had accumulated in the cell (data not shown).
Because we had not been able to detect activation of any of several well-established response pathways launched by mammalian cells against viral infection, we decided to investigate the cellular distribution of FHV proteins in BHK21 cells transfected with wt RNA1 and RNA2 in order to determine whether there were any obvious differences compared to Drosophila cells that could provide hints about the mechanism underlying poor translation of viral proteins in the absence of B2. However, the distribution of FHV proteins in BHK21 cells reflected what is observed in Drosophila cells (Fig. 9): RdRp colocalized with mitochondria, which were clustered in the perinuclear region, whereas coat protein had the same reticular appearance that is observed in Drosophila cells. There was significant overlap between the fluorescence signals for coat protein and B2, suggesting close proximity or colocalization of the two proteins, which also was previously observed in Drosophila cells. While we did not detect the ribbon-like structures of B2 that are seen woven between mitochondria in insect cells (18), these structures were detectable in BHK21 cells at later times following transfection of the viral RNAs (not shown). Taken together, these results did not provide further insight into the question at issue.
Fig 9.

Confocal immunomicroscopy showing subcellular distribution of FHV proteins in BHK21 cells 15 h posttransfection with wt FHV RNAs. (a) Cells were stained for mitochondria with MitoTracker (red) and for RdRp with anti-RdRp antibodies (green). Note that mitochondria cluster near the nucleus (blue), and that red signal is surrounded by a ring of green fluorescence representing RdRp on the outer membrane of the organelles. Mitochondrial distribution in an uninfected cell is shown in the bottom right corner. (b and c) Cells stained with antibodies for detection of B2 and capsid protein (CP), respectively. The cell in the upper left corner was transfected with RNA1 only and shows staining for B2 but not CP. (d) Overlay of panels b and c. B2 is shown in green, CP in red. Regions where the two proteins colocalize appear yellow. All images represent single optical slices and scale bars represent 10 micrometers.
Absence of B2 causes cytoplasmic aggregation of viral RNA.
We next examined the cellular distribution of viral RNAs to determine whether there were any differences in the location of RNA2 in the presence or absence of B2. The analysis was done by fluorescence in situ hybridization (FISH) using probes complementary to positive-sense RNA2 and positive-sense RNA1. The latter was designed to exclude the region that gives rise to the subgenomic RNA3, allowing specific detection of RNA1. The FISH protocol involved a protease digestion step that removed any potential protein bound to the RNA of interest (for details, see Materials and Methods).
Given that viral RNAs are synthesized on the outer membrane of mitochondria, we expected to observe progeny RNA in close proximity to these organelles, which tend to cluster in the perinuclear region of infected cells. Unexpectedly, in the vast majority of cells (95%) that were transfected with the wt RNAs, both RNA1 and RNA2 were distributed throughout the peripheral region of the cytosol and concentrated near the cell surface (Fig. 10a to d). Overlaying the fluorescence signal associated with RNA1 and RNA2 revealed a nonuniform, reticular pattern, likely caused by associations with cellular membranes and/or cytoskeletal elements. Although both RNAs appeared to be associated with the same structures, only a subpopulation displayed significant colocalization. Instead, the RNAs were spread out across closely associated regions of alternating signal intensity. Interestingly, progeny positive-sense RNA was consistently excluded from a large perinuclear region, representing the area populated by the mitochondria and RdRp. In an attempt to visualize replication complexes on mitochondria, we employed probes complementary to the negative-sense strands of RNA1 and RNA2 but were unable to detect appreciable fluorescence signal. Similarly, we were unable to detect dsRNA using a specific antibody in indirect immunofluorescence analysis (data not shown). Taken together, our results indicated that newly synthesized RNAs are rapidly transported away from the replication site to cellular locations, where they probably serve their roles as the substrates for translation and packaging into progeny particles.
Fig 10.

