

Abstract
In most retroviruses, plasma membrane (PM) association of the Gag structural protein is a critical step in viral assembly, relying in part on interaction between the highly basic Gag MA domain and the negatively charged inner leaflet of the PM. Assembly is thought to begin with Gag dimerization followed by multimerization, resulting in a hexameric lattice. To directly address the role of multimerization in membrane binding, we fused the MA domains of Rous sarcoma virus (RSV) and HIV-1 to the chemically inducible dimerization domain FK506-binding protein (FKBP) or to the hexameric protein CcmK4 from cyanobacteria. The cellular localization of the resulting green fluorescent protein (GFP)-tagged chimeric proteins was examined by fluorescence imaging, and the association of the proteins with liposomes was quantified by flotation in sucrose gradients, following synthesis in a reticulocyte extract or as purified proteins. Four lipid compositions were tested, representative of liposomes commonly reported in flotation experiments. By themselves, GFP-tagged RSV and HIV-1 MA proteins were largely cytoplasmic, but both hexamerized proteins were highly concentrated at the PM. Dimerization led to partial PM localization for HIV-1 MA. These in vivo effects of multimerization were reproduced in vitro. In flotation analyses, the intact RSV and HIV-1 Gag proteins were more similar to multimerized MA than to monomeric MA. RNA is reported to compete with acidic liposomes for HIV-1 Gag binding, and thus we also examined the effects of RNase treatment or tRNA addition on flotation. tRNA competed with liposomes in the case of some but not all lipid compositions and ionic strengths. Taken together, our results further underpin the model that multimerization is critical for PM association of retroviral Gag proteins. In addition, they suggest that the modulation of membrane binding by RNA, as previously reported for HIV-1, may not hold for RSV.
INTRODUCTION
Particle assembly and budding are critical steps in the retrovirus life cycle. For most retroviruses, the structural protein Gag assembles into a budding virus particle on the inner leaflet of the cellular plasma membrane (PM). The matrix domain (MA) at the N terminus of Gag mediates the PM interaction. Retroviral MAs contain as many as three membrane binding signals that provide membrane interactions as well as specificity. First, most contain a highly basic patch that interacts electrostatically with the negatively charged inner leaflet of the PM (1). The inner leaflet of the PM derives its net negative charge from phosphatidylserine (PS) and to a lesser extent phosphatidylinositols (PIPs) (2). Second, most retroviral MAs are myristoylated, a modification that is required for membrane interaction (3). However, the Gag proteins of some retroviruses, such as Rous sarcoma virus (RSV) and equine infectious anemia virus (EIAV), are not myristoylated.
Third, at least some retroviral MAs have phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] binding pockets. Examples include human immunodeficiency virus type 1 (HIV-1) and HIV-2, Mason-Pfizer monkey virus (MPMV) (4), and EIAV (5). Other viruses, such as RSV, murine leukemia virus (MLV), and human T-lymphotropic virus (HTLV), apparently do not (6, 7). In vivo, HIV-1 and MLV are sensitive to PI(4,5)P2 depletion mediated by polyphosphoinositide 5-phosphatase IV (5PtaseIV), in that plasma membrane localization and viral particle release are decreased (8–12). HTLV and EIAV are less sensitive to PI(4,5)P2 depletion (5, 11). However, EIAV virus-like particle (VLP) release is decreased by expression of synaptojanin 2, a polyphosphoinositide phosphatase with broad specificity (5). In vivo, RSV does not respond to 5PtaseIV depletion under the same conditions that HIV-1 does (8), but under different conditions, RSV Gag also is sensitive to 5PtaseIV (13).
In contrast with some in vivo results, in liposome binding assays in vitro most or all retroviral Gag proteins bind more tightly to membranes in the presence of PI(4,5)P2 as well as other PIPs than to membranes without PIPs (8–10, 14, 15). In such assays, it is challenging to ascertain to what degree PIP-enhanced binding is due to electrostatic interactions and to what degree it is due to specific recognition of the PIP head group. In solution with short-chain versions of PI(4,5)P2, HIV-1 MA interacts not only with the phosphorylated inositol head group but also with the 2′ acyl chain (7). This observation led to a model for HIV-1 MA and Gag in vivo, in which the typically unsaturated, long 2′ acyl chain “flips out” of the hydrophobic core of the lipid bilayer and binds instead to a hydrophobic groove in MA (7, 16). Similar observations recently have been reported for short-chain versions of the abundant inner leaflet phospholipids PS and phosphatidyl choline (PC) (17). Recently we demonstrated that HIV-1 Gag binding to liposomes is strongly modulated by the membrane cholesterol content and by the degree of acyl chain unsaturation, implying that the protein can somehow sense the hydrophobic environment of the membrane (15).
Assembly of the virus particle is driven by Gag-Gag interactions, ultimately resulting in the formation of an incomplete hexameric lattice in the immature virus particle just before its release from the cell (18, 19). Gag multimerization primarily is dependent on interactions of the capsid (CA) portion of Gag, plus short immediately adjoining N-terminal and C-terminal sequences (20–23). The CA-CA contacts that hold the immature Gag lattice together have not been elucidated at a molecular level, although an 8-Å model recently has been suggested based on electron cryotomographic reconstructions of the MPMV lattice (24). It is unknown if Gag hexamers form in the cytoplasm and are an intermediate in assembly or, alternatively, if the hexameric lattice grows by addition of monomers or dimers.
Gag dimers are inferred to be a critical building block of the Gag lattice, as evidenced by diverse studies. For example, in vitro assembly requires a nucleic acid (such as a DNA oligonucleotide) that is long enough to bind to two NC domains, or about 16 nucleotides (nt) in the case of RSV (25–27). The function of NC can be replaced by a coiled-coil domain that forms dimers (leucine zipper) (28–32), and in some cases disulfide-mediated cross-linking near the Gag C terminus also can replace NC (29).
While the role of Gag multimerization in particle assembly has been studied extensively, little is known about how multimerization affects Gag membrane binding. While MA is dispensable for assembly of particles in vitro (33–36), in vivo assembly of Gag at the PM requires a membrane binding domain: either its own MA or part of it (9, 37), or the MA from another retrovirus (11, 38–40), or a membrane binding domain from a cellular protein (8, 41, 42). In an extreme example, a deletion of Gag MA that removed the entire highly basic region (HBR), including the PI(4,5)P2 binding pocket, but left the N-terminal myristoylation site was found to only modestly reduce the number of released extracellular particles (43, 44). In contrast, a Gag mutation that prevented myristoylation abrogated membrane binding and VLP release, although some particles were assembled in the cytoplasm (45). In summary, while a membrane binding domain is not required for Gag assembly, it is required for targeted assembly at the PM of cells.
Both RSV and HIV-1 Gag MA domains can interact with RNA (46–52). At least in HIV-1, MA-RNA interaction can regulate membrane binding (14). RSV MA binds RNA much more weakly than does HIV-1 MA (K. Musier-Forsyth, personal communication), and hence any role that RNA may play in Gag membrane binding in vivo may not be common to all retroviruses.
Little is known about the interplay between Gag-membrane interaction and Gag-Gag interactions. Previously, we showed that dimerization of RSV MA and HIV-1 MA increases membrane binding by 1 order of magnitude (53, 54). We have now expanded on this observation with in vivo and in vitro assays of MA proteins that were engineered to be monomeric, dimeric, or hexameric. We have also investigated the effect that RNA has on the binding of RSV and HIV-1 Gag to membranes in vitro. Overall, the results show that while MA dimerization is sufficient to increase membrane binding under some conditions, the increase is most dramatic upon MA hexamerization.
MATERIALS AND METHODS
DNA vectors.
The MA multimers used in this study are shown schematically in Fig. 1. All DNA constructs were cloned using common subcloning techniques and were propagated in Escherichia coli DH5a cells. RSV and HIV-1 MA were PCR amplified and ligated at an AatII restriction site (added with primers) to either FK506-binding protein (FKBP) (Clontech) or Ccmk4 with a DVGSGS or DVTRPEL linker, respectively. The Ccmk4 plasmid was a gift from Owen Pornillos (55). RSV and HIV-1 MA-FKBP and MA-Ccmk4 fusion proteins were PCR amplified and cloned into pET3x for reticulocyte expression using sites NdeI and KpnI (in the backbone of the vector and added to the MA chimeras by PCR). The resulting fusion peptides have the following amino acid sequences at their linker region: RSV MA-Linker-FKBP, SCY-DVGSGS-MASR; RSV MA-Linker-Ccmk4, SCY-DVTRPEL-MASA; HIV MA-Linker-FKBP, QNY-DVGSGS-MASR; and HIV MA-Linker-Ccmk4, QNY-DVTRPEL-MASA. The same chimeras were fluorescently tagged by cloning into pEGFP-N1 (Clontech) using sites EcoRI and AgeI (in the backbone of the vector and added to the insert by PCR). RSV MA and super M-MA (SM-MA) were cloned into pET3x and pEGFP in the same way. The plasmid expressing HIV-1 MA (myr-MA) was previously described (54, 56). RSV GagΔPR and HIV-1 GagΔp6 (henceforth referred to as RSV Gag and HIV-1 Gag) were previously described in reference 8. Super M Gag was a gift from John Wills (57).
Fig 1.
