

Abstract
The herpes simplex virus host shutoff RNase (VHS-RNase) is the major early block of host responses to infection. VHS-RNase is introduced into cells during infection and selectively degrades stable mRNAs made before infection and the normally short-lived AU-rich stress response mRNAs induced by sensors of innate immunity. Through its interactions with pUL47, another tegument protein, it spares from degradation viral mRNAs. Analyses of embedded motifs revealed that VHS-RNase contains a nuclear export signal (NES) but not a nuclear localization signal. To reconcile the potential nuclear localization with earlier studies showing that VHS-RNase degrades mRNAs in polyribosomes, we constructed a mutant in which NES was ablated. Comparison of the mutant and wild-type VHS-RNases revealed the following. (i) On infection, VHS-RNase is transported to the nucleus, but only the wild-type protein shuttles between the nucleus and cytoplasm. (ii) Both VHS-RNases localized in the cytoplasm following transfection. On cotransfection with pUL47, a fraction of VHS-RNase was translocated to the nucleus, suggesting that pUL47 may enable nuclear localization of VHS-RNase. (iii) In infected cells, VHS-RNase lacking NES degraded the short-lived AU-rich mRNAs but not the stable mRNAs. In transfected cells, both wild-type and NES mutant VHS-RNases effectively degraded cellular mRNAs. Our results suggest that the stable mRNAs are degraded in the cytoplasm, whereas the AU-rich mRNAs may be degraded in both cellular compartments. The selective sparing of viral mRNAs may take place during the nuclear phase in the course of interaction of pUL47, VHS-RNase, and nascent viral mRNAs.
INTRODUCTION
The virion host shutoff (VHS) RNase is a late (γ2) tegument protein carried into the cell during infection. It plays a key role in blocking host responses to infection capable of curtailing viral replication (1–4). It functions as an endoribonuclease with the specificity of RNase A (5). VHS-RNase binds to eIF4H and cleaves stable mRNAs in polyribosomes 3′ to the 5′ cap structure. The RNA is then degraded processively 5′ to 3′ (6). The main target of the VHS-RNase are the short-lived stress response mRNAs characterized by the presence AU-rich elements in their 3′ untranslated regions (3′ UTR) targeted by tristetraprolin (TTP) (7). The VHS-RNase binds to tristetraprolin at the AU-rich elements and cleaves the RNA 5′ to the AU-rich element. The 3′ portion of the cleaved RNA is rapidly degraded 5′ to 3′. The 5′ portion of the cleaved AU-rich mRNA lingers for many hours in the cytoplasm, possibly because of a dearth in enzymes capable of cleaving the RNA 3′ to 5′ (8). Both the stable and the AU-rich short-lived mRNAs are also deadenylated in this process. It is noteworthy that on transfection, VHS-RNase degrades its own mRNA as well as the mRNAs of cotransfected genes (9).
In infected cells, VHS is regulated by several viral proteins. Thus, late in infection, VHS is completely neutralized by two late proteins, VP16 and VP22, prior to packaging in virions (9, 10). Early in infection, the function of VHS is regulated by pUL47, a tegument protein (11). Recent studies have shown that on entry into cells infected with wild-type virus, VHS-RNase targets mRNAs made before infection and AU-rich mRNAs, but largely spares viral mRNAs made after infection (12). The selective sparing of stable mRNAs made after infection has been shown to be due to the interaction of VHS with pUL47 (11). pUL47 has no effect on the degradation of AU-rich mRNAs. It has been reported to shuttle between the nucleus and cytoplasm and to bind poly(A) binding protein (13, 14).
The fundamental question posed by the present study was to define the subcellular compartment in which the VHS-RNase newly introduced into the infected cell functions. To resolve this question, we examined the coding sequence of VHS for nuclear localization and nuclear export motifs. We did not identify an obvious nuclear localization motif but we did uncover a typical motif for nuclear export signal (NES) as well as an EADD motif. The NES motif is embedded the codon sequence 30PIAVDLWNVMYTLVVKYQRR49. The EADD motif is embedded in the codon sequence 186INSGQLEADDACANLYHTNT205. The EADD motif has been reported previously as a catalytic motif is required for RNase activity (15). This motif is present in a number of cellular and phage nucleases (e.g., T5 5′ exonuclease, TaqI DNA polymerase, T4 RNase H, human FEN-1) (16–21).
This report focuses on the functions of VHS in which EADD and NES motifs were independently mutagenized by codon substitutions. The key findings are as follows. (i) In infected cells, VHS lacking the NES motif localizes in the nucleus, does not shuttle between the nucleus and the cytoplasm, and does not degrade stable mRNAs but does degrade AU-rich mRNAs. (ii) In transfected cells, VHS lacking the NES motif localizes in the cytoplasm and is effective in degrading stable mRNAs. UL47 protein enables VHS lacking the NES motif to localize in the nucleus. (iii) VHS lacking the EADD motif is not enzymatically active.
MATERIALS AND METHODS
Cells and viruses.
HEp-2, Vero, and HEK 293 cell lines were obtained from the American Type Culture Collection (Manassas, VA). All cell lines were grown in Dulbecco's modified Eagle medium supplemented with 5% fetal calf serum (HEp-2 cells) or 5% newborn calf serum (Vero cells) or 10% fetal bovine serum (HEK 293 cells). The limited-passage herpes simplex virus 1 strain F [HSV-1(F)] is the prototype HSV-1 strain used in this laboratory (22). The bacterial accessory chromosome (BAC) encoding the HSV-1(F) DNA was reported elsewhere (11).
Transient gene expression in transfected cells.
The procedures for transfection were described elsewhere (9).
Antibodies.
Mouse monoclonal antibody against β-actin and rabbit polyclonal antibody against SP1 were purchased from Sigma (St. Louis, MO) and Upstate Biotechnology (Billerica, MA), respectively. Mouse polyclonal antibodies against VHS, ICP8, ICP0, and US11 and rabbit polyclonal antibodies against VHS and UL47 were described elsewhere (8, 11, 23–25). All the antibodies were used at a dilution of 1:1,000.
Plasmid construction.
The plasmids construction was reported previously (9). Briefly, the two mutant UL41 open reading frames (ORFs) with E192A, D194A, and D195A (VHSm) or L35A and L42A (VHS-NESm) mutations were obtained by use of a QuikChange XL site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The wild-type and two mutant UL41 ORFs were subcloned in the multiple cloning site of the pCMS-EGFP vector (Clontech, Palo Alto, CA), which also expresses enhanced green fluorescent protein [EGFP] under the control of simian virus 40 early promoter, or pcDNA3.1(+), respectively. The resulting plasmids were named pEGFP-pVHSWT, pEGFP-pVHSNESm, and pEGFP-pVHSm, or pEGFP, pVHSWT, pVHSNESm, and pVHSm, respectively.
Immunoblot analyses.
Total or fractionated cell lysates were electrophoretically separated on denaturing polyacrylamide gels and transferred nitrocellulose membrane. The membranes were blocked in phosphate-buffered saline (PBS) containing 5% nonfat dry milk and reacted at 4°C overnight with primary antibody in TBST (0.01 M Tris HCl, 0.15 M NaCl, 0.05%[vol/vol] Tween 20) plus 1% bovine serum albumin (BSA), rinsed, and reacted with alkaline phosphatase- or peroxidase-conjugated secondary antibodies (Sigma). The membranes were rinsed and developed with either 5-bromo-4-chloro-3-indolylphosphate (BCIP) plus nitroblue tetrazolium (Denville Scientific, Metuchen, NJ) or ECL Western blotting detection reagents (Amersham Biosciences) according to the manufacturer's instructions. The membranes were probed for VHS, ICP0, ICP8, US11, or SP1 and β-actin with the antibodies listed above.
GST pull-down assay.