Detection of FHV RNAs in transfected BHK21 cells by fluorescence in situ hybridization (FISH). BHK21 cells were transfected with wt RNA2 and either wt or mutant RNA1 and processed for FISH 15 h later. Fluorescently labeled probes were directed against positive-strand RNA1 and RNA2. (a to d) Cells transfected with wt RNA2 and wt RNA1. (e to m) Cells transfected with wt RNA2 and R1ΔB2. (n to p) Cells transfected with wt RNA2 and RNA1 encoding B2 mutant K47A. (q to s) Cells transfected with wt RNA2 and RNA1 encoding B2 mutant R36D/K47D/K62D. Probes against RNA1 (FITC, green) did not hybridize with the region giving rise to subgenomic RNA3. RNA2 probes were labeled with Cy3 (red). Merged images show an overlay of RNA1 and RNA2 signals (yellow), with boxed areas magnified approximately 5-fold (d and h) or 2-fold (l and m). All images represent single optical slices and scale bars represent 10 μm.
A dramatic difference in RNA distribution was observed when wt RNA1 was replaced with RNA1ΔB2 (Fig. 10e to m). Most notably, while roughly half of the cells (49%) displayed normal positive-sense RNA1 distribution, positive-sense RNA2 was almost exclusively detected in disorganized clusters (82% of cells) that were located primarily in the perinuclear region near the replication sites (Fig. 10f to h and j to m). In some cases, the RNA2 masses were observed in structures at the extreme periphery or outside cells, possibly as the result of exocytic activity, although this has yet to be studied in detail (data not shown). In other cells, both positive-sense RNA1 and positive-sense RNA2 were detected in overlapping foci, although they appeared to contain significantly more RNA2 than RNA1 (Fig. 10l and m). At no point was RNA1 detected in the clustered distribution in the absence of RNA2, nor was clumped RNA1 observed with a normal RNA2 distribution.
We next performed FISH analysis on cells expressing two of the B2 mutants depicted in Fig. 8 to determine whether there was a correlation between their effect on coat protein translation and cellular distribution of the viral RNAs. Indeed, in the presence of the K47A mutant of B2, which had no effect on coat protein synthesis (Fig. 8, lane 3), the RNA distribution was indistinguishable from that observed in the presence of wild-type B2 (Fig. 10n to p). In contrast, the R36D/K47D/K62D triple mutant, in whose presence coat protein synthesis was drastically reduced (Fig. 8, lane 9), produced an RNA pattern analogous to that of ΔB2 (Fig. 10q to s).
In summary, we conclude that accumulation of viral RNAs in cytoplasmic granules interfered with their ability to serve as substrates for translation, thereby explaining the drastic reduction in coat protein synthesis and the milder effect on synthesis of RdRp. B2, by virtue of its RNA binding ability, appears to play a critical role in preventing the formation of these granules via a mechanism that transcends its role as an inhibitor of RNA interference. That the clustering of RNA in the absence of B2 was significantly more pronounced for RNA2 than RNA1 is intriguing and requires further investigation.
DISCUSSION
The study described here was prompted by our efforts to generate suitable quantities of an FHV mutant that lacked the ability to synthesize protein B2. Such a mutant can, in principle, be generated in Drosophila cells if one inactivates the RNAi pathway, e.g., by knockdown of Dicer-2 or Argonaute-2, but this is an impractical approach. Given an earlier report that B2 is not required for FHV RNA replication in BHK21 cells (23), we decided to use this cell line to generate the mutant virus. However, while FHV RNA replication was indeed robust in the absence of B2, the yield of progeny virus was negligible, and we traced this back to very low levels of coat protein despite the presence of large amounts of RNA2. These surprising results were inconsistent with effects caused by activation of mammalian antiviral pathways in response to dsRNA, such as activation of PKR, phosphorylation of eIF2α, global translational shutdown or activation of 2′-5′ oligoadenylate synthetase, and degradation of viral RNAs by RNaseL (35). Indeed, we did not detect any evidence that these pathways were operational. Moreover, there was no indication of the formation of stress granules, which could have explained translational arrest of FHV RNAs, although it would have still been unclear why the effect appeared to be more selective for RNA2. In addition, extensive analyses using reporter systems based on firefly luciferase, green fluorescent protein (GFP), and coat protein using replicating and nonreplicating versions of RNA2 excluded the possibility that B2 was directly involved in translation of RNA2, e.g., in a manner analogous to that of alfalfa mosaic virus coat protein or reovirus protein NSP3 (36–39 and data not shown).