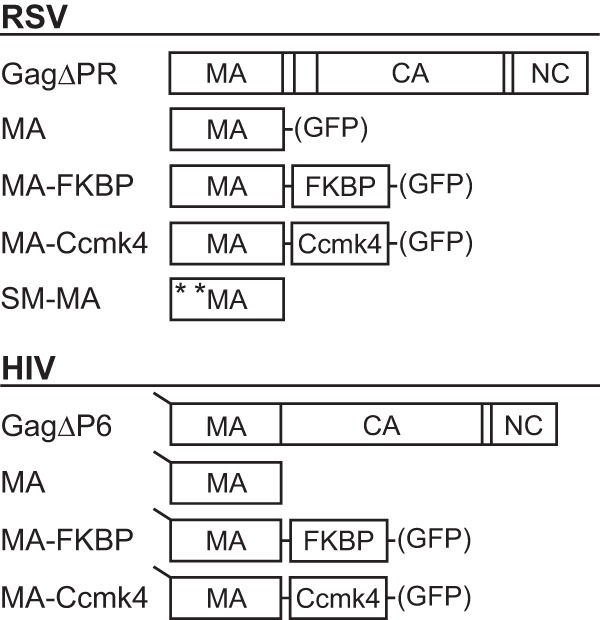
Schematic representations of proteins. RSV (top) and HIV-1 (bottom) proteins used in liposome flotation assays. GFP-tagged versions of the proteins expressed in transfected cells are denoted by GFP in parentheses. All HIV-1 proteins used were myristoylated, as denoted by the line at the N terminus. The mutations E25K and E70K of Super-M (SM) RSV MA are shown by asterisks.
Cells, transfection, and imaging.
Cell cultures and transfections were performed as previously described (8, 54). QT6 (quail) fibroblasts were maintained in Dulbecco modified Eagle medium supplemented with 5% fetal bovine serum, 5% NuSerum (BD Biosciences), 1% heat-inactivated chick serum, standard vitamins, l-glutamine, penicillin, and streptomycin. In brief, QT6 cells were seeded onto glass coverslips 24 h prior to transfection. At 50% confluence, cells were transfected with 1 μg of DNA per ml of medium with FuGENE HD (Roche) according to the manufacturer's instructions. For cells expressing MA-FKBP-GFP, medium was replaced 16 to 18 h posttransfection with medium with (dimer) or without (monomer) 100 nM homodimerization reagent AP20187 (here referred to as BB) (Clontech).
Cells were imaged as previously described (8). Briefly, at 20 to 24 h posttransfection, cells were fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS) for 15 min and mounted on glass slides with Fluoro-Gel (Electron Microscopy Sciences). Slides were viewed on an Ultraview spinning disc confocal microscope (Perkin-Elmer) with a Nikon 100× Plan-Apochromat oil objective lens. Image analysis was performed using ImageJ software (v1.40g). The strength of fluorescence at the plasma membrane and the cytoplasm was measured at least two representative locations for each cell using the plot profile function. Cells with low and high expression levels were included. Background was subtracted, and the ratio of plasma membrane to cytoplasmic signal was quantified. Outliers were identified and removed using the Q test.
Liposome binding and velocity sedimentation.
Large unilamellar vesicles (LUVs) were prepared by the rapid solvent exchange (RSE) method as previously described (15, 58, 59). Briefly, lipids in chloroform were mixed in glass tubes at the stated molar ratios. Excess chloroform was removed by evaporation under a stream of nitrogen to a final approximate volume of 50 μl. Buffer (20 mM HEPES, pH 7.0) was added, and the mixture was vortexed under vacuum for 90 s and sealed under argon gas, resulting in a final hydrated lipid mixture at 10 mg/ml. LUVs were prepared by extruding the lipid mixture at least 41 times through a 100-nm polycarbonate filter (Avanti) on a mini-extruder block heated to 45°C. LUVs were used within 10 days of preparation.
LUV binding was performed as previously described (8, 9, 15, 53, 54). The reported values for the flotations are the averages of no fewer than four replicates, and the error bars represent the standard deviations from the means. Briefly, radioactively labeled protein was prepared by translation in the TNT-coupled T7 rabbit reticulocyte reaction (Promega) in the presence of [35S]methionine-cysteine (ExPRE35S35 protein labeling mix; Perkin-Elmer). Five microliters of reticulocyte reaction mixture was combined with 15 μl binding buffer (20 mM HEPES, pH 7.0) and 50 μg LUVs (to a concentration of 8.5 mg/ml). The reaction mixture was incubated for 10 min at 22°C. The reaction mixture was than combined with 75 μl 67% sucrose (20 mM HEPES, pH 7.0). Next, 80 μl of the resulting sucrose and reaction mixture was placed in a TLA-100 tube followed by 120 μl 40% sucrose and 40 μl 4% sucrose. All sucrose solutions were made with binding buffer (wt/wt). Centrifugation was performed at 90,000 rpm in a TLA-100 rotor (Beckman) for 1 h. Purified protein flotations were performed with 10 μg of protein in place of the reticulocyte reaction mixture. The binding buffer and sucrose were supplemented with NaCl to a final concentration of 50 mM or 150 mM. For flotations with tRNA, 10 μg Escherichia coli tRNA (Roche) was added to the 25-μl binding reaction to a final approximate concentration of 15 μM.
Dimerization of reticulocyte-generated MA-FKBP was performed by incubating the reaction at 30°C for 90 min. The reaction mixture was incubated for an additional 30 min following the addition of 200 nM homodimerization reagent. Dimerization of purified MA-FKBP was achieved by adding 10 μM homodimerization reagent to the previously described reaction mixture, which was allowed to incubate for 30 min before the addition of sucrose.
Velocity sedimentation was performed with an eight-step sucrose gradient (20 mM Tris, pH 8.0, and 150 mM NaCl), 10 to 30% (wt/wt), overlaid with approximately 50 μg purified protein in storage buffer (20 mM Tris [pH 8.0], 150 to 350 mM NaCl, 30% glycerol, 10 mM dithiothreitol [DTT]). Gradients were centrifuged in an SW-60 (Beckman) rotor at 45,000 rpm for 20 h. Fourteen 270-μl fractions were taken, and 40 μl of each was subjected to SDS-PAGE on a 17.5% SDS gel. Gels were Coomassie blue stained for 16 h, destained, and imaged.
Protein purification.
Proteins were purified as previously described (15, 53, 54) with some modifications. Briefly, E. coli BL21 cultures were grown to an optical density at 600 nm (OD600) of 0.4, IPTG (isopropyl-β-d-thiogalactopyranoside) was added to a final concentration of 0.5 mM, and cells were collected 4 h later. Pelleted cells were resuspended in lysis buffer (20 mM Tris-HCl [pH 8.0], 500 mM NaCl, 10 mM DTT, protease inhibitor [Complete Mini tablet; Roche]), lysed by sonication, and cleared by centrifugation in a TLA-110 (Beckman) rotor at 90,000 rpm for 45 min. Polyethylenimine (PEI) was added to a final concentration of 0.3% to precipitate nucleic acid, which was spun down and removed. Ammonium sulfate was added to 30% saturation to precipitate the protein, followed by centrifugation. The pellet was resuspended in binding buffer (20 mM Tris [pH 8.0], 100 mM NaCl, 10 mM DTT). The protein was further purified by cation exchange chromatography (HiTrap SP; Amersham Pharmacia). All purified proteins were concentrated to 2 to 5 mg/ml (Ultracel −10K; Millipore), aliquoted, and stored at −80°C in storage buffer (10 to 20 mM Tris [pH 8.0], 150 to 330 mM NaCl, 10 mM DTT, 30% glycerol). HIV-1 MA (myr-MA) was purified as described previously (15, 54, 56). BL21 cultures were supplemented with myristic acid (10 mg/liter; Sigma) 1 h before induction with IPTG. The MA protein was purified from the supernatant by nickel-nitrilotriacetic acid (Ni-NTA) resin (Qiagen) followed by cation exchange chromatography (HiTrap SP; Amersham Pharmacia). Mass spectrometry confirmed that the protein was myristoylated.
RESULTS
We examined the effects of MA dimerization and hexamerization on membrane interaction in vivo and vitro. Chimeric MA proteins that can be induced to dimerize were created by fusion of MA with the FK506-binding protein (FKBP) (60–62) (Fig. 1). Similarly, hexameric MAs were created by fusion with the cyanobacterial carboxysome shell protein Ccmk4 (55, 63). The chimeras were designed so that the MA moieties were oriented with their membrane-binding domains all facing in the same direction, similar to their arrangement in the Gag lattice. For in vivo visualization, the chimeras were further fused to GFP (Fig. 1). MA dimers and hexamers were compared with MA monomers and with full-length Gag in a standard liposome (also referred to as large unilamellar vesicle [LUV]) flotation assay, using radiolabeled proteins generated in a reticulocyte extract, or in some cases using purified proteins. We selected four representative types of liposomes, corresponding to what we and others have used previously to study Gag-membrane interactions (8, 9, 15, 53).
Effects of MA dimerization and hexamerization on subcellular localization.
Reports of HIV-1 MA cellular localization vary from cytoplasmic (54, 64–66) to partially membrane associated (67, 68), while fluorescence of RSV MA-GFP is cytoplasmic and nuclear (69) owing to a nuclear localization signal (NLS) in the latter (69, 70). To determine the effect of dimerization on cellular localization of MA, we treated transfected cells expressing chimeric MA proteins with the dimerization chemical AP20187 (here referred to as BB). The amount of BB required to induce a maximal change in MA-FKBP localization, 100 nM, was determined empirically in a series of concentration tests (data not shown).