The procedures were performed as previously described (9). Briefly, HEK 293 cells were transiently transfected with a plasmid encoding full-length UL47 tagged with Myc at the N terminus for 48 h. Cells were lysed in glutathione S-transferase (GST) lysis buffer (20 mM Tris [pH 8.0], 1 mM EDTA, 1% Nonidet P-40, 200 mM NaCl, 0.1 mM sodium orthovanadate, 10 mM sodium fluoride [NaF], 2 mM dithiothreitol [DTT], protease inhibitor mixture [Complete protease mixture; Roche Diagnostics, Indianapolis, IN]) on ice for 1 h. Insoluble material was pelleted by centrifugation at maximum speed in a 5415C centrifuge (Eppendorf, Boulder, CO) for 10 min at 4°C. The supernatant fluids precleared with 50 μl of a 50% slurry of glutathione beads for 3 h at 4°C were incubated overnight at 4°C with equal amounts of a 50% slurry of glutathione beads bound to GST alone or GST fused to the VHS wild type (VHS-WT), VHSm, or VHS-NESm. The beads were pelleted by centrifugation and rinsed five times with GST buffer. The proteins bound to the beads were solubilized in 50 μl of SDS gel-loading buffer (2% SDS, 5% 2-mercaptoethanol, 50 mM Tris [pH 6.8], 2.75% sucrose), heated to 95°C for 5 min, resolved by PAGE, transferred to nitrocellulose membrane, and immunoblotted with the mouse anti-Myc antibody.
Northern blot analyses.
The Northern blot analyses were carried out as described earlier with minor modifications (11). Briefly, 10 μg of total RNA were loaded onto denaturing formaldehyde gel and probed with random hexanucleotide-primed 32P-labeled fragment of the indicated genes (the glyceraldehyde-3-phosphate dehydrogenase [GAPDH], IEX-1 EGFP, and VHS genes; see Fig. 3 and 4) upon transfer onto a nylon membrane. Prehybridization and hybridization were performed with ULTRAhyb buffer (Ambion, Austin, TX) supplemented with 200 μg of denatured salmon sperm DNA per milliliter (Stratagene, La Jolla, CA). The membranes were prehybridized for 2 h at 42°C and then overnight after the addition of the 32P-labeled probe. The membranes were rinsed as suggested by the manufacturer of the ULTRAhyb buffer and exposed to film for signal detection.
Fig 3.
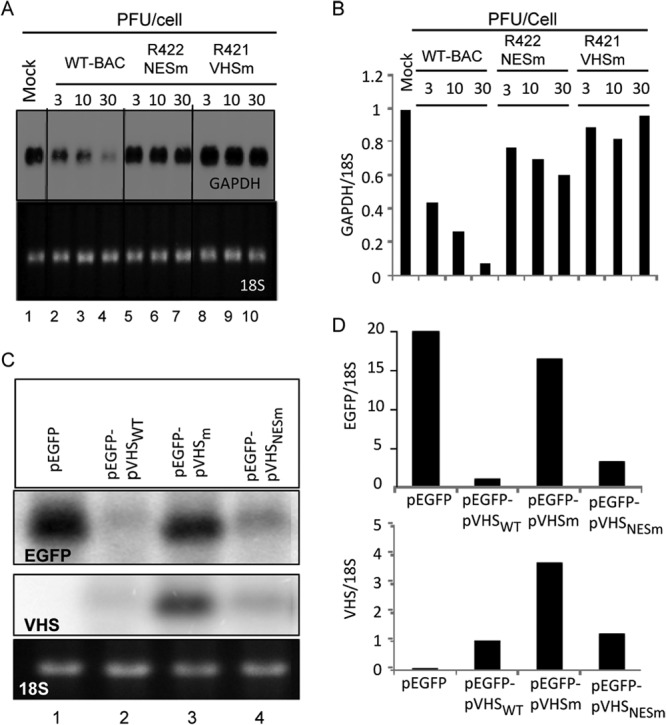
RNase activity of infected and transfected VHS mutants. HEp-2 cells were mock infected or exposed to 3, 10, or 30 PFU of HSV-1(WT-BAC), R422 (NESm), or R421 (VHSm) per cell for 1 h. The inocula were then replaced with fresh complete medium, and the cell cultures were incubated at 37°C for two additional hours. Transcription was then stopped by the addition of 10 μg of actinomycin D (ActD) per ml (time zero). Cells were harvested at 2 h after exposure to ActD. (A) GAPDH mRNA was analyzed by Northern blotting as described in Materials and Methods. The ethidium bromide-stained 18S rRNA was used as a loading control. (B) The intensities of the bands at various dose points were quantified with the aid of a General Dynamics Phosphorimager, and the residual mRNAs were normalized with respect to 18S rRNA and plotted relative to the RNA levels present in the mock-infected cells. (C) HEK 293 cells were transfected with 1 μg of pEGFP, pEGFP-pVHSWT, pEGFP-pVHSNESm, or pEGFP-pVHSm. The cells were collected 24 h after transfection. EGFP and VHS mRNAs were analyzed by Northern blotting. The ethidium bromide-stained 18S rRNA was used as a loading control. (D) The intensities of the bands were quantified with the aid of a General Dynamics Phosphorimager, and the residual mRNAs were normalized with respect to the 18S rRNA and plotted relative to the RNA levels present in the pEGFP-pVHSWT transfected cells.
Fig 4.

Effects of VHS mutants on AU-rich mRNAs. (A) Experimental design diagram. HEp2 cells were mock infected or exposed (10 PFU/cell) to HSV-1(WT/BAC), R422 (NESm) virus, or R421 (VHSm) virus for 1 h. The inocula were then replaced with fresh complete medium, and cells were harvested at the indicated times. (B) IEX-1 mRNA was analyzed by Northern blotting. The ethidium bromide-stained 18s rRNA was used as a loading control. (C) The value of the truncated IEX-1 mRNA obtained by densitometric scans was normalized with respect to 18S rRNA and plotted relative to the amounts in mock-infected cells at 3 h after the start of the experiment.
Immunofluorescence staining.
The transfected or infected cells were fixed in 4% paraformaldehyde for 10 min and blocked with 20% horse serum containing 1% BSA and 0.25% Triton X-100 for 1 h at room temperature and immunostained at 4°C overnight. The antibodies used in these studies were the VHS rabbit polyclonal antibody, ICP8 mouse polyclonal antibody, VHS mouse polyclonal antibody, and UL47 rabbit polyclonal antibody, all of which were used at a dilution of 1:1,000 in 10% horse serum containing 0.5% BSA and 0.125% Triton X-100. For transfected cells, the secondary antibodies were Alexa Fluor 594-conjugated goat anti-rabbit and Alexa Fluor 488-conjugated goat anti-mouse (Molecular Probes). For infected cells, the secondary antibodies were fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit (Sigma) and Texas goat anti-mouse (Molecular Probes), respectively. All the secondary antibodies were diluted 1:1,000 in 10% horse serum containing 0.5% BSA and 0.125% Triton X-100. The immunostained cells were examined with the aid of a Zeiss confocal microscope equipped with software provided by Zeiss.
Subcellular fractionation.
HEK 293 cells were transfected with 1 μg of the empty plasmid pcDNA3.1(+), pVHSWT, pVHSm, or pVHSNESm, respectively. The cells were collected 24 h after transfection. Cell pellets were resuspended in 0.6 ml of ice cold buffer 1 (150 mM NaCl, 50 mM HEPES [pH 7.4], 25 μg/ml digitonin,10 μl/ml protease inhibitor) and incubated for 10 min at 4°C end over end and then centrifuged at 4,600 rpm for 5 min to pellet cells. The supernatants represented the cytosol-enriched fraction. The pellets were then washed with 1 ml of ice-cold PBS and centrifuged as described above to remove any remaining digitonin extract. The pellets were resuspended in 0.6 ml of ice-cold buffer 2 (150 mM NaCl, 50 mM HEPES [pH 7.4], 1% [vol/vol] NP-40, 10 μl/ml protease inhibitor) and incubated for 30 min on ice. The samples were centrifuged at 8,700 rpm for 5 min to pellet nuclei and debris. The supernatants represented the extract comprising membrane bound organelles, such as the endoplasmic reticulum (ER), the Golgi body, mitochondria, and some nuclear lumenal proteins. The pellets were washed again with 1 ml of ice-cold PBS and centrifuged at 8,700 rpm for 5 min to remove any remaining NP-40 extract. The pellets were then resuspended in 0.4 ml of ice-cold buffer 3 (150 mM NaCl, 50 mM HEPES [pH 7.4], 0.5% [wt/vol] sodium deoxycholate, 0.5% [wt/vol] SDS, 1 mM DTT, 10 μl/ml protease inhibitor) and incubated at 4°C for 30 min followed by sonication twice (10 s, 20% amplification). The samples were then centrifuged at 13,000 rpm for 10 min. The supernatant contained the extract comprising nuclear membranes and nuclear proteins.
