

Abstract
The cellular protein IFI16 colocalizes with the herpes simplex virus 1 (HSV-1) ubiquitin ligase ICP0 at early times of infection and is degraded as infection progresses. Here, we report that the factors governing the degradation of IFI16 and its colocalization with ICP0 are distinct from those of promyelocytic leukemia protein (PML), a well-characterized ICP0 substrate. Unlike PML, IFI16 colocalization with ICP0 was dependent on the ICP0 RING finger and did not occur when proteasome activity was inhibited. Expression of ICP0 in the absence of infection did not destabilize IFI16, the degradation occurred efficiently in the absence of ICP0 if infection was progressing efficiently, and IFI16 was relatively stable in wild-type (wt) HSV-1-infected U2OS cells. Therefore, IFI16 stability appears to be regulated by cellular factors in response to active HSV-1 infection rather than directly by ICP0. Because IFI16 is a DNA sensor that becomes associated with viral genomes during the early stages of infection, we investigated its role in the recruitment of PML nuclear body (PML NB) components to viral genomes. Recruitment of PML and hDaxx was less efficient in a proportion of IFI16-depleted cells, and this correlated with improved replication efficiency of ICP0-null mutant HSV-1. Because the absence of interferon regulatory factor 3 (IRF3) does not increase the plaque formation efficiency of ICP0-null mutant HSV-1, we speculate that IFI16 contributes to cell-mediated restriction of HSV-1 in a manner that is separable from its roles in IRF3-mediated interferon induction, but that may be linked to the PML NB response to viral infection.
INTRODUCTION
Intrinsic and innate immunity mechanisms are highly important arms of cellular defense against viral infections. A prominent aspect of innate immunity involves interferon (IFN)-related signaling mechanisms that activate expression of IFN-stimulated genes (ISGs), several of which have antiviral activity. While many sensors that initiate these pathways reside on the cell surface or in the cytoplasm (reviewed in reference 1), a prominent strand of recent research involves sensors of “foreign” DNA within the nucleus, notably IFI16 (2, 3). IFI16 is a member of the IFN-inducible p200 family of proteins which has both human and murine homologues (4). All members of the p200 protein family contain at least one copy of a 200-amino-acid motif located toward its C terminus which is involved in DNA binding and protein-protein interactions (4, 5). Some family members, including IFI16, also have an amino-terminal Pyrin domain. Pyrin domains are thought to be involved in regulation of cytokine responses, supporting a role for IFI16 in innate immunity (5–7). A model emerging from recent work proposes that IFI16 signals to STING, a cytoplasmic protein that is required for the IFN response to pathogen DNA, which in turn activates TBK1-mediated phosphorylation of interferon regulatory factor 3 (IRF3) and subsequent transcriptional activation of the beta IFN (IFN-β) gene (8).
Several recent studies indicate that IFI16 acts as a sensor of viral DNA in a number of herpesvirus infections, including Kaposi's sarcoma-related herpesvirus (KSHV) (9, 10), Epstein-Barr virus (EBV) (11), human cytomegalovirus (HCMV) (12), and of particular relevance to this study, herpes simplex virus 1 (HSV-1) (3, 13, 14). The studies reported here were initiated to investigate whether IFI16 might be involved in the recruitment of promyelocytic leukemia protein nuclear body (PML NB) proteins to HSV-1 genomes that occurs soon after the viral genomes have entered the nucleus (15). Several PML NB components rapidly accumulate in novel PML NB-like foci that are closely associated with HSV-1 DNA, and this process appears to correlate with the restrictive effects of the PML NB proteins on HSV-1 infection (15–18). Although the properties of some of these proteins that are required for their recruitment have been explored, and notably it has been found that SUMO modification or noncovalent SUMO interactions are important (16), the signals that initiate the response remain unknown. Here, we report that although IFI16 is not absolutely required for the response of PML NB components to HSV-1 genomes, their recruitment appears to be delayed or defective in a proportion of IFI16-depleted cells. Depletion of IFI16 also allowed increased replication efficiency of an HSV-1 mutant that does not express the viral ubiquitin ligase ICP0.
During the course of this work, two important papers were published that report that IFI16 is degraded during HSV-1 infection by a proteasome- and apparently ICP0-dependent mechanism (13, 14). The RING finger-mediated E3 ubiquitin ligase activity of ICP0 induces the degradation of a number of cellular proteins, and this activity in general is required for efficient HSV-1 infection (reviewed in reference 19). One of the most prominent substrates of ICP0 is PML, the core component of PML NBs (20, 21). ICP0 also degrades or dysregulates other PML NB components such that they can no longer be recruited to the vicinity of HSV-1 genomes (15, 18). Therefore, we set out to compare the effects of ICP0 on IFI16 and PML in terms of their cellular localization during the initial stages of infection and the mechanism and efficiency of their degradation. We found that, like PML, IFI16 is recruited very rapidly to sites associated with HSV-1 genomes at the earliest stages of infection and that this response is inhibited by ICP0. However, IFI16 was degraded less efficiently than PML during infection, and ICP0 expression in the absence of infection was unable to degrade IFI16. Furthermore, we found that IFI16 degradation could proceed normally in the complete absence of ICP0 as long as the progress of ICP0-null mutant HSV-1 infection was efficient. Therefore, ICP0 is neither sufficient nor necessary for HSV-1-induced degradation of IFI16. Accordingly, we propose that the degradation is an indirect consequence of the ability of ICP0 to stimulate efficient progression of infection. These data provide important insights into the potential role of IFI16 and its stability during HSV-1 infection.
MATERIALS AND METHODS
Cells and viruses.
HSV-1 strain 17+ was the wild type (wt) strain used, from which the ICP0-null mutant dl1403 was derived (22). A derivative of the wt virus expressing enhanced yellow fluorescent protein (EYFP)-linked ICP4, and viruses expressing ICP0 mutant proteins FXE (ΔRING), D13 (with mutations of residues 633 to 680 [Δ633-680]), and E52X (Δ594-775) have been described previously (23–25). Virus dl0Y4, a derivative of dl1403 that expresses EYFP-linked ICP4, was constructed in a manner analogous to that of virus dl0C4 (26). Viruses in1863 and dl1403/CMVlacZ are wt and ICP0-null mutant strains that contain the lacZ gene under the control of the HCMV promoter/enhancer inserted into the tk gene (kindly provided by Chris Preston). All viruses were grown in BHK cells and titrated in U2OS cells, in which ICP0 is not required for efficient HSV-1 replication (27). Human diploid fibroblasts (HFs), telomerase-immortalized HFs (HFTs, a gift from Chris Boutell), U2OS cells, and HEK-293T cells were grown in Dulbecco's modified Eagles' medium supplemented with 10% fetal calf serum (FCS). BHK cells were grown in Glasgow modified Eagles' medium supplemented with 10% newborn calf serum and 10% tryptose phosphate broth. HepaRG cells (28) were grown in William's medium E supplemented with 10% fetal bovine serum Gold (PAA Laboratories Ltd.), 2 mM glutamine, 5 μg/ml insulin, and 500 nM hydrocortisone. Derivatives of HepaRG cells that express ICP0 and mutants thereof in an inducible manner were described previously (17, 29). HepaRG cells that express HCMV IE1 have also been described previously (30). All cell growth media were supplemented with 100 units/ml penicillin and 0.1 mg/ml streptomycin. Lentivirus transduced cells were maintained with continuous antibiotic selection, as appropriate.
Lentivirus transductions and induction of protein expression.
A lentivirus vector expressing an anti-IFI16 short hairpin RNA (shRNA) based on that described previously (9) was constructed by replacement of the shLuci control shRNA in a pLKO.1puro vector (31). Lentivirus transduction, selection of transduced cells, and maintenance of cell lines were as described previously (32). Selection during routine culture was done using puromycin at 500 ng/ml. The antibiotic was omitted from cells seeded for and during experimentation.