By mutational analysis of B2, we established that its dsRNA binding capacity was critical for maintaining the viral RNAs in a state in which they functioned as efficient substrates for translation. We began the analysis with a focus on the potential role of the C-terminal 32 amino acids of B2, which are not included in the crystal structure but are known to be dispensable for binding to dsRNA (7). Interestingly, deleting this region of the protein had no effect on viral growth or yield in BHK21 cells or Drosophila cells, and there were no appreciable differences in specific infectivity compared to the wild-type virus when tested in Drosophila cell culture. These results contrast with a previous report that deletion of the C-terminal 17 amino acids or point mutations in this region result in a significant decrease in the ability of B2 to suppress RNAi (34). However, these observations were made in Sf21 cells using a reporter assay and were not investigated in the context of FHV, potentially explaining the discrepancy with our results.
Various single-site mutations in B2 at positions known to be critical for interaction with dsRNA revealed that interfering with the RNA binding function led to a drastic decrease not only of coat protein synthesis but of B2 as well. The effect on RdRp accumulation, on the other hand, was much less pronounced. It is noteworthy that none of the mutations in B2 significantly affected RNA replication, given that each mutation was accompanied by a mutation in the C terminus of RdRp. The C-terminal region of RdRp is predicted to be highly unstructured; therefore, it may not be surprising that it tolerated amino acid substitutions well. That said, we noted that the presence of the mutations coincided with a slightly reduced level of RdRp relative to that generated by wt RNA1 or RNA1ΔB2 (Fig. 8). Recall that RNA1ΔB2 also encodes wt RdRp (Table 1). Therefore, it is possible that mutations in the C terminus have subtle effects on synthesis or stability of the polymerase, but this needs to be further investigated. That the C terminus does play an important role was also suggested by the fact that of several additional mutants not included in this report, the one that led to inadvertent deletion of the 56 C-terminal amino acids of RdRp completely inactivated the enzyme (C44D in B2 introduced a stop codon after amino acid 942 in the RdRp ORF). Thus, although potentially unstructured, the C terminus nevertheless appears to be critical. Analogous observations regarding the function of the C terminus of RdRp have been made previously (2).
Results indicating that interaction of B2 with dsRNA was an important factor in coat protein and B2 synthesis motivated us to investigate the cellular distribution of viral progeny RNA by FISH. RNA1 probes were designed such that they did not hybridize to the region that encompasses the subgenomic RNA3, allowing us to draw specific conclusions about the location of RNA1. Since viral RNA replication occurs on the outer membrane of mitochondria and they tend to cluster in the perinuclear region of infected cells, particularly at later times in infection, we expected to observe wild-type progeny RNAs in this general location of the cell. Surprisingly, the RNAs were detected in the periphery of the cytoplasm and often were concentrated near the cell surface. Indeed, they appeared to be specifically excluded from the region surrounding the replication sites. Unfortunately, it was not possible to visualize these replication sites in parallel using indirect immunofluorescence (IF) analysis and antibodies against RdRp, because the IF and FISH protocols are not compatible. We are currently developing strategies to solve this problem. We attempted to identify replication sites using probes specific for negative-sense RNA1 and RNA2 or, separately, an antibody specific for dsRNA, but we were unable to detect any appreciable signal in either case. This was curious but might be explained by the fact that FHV RNA replication complexes are sequestered in spherules formed by invagination of the outer mitochondrial membrane (4). The narrow neck that connects the spherules to the cytoplasm may present steric hindrance for antibodies or RNA probes to gain access to their targets as well as interfere with secondary interactions required for their visualization.