Sucrose gradient fractionation.
The procedures used were those described previously (26) with some modification. HEp-2 cell monolayers were either mock infected or exposed to 20 PFU of HSV-1 wild-type virus rederived from BAC [HSV-1(WT-BAC)] or VHS-NESm (R422) virus per cell. After 1 h, the inocula were replaced with fresh medium, and the cells were incubated at 37°C for additional 2 h. Cells were harvested by trypsinization, and the cell pellets were rinsed twice with ice-cold PBS and resuspended in 0.5 ml of buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 0.25 M sucrose, and protease inhibitor cocktail [Roche Diagnosis]) and incubated on ice for 10 min and disrupted by 20 strokes in a homogenizer. The cell lysates were layered on top of an 11-ml linear 10 to 30% (wt/vol of H2O) sucrose gradient. Samples were centrifuged at 27,500 rpm in a Beckman SW41 ultracentrifuge rotor for 3 h. Fraction of 1 ml each were collected from the top of the gradient. The pellet (nuclear fraction) was suspended in 1 ml of lysis buffer (20 mM Tris [pH 8.0], 1 mM EDTA, 0.5% NP-40, 0.4 M NaCl, 2 mM DTT, 0.1 mM sodium othovanadate [Na3VO4], 10 mM NaF) and sonicated twice for 10 s at 20% amplification. Volumes (150 μl) from each fraction were diluted with 50 μl of 4× loading buffer, subjected to electrophoresis in a denaturing gel, and then probed with anti-VHS and anti-SP1 antibodies.
Construction of VHS mutant viruses.
The 1.47-kb ORF of VHS-NESm and VHSm mutants plus their upstream and downstream flanking sequences (2.2 kb) were subcloned into pBlueScript KS(+). The resulting plasmids were named RB-NESm and RB-VHSm, respectively. These plasmids were then digested with XhoI and XbaI, and the 3.7-kb fragment containing UL41 ORF and flanking sequences was cloned into pKO5. The resulting plasmids were named KO-NESm and KO-VH5m, respectively. The competent Escherichia coli RR1 strain harboring BAC903 (ΔUL41) (11) was electroporated with pKO-NESm and pKO-VHSm, respectively, and incubated at 43°C on LB plates containing 25 μg/ml zeocin (Zeo) and 20 μg/ml chloramphenicol. The colonies were diluted and plated on LB plates containing chloramphenicol (20 μg/ml) and 5% sucrose. Colonies grown on the sucrose plates were screened with PCR or by colony hybridization. PCR analyses verified that the recombinants BAC919 and BAC920 contained NESm and VHSm, respectively. Plasmids DNAs isolated from E. coli were transfected into Vero cells. The resulting viruses were plaque purified three times.
RESULTS
Construction of recombinant viruses.
Mutagenesis by codon substitution in NES and EADD motifs is described in Materials and Methods. The EADD motif in the sequence 186INSGQLEADDACANLYHTNT205 was replaced by AAAA. In the NES motif (30PIAVDLWNVMYTLVVKYQRR49), the two lysine codons were replaced with alanine codons. The expression plasmids and the recombinant viruses were prepared as described in Materials and Methods. The viruses constructed for these studies were R421 expressing VHS with a mutation in the EADD motif (VHSm) and R422 with a mutation in the NES motif (VHS-NESm). These were derived from the wild-type parent, HSV-1(F) cloned as a bacterial accessory chromosome (BAC). HSV-1(F) and HSV-1(WT-BAC) were used interchangeably as wild-type controls.
Replication of the VHS mutant viruses.
In this series of experiments, HEp-2 cells were exposed to 1 PFU of wild-type or mutant viruses per cell. The cultures were harvested at 3, 7, 24, or 48 h after infection, and the virus titers were determined on Vero cells. We tested two wild-type parent and two mutant viruses, i.e., HSV-1(F), HSV-1(WT-BAC), R421, and R422, respectively. The results (Fig. 1) show that the patterns of replication of HSV-1(F) and HSV-1(WT-BAC) were similar and differed approximately 2-fold with respect to yields at 24 h and 48 h. The yields of mutant viruses were generally lower. Thus, the yield of the R421 mutant was 10-fold lower than that of its parent, HSV-1(WT-BAC). The yield of R422 mutant was intermediate between those of the wild-type and R421 mutant viruses.
Fig 1.
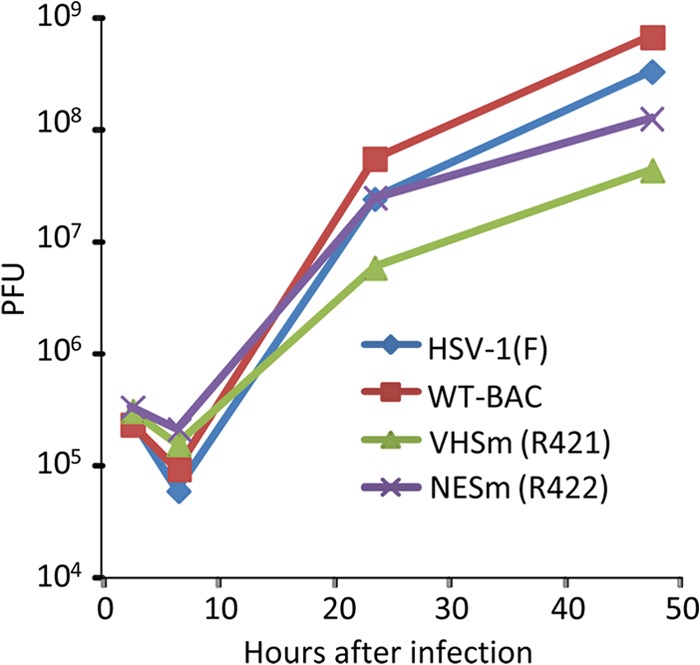
Replication of wild-type viruses encoding wild-type and mutant VHS-RNases. HEp-2 cells were exposed to the indicated viruses at a multiplicity of 1 PFU per cell. The inocula were replaced 1 h after exposure to virus. The cells were harvested at 3, 7, 24, and 48 h after infection. Viral progeny was titrated on Vero cells. WT-BAC, HSV-1(WT-BAC).
Accumulation of proteins representative of various kinetic classes in cells infected with wild-type or mutant viruses.
In this series of experiments, HEp-2 and Vero cells were exposed to 1 PFU of wild-type or mutant viruses per cell. The cells were harvested at 3, 7, or 21 h after infection, solubilized, transferred to a nitrocellulose sheet, and probed with antibodies to ICP0, ICP8, VHS, US11, or β-actin. The results (Fig. 2) show that the patterns of accumulation of viral proteins in cells infected with HSV-1(WT-BAC) or HSV-1(F) were intrinsically similar. The accumulation of the proteins encoded by mutant viruses was delayed in HEp-2 cells and to a much lesser extent in Vero cells. We estimate the delay to be 2 to 4 h on the basis of the differences in the patterns of accumulation of viral proteins at 3 and 7 h after infection.
Fig 2.