Virus plaque assays.
Cells were seeded for plaque assays into 24-well dishes at 1 × 105 cells per well and infected the following day with appropriate sequential 3-fold dilutions of in1863 or dl1403/CMVlacZ. After virus adsorption, the cells were overlaid with medium containing 1% human serum and were stained for β-galactosidase-positive plaques 24 h later (33).
Infections and Western blot analysis.
Cells were seeded into 24-well dishes at 1 × 105 cells per well. After the relevant experimental manipulations, the cells were washed twice with phosphate-buffered saline (PBS) before harvesting in SDS-PAGE loading buffer. Proteins were resolved on 7.5% SDS gels and then transferred to nitrocellulose membranes by Western blotting. The following antibodies were used: anti-actin monoclonal antibody (MAb) AC-40 (Sigma-Aldrich), anti-tubulin MAb T4026 (Sigma-Aldrich), anti-PML rabbit polyclonal antibody (rAb) A301-167A (Bethyl Laboratories) or MAb 5E10 (34), and anti-IFI16 MAb ab55328 (Abcam). Anti-ICP0 MAb 11060 and anti-UL42 MAb Z1F11 were used as described previously (31). Anti-VP5 MAb DM165 was a gift from Frazer Rixon.
Immunofluorescence and confocal microscopy.
Cells on 13-mm glass coverslips were fixed with formaldehyde and prepared for immunofluorescence using standard methods. PML was detected with rAb ABD-030 (Jena Bioscience) or MAb 5E10, hDaxx with rAb 07-471 (Upstate), ICP0 with MAb 11060 or rAb r190, and IFI16 with MAb 55328 (Abcam). Note that anti-IFI16 MAb sc8023 available from Santa Cruz works well for Western blotting and for immunofluorescence assays of uninfected cells, but it recognizes the component of IFI16 that is recruited to HSV-1 genomes poorly. The secondary antibodies used were Alexa 555 conjugated goat anti-mouse IgG and Alexa 633 conjugated goat anti-rabbit IgG (Invitrogen). The samples were examined using a Zeiss LSM 710 confocal microscope, with 488-nm, 561-nm, and 633-nm laser lines, scanning each channel separately under image capture conditions that eliminated channel overlap. The images were exported as tagged image format (tif) files, minimally adjusted using Photoshop, and then assembled into figures using Illustrator.
RESULTS
ICP0 RING finger and proteasome activity are required for colocalization of ICP0 with IFI16 during HSV-1 infection.
The following experiments were designed to compare the intracellular localization of IFI16 and PML with respect to ICP0 during HSV-1 infection and, in particular, to identify regions of ICP0 that might be involved in their colocalization. IFI16 is diffusely localized in the general nucleoplasm and also sometimes within nucleoli in uninfected cells (4, 9, 14), while during the early stages of HSV-1 infection, some of the protein relocalizes to ICP0 foci (13, 14). We confirmed these observations (Fig. 1A), but surprisingly, we then found that colocalization of IFI16 with RING finger deletion mutant ICP0 was defective both during infection of HFs (Fig. 1B) and when RING finger mutant ICP0 was expressed by itself in ICP0-inducible HepaRG cells (data not shown). The results at the 4-h time point are shown in Fig. 1B to guard against the possibility that any colocalization might be delayed in the mutant infection, but similar results were also obtained at the 2-h time point (data not shown). This is in complete contrast to the situation with PML, which colocalizes even more strongly and more stably with RING finger mutant ICP0 than with the wt protein (35). Even more surprising was the observation that IFI16 recruitment into wt ICP0 foci was inhibited by the proteasome inhibitor MG132 (Fig. 1C), and although good colocalization could be seen in a small proportion of cells at 6 h after infection (Fig. 1C), this is again in contrast to the behavior of PML (21). The latter result suggests that it is not the RING finger of ICP0 per se that is required for colocalization with IFI16, but rather that there is some connection with ICP0-mediated protein degradation activity. Whatever the explanation of these results, it is clear that the localization behavior of IFI16 with respect to ICP0 is readily distinguishable from that of PML. We therefore went on to compare the degradation of the two proteins.
Fig 1.
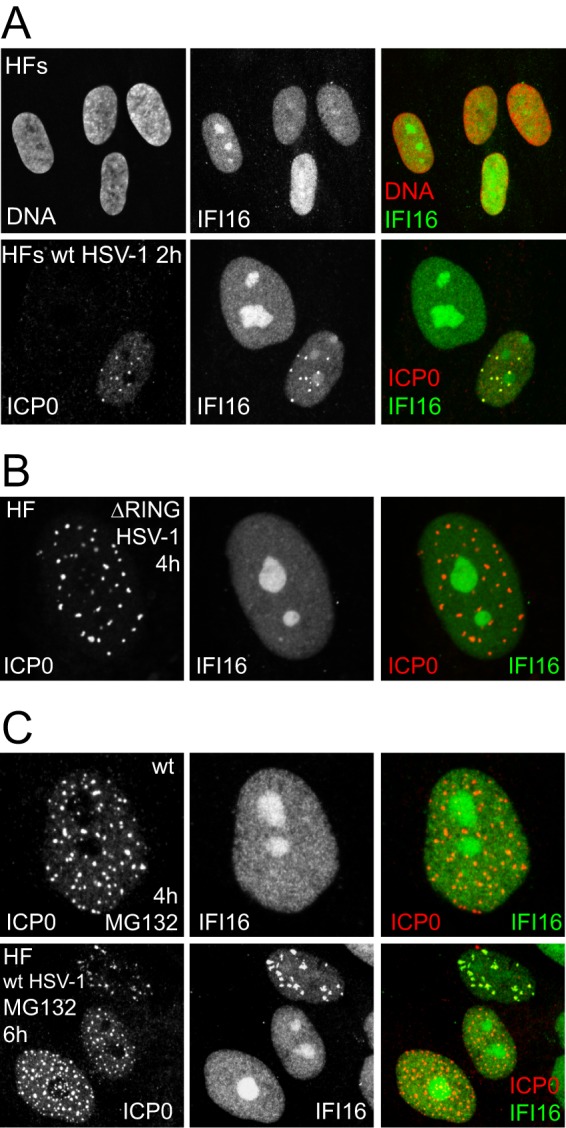
Localization of IFI16 in uninfected and infected HFs. (A) Upper row, Uninfected HFs were stained for IFI16 and DNA (Topro-3); lower row, HFs were infected with wt HSV-1 at an MOI of 4 and stained 2 h later for IFI16 (Abcam ab55328) and ICP0. (B) HFs were infected with HSV-1 expressing a RING finger deletion mutant ICP0 at an MOI of 4 and stained 4 h later for IFI16 and ICP0. (C) HFs were infected with wt HSV-1 at an MOI of 4 in the presence of 3 μM MG132 and stained 4 h and 6 h later for IFI16 and ICP0.
IFI16 is degraded less efficiently than PML during HSV-1 infection.
Two recent papers report that IFI16 is degraded in an apparently ICP0-dependent manner during HSV-1 infection (13, 14). We set out to investigate this activity in more detail, and in particular to compare IFI16 degradation with that of PML, a well-characterized ICP0 substrate. Initially, we confirmed that IFI16 is indeed degraded in HSV-1-infected HFs in a proteasome-dependent manner (Fig. 2A) and that this also occurs in infected HepaRG cells (Fig. 2B). However, the rates of IFI16 degradation are significantly lower than those of PML, at least in terms of the proportions of the endogenous levels of the proteins. Previous work found that degradation of IFI16 did not occur in cells infected with a virus expressing a RING finger mutant of ICP0 (14). To extend these data, we compared the degradation rates of PML and IFI16 in HepaRG cells infected with viruses expressing wt and mutant forms of ICP0 lacking the entire C-terminal quarter of the protein (Δ594-775) or a region within this portion (Δ633-680). At the multiplicities of infection used, both of these deletions reduce the rate at which PML is degraded, yet neither affected the relatively lower rate of degradation of IFI16 (Fig. 2C). Therefore, IFI16 is degraded relatively slowly compared to the rate for PML, and the characteristics of ICP0 required for IFI16 degradation differ from those of PML as well.