The current results lead us to conclude that upon synthesis and release from RdRp, progeny RNA is rapidly transported away from the replication site toward peripheral regions of the cell. This transport may involve the cytoskeletal network, as both microtubules and actin filaments are known to play a critical role in transport of cellular and viral mRNAs (40–42). Indeed, microtubule-dependent transport of progeny RNA away from the site of replication was recently reported for vesicular stomatitis virus (VSV), a negative-strand RNA virus (43).
Preliminary analyses of FHV-infected Drosophila cells indicate that the distribution of viral RNA is analogous to that described here for BHK21 cells. In addition, in both Drosophila and BHK21 cells, this distribution is independent of the coat protein, excluding the possibility that coat protein is required for RNA transport or that the RNA is transported inside assembled particles. The apparent movement of progeny RNA from the replication site to the site of translation and assembly in the periphery of the cell presents a departure from our previous model, in which we proposed that coat protein travels from the site of translation to the site of replication in order to retrieve the RNAs for assembly (32). In light of the current data, the latter scenario appears to be less likely.
The distribution of viral RNAs in the absence of B2 was strikingly different in the vast majority of cells (82%). Although the pattern of RNA1 remained largely unchanged, RNA2 was detected in distinct foci that differed in size, shape, and number per cell. Moreover, these foci were located in the space devoid of RNA signal in the presence of B2 and occupied by replication complexes. In some cells, RNA1 formed similar foci that colocalized with RNA2, but their fluorescent signal was consistently weaker. As already indicated, we do not believe that these foci represent canonical stress granules, since we did not detect the presence of stress granule markers TIA-1 and G3BP. In addition, there was no evidence of phosphorylated eIF2α, which is typically linked to stress granule formation (44). We have not yet investigated whether the observed structures represent P-bodies. P-bodies serve to process RNAs for decay, and they usually have a uniform size and shape, two features that are not consistent with our observations (44, 45). Numerous other types of cytoplasmic ribonucleoprotein bodies have been described in the literature (45), including, most recently, so-called D2 bodies in Drosophila cells. The latter represent cytoplasmic foci containing Dicer-2, the dsRNA binding protein R2D2, as well as siRNAs and possibly other components (46). Whether D2 bodies also exist in mammalian cells is not yet known. Clearly, further characterization of the RNA granules detected in our study, including their composition and dynamic behavior, is of high priority.
The cellular distribution of viral RNAs in the absence of B2 provided an explanation for the observed phenotype at the protein level. The relatively normal pattern of RNA1 produced few changes in the level of RdRp, whereas aggregation of RNA2 in cytoplasmic granules led to a drastic decrease in coat protein synthesis. Evidently, RNA2 is translationally inactive in these granules, and this is supported by our analyses indicating that association of RNA2 with polyribosomes is severely reduced. Absence of RNA2-containing granules in the presence of B2 must be a result of interactions between B2 and RNA2 based on the inability of B2 mutants with reduced RNA binding ability to prevent their formation. Since B2 binds dsRNA but not ssRNA (7), there have to be local double-stranded regions in progeny RNA2 that permit complex formation with B2, and it is possible that multiple B2 dimers bind the RNA in order to protect it from entrapment in cytoplasmic inclusions. Because we also observed a drastic reduction in synthesis of the B2 RNA binding mutants, we hypothesize that subgenomic RNA3 becomes trapped in RNA granules as well. Confirmation of this hypothesis will have to await development of an experimental system that allows specific detection of RNA3, and we are currently in the process of designing suitable probes to that end. Interestingly, it was recently shown that the cellular decapping activators LSm1, Pat1, and Dhh1 are involved in FHV RNA replication in yeast and control the ratio of FHV RNA1 to subgenomic RNA3 (47). These decapping activators are known P-body components, and they may be poised for additional roles in the absence of a functional B2 protein.