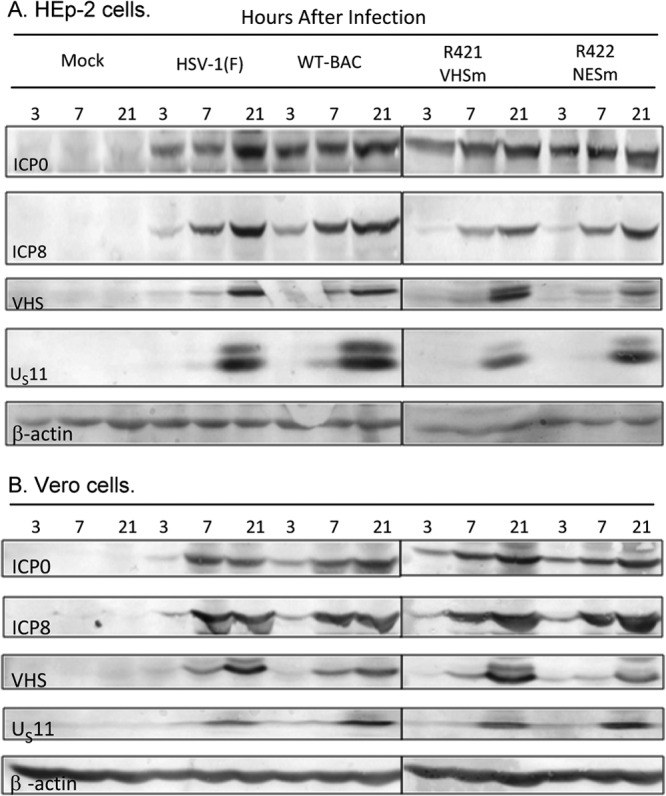
Expression of viral genes carried by wild-type and mutant viruses. HEp-2 and Vero cells were mock infected (Mock) or infected with the indicated viruses at a multiplicity of 1 PFU per cell for 1 h. Cells were collected at 3, 7, and 21 h postinfection, and equal amounts of total proteins were electrophoretically separated on a 10% denaturing polyacrylamide gel, transferred to a nitrocellulose sheet, and probed with antibodies against ICP0, ICP8, VHS, US11, and β-actin.
Analyses of the ability of mutant viruses to degrade stable mRNAs.
Two series of experiments were done to measure RNase activity of the mutant derived in this study. In the first series, we measured the activity of the mutant VHS-RNases in infected cells. Specifically, HEp-2 cells were mock infected or exposed for 1 to 3 h to 10 or 30 PFU of HSV-1(WT-BAC) or the mutant virus R421 or R422 per cell. At 1 h after exposure to virus, the inocula were replaced with fresh medium containing actinomycin D (ActD; 10 μg/ml). The cells were harvested 2 h after the addition of ActD. RNA extracted from the cells was analyzed by Northern blotting for GAPDH (Fig. 3A). The bands representing GAPDH were scanned with the aid of a General Dynamics Phosphorimager and normalized with respect to 18s rRNA. The results shown in Fig. 3B indicate that in contrast to the HSV-1(WT-BAC) parent, the mutants had very little RNase residual activity that was virus dose dependent.
In the second series of experiments, HEK 293 cells were transfected with 1 μg of pEGFP alone or plasmids containing the open reading frames of pEGFP with pVHSWT, pVHSm, or pVHSNESm, respectively. The cells were collected 24 h after transfection. EGFP and VHS mRNAs were analyzed by Northern blotting and normalized with respect to the 18S rRNA used as a loading control. The Northern blots shown in Fig. 3C were scanned, and the results are shown in Fig. 3D. As expected, the levels of EGFP mRNA were reduced dramatically in cells transfected with the plasmid pEGFP-pVHSWT and to only a slightly lesser extent in cells transfected with the pEGFP-pVHSm plasmid. In contrast, decreases in EGFP mRNA levels were greater in cells transfected with plasmid pEGFP-pVHSNESm. Concurrently, the levels of VHS mRNA recovered from cells transfected with pEGFP-pVHSm were significantly higher than those in cells transfected with the pEGFP-pVHSWT or pEGFP-pVHSNESm plasmid.
We conclude that there is a striking difference in the activities of VHSNESm in the context of infection or transfection. In infection, VHSNESm delivered by the virus shows little activity against stable RNAs. In transfected cells, this mutant virus is nearly as effective in degrading EGFP or its own RNA as is wild-type virus.
Analyses of the ability of mutant viruses to degrade AU-rich mRNAs.
In this series of experiments, HEp-2 cells were mock infected or exposed for 1 h to 10 PFU of the parent virus HSV-1(WT-BAC) or the mutant viruses R421 or R422. The inocula were then replaced with fresh medium. The cells were harvested at 3 or 5 h after exposure to the virus. The RNAs were extracted, resolved in Northern blots, and hybridized to the IEX-1 mRNA probe as previously described (11). The amount of truncated IEX-1 RNA bracketed by the quadrangle shown in Fig. 4B were quantified with the aid of a General Dynamics Phosphorimager and normalized with respect to 18S rRNA at 3 h after infection. The results shown in Fig. 4C indicate that cells infected with wild-type parent virus or R422 encoding the VHSNESm mutant accumulated truncated mRNAs to virtually identical levels. Significantly lower levels of truncated IEX-1 RNA were detected in cells infected with the R421 mutant virus. We conclude that the R422 encoding the VHSNESm mutation is able to cleave IEX-1 mRNA in cells infected with the mutant virus.
Distribution of VHS-RNase in newly infected cells.
The presence of a NES in VHS raised the question of whether it plays a role in the distribution of VHS upon release of the protein into the newly infected cell. To clarify the distribution of VHS, we carried out several experiments.
In the first series of experiments, HEK 293 cells were transfected with 1 μg of the empty plasmid pcDNA3.1(+) or plasmids encoding pVHSWT, pVHSm, or pVHSNESm, respectively. The cells were harvested 24 h after transfection and fractionated into nuclear, membrane, and cytoplasmic fractions, as described in Materials and Methods. Typical results, shown in Fig. 5A, were as follows. As expected, no VHS was detected in extracts of cells transfected with the empty plasmid or plasmid encoding wild-type VHS. VHS was detected in nuclei of cells transfected with all mutants (lanes 6 and 12).
Fig 5.
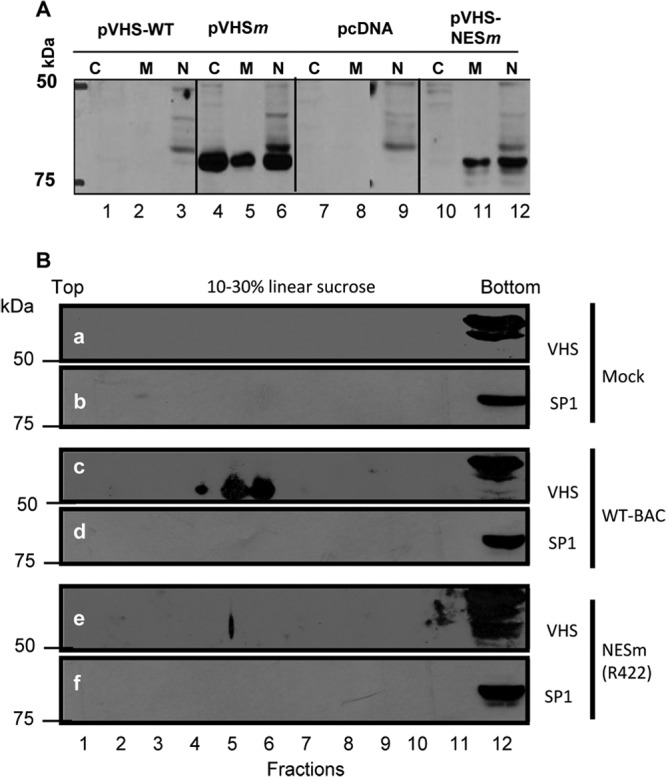
Subcellular localization of wild-type and mutant VHS-RNases. (A) HEK 293 cells were transfected with 1 μg of pcDNA3.1(+), pVHS-WT, pVHSm, or pVHS-NESm. The cells were collected 24 h after transfection and fractionated into nuclear, membrane, and cytoplasmic fractions as described in Materials and Methods. (B) HEp-2 cells were mock infected or exposed (20 PFU/cell) to HSV-1(WT-BAC) or R422 (VHS-NESm) virus for 1 h. The inocula were then replaced with fresh complete medium, and cells were harvested at 2 h postinfection. The nuclear and cytoplasmic fractions were centrifuged through a linear sucrose gradient. The fractions were solubilized, electrophoretically separated on liner sucrose density gradients, and probed with antibodies to VHS and the SP1 nuclear marker protein.