Fig 2.
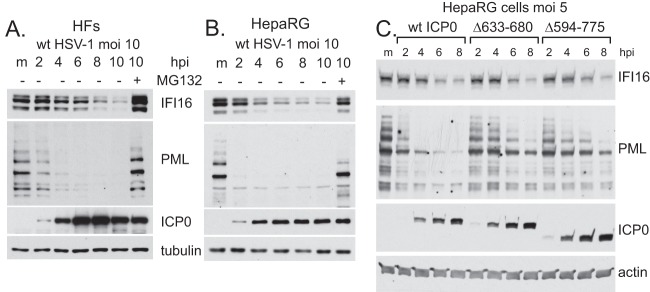
IFI16 is degraded more slowly than PML in wt HSV-1-infected HFs and HepaRG cells. HFs (A) and HepaRG cells (B) were mock infected (m) or infected with wt HSV-1 (MOI, 10) in the absence or presence of MG132 (3 μM) and harvested at 2, 4, 6, 8, and 10 h later. Whole-cell extracts were analyzed for IFI16, PML, ICP0, and tubulin. (C) The C-terminal region of ICP0 is required for maximal rates of degradation of PML but not IFI16. HepaRG cells were mock infected (m) or infected with wt HSV-1 or ICP0 mutants Δ633-680 and Δ594-775 (all at an MOI of 5), and samples taken at 2, 4, 6, and 8 h postinfection (hpi) were analyzed for IFI16, PML, ICP0, and actin.
ICP0 is not of itself sufficient to induce efficient degradation of IFI16.
We next tested whether ICP0 by itself, in the absence of all other viral factors, was sufficient to induce degradation of IFI16. We used a previously characterized HepaRG cell line that expresses ICP0 in an inducible manner and in amounts that are equivalent to those at the early stages of infection and sufficient to fully complement the plaque formation defect of ICP0-null mutant HSV-1 (17). In contrast to results for PML, no degradation of IFI16 was observed after a 24-h induction (Fig. 3A). The degradation of PML in this system is dependent on the ICP0 RING finger (Fig. 3A). Given that IFI16 degradation occurs during infection with an HSV-1 mutant that expresses high levels of ICP0 but no other IE proteins (13, 14), it is possible either that the amount of ICP0 expressed in the induced cells is insufficient to degrade IFI16 efficiently or that some other factor that is dependent on virus infection is also involved. To explore these possibilities, control and ICP0-inducible cells were induced and then infected with either wt or ICP0-null mutant HSV-1. As before, ICP0 induction in uninfected cells caused degradation of PML but not IFI16 and, as expected, infection of induced cells with wt HSV-1 led to a decrease in IFI16 stability (Fig. 3B). Infection of the induced cells with ICP0-null mutant HSV-1, however, also caused a decrease in IFI16 at later times of infection (Fig. 3B). Note that the induced ICP0 expression enabled equal rates of expression of UL42 (the viral DNA polymerase accessory subunit, a member of the early class of viral proteins) in mutant- and wt-infected cells. Degradation of IFI16 in the mutant-infected cells could not be explained simply by the increased time for which ICP0 was expressed, because an analogous time course of induction beyond 24 h in uninfected cells had no effect on IFI16 (Fig. 3C). IFI16 degradation did not occur during ICP0-null mutant HSV-1 infection of these cells if ICP0 expression was not induced (Fig. 3D). This is consistent with previous data (14), but note that in the absence of complementation, expression of UL42 was greatly diminished compared to that seen in wt infection at this multiplicity of infection (MOI). Therefore, the progression of the mutant virus infection is inefficient; this issue becomes a major point later in the paper.
Fig 3.

ICP0 expression in the absence of HSV-1 infection is insufficient to mediate IFI16 degradation. (A) Control HepaRG cells expressing the tetracycline repressor and derivates in which inducible expression of either wt or RING finger deletion mutant ICP0 (as indicated) were analyzed for IFI16, PML, ICP0, and tubulin at 24 h after induction of ICP0 expression with doxycycline (Dox; 100 ng/ml). (B) Control or wt ICP0-inducible cells were treated with doxycycline and infected with either wt or ICP0-null mutant HSV-1 (MOI, 10) as indicated. Samples were harvested at 2, 4, 6, and 8 h after infection and analyzed for IFI16, PML, ICP0, UL42, and actin. (C) Control or ICP0-inducible cells were treated with doxycycline (100 ng/ml) for 24 h, and the inducible cells were further incubated for 2, 4, 6, and 8 h. Samples were analyzed for IFI16, ICP0, and actin. (D) Control or ICP0-inducible cells were infected (MOI, 10) with wt or ICP0-null mutant HSV-1 in the absence of induction, and samples were harvested at the indicated time points after infection for analysis of IFI16, ICP0, UL42, and actin. infn, infection.
The results of Fig. 3 open an avenue to explore more closely the properties of ICP0 that are required for IFI16 degradation because a more extensive collection of ICP0 mutants is available in the cell line system than in the virus itself (17, 29). Degradation of IFI16 was equally efficient in cells expressing the wt and ICP0 truncation mutants 1-594 (i.e., containing residues 1 to 594 only) (consistent with Fig. 1C) and 1-547 (Fig. 4A) after infection with ICP0-null mutant HSV-1. Degradation of IFI16 was, however, much less efficient in dl1403-infected cells expressing ICP0 mutant 1-396nls (ICP0 residues 1-396 with an added nuclear localization signal) (Fig. 4B), thereby apparently identifying ICP0 sequences between residues 397 and 546 as being important for this activity. An important caveat to this conclusion is that under the high-MOI infection conditions used, dl1403 infection proceeded with equal efficiency in cells expressing wt, 1-547, and 1-594 ICP0 proteins, but progressed less rapidly in cells expressing ICP0 1-396nls. Therefore, it was possible that the difference in the rates of IFI16 degradation reflects the efficiency of infection progression rather than a specific requirement for these ICP0 sequences.
Fig 4.
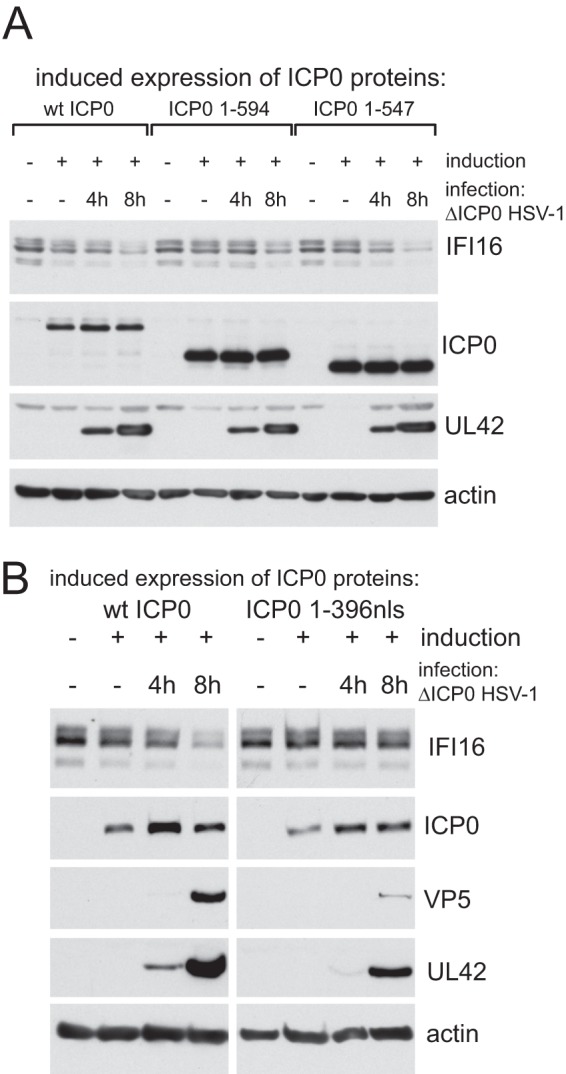
Investigation of the properties of ICP0 required for efficient degradation of IFI16. Expression of ICP0 was induced in cells expressing wt or mutant proteins as indicated and then infected with ICP0-null mutant HSV-1 (MOI, 10) as shown. Samples were harvested at 4 and 8 h after infection and compared with uninduced controls for levels of IFI16, ICP0, UL42, and actin (and VP5 in the experiment shown in panel B). For each protein in panel B, the wt and 1-396nls panels show the same exposure of the samples analyzed on the same gel.