One of the most intriguing results was the observation that absence of B2 did not affect RNA1 to the same extent as RNA2. We can currently only speculate what the reasons for this differential behavior might be, but we suspect that it is a reflection of where or when during infection the two RNAs are translated and/or how efficiently they assemble translation initiation complexes. Additional studies are required to tease out the mechanism underlying differential translational inactivation of the two genomic RNA segments. Furthermore, it will be important to determine whether the observed translational arrest is permanent or whether it can be reversed if B2 is provided subsequent to granule formation. The novel function of B2 described here may also be required for FHV infection of Drosophila cells, but this will be more difficult to investigate due to the vital upstream function of B2 as an inhibitor of RNA silencing.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health to A.S. (GM053491) and J.-L.I. (PO1 AI070167) and the French National Research Agency to J.-L.I. (ANR Darvis). J.E.P. was the recipient of a Ruth L. Kirschstein NRSA fellowship.
Footnotes
Published ahead of print 2 October 2013
REFERENCES
- 1.Venter PA, Schneemann A. 2008. Recent insights into the biology and biomedical applications of Flock House virus. Cell Mol. Life Sci. 65:2675–2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller DJ, Ahlquist P. 2002. Flock house virus RNA polymerase is a transmembrane protein with amino-terminal sequences sufficient for mitochondrial localization and membrane insertion. J. Virol. 76:9856–9867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller DJ, Schwartz MD, Ahlquist P. 2001. Flock house virus RNA replicates on outer mitochondrial membranes in Drosophila cells. J. Virol. 75:11664–11676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kopek BG, Perkins G, Miller DJ, Ellisman MH, Ahlquist P. 2007. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 5:e220. 10.1371/journal.pbio.0050220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flenniken M, Kunitomi M, Tassetto M, Andino R. 2010. The antiviral role of RNA interference, p 367–388 In Asgari S, Johnson K. (ed), Insect Virology Caister Academic Press, Norfolk United Kingdom [Google Scholar]
- 6.Li H, Li WX, Ding SW. 2002. Induction and suppression of RNA silencing by an animal virus. Science 296:1319–1321 [DOI] [PubMed] [Google Scholar]
- 7.Chao JA, Lee JH, Chapados BR, Debler EW, Schneemann A, Williamson JR. 2005. Dual modes of RNA-silencing suppression by Flock House virus protein B2. Nat. Struct. Mol. Biol. 12:952–957 [DOI] [PubMed] [Google Scholar]
- 8.Galiana-Arnoux D, Dostert C, Schneemann A, Hoffmann JA, Imler JL. 2006. Essential function in vivo for Dicer-2 in host defense against RNA viruses in Drosophila. Nat. Immunol. 7:590–597 [DOI] [PubMed] [Google Scholar]
- 9.Eckerle LD, Ball LA. 2002. Replication of the RNA segments of a bipartite viral genome is coordinated by a transactivating subgenomic RNA. Virology 296:165–176 [DOI] [PubMed] [Google Scholar]
- 10.Eckerle LD, Albarino CG, Ball LA. 2003. Flock House virus subgenomic RNA3 is replicated and its replication correlates with transactivation of RNA2. Virology 317:95–108 [DOI] [PubMed] [Google Scholar]
- 11.Lindenbach BD, Sgro JY, Ahlquist P. 2002. Long-distance base pairing in flock house virus RNA1 regulates subgenomic RNA3 synthesis and RNA2 replication. J. Virol. 76:3905–3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhong W, Rueckert RR. 1993. Flock house virus: down-regulation of subgenomic RNA3 synthesis does not involve coat protein and is targeted to synthesis of its positive strand. J. Virol. 67:2716–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friesen PD, Rueckert RR. 1981. Synthesis of Black Beetle Virus Proteins in Cultured Drosophila Cells: Differential Expression of RNAs 1 and 2. J. Virol. 