The fractionation of cell lysates into nuclear and cytoplasmic fraction requires pelleting the nuclei. Earlier studies (unpublished) suggested that VHS entering newly infected cells might be poorly soluble. To test the hypothesis that the cofractionation of VHS with nuclei may be an artifact, in the second series of experiments, HEp-2 cells were mock infected or infected with HSV-1(WT-BAC) or R422 mutant virus for 1 h at a multiplicity of 20 PFU per cell. The inocula were then replaced with fresh medium. At 3 h after exposure to virus, the cells were harvested, lysed, and centrifuged through a linear sucrose density gradient. The fractions collected from the gradient were solubilized, and the contents were separated by electrophoresis on denaturing gels and probed with antibody to VHS or to the nuclear marker SP1. The results shown in Fig. 5B indicate that the VHS encoded by wild-type virus bands separately from the pellet containing nuclei (Fig. 5B, band c). VHS encoded by the R422 mutant was at the bottom of the gradient with nuclear proteins (Fig. 5B, band e). Note that the nuclei contained an antigen that formed a doublet migrating more slowly than VHS and cross-reacted with the anti-VHS antibody. This antigen was present in mock-infected nuclei (Fig. 5B, band a) as well as in nuclear fraction of cells infected with wild-type and mutant viruses (Fig. 5B, bands c and e). On the basis of these studies, we conclude that fractionation of cells by separation of nuclei from cytoplasm by centrifugation does not yield reliable data on the localization of VHS.
The objective of the third series of experiments was to determine the localization of VHS-RNase by immunofluorescence. In the first study, Vero cells were mock transfected or transfected with 0.25 μg per well of a plasmid encoding EGFP alone or plasmids encoding both EGFP and VHSWT, EGFP and VHSNESm, or EGFP and VHSm, respectively. EGFP served as a transfection marker. At 24 h after transfection, the cells were fixed and reacted with corresponding antibodies as described in Materials and Methods. The results shown in Fig. 6 indicate that, as expected from earlier studies (9), no VHS-RNase or EGFP was detected in cells mock transfected or transfected with the plasmid encoding the wild-type VHS-RNase. EGFP and VHS were readily detected in cells transfected with plasmids encoding mutant VHS. Figure 6C and D each show five randomly selected cells showing the presence of both EGFP and mutant VHS. An examination of numerous fields revealed that in virtually 100% of cells exhibiting both VHS and EGFP, the VHS-RNase was localized in the cytoplasm.
Fig 6.
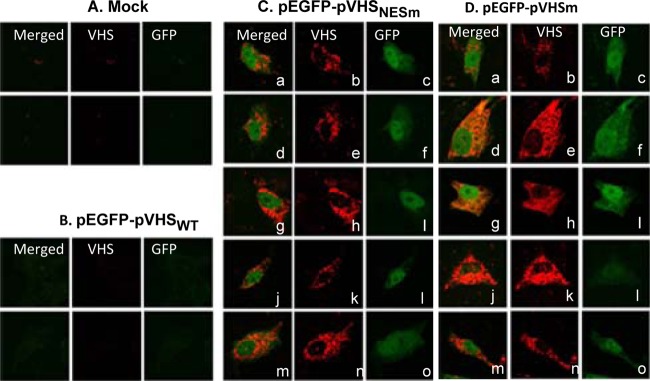
Subcellular localization of VHS in transfected cells. Vero cells were mock transfected or transfected (0.25 μg of DNA per well) with a plasmid encoding EGFP alone or plasmids encoding both EGFP and VHSWT, EGFP and VHSNESm, or EGFP and VHSm, respectively. Cells were fixed at 24 h after transfection and reacted with antibody to VHS-RNase. Images of transfected cells were collected with the aid of a Zeiss confocal microscope.
In the second study, HEp-2 cells were mock infected or exposed for 1 h to 20 PFU of HSV-1(WT-BAC) or viruses encoding VHS-NESm (R422), or VHSm (R421). The inocula were replaced with fresh medium after 1 h, and the cells were fixed and reacted with antibodies to VHS and ICP8 at 3 h after exposure to the viruses. ICP8 served as a marker of infection. The results (Fig. 7) were as follows. In culture infected with HSV-1(WT-BAC), 94.6% of the VHS-RNase in infected cells was localized in the cytoplasm, primarily in the perinuclear space as illustrated in Fig. 7D. In the remaining 5.4%, VHS localized in both the nucleus and the cytoplasm. In 91% of infected cells in cultures infected with R421 mutant virus, VHS was localized in the cytoplasm (Fig. 7C). In the remaining 9%, VHS-RNase was localized in both the nucleus and the cytoplasm. Finally, in cultures infected with the R422 mutant virus, 89.7% of infected cells contained VHS in the nucleus only. In only 10.3% of the cells, VHS-RNase localized in both the cytoplasm and the nucleus (Fig. 7B).
Fig 7.
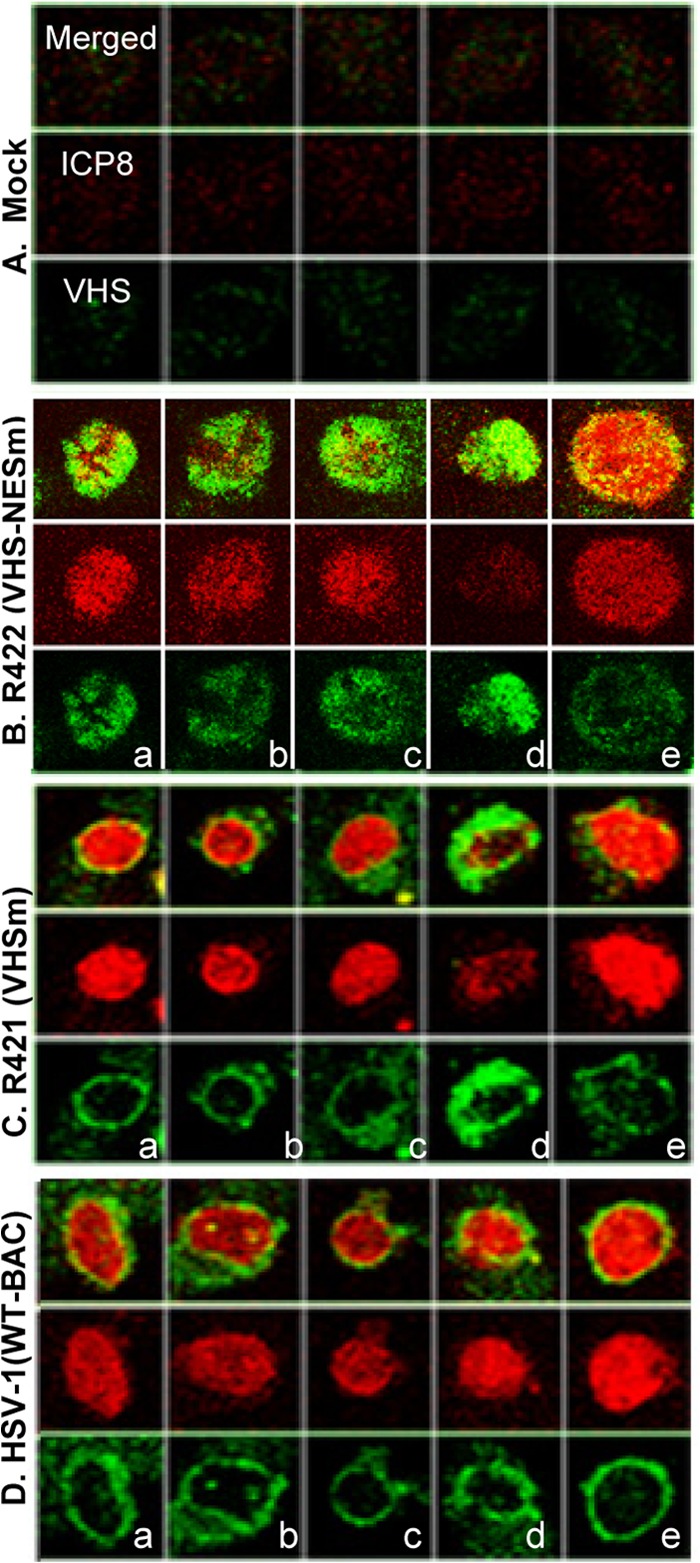
Subcellular localization of VHS in infected cells. HEp-2 cells were mock infected or exposed (20 PFU/cell) to wild-type HSV-1(WT-BAC), R422 (NESm) virus, or R421 (VHSm) virus for 1 h. The inocula were then replaced with fresh medium. The cells were fixed at 2 h after infection and reacted with antibodies to VHS and ICP8. ICP8 was used as a marker of infection.