To explore this further, we tested whether inhibition of DNA replication in cells infected with wt HSV-1 might affect the results. Degradation of IFI16 was not affected by treatment with acycloguanosine (ACG) at 50 μM using an MOI of 5 (Fig. 5A), indicating that DNA replication is not necessary for the effect. However, IFI16 was more stable in the presence of ACG under more stringent conditions (ACG at 100 μM and cells infected at an MOI of 2) (Fig. 5B). These conditions reduced the expression of not only the late protein VP5 but also UL42 and ICP0; therefore, these data are also consistent with the hypothesis that the rate of IFI16 degradation is proportional to viral infection progression and/or the involvement of viral proteins in addition to ICP0.
Fig 5.
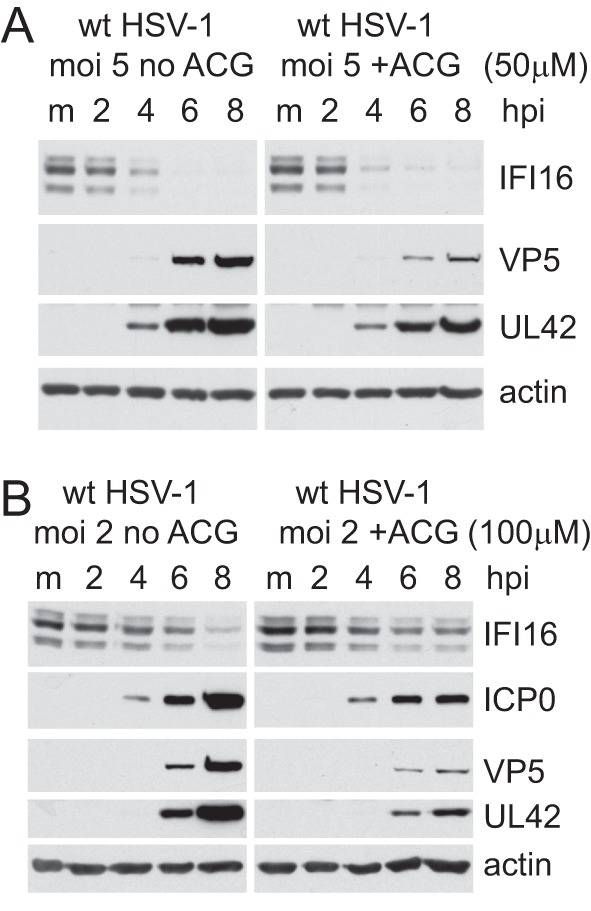
Effect of inhibition of DNA replication on the rate of IFI16 degradation. (A) HFs were infected at an MOI of 5 in the presence and absence of ACG (50 μM). Samples were harvested at 2, 4, 6, and 8 h after infection and analyzed for IFI16, VP5, UL42 and actin in comparison with the mock-infected control (m). (B) An experiment similar to that shown in panel A, except that infection was at an MOI of 2 and ACG was used at 100 μM.
ICP0 is not required for degradation of IFI16 during HSV-1 infection.
Although there are several potential explanations for the results presented thus far, before proceeding further it had become necessary to reexamine whether the degradation of IFI16 during HSV-1 infection was truly and directly ICP0 dependent. This presents some tricky experimental challenges because the absence of functional ICP0 causes a severe replication defect. Therefore, under normal conditions at an equalized MOI, viral gene expression and the cellular changes that occur during a wt infection are, at best, greatly delayed in mutant infected cells. It is necessary to use the mutant virus at a much higher multiplicity than that of the wt in order to ensure that the infections are progressing equally efficiently and therefore rule out the possibility that degradation of IFI16 correlates with infection progression (i.e., changes in the cell that occur as the cells become heavily infected) rather than occurring as a direct consequence of the ubiquitin ligase activity of ICP0. Accordingly, we compared the rates of IFI16 degradation in cells infected at an MOI of 2 with the wt virus and at an MOI of 50 with the mutant. This experiment was performed with HepaRG cells because they are slightly more permissive for the mutant infection than HFs, and thus it is easier to equalize infection rates. At these MOIs, infection progression was equivalent in the two infections (as judged by expression of UL42 and VP5), as was the rate of loss of IFI16 (Fig. 6A). In contrast, the overall level of PML remained stable in the mutant virus infection. Therefore, ICP0 is neither sufficient (Fig. 3) nor absolutely required for IFI16 degradation.
Fig 6.
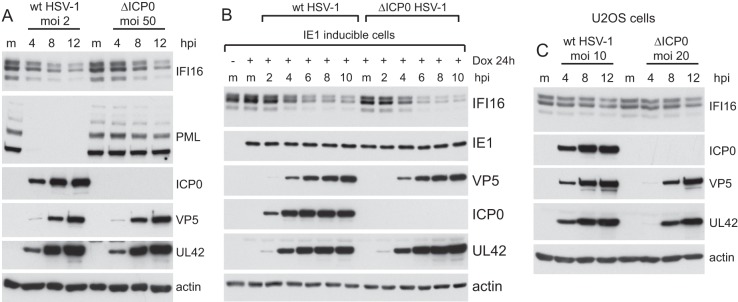
ICP0 is not essential for degradation of IFI16 during HSV-1 infection. (A) HepaRG cells were infected with wt HSV-1 (MOI, 2) or ICP0-null mutant dl1403 (ΔICP0) (MOI, 50), and samples were harvested at 4, 8, and 12 h after infection and analyzed for IFI16, PML, ICP0, VP5, UL42, and actin in comparison to mock-infected controls (m). (B) HepaRG cells that express HCMV protein IE1 in an inducible manner were left untreated (left most lane) or treated with 100 nM doxycycline (Dox) for 24 h. The induced cells were either mock infected (m) or infected with wt or ICP0-null mutant HSV-1 (ΔICP0) at an MOI of 10, and samples were harvested at 2, 4, 6, 8, and 10 h after infection, as indicated. Whole-cell extracts were analyzed by Western blotting for IFI16, IE1, VP5, ICP0, UL42, and actin. (C) U2OS cells were infected with wt HSV-1 (MOI, 10) or ICP0-null mutant dl1403 (ΔICP0) (MOI 20), and samples were harvested at 4, 8, and 12 h after infection and analyzed for IFI16, ICP0, VP5, UL42, and actin in comparison to mock-infected controls (m).
The weakness of the above-described experiment is the difference in wt and mutant MOIs used; it could be argued that the rate of IFI16 degradation is influenced by the number of viral genomes (or other virion-associated factors) entering the nucleus. To circumvent this problem, we investigated the rate of degradation of IFI16 in HepaRG cells expressing HCMV protein IE1 (also called IE72), which substantially complements ICP0-negative HSV-1 (30). Infection of these cells with wt and mutant virus at an MOI of 10 resulted in very similar rates of viral gene expression, and also of IFI16 degradation (Fig. 6B). IE1 expression by itself did not affect IFI16 stability (compare the induced and uninduced, uninfected lanes), and there is no evidence that IFI16 is degraded during HCMV infection (12). These results confirm that ICP0 is not required for IFI16 degradation during HSV-1 infection.