The fluorescence from RSV MA-GFP and MA-FKBP-GFP monomers (−BB) were cytoplasmic and nuclear, as expected (Fig. 2A) (69). Inducing dimerization (+BB) did not significantly affect this result. In contrast, the RSV MA-Ccmk4-GFP hexamer was strongly concentrated at the PM, with little fluorescence elsewhere in the cell. To provide a graphical representation of the images, we used the ImageJ plot profile function to count pixel intensity, which is correlated with the fluorescence signal, at the plasma membrane and in the cytoplasm for each chimera tested. Over 25 measurements were made for each chimera, with at least two measurements per cell. Ratios of the average fluorescence intensity at the PM and cytoplasm showed that for the RSV MA chimeras, hexamerization increased PM localization approximately 5-fold over that in the monomers and dimers (Fig. 2B).
Fig 2.
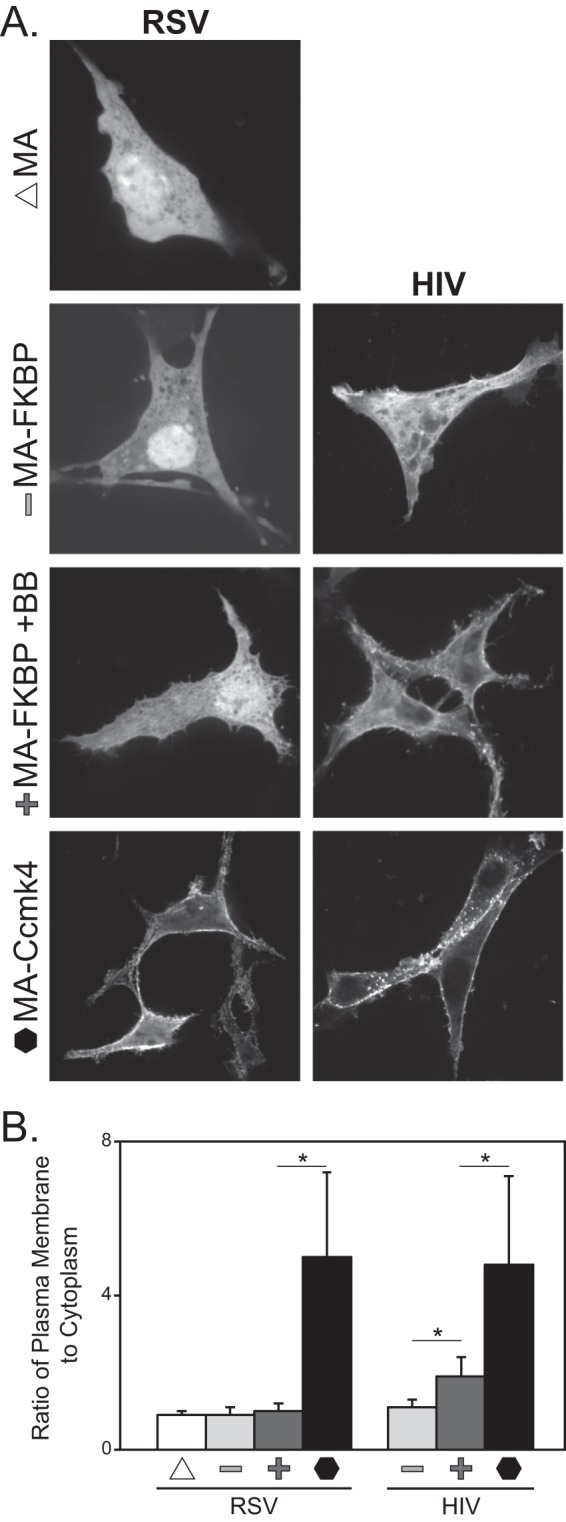
Effect of multimerization on RSV and HIV-1 MA-GFP localization. (A) QT6 cells were transfected with DNAs for either RSV (left) or HIV-1 (right) GFP-tagged MA multimers. MA and MA chimeras are indicated on the far left as MA (monomer; triangle), MA-FKBP (monomer; minus sign), MA-FKBP +BB (dimerized; plus sign), and MA-Ccmk4 (hexamer; hexagon). Images are of a single confocal section near the midbody of cells. Images are representative for each protein (n > 25). (B) Ratio of plasma membrane to cytoplasmic fluorescent signal. For each cell, at least two measurements of signal intensity, at representative locations, were taken as described in Materials and Methods, for a total of at least 50 measurements per protein. P values were determined using Student's t test for comparisons denoted by a horizontal line above two bars. *, P < 0.001.
For HIV-1, monomeric MA-FKBP-GFP was cytoplasmic while the dimerized MA-FKBP-GFP (+BB) was enriched somewhat at the PM. Similar to RSV MA-Ccmk4-GFP, the HIV-1 MA-Ccmk4-GFP hexamer was very strongly concentrated at the PM (Fig. 2A and B). These results for both viruses suggest that for the full-length Gag protein, stable binding to the PM most likely requires multimerization. For HIV-1, dimerization may be sufficient to drive Gag to the PM, but for RSV, dimerization evidently is not sufficient.
To investigate the possible cell type specificity of the effects of hexamerization, localization of both RSV and HIV-1 MA-Ccmk4-GFP was tested in 293T cells, yielding the same results as observed in QT6 cells (data not shown). As a further control to rule out the possibility that Ccmk4 itself might interact with membranes, we visualized the localization of Ccmk4-GFP. The fluorescence was diffuse and cytoplasmic (data not shown).
Effects of MA dimerization and hexamerization on liposome interaction.
We previously reported that dimerization of purified RSV and HIV-1 MA results in an increase in LUV association across a range of lipid concentrations (53, 54). In those studies, dimerization was achieved by fusing the MA domain to either a monomeric (mutant W184A/M185A [71]) or a dimeric (Q192A, [72]) version of the HIV-1 CA C-terminal domain (CACTD). In transfected cells, the GFP-tagged HIV-1 MACACTD(Q192A) was somewhat concentrated at the PM in 293T cells (54), while the similarly constructed RSV dimer was largely cytoplasmic in avian DF-1 cells (our unpublished results), fully consistent with the present results based on FKBP chimeras. We retested both of the CACTD chimeras in 293T cells and again observed similar localization. However, when these monomeric and dimeric proteins were translated in vitro in a rabbit reticulocyte system and subjected to liposome flotation, the proteins did not behave as reported previously for the purified proteins. Little difference in flotation between monomeric and dimeric MAs was observed, both for RSV MACACTD and HIV-1 MACACTD (data not shown). By comparison with MA and MA-FKBP proteins, we infer that the reticulocyte-generated MACACTD(W184A/M185A) monomers behave like dimers. Apparently, some component in the reticulocyte system affects dimerization or membrane binding for these chimeric proteins.
To address this discrepancy, we employed an independent approach to study the effect of MA dimerization, based on the chemically inducible dimerization domain FKBP (60, 73). When RSV MA by itself and monomeric RSV MA-FKBP were compared, similar flotation results were obtained for all LUV types tested, implying that in this assay MA and MA-FKBP in the absence of BB are functionally equivalent. Similar to the approach used in vivo, a series of flotations was carried out to find the optimal homodimerization reagent concentration to elicit dimerization of MA-FKBP, as measured by augmented liposome flotation (data not shown). Dimerization induced by 200 nM BB led to a 2- to 3-fold increase in flotation of RSV MA-FKBP (Fig. 3A and B) and up to a 2-fold increase for HIV-1 MA-FKBP (Fig. 3A and C). For the hexamerized MA-CcmK4 chimeras, association with liposomes was 3- to 5-fold higher than for the monomeric protein for RSV and about 2-fold higher for HIV-1. In summary, though differing in detail, LUV binding of these chimeric monomeric, dimeric, and hexameric MA species to four types of LUVs followed the same trends as observed in cells.
Fig 3.