37:876–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friesen PD, Rueckert RR. 1984. Early and late functions in a bipartite RNA virus: evidence for translational control by competition between viral mRNAs. J. Virol. 49:116–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallagher TM, Rueckert RR. 1988. Assembly-dependent maturation cleavage in provirions of a small icosahedral insect ribovirus. J. Virol. 62:3399–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Go EP, Wikoff WR, Shen Z, O'Maille G, Morita H, Conrads TP, Nordstrom A, Trauger SA, Uritboonthai W, Lucas DA, Chan KC, Veenstra TD, Lewicki H, Oldstone MB, Schneemann A, Siuzdak G. 2006. Mass spectrometry reveals specific and global molecular transformations during viral infection. J. Proteome Res. 5:2405–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aliyari R, Wu Q, Li HW, Wang XH, Li F, Green LD, Han CS, Li WX, Ding SW. 2008. Mechanism of induction and suppression of antiviral immunity directed by virus-derived small RNAs in Drosophila. Cell Host Microbe 4:387–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jovel J, Schneemann A. 2011. Molecular characterization of Drosophila cells persistently infected with Flock House virus. Virology 419:43–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Selling BH, Allison RF, Kaesberg P. 1990. Genomic RNA of an insect virus directs synthesis of infectious virions in plants. Proc. Natl. Acad. Sci. U. S. A. 87:434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu R, Maduro M, Li F, Li HW, Broitman-Maduro G, Li WX, Ding SW. 2005. Animal virus replication and RNAi-mediated antiviral silencing in Caenorhabditis elegans. Nature 436:1040–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ball LA, Amann JM, Garrett BK. 1992. Replication of nodamura virus after transfection of viral RNA into mammalian cells in culture. J. Virol. 66:2326–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price BD, Rueckert RR, Ahlquist P. 1996. Complete replication of an animal virus and maintenance of expression vectors derived from it in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 93:9465–9470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ball LA. 1995. Requirements for the self-directed replication of flock house virus RNA 1. J. Virol. 69:720–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneemann A, Zhong W, Gallagher TM, Rueckert RR. 1992. Maturation cleavage required for infectivity of a nodavirus. J. Virol. 66:6728–6734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneemann A, Marshall D. 1998. Specific encapsidation of nodavirus RNAs is mediated through the C terminus of capsid precursor protein alpha. J. Virol. 72:8738–8746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brody JR, Kern SE. 2004. Sodium boric acid: a Tris-free, cooler conductive medium for DNA electrophoresis. Biotechniques 36:214–216 [DOI] [PubMed] [Google Scholar]
- 27.Schneider PA, Schneemann A, Lipkin WI. 1994. RNA splicing in Borna disease virus, a nonsegmented, negative-strand RNA virus. J. Virol. 68:5007–5012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Venter PA, Dirksen A, Thomas D, Manchester M, Dawson PE, Schneemann A. 2011. Multivalent display of proteins on viral nanoparticles using molecular recognition and chemical ligation strategies. Biomacromolecules 12:2293–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venter PA, Krishna NK, Schneemann A. 2005. Capsid protein synthesis from replicating RNA directs specific packaging of the genome of a multipartite, positive-strand RNA virus. J. Virol. 79:6239–6248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marshall D, Schneemann A. 2001. Specific packaging of nodaviral RNA2 requires the N-terminus of the capsid protein. Virology 285:165–175 [DOI] [PubMed] [Google Scholar]