In another approach to investigate this issue, we examined the fate of IFI16 during infection of U2OS cells with wt and mutant viruses, a situation in which the infection efficiencies would be expected to be similar in the presence and absence of ICP0. Although plaque formation by wt and ICP0 mutant virus is equally efficient in U2OS cells (27), we found that the mutant viral gene expression was slightly delayed; therefore, the experiment was conducted using the wt at an MOI of 10 and the mutant at an MOI of 20. This experiment produced another surprising result, in that IFI16 was only marginally degraded during either infection, even at the 12-h time point, despite the high MOI used (Fig. 6C). This result identified another situation in which ICP0 appears insufficient for IFI16 degradation, even in the presence of an active infection. The strong implication is that cell-specific factors govern the stability of IFI16 in response to HSV-1 infection and that these are in some way activated during productive infection of HFs and HepaRG cells but defective in U2OS cells.
IFI16 is rapidly recruited to incoming parental HSV-1 genome sites.
The original motivation behind this study was to investigate the response of IFI16 to nuclear entry of the viral genome, whether it influences the response of PML NB components to HSV-1 infection, and its potential role in regulating ICP0-null mutant HSV-1 infection. The remainder of this paper concentrates on these topics. It has been reported that IFI16 colocalizes with HSV-1 DNA during the early stages of infection (13). We have extended these results by examining the behavior of IFI16 in cells at the periphery of developing wt and ICP0-null mutant HSV-1 plaques, which enables cells in the very earliest stages of infection to be examined (15, 36). The top row of Fig. 7A shows the separated and merged channels of an image of an ICP0-null mutant plaque, detecting ICP4, IFI16, and PML. A proportion of cells close to the plaque show early viral replication compartments (identified by ICP4) in a typical asymmetric pattern near the edge of the nucleus (e.g., the cell indicated by a long arrow). In these cells, both PML and IFI16 have been recruited into foci that are closely associated with ICP4. The middle row of Fig. 7A shows an example of this type of cell at a higher magnification. Cells were also readily identified close to a developing plaque in which IFI16 and PML are in asymmetric foci similar to those that form in association with ICP4 foci (that represent viral genomes [15]), but in which ICP4 is below the level of detection (Fig. 7A, top row, the cell indicated by the shorter arrow). This indicates that recruitment of IFI16 to HSV-1 genomes is a very rapid event which occurs before abundant IE gene expression has taken place. At later times of infection in some cells, once viral replication compartments have developed extensively, IFI16 can form tangled thread-like structures which partially colocalize with PML (Fig. 7A, bottom row).
Fig 7.
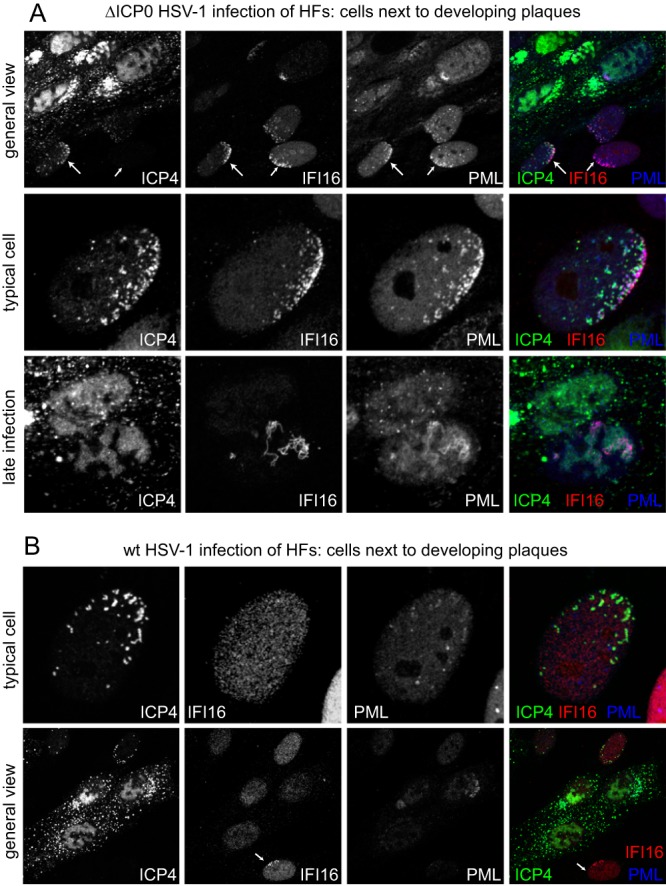
Recruitment of IFI16 to sites associated with HSV-1 genomes in the presence and absence of ICP0. (A) HFs were infected with dl0Y4 (ICP0-null mutant HSV-1 expressing EYFP-linked ICP4) at a low MOI, and viral plaques were imaged the following day. ICP4 was detected by EYFP autofluorescence, and IFI16 by staining with MAb ab55328 (Abcam). PML was detected using rAb ABD-030. Secondary antibodies were Alexa 663-linked anti-mouse and Alexa 555-linked anti-rabbit IgG. Top row, a view of the edge of a plaque, illustrating cells in which PML and IFI16 have been recruited to ICP4 foci and others that have asymmetric accumulations of IFI16 and PML prior to the accumulation of detectable levels of EYFP-ICP4; middle row, a high magnification of a typical cell exhibiting recruitment of IFI16 and PML into ICP4-associated foci; bottom row, a detail of highly infected cells, showing an example in which IFI16 and PML form thread-like structures. (B) An experiment similar to that shown in the top two rows of part A, but using vEYFP-ICP4 (wt HSV-1 expressing EYFP-linked ICP4).
During wt HSV-1 infection, cells with the characteristic pattern of peripheral ICP4 foci do not exhibit recruitment of IFI16 (Fig. 7B, upper row), indicating that the presence of ICP0 inhibits the response of IFI16 to viral genomes at the early stages of infection. Nonetheless, careful examination of developing wt HSV-1 plaques revealed the existence of cells close to developing plaques in which ICP4 expression was undetectable yet relocation of IFI16 into asymmetric foci was well marked (Fig. 7B, lower row, arrowed cell). These again are most likely to be cells in the very early stages of infection that are responding to the entry of viral genomes into the nucleus and in which recruitment has occurred before sufficient ICP0 has been expressed to disperse the recruited proteins.
Depletion of IFI16 enhances the ICP0-null mutant HSV-1 plaque formation.
We next investigated the consequence of depletion of IFI16 for HSV-1 replication. A lentivirus vector that expresses a previously characterized anti-IFI16 shRNA (9) was constructed. This caused an efficient reduction in IFI16 expression in transduced HFs (Fig. 8A). Compared to the parental HFs, and HFs transduced with a control shRNA (shLuci), the IFI16-depleted cells were infected with equal efficiency by wt HSV-1 (Fig. 8B) but there was an approximately 7-fold enhancement in ICP0-null mutant plaque formation efficiency (Fig. 8C). Similar results were obtained using telomerase-immortalized HFs (HFTs) as the parental cell line (data not shown). Therefore, IFI16 is involved in mechanisms that restrict HSV-1 infection in the absence of ICP0. IFI16 has been linked to several functions, one of which is the induction of IFN-β expression through STING, TBK1, and IRF3 (3, 8, 14). While IFN-β induction is a physiologically important aspect of control of HSV-1 infection, defective signaling through IRF3 cannot explain the increase in plaque formation of ICP0-null mutant HSV-1 in IFI16-depleted cells, as removal of IRF3 itself does not affect mutant virus plaque formation (37). Therefore, some other aspect of IFI16 must be involved in the increase of ICP0 mutant plaque formation in shIFI16 cells.
Fig 8.