We conclude that wild-type VHS and VHSm proteins brought into cells during infection are transported initially into the nucleus and then accumulate in the cytoplasm. The NES mutation disables the VHSNESm mutant from being translocated to the cytoplasm.
VHS shuttles between the nucleus and cytoplasm.
The observation that VHS localizes initially in the nucleus and is then translocated to the cytoplasm raises the question of whether this is a one-time translocation or whether VHS shuttles between the nucleus and cytoplasm. To resolve this question, HEp-2 cells were exposed to 20 PFU of wild-type HSV-1(WT-BAC) per cell. After 1 h, the inoculum was replaced with fresh medium with or without leptomycin B (LMB; 10 μM). The cells were fixed 2 h after exposure to virus and reacted with antibodies to VHS and ICP8. ICP8 served as a marker of infection. The results shown in Fig. 8 were as follows. In the absence of LMB (Fig. 8A), VHS was found in the perinuclear space (93% of cells). In the presence of LMB (Fig. 8B), VHS-RNase was present in 46.4% of infected cells in the cytoplasm only and in 53.6% of cells in both the nucleus and cytoplasm. We conclude that WT-VHS shuttles between the nucleus and cytoplasm.
Fig 8.
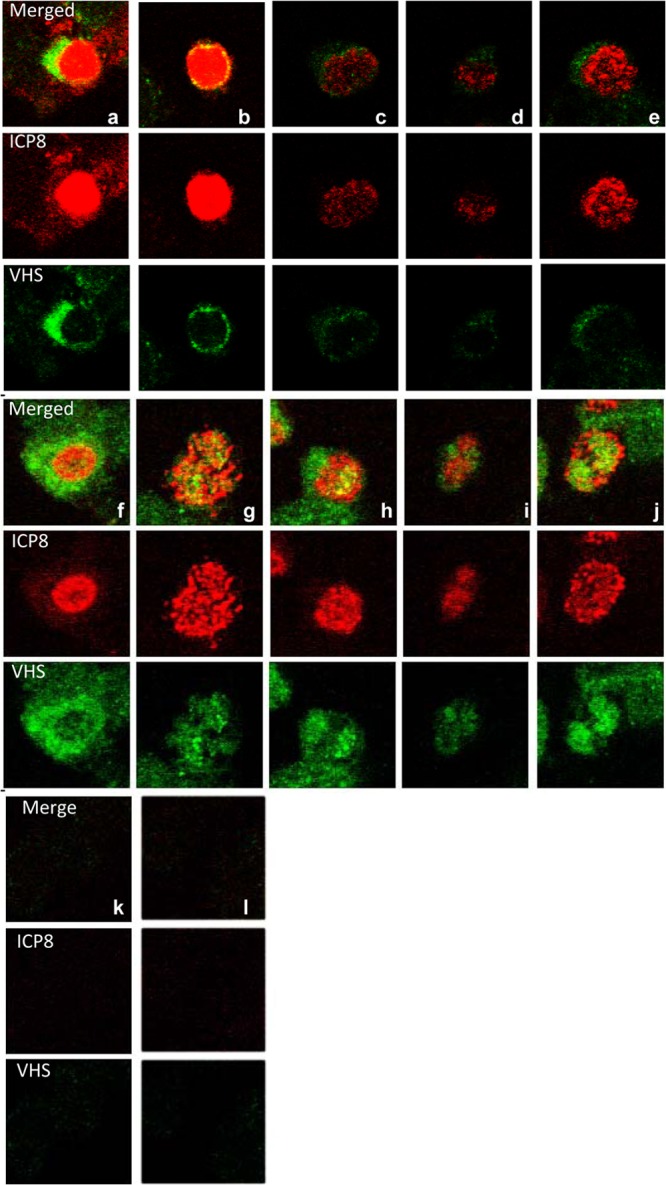
Wild-type VHS shuttles between the nucleus and cytoplasm. HEp-2 cells were mock infected or exposed (20 PFU/cell) to HSV-1(WT-BAC) virus for 1 h. The inocula were then replaced with fresh complete medium with or without leptomycin B (10 μM). The cells were fixed at 2 h after exposure to the virus and fixed and reacted with antibodies to VHS and ICP8. The images were collected with the aid of a Zeiss confocal microscope.
Role of UL47 in the localization of VHS.
In an earlier report, we showed that in wild-type virus-infected cells VHS degrades mRNA made before infection and short-lived stress response mRNAs characterized by the presence of AU-rich elements in their 3′ UTR (12). At the same time, VHS largely spares viral mRNAs as a consequence of the interaction of VHS with pUL47, another HSV tegument protein (11). The question arose as to the role of UL47 in the subcellular localization of VHS. To resolve this question, two experiments were done. The objective of the first series of experiments was to verify that the mutant VHS proteins retained their ability to interact with pUL47. Thus, HEK 293 cells were transfected with a plasmid encoding UL47 tagged with Myc at its N terminus (UL47-N-Myc). Soluble cell extracts prepared 48 h after transfection (Fig. 9A, lane 1) was reacted with GST (lanes 2), GST-VHSWT (lanes 3), GST-VHSm (lanes 4), or GST-VHSNESm (lanes 5) bound to glutathione-agarose beads. The proteins bound to the beads were subjected to electrophoresis on a denaturing polyacrylamide gel, transferred to a nitrocellulose membrane, and reacted with antibody against Myc. The results (Fig. 9A) show that the mutant VHS proteins retained the ability of wild-type VHS to bind pUL47.
Fig 9.
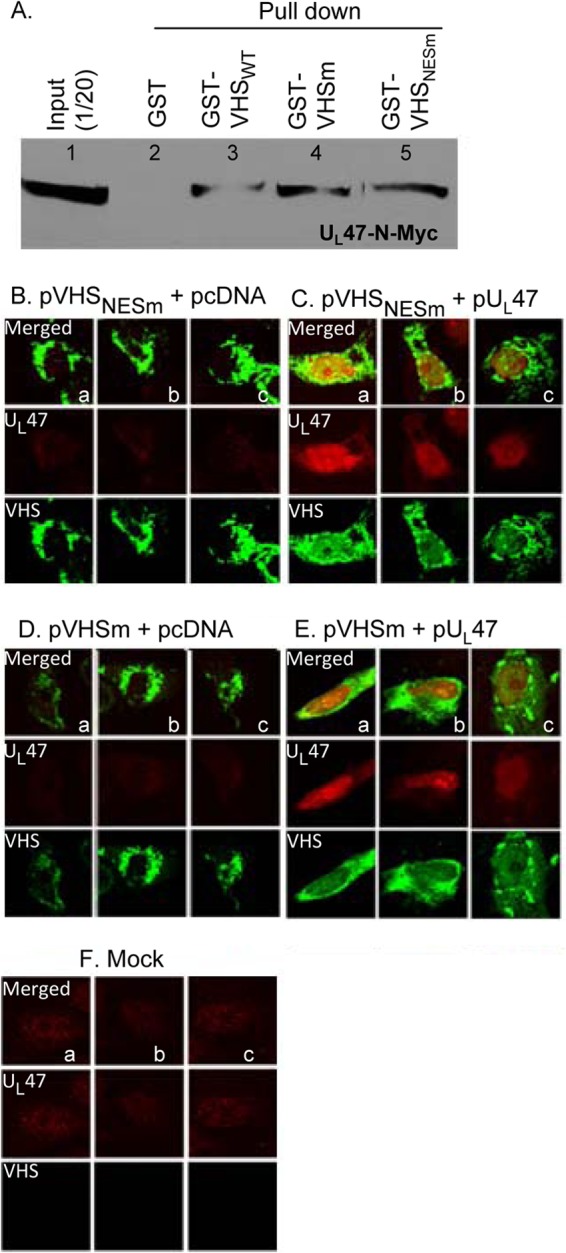
UL47 protein enables nuclear localization of VHS. (A) HEK 293 cells were transfected with a plasmid encoding UL47 tagged with Myc at the N terminus (UL47-N-Myc). Soluble cell extracts prepared 48 h after transfection were reacted with GST (lanes 2), GST-VHSWT (lanes 3), GST-VHSm (lanes 4), or GST-VHSNESm (lanes 5) bound to glutathione-agarose beads. The proteins bound to the beads were subjected to electrophoresis on a denaturing polyacrylamide gel, transferred to a nitrocellulose membrane, and reacted with antibody against Myc. (B) Vero cells were mock transfected or doubly transfected with pVHS mutants and pcDNA or pUL47. Cells were fixed and reacted with antibodies to VHS (green) or pUL47 (red) at 24 h after infection. The images were captured with the aid of a Zeiss confocal microscope.