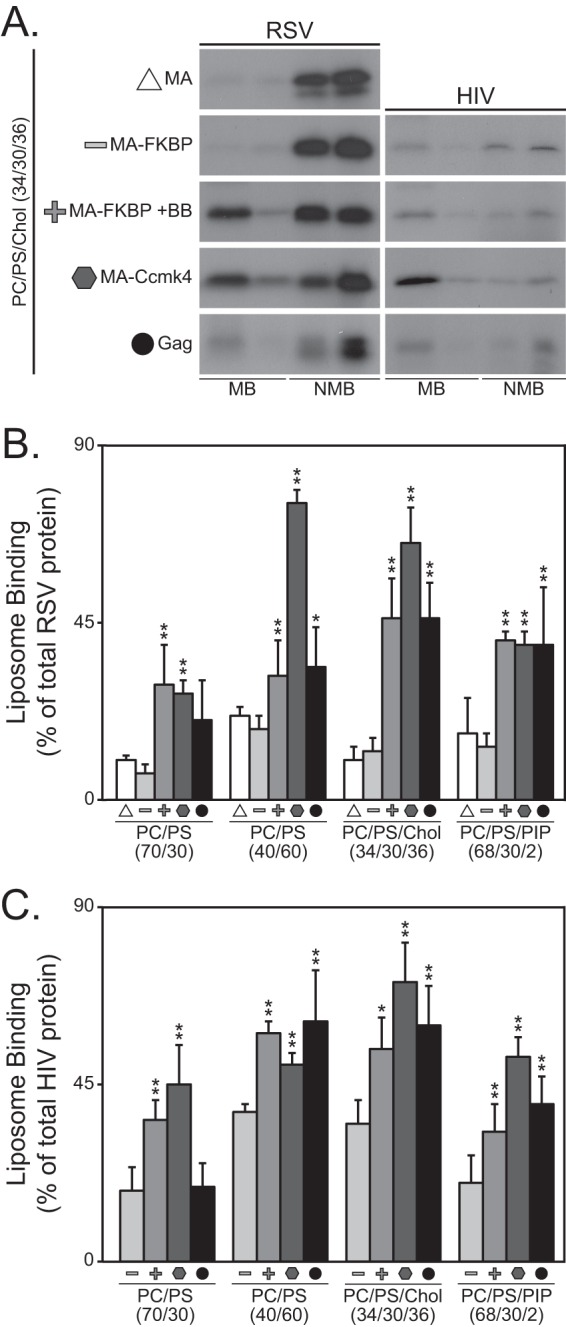
Flotation analysis of RSV and HIV-1 MA multimers. Liposomes were made with the following compositions: PC/PS (70/30), PC/PS (40/60), PC/PS/cholesterol (Chol) (34/30/36), and PC/PS/PI(4,5)P2 (68/30/2). All proteins were radiolabeled in reticulocyte translation reactions. (A) Representative flotation results of MA multimers to PC/PS/Chol liposomes. Symbols denoting multimeric state are the same as for Fig. 2. The radioactive protein was scored either as membrane bound (MB) or as non-membrane bound (NMB). (B and C) Percentages of total protein found associated with liposomes for RSV (B) and HIV-1 (C) MA multimers. Error bars represent standard deviations from the means. P values for Student's t test for comparison of MA-FKBP monomer to MA-FKBP dimer, MA-Ccmk4 hexamer, and Gag (*, P < 0.05; **, P < 0.005).
We also compared flotation of full-length RSV and HIV-1 Gag proteins with flotation of the chimeric MA species. Previous work by us and by others had shown that Gag binding to liposome membranes is augmented by high concentrations of PS, by cholesterol, and by low concentrations of PIPs (8, 9, 15, 74). These findings were qualitatively recapitulated in the present study (Fig. 3). In comparisons of Gag with the different MA multimers, Gag behaved like the MA multimers, but with some variation. For example, flotation of HIV-1 Gag with LUVs made with low PS was consistently not higher than flotation of monomeric MA (Fig. 3C). Also, flotation of the RSV MA-FKBP monomer was not boosted by cholesterol or PIP2, and similarly flotation of the HIV-1 MA-FKBP monomer was not boosted by PIP2. To explain these results, we speculate that the observed high binding of Gag to LUVs with cholesterol or PIPs may reflect Gag multimerization. Expressed in another way, membranes that support strong binding of Gag may promote Gag multimerization.
Membrane association of purified MA multimers.
To further investigate the effect of MA multimerization on membrane association, we purified RSV MA, RSV MA-FKBP, RSV MA-Ccmk4, and HIV-1 myr-MA after expression in E. coli. First, to confirm that these proteins were in their predicted multimeric state, sucrose gradient velocity sedimentation was performed. The putative MA-FKBP dimer sedimented about 1.3-fold farther than the monomer, and the MA-CcmK4 hexamer sedimented about 2.6 times farther than expected of an MA-CcmK4 monomer (Fig. 4A). Although the low resolution of this technique does not allow detailed estimates of size, we interpret the sedimentation behavior to be consistent with a majority of the protein moving as dimers or hexamers, respectively. Second, the proteins were submitted to flotation analyses like those for radioactive proteins translated in vitro. The LUV binding was in agreement with that found for the reticulocyte-translated proteins. For example, the RSV MA-Ccmk4 hexamer associated with all liposome types significantly more than did the MA-FKBP monomer (Fig. 4B). Similarly, LUV association of the RSV MA monomer, dimer, and hexamer was strengthened by high PS, by cholesterol, and by PIP2, as found for the in vitro-translated proteins. We conclude that the effects of multimerization and of membrane composition on membrane binding are not substantially altered by the vast excess of rabbit protein present in the in vitro translation mixture.
Fig 4.
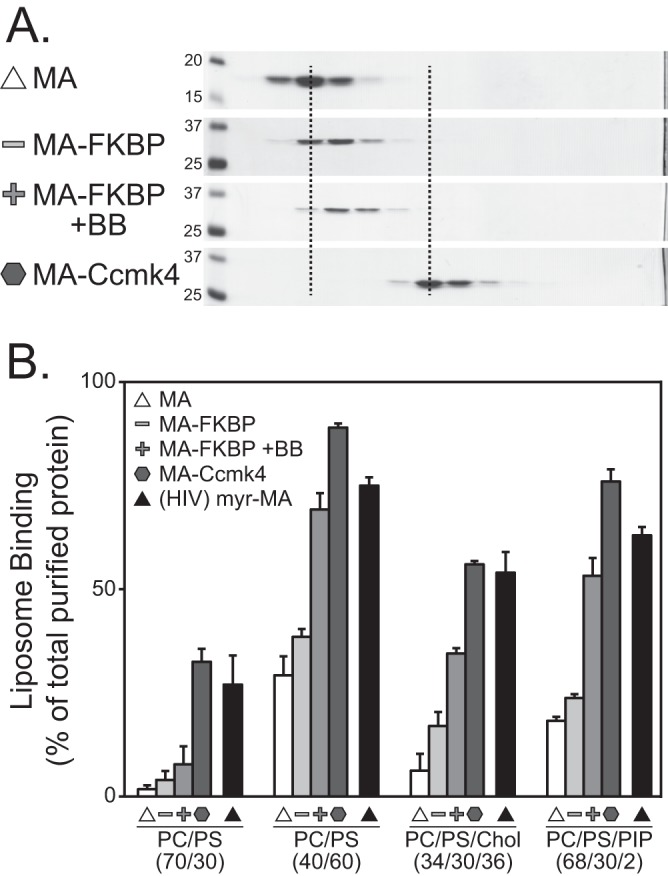
Flotation analysis of purified RSV MA multimers. (A) Velocity sedimentation of RSV MA, MA-FKBP, MA-FKBP +BB, and MA-Ccmk4 proteins. Molecular masses (in kDa) are indicated for the MA and MA-FKBP gels; all other gels were run identically. Dotted lines are added to more easily show the shift of MA monomers to dimer and hexamer. (B) Percentages of total protein found associated with four liposome types. Symbols indicate protein types as for Fig. 2. All MA proteins are RSV except HIV-1 myr-MA, which is indicated by the black triangle. Error bars represent standard deviations from the means.
Previously, we reported that the relative binding constants of monomeric RSV MACACTD and monomeric HIV-1 MACACTD were of similar magnitude, and the same was observed for the dimeric versions of these proteins (53, 54). In contrast, in the present study RSV MA and MA-FKBP on the one hand and HIV-1 myr-MA on the other showed significant differences in the flotation assays. Perhaps this discrepancy arises from the different salt concentrations used in the analyses, 75 mM NaCl in the earlier work (53, 54) and 150 mM NaCl here.
Effect of RNA on Gag and MA membrane interaction.
RNase treatment of a reticulocyte extract in which HIV-1 Gag has been synthesized increases Gag binding to liposomes, suggesting that RNA competes for Gag-membrane interactions (14). To determine if this is a general principle of retroviral Gag- and MA-membrane interaction, we tested the effect of RNase on RSV MA monomer and RSV Gag compared with HIV-1 MA-FKBP monomer and HIV-1 Gag. RNase treatment for RSV MA, MA-FKBP monomer, and Gag had little effect on membrane binding of these proteins. Enhanced binding was observed only for RSV MA monomers to LUVs with high levels of PS (Fig. 5A). In contrast, the LUV binding of HIV-1 MA-FKBP monomer and of HIV-1 Gag was augmented by RNase treatment (Fig. 5B), as reported previously (11, 14). At low PS, the flotation levels for the HIV-1 proteins increased about 3-fold. At high PS and with the addition of cholesterol, flotation of HIV-1 MA-FKBP monomer increased about 2-fold, but that of HIV-1 Gag increased only modestly. The small effect of RNase on HIV-1 Gag for these lipid mixtures might be explained by saturation of binding. In our hands, RNase also led to about a 2-fold stimulation of flotation of HIV-1 MA-FKBP monomer and of HIV-1 Gag to PIP2-containing liposomes, in agreement with the results of Chukkapalli et al. (14). In summary, these results suggest that at equivalent protein, salt, and lipid concentrations, RNA does not inhibit the LUV binding of RSV MA and Gag, while it does inhibit LUV binding of HIV-1 MA and Gag.
Fig 5.