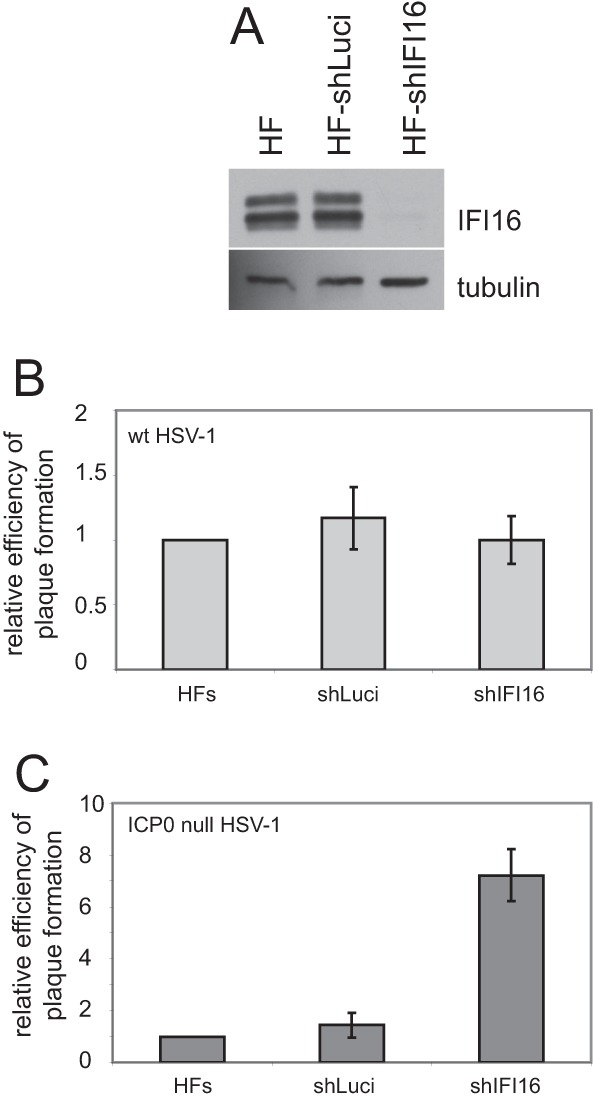
Depletion of IFI16 increases the plaque formation efficiency of ICP0-null mutant but not wt HSV-1. (A) Western blot analysis of nontransduced HFs and HFs expressing control (shLuci) or IFI16-specific (shIFI16) shRNAs. (B and C) Relative plaque formation of wt (B) and ICP0-null mutant (C) HSV-1 in nontransduced, shLuci (control), or shIFI16 HFs, as indicated. The data from several independent experiments were averaged and then plotted ± standard deviations. Calculations were as described in reference 32.
IFI16 contributes to the efficiency of recruitment of PML NB proteins to HSV-1 genome-associated sites.
The rapid recruitment of PML NB components to sites associated with HSV-1 genomes is a cellular response which is regulated in part by SUMO modification pathways (16). The cellular factors that initiate this response are, however, yet to be characterized. Because IFI16 is a nuclear sensor of pathogen DNA (2, 3), it is conceivable that it is involved in the response of PML NB components to HSV-1 genomes. Therefore, we compared the recruitment of PML and other PML NB proteins to ICP4-defined HSV-1 genome loci in ICP0-null mutant-infected control and IFI16-depleted cells. In control cells, recruitment of both PML (Fig. 9A, top row) and hDaxx (Fig. 9B, top row) was extensive in every cell examined. At first sight, similar recruitment also occurred in the shIFI16 cells, with the possible exception for a tendency for a greater proportion of PML or hDaxx foci to remain unassociated with ICP4 and spread throughout the nucleoplasm (Fig. 9A and B, middle rows). However, an examination of a large number of cells revealed occasional examples (approximately 10% of cells with discrete small foci of ICP4 near the nuclear periphery) in which PML and hDaxx remained in distinct foci that were rarely associated with ICP4 (Fig. 9A and B, lower rows). These cells are reminiscent of those in which recruitment of PML has been inhibited by mutations in SUMO conjugation or SUMO interaction sites (16). This assay is not amenable to precise quantification because of the inherent variability between infected cells in terms of stage of infection, number of ICP4 foci, and distinctness of the recruited foci. Therefore, conclusions must be treated cautiously, but it is safe to say that cells such as those in the lower rows of Fig. 9A and B were never observed in the controls. Similar results were obtained in three independent isolates of IFI16-depleted cells.
Fig 9.
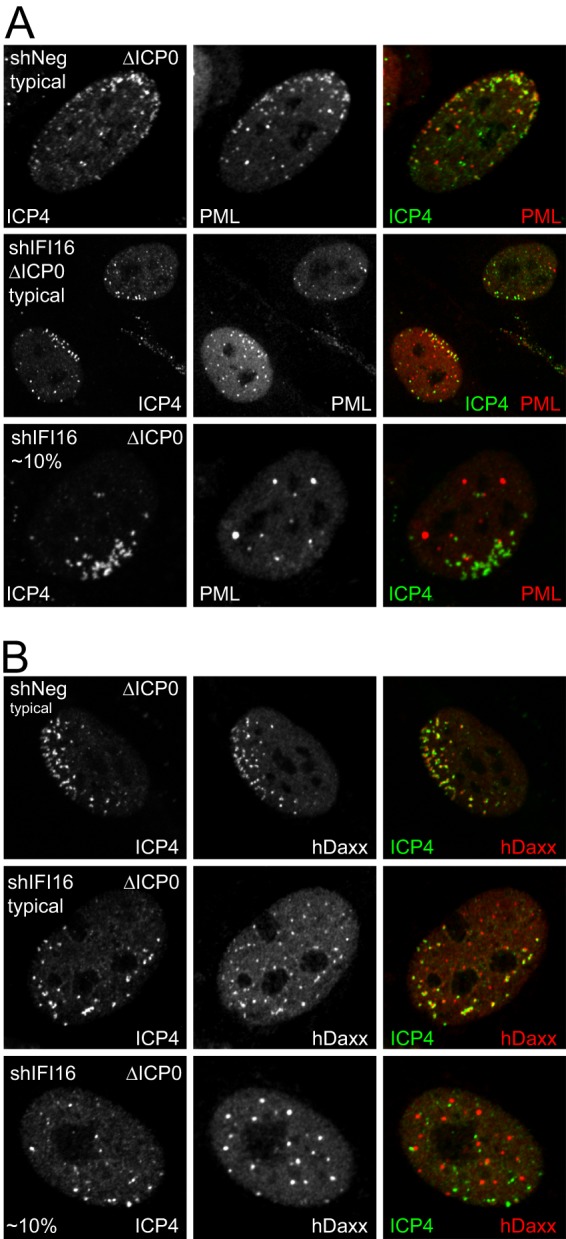
Recruitment of PML and hDaxx to HSV-1 genomes is diminished in a proportion of IFI16-depleted cells. HF-shNeg and HF-shIFI16 were infected with dl0Y4 at a low MOI, and plaques were examined by ICP4 autofluorescence and staining for either PML (A) or hDaxx (B), using an Alexa 633-linked anti-rabbit secondary antibody. Typical cells with asymmetric ICP4 foci are shown for the control and shIFI16 cells, as indicated (top two rows of each panel). Examples of shIFI16 cells in which recruitment of the PML NB proteins is poor are shown in the bottom row of each panel.