In the second series of experiments, Vero cells were mock transfected or cotransfected with plasmids encoding pVHS mutants and pcDNA (empty vector) or pUL47. Cells were fixed at 24 h posttransfection and reacted with mouse antibody against VHS-RNase (Fig. 9, green) or rabbit antibody against pUL47 protein (red). The confocal microscopy images are shown in Fig. 9B to F. As expected on the basis of the results shown in Fig. 6, in cells transfected with the plasmids encoding mutant VHS and pcDNA, VHS localized in the cytoplasm (Fig. 6B and D). In each case, VHS-RNase localized exclusively in the cytoplasm in 100% of the cells. Cells were cotransfected with plasmids encoding VHSm and pUL47 (Fig. 6E); for 29% of the cells expressing both proteins, the VHS-RNase was present in the cytoplasm only, and for 71%, the VHS-RNase was present in both nucleus and cytoplasm. Finally, in cells transfected with the plasmids encoding VHSNESm and pUL47 (Fig. 6C) and which expressed both proteins, 19% contained the VHS-RNase only in the cytoplasm and 81% contained the VHS-RNase in both the nucleus and cytoplasm.
DISCUSSION
From the instant of the interaction of HSV with cells targeted for infection, a series of innate immunity signaling pathways activate the transcription of stress response mRNAs that, if fully expressed, would block viral replication (27–31). One of the main components of the armamentarium to defeat the early innate immune responses to infection is the VHS-RNase, a tegument protein encoded by UL41, a late (γ2) viral protein that is introduced into the infected cells along with the capsid containing viral DNA and approximately another 19 tegument proteins (4, 32, 33).
Our current understanding of the function of VHS is as follows.
(i) VHS degrades mRNAs in polyribosomes in two ways. It degrades stable mRNAs by binding to the CAP structure via eIF4H. The mRNA is deadenylated and cleaved 3′ to the CAP structure and then it is degraded processively 5′ to 3′ (6). VHS-RNase also targets short-lived stress response mRNAs induced by the innate immune responses to the virus. Characteristically, these mRNAs have AU-rich elements in the 3′ UTR (7). One of the host proteins induced by the presence of AU-rich elements is TTP (7). TTP binds the AU-rich elements, sequesters them, and ultimately enables their degradation (34–37). VHS-RNase binds to TTP and cleaves the mRNAs at the AU-rich elements. While the 3′ cleaved portion is rapidly degraded 5′ to 3′, the 5′ portion lingers in the cytoplasm for many hours (8).
(ii) VHS-RNase is tightly regulated. Late in infection, it is neutralized by two tegument proteins, VP16 and VP22 (9, 10). This enables the accumulation and packaging of VHS into virions. On entry into cells, it binds first the tegument proteins pUL47 and later ICP27, an α protein made immediately after infection (11, 38). Recent studies have shown that in wild-type virus-infected cells, VHS degrades stable mRNAs made before infection but spares the viral mRNAs made after infection. The selective sparing of viral mRNA was mapped to the physical interaction of the tegument protein pUL47 with VHS-RNase (11). Studies reported elsewhere have shown that pUL47 interacts with the poly(A) binding protein and shuttles between the nucleus and cytoplasm (14).
The studies described in this report were initiated on the basis of an apparent contradiction. Studies carried out to date have shown that VHS-RNase degrades mRNA in polyribosomes (39). Yet the VHS-RNase contained a putative NES, and fractionation of newly infected cells by separation of cytoplasm and nucleus by centrifugation suggested, erroneously, as it turned out, that a significant portion of VHS-RNase localized in the nucleus.
The studies reported here show the following.
(i) VHS-RNase released into newly infected cells is transported to the nucleus. This conclusion is based on the observation that VHS-RNase in which NES was ablated localized in the nucleus.
(ii) The studies based on short-term exposure of infected cells with wild-type virus to LMB suggest that VHS-RNase shuttles between the nucleus and cytoplasm. The NES is required for egress into the cytoplasm. The observation that in cells transfected with a plasmid encoding VHS-RNase the protein accumulates in the cytoplasm suggests that the VHS-RNase does not have a nuclear localization signal and therefore must depend on other proteins for import into the nucleus. The observation that the VHS-RNase is partially localized in the nucleus in cells transfected with plasmids encoding VHS-RNase and pUL47 suggests that this protein may be involved in translocation of VHS-RNase to the nucleus.
(iii) VHS-RNase lacking the NES degraded stable mRNA on transfection when it accumulated in the cytoplasm. In contrast to wild-type VHS-RNase, the enzyme lacking NES did not degrade the same mRNA. One significant difference between the two is that while the wild-type VHS-RNase shuttles between the nucleus and cytoplasm, the VHS-RNase devoid of NES does not shuttle. It is conceivable and consistent with available data that degradation of stable mRNAs by VHS-RNase is restricted to the cytoplasm.
(iv) In contrast to the stable mRNAs, the stress response AU-rich mRNAs were degraded almost to the same extent in wild-type and ΔNES mutant virus-infected cells. Since earlier studies have shown that AU-rich mRNAs were also degraded in polyribosomes (39), the present results, at face value, suggest that the AU-rich mRNAs can be degraded by VHS-RNase in either the nucleus or cytoplasm.
The fundamental question posed by the present study is, given that the degradation of stable mRNAs is constrained to the cytoplasm, why does VHS-RNase shuttle between the nucleus and cytoplasm? A trivial explanation is that VHS-RNase transports mRNA as cargo prior to degradation in the cytoplasm. A more likely explanation is that VHS resident in the cytoplasm degrades mRNAs indiscriminately. The conclusion is supported by the observation that in cotransfection studies, wild-type VHS-RNase degrades both its own mRNA and that of the cotransfected gene. The mRNA of VHS-RNase is readily detected in infected cells to attain selectivity. VHS-mRNase and the UL47 protein must assemble with the newly synthesized and processed mRNA in the nucleus prior to its transport to the cytoplasm.
Finally, the original reports that led to the discovery of VHS were based on studies of infected cells (1–3). Subsequent studies took on two distinct directions. One set provided valuable information on the range of substrates of VHS-RNase and its properties (7). Other studies based on analyses of infected cells revealed that in fact VHS interacts with and is regulated by numerous viral proteins that shape both the duration of its function and its substrate in the infected cells. Thus, the RNase activity is neutralized by VP22 and VP16 late in infection and directed to specific substrates by pUL47, by the host protein TTP, and possibly also by ICP27 (9–11, 38, 40–41). The incongruity between the results obtained from studies of VHS-RNase in vitro or in the context of transient expression and studies of infected cells emerged only recently. Thus, the results showing that cotransfection of VHS-RNase with a viral gene leads to degradation of the viral mRNA is obviously misleading in light of the evidence that in infected cells viral mRNA degradation by VHS-RNase is largely interdicted by pUL47. This report goes a step further in that it shows that a specific mutation enables the VHS-RNase to degrade stable mRNAs in transfected cells but not in the context of infected cells. Obviously, both sets of studies have an intrinsic value, but only one properly describes the role on VHS in the biology of the infected cell.
ACKNOWLEDGMENTS
These studies were aided by National Cancer Institute grant CA115662.
We thank Weiran Zhang for providing technical assistance.