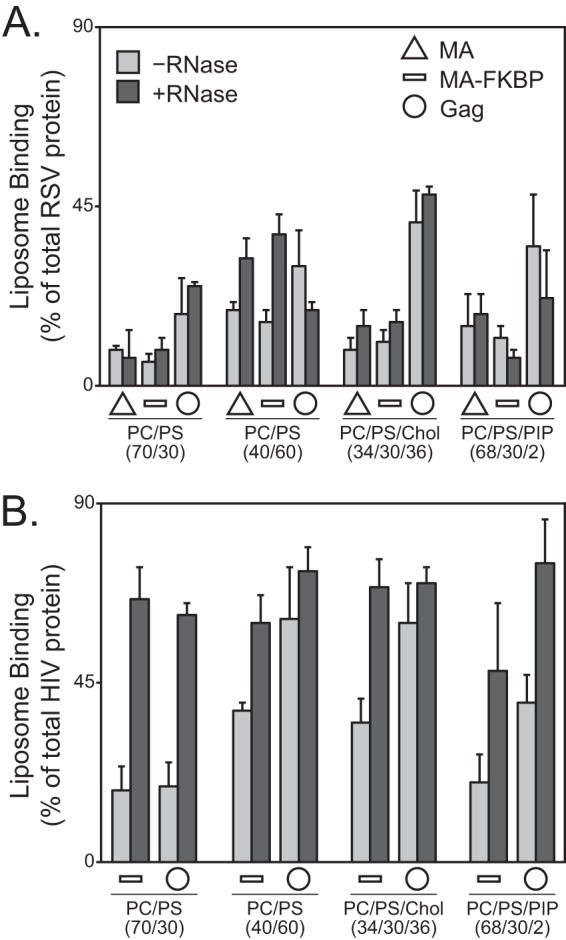
Effect of RNA on liposome binding of reticulocyte-generated RSV MA or RSV Gag. Liposome binding of RSV (A) and HIV-1 (B) MA (triangle; not done for HIV-1), MA-FKBP monomer (dash), and Gag (circle) protein to liposomes in the absence of (light gray) or presence of (dark gray) RNase. Error bars represent standard deviations from the means.
RSV MA has a net surface charge of +3, while HIV-1 MA has net surface charge of +6 (1). We speculated that this difference might underlie the observed differences in RNase sensitivity. To explore this idea, we tested the binding of a mutant RSV MA called Super-M (SM) to liposomes, with and without RNase treatment. SM-MA carries the two mutations E25K and E70K (57, 75), resulting in a net change in charge of +4, leading to a total surface charge of +7. SM-Gag is known to bind more rapidly to the PM and apparently as a consequence to bypass trafficking into the nucleus (57, 75). SM-MA floated with LUVs approximately 2-fold more extensively than did wild-type (WT) MA when the liposomes had low levels of PS or included cholesterol or included PIP2 (Fig. 6). Similar to what was observed for HIV-1 MA-FKBP, RNase increased the binding of SM-MA to three of the four LUV types tested. From these results, we conclude that in this experimental setting RNA plays less of a role in membrane interaction for WT RSV than for HIV-1 and that this difference is accounted for at least in part by surface charge density.
Fig 6.
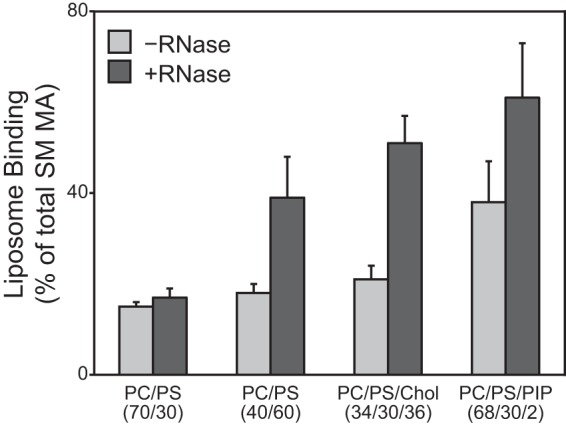
Effect of RNase on Super-M MA. Liposome binding of RSV SM MA to liposomes without (light gray) or with (dark gray) RNase. Error bars represent standard deviations from the means.
RNA-membrane competition with purified RSV MA and HIV-1 MA.
Considering the basic nature of both RSV and HIV-1 MA, it seemed surprising that RNase treatment of the reticulocyte extract led to different results for the WT RSV and HIV-1 proteins. Therefore, we decided to test for RNA competition in another way, using flotation analysis of purified RSV MA and HIV-1 myr-MA proteins in the presence or absence of purified tRNA. For a 25-μl binding reaction volume, tRNA was added to a final concentration of 15 μM. At 150 mM NaCl, little or no tRNA effect on flotation of either protein was observed (data not shown). But at 50 mM NaCl, consistent with flotations of RNase-treated reticulocyte translation reaction mixtures, addition of tRNA decreased the binding of HIV myr-MA to all LUVs tested (Fig. 7B). This decrease was greatest for membranes with low levels of PS. For RSV MA at this salt concentration, addition of tRNA also led to greatly reduced flotation for LUVs with low levels of PS. But unlike for HIV-1 myr-MA, for RSV MA the addition of tRNA did not significantly reduce binding to LUVs with PIP2. The effect of tRNA on protein binding to LUVs prepared with high levels of PS or cholesterol was modest for both HIV-1 and RSV MA. These results show that RSV MA binding to membranes with high PS, with cholesterol, or with 2% PIP2 is hardly perturbed by tRNA.
Fig 7.
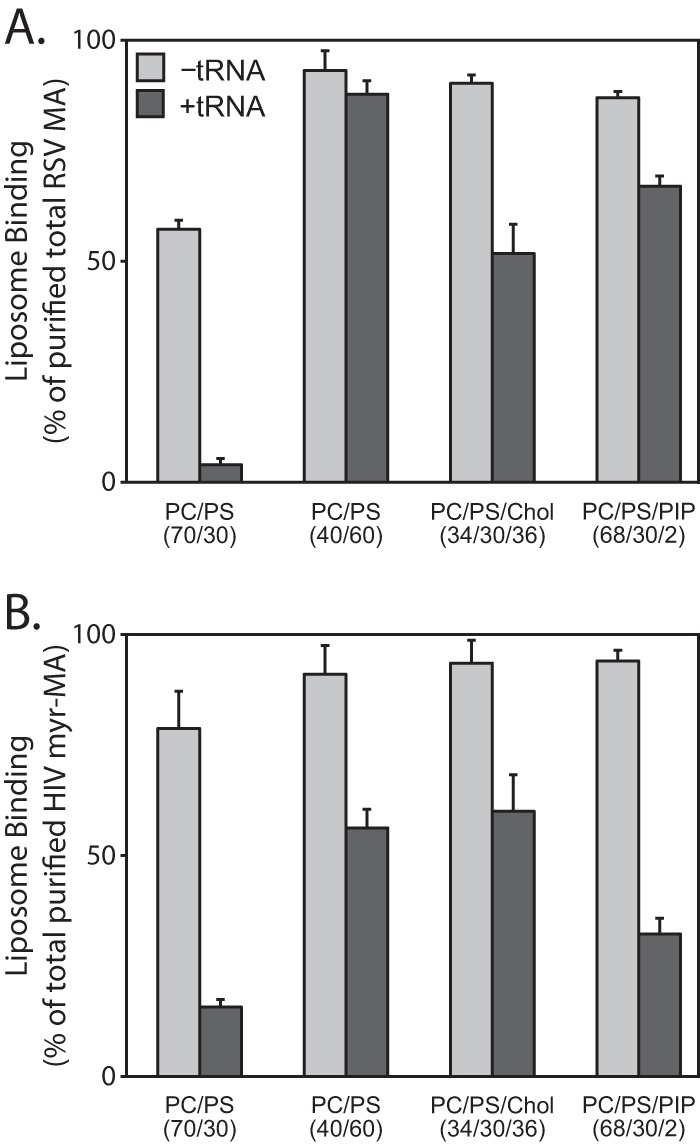
RNA inhibition of purified RSV and HIV-1 MA proteins. Liposome binding of purified RSV MA (A) and HIV-1 myr-MA (B) proteins to liposomes in the absence (light gray) or presence (dark gray) of tRNA. Error bars represent standard deviations from the means.
In published assays measuring HIV MA or Gag membrane binding, the addition of PI(4,5)P2 was found to partially (14) or fully (74) relieve nucleic acid competition. In addition, Jones et al. showed that inositol phosphates can relieve MA inhibition of NC-mediated tRNA annealing (76). In our reticulocyte reaction flotation assay, PI(4,5)P2 only modestly increased membrane binding for HIV Gag but not for HIV MA (Fig. 5B). In addition, in the presence of tRNA, liposomes containing PI(4,5)P2 only modestly enhanced membrane binding of purified HIV myr-MA (Fig. 7B). We predict that the differences between our results and these published results may reside in differences in experimental methods. For example, we used liposomes containing 2% PI(4,5)P2, while Chukkapalli et al. (14) and Alfadhli et al. (74) used liposomes containing 7.5% PI(4,5)P2 and 10% PI(4,5)P2, respectively. Additional differences are in the methods for preparation of liposomes and in lipid acyl chain saturation, which we have previously reported to be a factor in protein recognition of membranes (15).