The recruitment of PML NB proteins to viral genome sites is obviously a rapid and dynamic process which, as Fig. 9 suggests, may be delayed in a proportion of IFI16-depleted cells. The process is so rapid under normal circumstances that, in addition to the common cells in which PML NB component recruitment to ICP4 foci is visible, examples of apparent recruitment prior to detectable accumulation of ICP4 are readily observed (Fig. 7). To investigate whether the frequency of this type of cell was influenced by depletion of IFI16, entire plaques in control and depleted ICP0-null mutant-infected cells were examined by capturing arrays of images at high resolution, so that the detail in individual cells could be assessed (by zooming in to the composite image) (Fig. 10, color images). Image capture conditions were set so that ICP4 intensity was saturated in the more heavily infected cells, and identical settings were used for all plaques and images. Sixteen plaques of similar size were examined for each cell type, and the total numbers of cells with characteristic asymmetric hDaxx staining but without detectable ICP4 were counted (for examples, see the hDaxx and ICP4 greyscale images below the control plaque; corresponding cells are indicated by arrows). A total of 84 such examples were detected in the 16 control plaques, while only 9 were found in the plaques in the shIFI16 cells. The greyscale images in Fig. 10 (right) show a detail of the shIFI16 plaque (with corresponding cells indicated by arrows); one is cell positive for recruitment of hDaxx, while in the other (lower right), this is less obvious. These results indicate that depletion of IFI16 reduces the efficiency with which the PML NB protein recruitment response occurs, particularly during the earliest stages of infection.
Fig 10.

Evidence for a reduced rate of response of hDaxx to the entry of HSV-1 genomes into the nucleus in IFI16-depleted cells. Control cells (left panels) and IFI16-depleted HFT cells (right panels) were infected with dl0Y4 at a low MOI, and plaques were examined by ICP4 autofluorescence and staining for hDaxx using an Alexa 633-linked anti-rabbit secondary antibody. The color images are a composite of arrays of 9 (control sample) or 16 (shIFI16 sample) images taken using a ×40 objective lens. Individual cells surrounding the core of the plaques and displaying clear asymmetric patterns of hDaxx staining were examined under zoom and scored for the presence of detectable ICP4 by switching off the red channel. The greyscale images below the color ones show the hDaxx and ICP4 channels of selected regions of the plaques. Other details and explanations are given in the text.
DISCUSSION
We report here that the characteristics of the degradation of IFI16 and PML and their relationship to ICP0 during HSV-1 infection are entirely distinguishable. Whereas ICP0 is both necessary and sufficient for PML degradation, through both SUMO-dependent and -independent mechanisms (20, 29), ICP0 is neither sufficient nor necessary for the degradation of IFI16 during the course of HSV-1 infection. Previous studies found that IFI16 is degraded during high-multiplicity infection with virus d106, which expresses very high levels of ICP0 but no other IE proteins (13, 14). Because d106 should express only low levels of other viral proteins, those studies are in apparent contrast to the results presented here. It is possible that degradation of IFI16 can occur through both ICP0-independent and ICP0-stimulated mechanisms, the latter of which nonetheless requires additional virus infection-specific events, but our data clearly demonstrate that IFI16 can be degraded normally in the absence of ICP0 as long as the lytic cycle is progressing efficiently.
Our observation that ICP0 is insufficient for IFI16 degradation, while infection of ICP0-expressing cells with ICP0-null mutant HSV-1 triggered the degradation (Fig. 3), initially posed the question of whether the presence of the viral genome is an important factor. This was a plausible hypothesis given the available information on IFI16 recognition of foreign DNA (2, 3, 13, 14). This appears not to be the case, however, as virus in1374 (an HSV-1 mutant with lesions in ICP0, ICP4, and VP16 and which therefore expresses other viral proteins poorly [38]) did not destabilize IFI16 in cells induced to express ICP0 (data not shown). The poor degradation of IFI16 in U2OS cells even at late times of a high-MOI wt virus infection (Fig. 6C) is also consistent with the conclusion that neither ICP0 nor the mere presence of the viral genome are dominant factors that directly govern IFI16 stability.
An obvious question that this study does not address is the mechanism of degradation of IFI16 in the absence of ICP0 during HSV-1 infection. These data do not exclude a potential role for other viral proteins whose expression is dependent on ICP0. For example, it is conceivable that viral kinases might phosphorylate IFI16 such that it then becomes a substrate for cellular ubiquitin ligases. Alternatively, the stress of the developing infection might trigger entirely cell-based mechanisms that could destabilize IFI16. Either way, cellular factors must play an essential role in IFI16 degradation, a conclusion that is consistent with the fact that the efficiency of IFI16 degradation varies with cell type.
Despite the lack of requirement for ICP0 for IFI16 degradation, it is definitely the case that ICP0 affects the response of IFI16 to HSV-1 even at very early times of infection. Like PML and PML NB components such as hDaxx, IFI16 is recruited very rapidly to viral genome sites even before viral proteins can be detected by immunofluorescence (Fig. 7). This recruitment remains obvious as ICP4 accumulates in cells infected with ICP0-null mutant HSV-1 but is substantially inhibited in wt HSV-1 infections. Therefore, like that of PML and other PML NB components, recruitment of IFI16 to viral genomes is inhibited in the presence of ICP0. Because this inhibition occurs before the abundant accumulation of viral proteins, it is likely to occur prior to substantial degradation of IFI16. Note that ICP0 also inhibits the recruitment of hDaxx to viral genomes but does not affect its stability (18), indicating that recruitment inhibition of a given protein does not necessarily require its degradation.
The initial motivation behind this study was to test whether the DNA-sensing activity of IFI16 in the nucleus was involved in the recruitment of PML and other PML NB proteins to sites associated with HSV-1 genomes. Our results indicate that in most cells, depletion of IFI16 did not eliminate the recruitment response and therefore IFI16 is not a dominant factor. Nonetheless, PML and hDaxx recruitment was defective in a small proportion of IFI16-depleted HFs, and more generally, the rate of response of the PML NB components to the viral genomes appeared to be reduced in IFI16-depleted cells. This conclusion comes from the greatly reduced frequency of cells in which hDaxx had been redistributed into characteristic nuclear peripheral foci prior to detectable accumulation of ICP4. Although this is a subtle phenotype that can only be recognized by careful observation, the difference between the control and IFI16-depleted cells was reproducible in several independent cell isolates and it could be used in a blinded experiment to identify whether control or depleted cells were being examined. We do not know what distinguishes the approximately 10% of IFI16-depleted cells that show little or no recruitment of PML and hDaxx to ICP4 foci from the bulk of the population. Possibly this phenotype occurs only in the most highly depleted cells, but this explanation cannot be tested. The mechanism of any potential connection between IFI16 and the PML NB component response to HSV-1 genomes remains to be investigated, but given the nuclear location of IFI16, PML NBs, and the viral genomes, it is more likely to be a nuclear pathway than STING-dependent signaling events involving the cytoplasm. Although IFI16 has been linked to the DNA damage response pathway (39, 40), we did not detect any difference in the formation of γH2AX foci in response to HSV-1 infection in IFI16-depleted cells (data not shown).
Our results strengthen the emerging picture that IFI16 responds to the entry of the HSV-1 genome into the nucleus and is involved in antiviral activities. It is a speculation, although no more than that, that the reduced response of PML NB components to the viral genome that occurs in a proportion of IFI16-depleted cells decreases host restriction and therefore increases the probability of progression into lytic infection in a sufficient proportion of cells to explain the improvement in plaque formation of ICP0-null mutant HSV-1. On the other hand, the evidence to date indicates that IFI16 is involved in IFN-related signaling, sequentially through STING and then IRF3 and/or NF-κB, to induce IFN-β expression in response to viral infection (2, 3, 8, 14). Indeed, depletion of IFI16 reduces induction of IFN in response to HSV-1 infection (3, 14). It has been proposed that ICP0 inhibits this pathway through the induction of IFI16 degradation (3, 14). While in no way calling into question the accepted role of IFI16 in the innate immunity signaling pathway, the results presented here raise some important considerations. First, although ICP0 may well inactivate the effects of IFI16, this appears not to be through its direct targeted degradation. Second, removal of IRF3 does not increase the efficiency of ICP0-null mutant HSV-1 plaque formation (37); therefore, the restrictive effects of IFI16 cannot be mediated through an IRF3-related pathway in cultured cells.