Footnotes
Published ahead of print 9 October 2013
REFERENCES
- 1.Strom T, Frenkel N. 1987. Effects of herpes simplex virus on mRNA stability. J. Virol. 61:2198–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Read GS, Frenkel N. 1983. Herpes simplex virus mutants defective in the virion-associated shutoff of host polypeptide synthesis and exhibiting abnormal synthesis of alpha (immediate early) viral polypeptides. J. Virol. 46:498–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwong AD, Frenkel N. 1987. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. U. S. A. 84:1926–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barzilai A, Zivony-Elbom I, Sarid R, Noah E, Frenkel N. 2006. The herpes simplex virus type 1 vhs-UL41 gene secures viral replication by temporarily evading apoptotic cellular response to infection: vhs-UL41 activity might require interactions with elements of cellular mRNA degradation machinery. J. Virol. 80:505–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taddeo B, Roizman B. 2006. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J. Virol. 80:9341–9345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng P, Everly DN, Jr, Read GS. 2001. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J. Virol. 75:10272–10280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esclatine A, Taddeo B, Evans L, Roizman B. 2004. The herpes simplex virus 1 UL41 gene-dependent destabilization of cellular RNAs is selective and may be sequence-specific. Proc. Natl. Acad. Sci. U. S. A. 101:3603–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taddeo B, Zhang W, Roizman B. 2006. The U(L)41 protein of herpes simplex virus 1 degrades RNA by endonucleolytic cleavage in absence of other cellular or viral proteins. Proc. Natl. Acad. Sci. U. S. A. 103:2827–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taddeo B, Sciortino MT, Zhang W, Roizman B. 2007. Interaction of herpes simplex virus RNase with VP16 and VP22 is required for the accumulation of the protein but not for accumulation of mRNA. Proc. Natl. Acad. Sci. U. S. A. 104:12163–12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lam Q, Smibert CA, Koop KE, Lavery C, Capone JP, Weinheimer SP, Smiley JR. 1996. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J. 15:2575–2581 [PMC free article] [PubMed] [Google Scholar]
- 11.Shu M, Taddeo B, Zhang W, Roizman B. 2013. Selective degradation of mRNAs by the HSV host shutoff RNase is regulated by the UL47 tegument protein. Proc. Natl. Acad. Sci. U. S. A. 110:E1669–E1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taddeo B, Zhang W, Roizman B. 2013. The herpes simplex virus host shutoff RNase degrades cellular and viral mRNAs made before infection but not viral mRNA made after infection. J. Virol. 87:4516–4522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donnelly M, Verhagen J, Elliott G. 2007. RNA binding by the herpes simplex virus type 1 nucleocytoplasmic shuttling protein UL47 is mediated by an N-terminal arginine-rich domain that also functions as its nuclear localization signal. J. Virol. 81:2283–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobrikova E, Shveygert M, Walters R, Gromeier M. 2010. Herpes simplex virus proteins ICP27 and UL47 associate with polyadenylate-binding protein and control its subcellular distribution. J. Virol. 84:270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everly DN, Jr, Feng P, Mian IS, Read GS. 2002. mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: genetic and biochemical evidence that Vhs is a nuclease. J. Virol. 76:8560–8571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ceska TA, Sayers JR. 1998. Structure-specific DNA cleavage by 5′ nucleases. Trends Biochem. Sci. 23:331–336 [DOI] [PubMed] [Google Scholar]
- 17.Ceska TA, Sayers JR, Stier G, Suck D. 1996. A helical arch allowing single-stranded DNA to thread through T5 5′-exonuclease. Nature 382:90–93 [DOI] [PubMed] [Google Scholar]
- 18.Hosfield DJ, Mol CD, Shen B, Tainer JA. 1998. Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: coupling DNA and PCNA binding to FEN-1 activity. Cell 95:135–146 [DOI] [PubMed] [Google Scholar]
- 19.Hwang KY, Baek K, Kim HY, Cho Y. 1998. The crystal structure of flap endonuclease-1 from Methanococcus jannaschii. Nat. Struct. Biol. 5:707–713 [DOI] [PubMed] [Google Scholar]
- 20.Kim Y, Eom SH, Wang J, Lee DS, Suh SW, Steitz TA. 1995. Crystal structure of Thermus aquaticus DNA polymerase. Nature 376:612–616 [DOI] [PubMed] [Google Scholar]
- 21.Mueser TC, Nossal NG, Hyde CC. 1996. Structure of bacteriophage T4 RNase H, a 5′ to 3′ RNA-DNA and DNA-DNA exonuclease with sequence similarity to the RAD2 family of eukaryotic proteins. Cell 85:1101–1112 [DOI] [PubMed] [Google Scholar]
- 22.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2:357–364 [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Patenode C, Roizman B. 2011. US3 protein kinase of HSV-1 cycles between the cytoplasm and nucleus and interacts with programmed cell death protein 4 (PDCD4) to block apoptosis. Proc. Natl. Acad. Sci. U. S. A. 108:14632–14636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roller RJ, Roizman B. 1992. The herpes simplex virus 1 RNA binding protein US11 is a virion component and associates with ribosomal 60S subunits. J. Virol. 66:3624–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalamvoki M, Roizman B. 2010. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc. Natl. Acad. Sci. U. S. A. 107:17721–17726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugiura T, Yamaguchi A, Miyamoto K. 2008. A cancer-associated RING finger protein, RNF43, is a ubiquitin ligase that interacts with a nuclear protein, HAP95. Exp. Cell Res. 314:1519–1528 [DOI] [PubMed] [Google Scholar]
- 27.He B, Gross M, Roizman B. 1997. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 94:843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esclatine A, Taddeo B, Roizman B. 2004. The UL41 protein of herpes simplex virus mediates selective stabilization or degradation of cellular mRNAs. Proc. Natl. Acad. Sci. U. S. A. 101:18165–18170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Lint AL, Murawski MR, Goodbody RE, Severa M, Fitzgerald KA, Finberg RW, Knipe DM, Kurt-Jones EA. 2010. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J. Virol. 84:10802–10811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sciortino MT, Suzuki M, Taddeo B, Roizman B. 2001. RNAs extracted from herpes simplex virus 1 virions: apparent selectivity of viral but not cellular RNAs packaged in virions. J. Virol. 75:8105–8116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sciortino MT, Taddeo B, Poon AP, Mastino A, Roizman B. 2002. Of the three tegument proteins that package mRNA in herpes simplex virions, one (VP22) transports the mRNA to uninfected cells for expression prior to viral infection. Proc. Natl. Acad. Sci. U. S. A. 99:8318–8323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwong AD, Kruper JA, Frenkel N. 1988. Herpes simplex virus virion host shutoff function. J. Virol. 62:912–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwong AD, Frenkel N. 1989. The herpes simplex virus virion host shutoff function. J. Virol. 63:4834–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hudson BP, Martinez-Yamout MA, Dyson HJ, Wright PE. 2004. Recognition of the mRNA AU-rich element by the zinc finger domain of TIS11d. Nat. Struct. Mol. Biol. 11:257–264 [DOI] [PubMed] [Google Scholar]
- 35.Lai WS, Kennington EA, Blackshear PJ. 2003. Tristetraprolin and its family members can promote the cell-free deadenylation of AU-rich element-containing mRNAs by poly(A) ribonuclease. Mol. Cell. Biol. 23:3798–3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lai WS, Carballo E, Thorn JM, Kennington EA, Blackshear PJ. 2000. Interactions of CCCH zinc finger proteins with mRNA: binding of tristetraprolin-related zinc finger proteins to Au-rich elements and destabilization of mRNA. J. Biol. Chem. 275:17827–17837 [DOI] [PubMed] [Google Scholar]
- 37.Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. 1999. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell. Biol. 19:4311–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taddeo B, Zhang W, Roizman B. 2010. Role of herpes simplex virus ICP27 in the degradation of mRNA by virion host shutoff RNase. J. Virol. 84:10182–10190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taddeo B, Zhang W, Roizman B. 2009. The virion-packaged endoribonuclease of herpes simplex virus 1 cleaves mRNA in polyribosomes. Proc. Natl. Acad. Sci. U. S. A. 106:12139–12144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esclatine A, Taddeo B, Roizman B. 2004. Herpes simplex virus 1 induces cytoplasmic accumulation of TIA-1/TIAR and both synthesis and cytoplasmic accumulation of tristetraprolin, two cellular proteins that bind and destabilize AU-rich RNAs. J. Virol. 78:8582–8592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corcoran JA, Hsu WL, Smiley JR. 2006. Herpes simplex virus ICP27 is required for virus-induced stabilization of the ARE-containing IEX-1 mRNA encoded by the human IER3 gene. J. Virol. 80:9720–9729 [DOI] [PMC free article] [PubMed] [Google Scholar]