DISCUSSION
We have studied the effect of multimerization on the binding of RSV and HIV-1 MA to membranes in vivo and vitro. For HIV-1, dimerization of MA-GFP increased fluorescence at the PM, as reported previously (54), while hexamerization led to highly concentrated PM fluorescence. For RSV, dimerization of MA-GFP did not lead to significant PM localization, but hexamerization resulted in highly concentrated PM fluorescence, as for HIV-1. Liposome flotation analyses of MA multimers translated in vitro in a reticulocyte extract largely mirrored what was observed in transfected cells. For both viruses, binding of full-length Gag protein to liposomes resembled binding by dimeric or hexameric MA. In their interactions with liposomes, RSV and HIV-1 MA proteins in the reticulocyte translation mixtures differed in their response to RNase treatment. As had been shown previously for HIV-1 Gag (14), RNase increased the flotation of HIV-1 Gag and MA-FKBP but had little effect on RSV Gag, MA, and MA-FKBP.
Our interpretations of the imaging and liposome flotation results have several theoretical limitations, although these are unlikely to affect the conclusions in a substantive way. First, we have not directly measured the extent of dimerization mediated by the binding of BB to the FKBP domain in cells or in the reticulocyte extracts. The optimal concentration of BB was determined empirically and was similar to values reported in the literature. But even at optimal concentrations, on a statistical basis, some of the BB reagent will bind to only one FKBP domain and some domains will have no reagent bound. Second, in the case of HIV-1 MA and MA-FKBP, we have not independently verified the assumption that in the absence of BB these proteins are monomeric. HIV-1 MA can trimerize, both in solution (56, 77) and in crystals (78). However, according to the published data for solution, one-half of the MA is trimerized at 70 μM, a concentration that we estimate is considerably higher than that in the reticulocyte lysates. Third, the sedimentation velocity experiments are not precise enough to rule out that a small percentage of the MA-CcmK4 polypeptides are in lesser or greater states of multimerization.
In previous work on binding of full-length Gag to liposomes (8, 9, 15), it was assumed, explicitly or tacitly, that Gag remained monomeric, since the Gag concentration in the reticulocyte extract was presumed to be below the dissociation constant (Kd) for dimerization of CA (∼10 μM [71]) and also of Gag (∼5 μM [79]). Two scenarios could explain our present results showing that by liposome flotation both HIV-1 and RSV Gag behave more like MA multimers than MA monomers. The first is based on the presence of two highly basic domains in Gag, MA and NC, both of which may interact with membranes. Indeed it is known for HIV-1 Gag (in its nonmyristoylated form) that in solution the MA and NC domains are near each other, making the overall shape of Gag like that of a horseshoe (80, 81). Moreover, from low-angle neutron scattering analyses of HIV-1 Gag bound to a tethered membrane, it appears that the horseshoe shape is maintained, implying that both domains contact the membrane simultaneously under the relatively low ionic-strength conditions used (81). One domain, presumably NC, can be displaced by addition of nucleic acid or by increase of the ionic strength, leading to an extended Gag conformation. On the other hand, the possible function of HIV-1 NC in membrane binding is called into question by the observation that WT Gag and Gag missing the NC domain bind similarly to PC/PS liposomes (14).
The second possible explanation to account for the similarity of liposome binding by Gag and multimeric MA is that Gag multimerization is promoted by the RNA in the reticulocyte extract, and/or by membranes. For example, in preliminary experiments we confirmed earlier reports that after ultracentrifugation much of HIV-1 Gag was in the pellet, even in the absence of added liposomes (82). In contrast, under the same conditions, most of the RSV Gag remained in the supernatant. To address the questions implied by these observations, it will be important to carefully compare the properties of purified Gag with the properties of in vitro-translated Gag in crude extracts.
Comparisons and quantitative interpretations of the various published liposome binding experiments with Gag proteins, including the flotation experiments described here, are hampered by the diversity of experimental conditions used in different labs and even the same lab. For example, the ionic strength in the liposome-Gag mix before centrifugation as well as in the sucrose gradients has not been constant for the several published papers. Nor have the methods for liposome preparation and for centrifugation been identical. In our own work presented here, to facilitate processing of the large number of flotation experiments analyzed, we used very small (0.25-ml) sucrose gradients, similar to those described by Dalton et al. (53, 54). In an earlier publication, we used 4-ml gradients and longer centrifugation times, which may explain why some of the results are quantitatively, though not qualitatively, different. It would be very useful to develop a liposome binding assay that is more rapid and quantitative than flotation and that has a greater dynamic range.
Both in vivo (8–11, 13, 14) and in vitro (6–11, 15, 83), PIPs stimulate PM interaction of retroviral Gag or MA proteins, but these several observations are difficult to interpret in a unified manner. For example, on the one hand, the specificity of the HIV-1 MA binding pocket for short-chain PI(4,5)P2 is well established (7), and depletion of PI(4,5)P2, the major PIP species at the PM, compromises HIV-1 Gag PM association (8, 9, 11). But on the other hand, HIV-1 Gag shows only very modest specificity for PI(4,5)P2 by flotation analysis (8, 9). It remains to be established to what degree PIP stimulation of membrane binding in vitro is due only to electrostatics.
In the experiments described here, we observed that the addition of 2% PI(4,5)P2 to liposomes enhanced binding of HIV-1 Gag more than it did the binding of the monomeric or the multimeric chimeric species of MA. To explain this effect, we speculate that PI(4,5)P2 promotes multimerization of HIV-1 Gag. This hypothesis is based in part on the effects of the PIP head group analogs on in vitro assembly of HIV-1 Gag. A nonmyristoylated, MA-deleted version of Gag (missing residues 16 to 99) assembles into normal immature particles in the presence of nucleic acid (33). However, full-length nonmyristoylated HIV-1 Gag assembles into tiny aberrant particles. This defect can be corrected by addition of inositol hexakisphosphate (IP6) or IP5 (33). Tight interaction of IP6 with Gag requires both the MA and NC domains (79), and this interaction appears to promote a Gag monomer-trimer equilibrium. The authors of these studies interpreted the action of IP6 or IP5 in this in vitro system to mimic PIPs, perhaps PI(4,5)P2 or PI(3,4,5)P2. The hypothesis that PIP enhancement of liposome binding at least in part reflects a stimulation of Gag multimerization remains to be tested.
HIV-1 Gag binding to membranes in vitro is modulated by nucleic acid. Depletion of RNA from reticulocyte reactions by RNase treatment or by mutation of the two lysine residues in MA that are inferred to be RNA binding residues (14) results in enhanced flotation (11, 14, 83). We have extended the study of this RNase effect to HIV-1 and RSV MA proteins. It seems surprising that in contrast to HIV-1 Gag and MA, RSV Gag and MA hardly respond to RNase treatment of the reticulocyte lysate. The simplest explanation of these differences between the two viruses is based on the observation that the interaction of RSV MA with RNA is much weaker than that of HIV MA (K. Musier-Forsythe, personal communication). These differences may be grounded in part in the different surface potential of the MA domain, with HIV-1 MA having a net positive surface charge of +6 compared with +3 for RSV MA (1). The importance of surface charge in this context is suggested by the observation that the super M mutant form of RSV MA, with a surface charge of +7, was similar to HIV-1 MA in the effect of RNase treatment on liposome flotation. Alternatively or in addition, HIV-1 MA has been inferred to have a specific RNA binding surface that partially overlaps with the binding surface required for PIP2 interaction (84), which might be absent in RSV, even though RSV MA also can bind to RNA (52). It remains challenging to understand how RSV Gag is targeted to the PM despite both a lesser surface charge and a lack of myristoylation. We hypothesize that while both RSV and HIV-1 rely on multimerization for stable PM interaction, RSV requires higher order multimerization than does HIV-1.
For all retroviruses, surprisingly little is known about the multimeric state of Gag between the time it is synthesized as a monomer on cytoplasmic polysomes and the time it becomes associated with a budding virus particle at the PM. Prior to assembly, HIV-1 Gag becomes complexed with cellular proteins, for example, ABCE1 (also called HP68) (85, 86) or AP3 (87), and RSV Gag may enter similar complexes (88), but the stoichiometry of Gag molecules in these structures has not been established. In order to understand the mechanism of assembly, it will be important to determine the multimeric status of retroviral Gag proteins before they reach the plasma membrane. Two studies have addressed this issue. Based on in vivo cross-linking, Kutluay and Bieniasz (89) concluded that the most abundant HIV-1 species in the cytoplasm is monomeric. Fogarty et al. (90) came to a similar conclusion using multiphoton microscopy coupled with quantitative fluorescence fluctuation analysis of Gag-GFP (91, 92). With this technology, it is possible to estimate the actual molar concentration of Gag-GFP in the cytoplasm. In that experimental setting, PM binding of HIV-1 Gag-GFP and limited multimeric forms of Gag were observed only after the cytoplasmic concentration reached a critical level, approaching micromolar (L. Mansky, personal communication). What these techniques are not able to answer unambiguously is whether Gag multimers that form in the cytoplasm then quickly become PM bound or, alternatively, Gag molecules are recruited as monomers to assembly sites on the PM. More-sophisticated dynamic analyses will be needed to address these questions.
ACKNOWLEDGMENTS
This work was supported by USPHS grant CA20081-34 and its continuation as GM107013-35.
We thank the reviewers of an early version of the manuscript for their helpful comments.