Finally, this study provides another example of the inherent difficulties of distinguishing the direct and indirect effects of ICP0. It may be premature to conclude that a particular cellular protein that diminishes in quantity during wt but not ICP0-null mutant HSV-1 infection is a substrate of ICP0 unless steps are taken to ensure that the infections proceed through the lytic cycle at equivalent rates. Another example with some analogies to the current case is that of cyclins D1 and D3, in which effects that were initially attributed to ICP0 were later found to be ICP0 independent (41). A second important criterion is whether ICP0 is sufficient to induce degradation of a given protein in the absence of all other viral proteins. The inducible cell line system used here is particularly useful in this regard as, in contrast to transfections, it ensures that ICP0 is expressed in all cells in the population at physiological levels. Over the years, ICP0 has been linked to a wide variety of functions through comparisons of the phenotypes of wt and mutant viruses. Given the clear example of IFI16 presented here, it may be valuable to consider the possibility of indirect effects of ICP0 when interpreting such studies. Despite these qualifications, this study supports the overall conclusions of the previous publications (13, 14) which propose that IFI16 plays a role in regulating HSV-1 infection and that ICP0 has impacts on IFI16-related activities.
ACKNOWLEDGMENTS
This work was funded by the Medical Research Council and a University of Glasgow MRes studentship for G.A.
We thank Frazer Rixon, Chris Boutell, and Roel van Driel for gifts of reagents and Chris Boutell for helpful suggestions and comments on the manuscript.
Footnotes
Published ahead of print 2 October 2013
REFERENCES
- 1.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47 [DOI] [PubMed] [Google Scholar]
- 2.Unterholzner L, Bowie AG. 2011. Innate DNA sensing moves to the nucleus. Cell Host Microbe 9:351–353 [DOI] [PubMed] [Google Scholar]
- 3.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11:997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veeranki S, Choubey D. 2012. Interferon-inducible p200-family protein IFI16, an innate immune sensor for cytosolic and nuclear double-stranded DNA: regulation of subcellular localization. Mol. Immunol. 49:567–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, Jiang Z, Horvath G, Rathinam VA, Johnstone RW, Hornung V, Latz E, Bowie AG, Fitzgerald KA, Xiao TS. 2012. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 36:561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schattgen SA, Fitzgerald KA. 2011. The PYHIN protein family as mediators of host defenses. Immunol. Rev. 243:109–118 [DOI] [PubMed] [Google Scholar]
- 7.Ludlow LE, Johnstone RW, Clarke CJ. 2005. The HIN-200 family: more than interferon-inducible genes? Exp. Cell Res. 308:1–17 [DOI] [PubMed] [Google Scholar]
- 8.Tanaka Y, Chen ZJ. 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5:ra20. 10.1126/scisignal.2002521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B. 2011. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 9:363–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh VV, Kerur N, Bottero V, Dutta S, Chakraborty S, Ansari MA, Paudel N, Chikoti L, Chandran B. 2013. Kaposi's sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J. Virol. 87:4417–4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ansari MA, Singh VV, Dutta S, Veettil MV, Dutta D, Chikoti L, Lu J, Everly D, Chandran B. 2013. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J. Virol. 87:8606–8623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gariano GR, Dell'Oste V, Bronzini M, Gatti D, Luganini A, De Andrea M, Gribaudo G, Gariglio M, Landolfo S. 2012. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog. 8:e1002498. 10.1371/journal.ppat.1002498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson KE, Chikoti L, Chandran B. 2013. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J. Virol. 87:5005–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. U. S. A. 109:E3008–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everett RD, Murray J. 2005. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 79:5078–5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuchet-Lourenço D, Boutell C, Lukashchuk V, Grant K, Sykes A, Murray J, Orr A, Everett RD. 2011. SUMO pathway dependent recruitment of cellular repressors to herpes simplex virus type 1 genomes. PLoS Pathog. 7:e1002123. 10.1371/journal.ppat.1002123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Everett RD, Parsy ML, Orr A. 2009. Analysis of the functions of herpes simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. J. Virol. 83:4963–4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukashchuk V, Everett RD. 2010. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol. 84:4026–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boutell C, Everett RD. 2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J. Gen. Virol. 94:465–481 [DOI] [PubMed] [Google Scholar]
- 20.Cuchet-Lourenço D, Vanni E, Glass M, Orr A, Everett RD. 2012. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J. Virol. 86:11209–11222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72:6581–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stow ND, Stow EC. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 67:2571–2585 [DOI] [PubMed] [Google Scholar]
- 23.Everett RD. 1989. Construction and characterization of herpes simplex virus type 1 mutants with defined lesions in immediate early gene 1. J. Gen. Virol. 70:1185–1202 [DOI] [PubMed] [Google Scholar]
- 24.Everett RD, Sourvinos G, Orr A. 2003. Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J. Virol. 77:3680–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meredith M, Orr A, Elliott M, Everett R. 1995. Separation of sequence requirements for HSV-1 Vmw110 multimerisation and interaction with a 135-kDa cellular protein. Virology 209:174–187 [DOI] [PubMed] [Google Scholar]
- 26.Everett RD, Boutell C, Orr A. 2004. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J. Virol. 78:1763–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao F, Schaffer PA. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J. Virol. 69:6249–6258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. 2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. U. S. A. 99:15655–15660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boutell C, Cuchet-Lourenço D, Vanni E, Orr A, Glass M, McFarlane S, Everett RD. 2011. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 7:e1002245. 10.1371/journal.ppat.1002245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Everett RD, Bell AJ, Lu Y, Orr A. 2013. The replication defect of ICP0-null mutant herpes simplex virus 1 can be largely complemented by the combined activities of human cytomegalovirus proteins IE1 and pp71. J. Virol. 87:978–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Everett RD, Parada C, Gripon P, Sirma H, Orr A. 2008. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol. 82:2661–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80:7995–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jamieson DR, Robinson LH, Daksis JI, Nicholl MJ, Preston CM. 1995. Quiescent viral genomes in human fibroblasts after infection with herpes simplex virus type 1 Vmw65 mutants. J. Gen. Virol. 76:1417–1431 [DOI] [PubMed] [Google Scholar]
- 34.Stuurman N, de Graaf A, Floore A, Josso A, Humbel B, de Jong L, van Driel R. 1992. A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J. Cell Sci. 101:773–784 [DOI] [PubMed] [Google Scholar]
- 35.Maul GG, Everett RD. 1994. The nuclear location of PML, a cellular member of the C3HC4 zinc- binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 75:1223–1233 [DOI] [PubMed] [Google Scholar]
- 36.Everett RD, Murray J, Orr A, Preston CM. 2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 81:10991–11004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Everett RD, Young DF, Randall RE, Orr A. 2008. STAT-1- and IRF-3-dependent pathways are not essential for repression of ICP0-null mutant herpes simplex virus type 1 in human fibroblasts. J. Virol. 82:8871–8881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Preston CM, Nicholl MJ. 2005. Human cytomegalovirus tegument protein pp71 directs long-term gene expression from quiescent herpes simplex virus genomes. J. Virol. 79:525–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aglipay JA, Lee SW, Okada S, Fujiuchi N, Ohtsuka T, Kwak JC, Wang Y, Johnstone RW, Deng C, Qin J, Ouchi T. 2003. A member of the Pyrin family, IFI16, is a novel BRCA1-associated protein involved in the p53-mediated apoptosis pathway. Oncogene 22:8931–8938 [DOI] [PubMed] [Google Scholar]
- 40.Johnstone RW, Wei W, Greenway A, Trapani JA. 2000. Functional interaction between p53 and the interferon-inducible nucleoprotein IFI 16. Oncogene 19:6033–6042 [DOI] [PubMed] [Google Scholar]
- 41.Everett RD. 2004. Herpes simplex virus type 1 regulatory protein ICP0 does not protect cyclins D1 and D3 from degradation during infection. J. Virol. 78:9599–9604 [DOI] [PMC free article] [PubMed] [Google Scholar]