- 31.Deddouche S, Matt N, Budd A, Mueller S, Kemp C, Galiana-Arnoux D, Dostert C, Antoniewski C, Hoffmann JA, Imler JL. 2008. The DExD/H-box helicase Dicer-2 mediates the induction of antiviral activity in Drosophila. Nat. Immunol. 9:1425–1432 [DOI] [PubMed] [Google Scholar]
- 32.Venter PA, Marshall D, Schneemann A. 2009. Dual roles for an arginine-rich motif in specific genome recognition and localization of viral coat protein to RNA replication sites in flock house virus-infected cells. J. Virol. 83:2872–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han YH, Luo YJ, Wu Q, Jovel J, Wang XH, Aliyari R, Han C, Li WX, Ding SW. 2011. RNA-based immunity terminates viral infection in adult Drosophila in the absence of viral suppression of RNA interference: characterization of viral small interfering RNA populations in wild-type and mutant flies. J. Virol. 85:13153–13163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh G, Korde R, Malhotra P, Mukherjee S, Bhatnagar RK. 2010. Systematic deletion and site-directed mutagenesis of FHVB2 establish the role of C-terminal amino acid residues in RNAi suppression. Biochem. Biophys. Res. Commun. 398:290–295 [DOI] [PubMed] [Google Scholar]
- 35.Gantier MP, Williams BR. 2007. The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev. 18:363–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neeleman L, Olsthoorn RC, Linthorst HJ, Bol JF. 2001. Translation of a nonpolyadenylated viral RNA is enhanced by binding of viral coat protein or polyadenylation of the RNA. Proc. Natl. Acad. Sci. U. S. A. 98:14286–14291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guogas LM, Filman DJ, Hogle JM, Gehrke L. 2004. Cofolding organizes alfalfa mosaic virus RNA and coat protein for replication. Science 306:2108–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vende P, Piron M, Castagne N, Poncet D. 2000. Efficient translation of rotavirus mRNA requires simultaneous interaction of NSP3 with the eukaryotic translation initiation factor eIF4G and the mRNA 3′ end. J. Virol. 74:7064–7071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deo RC, Groft CM, Rajashankar KR, Burley SK. 2002. Recognition of the rotavirus mRNA 3′ consensus by an asymmetric NSP3 homodimer. Cell 108:71–81 [DOI] [PubMed] [Google Scholar]
- 40.Muller M, Heuck A, Niessing D. 2007. Directional mRNA transport in eukaryotes: lessons from yeast. Cell Mol. Life Sci. 64:171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mouland AJ, Xu H, Cui H, Krueger W, Munro TP, Prasol M, Mercier J, Rekosh D, Smith R, Barbarese E, Cohen EA, Carson JH. 2001. RNA trafficking signals in human immunodeficiency virus type 1. Mol. Cell. Biol. 21:2133–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amorim MJ, Bruce EA, Read EK, Foeglein A, Mahen R, Stuart AD, Digard P. 2011. A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J. Virol. 85:4143–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heinrich BS, Cureton DK, Rahmeh AA, Whelan SP. 2010. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog. 6:e1000958. 10.1371/journal.ppat.1000958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reineke LC, Lloyd RE. 2013. Diversion of stress granules and P-bodies during viral infection. Virology 436:255–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moser J, Fritzler M. 2010. Cytoplasmic ribonucleoprotein (RNP) bodies and their relationship to GW/P bodies. Int. J. Biochem. Cell Biol. 42:828–843 [DOI] [PubMed] [Google Scholar]
- 46.Nishida KM, Miyoshi K, Ogino A, Miyoshi T, Siomi H, Siomi MC. 2013. Roles of R2D2, a cytoplasmic D2 body component, in the endogenous siRNA pathway in Drosophila. Mol. Cell 49:680–691 [DOI] [PubMed] [Google Scholar]
- 47.Gimenez-Barcons M, Alves-Rodrigues I, Jungfleisch J, Van Wynsberghe PM, Ahlquist P, Diez J. 2013. The cellular decapping activators LSm1, Pat1, and Dhh1 control the ratio of subgenomic to genomic Flock House virus RNAs. J. Virol. 87:6192–6200 [DOI] [PMC free article] [PubMed] [Google Scholar]