Footnotes
Published ahead of print 9 October 2013
REFERENCES
- 1.Murray P, Li Z, Wang J, Tang C, Honig B, Murray D. 2005. Retroviral matrix domains share electrostatic homology: models for membrane binding function throughout the viral life cycle. Structure 13:1521–1531 [DOI] [PubMed] [Google Scholar]
- 2.van Meer G, Voelker DR, Feigenson GW. 2008. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9:112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Resh MD. 1999. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta 1451:1–16 [DOI] [PubMed] [Google Scholar]
- 4.Prchal J, Srb P, Hunter E, Ruml T, Hrabal R. 2012. The structure of myristoylated Mason-Pfizer monkey virus matrix protein and the role of phosphatidylinositol-(4,5)-bisphosphate in its membrane binding. J. Mol. Biol. 423:427–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandes F, Chen K, Ehrlich LS, Jin J, Chen MH, Medina GN, Symons M, Montelaro R, Donaldson J, Tjandra N, Carter CA. 2011. Phosphoinositides direct equine infectious anemia virus Gag trafficking and release. Traffic 12:438–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saad J, Ablan S, Ghanam R, Kim A, Andrews K, Nagashima K, Soheilian F, Freed E, Summers M. 2008. Structure of the myristylated human immunodeficiency virus type 2 matrix protein and the role of phosphatidylinositol-(4,5)-bisphosphate in membrane targeting. J. Mol. Biol. 382:434–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saad J, Miller J, Tai J, Kim A, Ghanam R, Summers M. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. U. S. A. 103:11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan J, Dick RA, Vogt VM. 2011. Rous sarcoma virus Gag has no specific requirement for phosphatidylinositol-(4,5)-bisphosphate for plasma membrane association in vivo or for liposome interaction in vitro. J. Virol. 85:10851–10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A. 2008. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient Gag membrane binding. J. Virol. 82:2405–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamard-Peron E, Juillard F, Saad J, Roy C, Roingeard P, Summers M, Darlix J, Picart C, Muriaux D. 2010. Targeting of murine leukemia virus Gag to the plasma membrane is mediated by PI(4,5)P2/PS and a polybasic region in the matrix. J. Virol. 84:503–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inlora J, Chukkapalli V, Derse D, Ono A. 2011. Gag localization and virus-like particle release mediated by the matrix domain of human T-lymphotropic virus type-1 Gag are less dependent on phosphatidylinositol-(4,5)-bisphosphate than those mediated by the matrix domain of human immunodeficiency virus type-1 Gag. J. Virol. 85:3802–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ono A, Ablan S, Lockett S, Nagashima K, Freed E. 2004. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. U. S. A. 101:14889–14894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nadaraia-Hoke S, Bann DV, Lochmann TL, Gudleski-O'Regan N, Parent LJ. 2013. Alterations in the MA and NC domains modulate phosphoinositide-dependent plasma membrane localization of the Rous sarcoma virus Gag protein. J. Virol. 87:3609–3615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chukkapalli V, Oh SJ, Ono A. 2010. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc. Natl. Acad. Sci. U. S. A. 107:1600–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dick RA, Goh SL, Feigenson GW, Vogt VM. 2012. HIV-1 Gag protein can sense the cholesterol and acyl chain environment in model membranes. Proc. Natl. Acad. Sci. U. S. A. 109:18761–18766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freed EO. 2006. HIV-1 Gag: flipped out for PI(4,5)P(2). Proc. Natl. Acad. Sci. U. S. A. 103:11101–11102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vlach J, Saad JS. 2013. Trio engagement via plasma membrane phospholipids and the myristoyl moiety governs HIV-1 matrix binding to bilayers. Proc. Natl. Acad. Sci. U. S. A. 110:3525–3530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, Krausslich HG. 2009. Structure and assembly of immature HIV. Proc. Natl. Acad. Sci. U. S. A. 106:11090–11095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. 2007. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 26:2218–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demirov DG, Freed EO. 2004. Retrovirus budding. Virus Res. 106:87–102 [DOI] [PubMed] [Google Scholar]
- 21.Keller PW, Johnson MC, Vogt VM. 2008. Mutations in the spacer peptide and adjoining sequences in Rous sarcoma virus Gag lead to tubular budding. J. Virol. 82:6788–6797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krausslich HG, Facke M, Heuser AM, Konvalinka J, Zentgraf H. 1995. The spacer peptide between human immunodeficiency virus capsid and nucleocapsid proteins is essential for ordered assembly and viral infectivity. J. Virol. 69:3407–3419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips JM, Murray PS, Murray D, Vogt VM. 2008. A molecular switch required for retrovirus assembly participates in the hexagonal immature lattice. EMBO J. 27:1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bharat TA, Davey NE, Ulbrich P, Riches JD, de Marco A, Rumlova M, Sachse C, Ruml T, Briggs JA. 2012. Structure of the immature retroviral capsid at 8 Å resolution by cryo-electron microscopy. Nature 487:385–389 [DOI] [PubMed] [Google Scholar]
- 25.Ma YM, Vogt VM. 2004. Nucleic acid binding-induced Gag dimerization in the assembly of Rous sarcoma virus particles in vitro. J. Virol. 78:52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma YM, Vogt VM. 2002. Rous sarcoma virus Gag protein-oligonucleotide interaction suggests a critical role for protein dimer formation in assembly. J. Virol. 76:5452–5462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu F, Joshi SM, Ma YM, Kingston RL, Simon MN, Vogt VM. 2001. Characterization of Rous sarcoma virus Gag particles assembled in vitro. J. Virol. 75:2753–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Accola MA, Strack B, Gottlinger HG. 2000. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J. Virol. 74:5395–5402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alfadhli A, Dhenub TC, Still A, Barklis E. 2005. Analysis of human immunodeficiency virus type 1 Gag dimerization-induced assembly. J. Virol. 79:14498–14506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crist RM, Datta SA, Stephen AG, Soheilian F, Mirro J, Fisher RJ, Nagashima K, Rein A. 2009. Assembly properties of human immunodeficiency virus type 1 Gag-leucine zipper chimeras: implications for retrovirus assembly. J. Virol. 83:2216–2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson MC, Scobie HM, Ma YM, Vogt VM. 2002. Nucleic acid-independent retrovirus assembly can be driven by dimerization. J. Virol. 76:11177–11185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Qian H, Love Z, Barklis E. 1998. Analysis of the assembly function of the human immunodeficiency virus type 1 gag protein nucleocapsid domain. J. Virol. 72:1782–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campbell S, Fisher R, Towler E, Fox S, Issaq H, Wolfe T, Phillips L, Rein A. 2001. Modulation of HIV-like particle assembly in vitro by inositol phosphates. Proc. Natl. Acad. Sci. U. S. A. 98:10875–10879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campbell S, Rein A. 1999. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J. Virol. 73:2270–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campbell S, Vogt VM. 1997. In vitro assembly of virus-like particles with Rous sarcoma virus Gag deletion mutants: identification of the p10 domain as a morphological determinant in the formation of spherical particles. J. Virol. 71:4425–4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gross I, Hohenberg H, Krausslich HG. 1997. In vitro assembly properties of purified bacterially expressed capsid proteins of human immunodeficiency virus. Eur. J. Biochem. 249:592–600 [DOI] [PubMed] [Google Scholar]
- 37.Perez-Caballero D, Hatziioannou T, Martin-Serrano J, Bieniasz PD. 2004. Human immunodeficiency virus type 1 matrix inhibits and confers cooperativity on Gag precursor-membrane interactions. J. Virol. 78:9560–9563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen BK, Rousso I, Shim S, Kim PS. 2001. Efficient assembly of an HIV-1/MLV Gag-chimeric virus in murine cells. Proc. Natl. Acad. Sci. U. S. A. 98:15239–15244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parent LJ, Wilson CB, Resh MD, Wills JW. 1996. Evidence for a second function of the MA sequence in the Rous sarcoma virus Gag protein. J. Virol. 70:1016–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reed M, Mariani R, Sheppard L, Pekrun K, Landau NR, Soong NW. 2002. Chimeric human immunodeficiency virus type 1 containing murine leukemia virus matrix assembles in murine cells. J. Virol. 76:436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. 2006. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 4:e435. 10.1371/journal.pbio.0040435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Urano E, Aoki T, Futahashi Y, Murakami T, Morikawa Y, Yamamoto N, Komano J. 2008. Substitution of the myristoylation signal of human immunodeficiency virus type 1 Pr55Gag with the phospholipase C-delta1 pleckstrin homology domain results in infectious pseudovirion production. J. Gen. Virol. 89:3144–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borsetti A, Ohagen A, Gottlinger HG. 1998. The C-terminal half of the human immunodeficiency virus type 1 Gag precursor is sufficient for efficient particle assembly. J. Virol. 72:9313–9317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reil H, Bukovsky AA, Gelderblom HR, Gottlinger HG. 1998. Efficient HIV-1 replication can occur in the absence of the viral matrix protein. EMBO J. 17:2699–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Carroll IP, Soheilian F, Kamata A, Nagashima K, Rein A. 2013. Elements in HIV-1 Gag contributing to virus particle assembly. Virus Res. 171:341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gudleski N, Flanagan JM, Ryan EP, Bewley MC, Parent LJ. 2010. Directionality of nucleocytoplasmic transport of the retroviral Gag protein depends on sequential binding of karyopherins and viral RNA. Proc. Natl. Acad. Sci. U. S. A. 107:9358–9363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hearps AC, Wagstaff KM, Piller SC, Jans DA. 2008. The N-terminal basic domain of the HIV-1 matrix protein does not contain a conventional nuclear localization sequence but is required for DNA binding and protein self-association. Biochemistry 47:2199–2210 [DOI] [PubMed] [Google Scholar]
- 48.Lochrie MA, Waugh S, Pratt DG, Jr, Clever J, Parslow TG, Polisky B. 1997. In vitro selection of RNAs that bind to the human immunodeficiency virus type-1 Gag polyprotein. Nucleic Acids Res. 25:2902–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parent LJ, Cairns TM, Albert JA, Wilson CB, Wills JW, Craven RC. 2000. RNA dimerization defect in a Rous sarcoma virus matrix mutant. J. Virol. 74:164–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Purohit P, Dupont S, Stevenson M, Green MR. 2001. Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA 7:576–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramalingam D, Duclair S, Datta SA, Ellington A, Rein A, Prasad VR. 2011. RNA aptamers directed to human immunodeficiency virus type 1 Gag polyprotein bind to the matrix and nucleocapsid domains and inhibit virus production. J. Virol. 85:305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steeg CM, Vogt VM. 1990. RNA-binding properties of the matrix protein (p19gag) of avian sarcoma and leukemia viruses. J. Virol. 64:847–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dalton A, Murray P, Murray D, Vogt V. 2005. Biochemical characterization of Rous sarcoma virus MA protein interaction with membranes. J. Virol. 79:6227–6238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dalton AK, Ako-Adjei D, Murray PS, Murray D, Vogt VM. 2007. Electrostatic interactions drive membrane association of the human immunodeficiency virus type 1 Gag MA domain. J. Virol. 81:6434–6445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. 2009. X-ray structures of the hexameric building block of the HIV capsid. Cell 137:1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. 2004. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl. Acad. Sci. U. S. A. 101:517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Callahan E, Wills J. 2000. Repositioning basic residues in the M domain of the Rous sarcoma virus Gag protein. J. Virol. 74:11222–11229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buboltz JT, Feigenson GW. 1999. A novel strategy for the preparation of liposomes: rapid solvent exchange. Biochim. Biophys. Acta 1417:232–245 [DOI] [PubMed] [Google Scholar]
- 59.Zhao J, Wu J, Heberle FA, Mills TT, Klawitter P, Huang G, Costanza G, Feigenson GW. 2007. Phase studies of model biomembranes: complex behavior of DSPC/DOPC/cholesterol. Biochim. Biophys. Acta 1768:2764–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kay JE. 1996. Structure-function relationships in the FK506-binding protein (FKBP) family of peptidylprolyl cis-trans isomerases. Biochem. J. 314(Part 2):361–385 [PMC free article] [PubMed] [Google Scholar]
- 61.Rollins CT, Rivera VM, Woolfson DN, Keenan T, Hatada M, Adams SE, Andrade LJ, Yaeger D, van Schravendijk MR, Holt DA, Gilman M, Clackson T. 2000. A ligand-reversible dimerization system for controlling protein-protein interactions. Proc. Natl. Acad. Sci. U. S. A. 97:7096–7101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schreiber SL. 1991. Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science 251:283–287 [DOI] [PubMed] [Google Scholar]
- 63.Kerfeld CA, Sawaya MR, Tanaka S, Nguyen CV, Phillips M, Beeby M, Yeates TO. 2005. Protein structures forming the shell of primitive bacterial organelles. Science 309:936–938 [DOI] [PubMed] [Google Scholar]
- 64.Ding L, Derdowski A, Wang JJ, Spearman P. 2003. Independent segregation of human immunodeficiency virus type 1 Gag protein complexes and lipid rafts. J. Virol. 77:1916–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ono A, Demirov D, Freed EO. 2000. Relationship between human immunodeficiency virus type 1 Gag multimerization and membrane binding. J. Virol. 74:5142–5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ono A, Freed EO. 1999. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J. Virol. 73:4136–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spearman P, Horton R, Ratner L, Kuli-Zade I. 1997. Membrane binding of human immunodeficiency virus type 1 matrix protein in vivo supports a conformational myristyl switch mechanism. J. Virol. 71:6582–6592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou W, Resh MD. 1996. Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J. Virol. 70:8540–8548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scheifele LZ, Garbitt RA, Rhoads JD, Parent LJ. 2002. Nuclear entry and CRM1-dependent nuclear export of the Rous sarcoma virus Gag polyprotein. Proc. Natl. Acad. Sci. U. S. A. 99:3944–3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Butterfield-Gerson KL, Scheifele LZ, Ryan EP, Hopper AK, Parent LJ. 2006. Importin-beta family members mediate alpharetrovirus Gag nuclear entry via interactions with matrix and nucleocapsid. J. Virol. 80:1798–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278:849–853 [DOI] [PubMed] [Google Scholar]
- 72.del Alamo M, Neira JL, Mateu MG. 2003. Thermodynamic dissection of a low affinity protein-protein interface involved in human immunodeficiency virus assembly. J. Biol. Chem. 278:27923–27929 [DOI] [PubMed] [Google Scholar]
- 73.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. 1993. Controlling signal transduction with synthetic ligands. Science 262:1019–1024 [DOI] [PubMed] [Google Scholar]
- 74.Alfadhli A, Still A, Barklis E. 2009. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J. Virol. 83:12196–12203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Callahan E, Wills J. 2003. Link between genome packaging and rate of budding for Rous sarcoma virus. J. Virol. 77:9388–9398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jones CP, Datta SA, Rein A, Rouzina I, Musier-Forsyth K. 2011. Matrix domain modulates HIV-1 Gag's nucleic acid chaperone activity via inositol phosphate binding. J. Virol. 85:1594–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morikawa Y, Zhang WH, Hockley DJ, Nermut MV, Jones IM. 1998. Detection of a trimeric human immunodeficiency virus type 1 Gag intermediate is dependent on sequences in the matrix protein, p17. J. Virol. 72:7659–7663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. 1996. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl. Acad. Sci. U. S. A. 93:3099–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Datta SA, Zhao Z, Clark PK, Tarasov S, Alexandratos JN, Campbell SJ, Kvaratskhelia M, Lebowitz J, Rein A. 2007. Interactions between HIV-1 Gag molecules in solution: an inositol phosphate-mediated switch. J. Mol. Biol. 365:799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Datta SA, Curtis JE, Ratcliff W, Clark PK, Crist RM, Lebowitz J, Krueger S, Rein A. 2007. Conformation of the HIV-1 Gag protein in solution. J. Mol. Biol. 365:812–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Datta SA, Heinrich F, Raghunandan S, Krueger S, Curtis JE, Rein A, Nanda H. 2011. HIV-1 Gag extension: conformational changes require simultaneous interaction with membrane and nucleic acid. J. Mol. Biol. 406:205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Spearman P, Ratner L. 1996. Human immunodeficiency virus type 1 capsid formation in reticulocyte lysates. J. Virol. 70:8187–8194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chukkapalli V, Inlora J, Todd GC, Ono A. 2013. Evidence in support of RNA-mediated inhibition of phosphatidylserine-dependent HIV-1 Gag membrane binding in cells. J. Virol. 87:7155–7159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alfadhli A, McNett H, Tsagli S, Bachinger HP, Peyton DH, Barklis E. 2011. HIV-1 matrix protein binding to RNA. J. Mol. Biol. 410:653–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dooher JE, Schneider BL, Reed JC, Lingappa JR. 2007. Host ABCE1 is at plasma membrane HIV assembly sites and its dissociation from Gag is linked to subsequent events of virus production. Traffic 8:195–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Klein KC, Reed JC, Tanaka M, Nguyen VT, Giri S, Lingappa JR. 2011. HIV Gag-leucine zipper chimeras form ABCE1-containing intermediates and RNase-resistant immature capsids similar to those formed by wild-type HIV-1 Gag. J. Virol. 85:7419–7435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dong X, Li H, Derdowski A, Ding L, Burnett A, Chen X, Peters TR, Dermody TS, Woodruff E, Wang JJ, Spearman P. 2005. AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell 120:663–674 [DOI] [PubMed] [Google Scholar]
- 88.Larson DR, Ma YM, Vogt VM, Webb WW. 2003. Direct measurement of Gag-Gag interaction during retrovirus assembly with FRET and fluorescence correlation spectroscopy. J. Cell Biol. 162:1233–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kutluay SB, Bieniasz PD. 2010. Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathog. 6:e1001200. 10.1371/journal.ppat.1001200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fogarty KH, Chen Y, Grigsby IF, Macdonald PJ, Smith EM, Johnson JL, Rawson JM, Mansky LM, Mueller JD. 2011. Characterization of cytoplasmic Gag-Gag interactions by dual-color z-scan fluorescence fluctuation spectroscopy. Biophys. J. 100:1587–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fogarty KH, Zhang W, Grigsby IF, Johnson JL, Chen Y, Mueller JD, Mansky LM. 2011. New insights into HTLV-1 particle structure, assembly, and Gag-Gag interactions in living cells. Viruses 3:770–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grigsby IF, Zhang W, Johnson JL, Fogarty KH, Chen Y, Rawson JM, Crosby AJ, Mueller JD, Mansky LM. 2010. Biophysical analysis of HTLV-1 particles reveals novel insights into particle morphology and Gag stochiometry. Retrovirology 7:75. 10.1186/1742-4690-7-75 [DOI] [PMC free article] [PubMed] [Google Scholar]