

Abstract
Host cell-mediated proteolytic cleavage of the human immunodeficiency virus type 1 (HIV-1) gp160 precursor glycoprotein into gp120 and gp41 subunits is required to generate fusion-competent envelope glycoprotein (Env) spikes. The gp120-directed broadly neutralizing monoclonal antibodies (bNabs) isolated from HIV-infected individuals efficiently recognize fully cleaved JRFL Env spikes; however, nonneutralizing gp120-directed monoclonal antibodies isolated from infected or vaccinated subjects recognize only uncleaved JRFL spikes. Therefore, as an immunogen, cleaved spikes that selectively present desired neutralizing epitopes to B cells may elicit cross-reactive neutralizing antibodies. Accordingly, we inoculated nonhuman primates (NHPs) with plasmid DNA encoding transmembrane-anchored, cleaved JRFL Env or by electroporation (EP). Priming with DNA expressing soluble, uncleaved gp140 trimers was included as a comparative experimental group of NHPs. DNA inoculation was followed by boosts with soluble JRFL gp140 trimers, and control NHPs were inoculated with soluble JRFL protein trimers without DNA priming. In the TZM-bl assay, elicitation of neutralizing antibodies against HIV-1 tier 1 isolates was robust following the protein boost. Neutralization of tier 2 isolates was detected, but only in animals primed with plasmid DNA and boosted with trimeric protein. Using the more sensitive A3R5 assay, consistent neutralization of both clade B and C tier 2 isolates was detected from all regimens assessed in the current study, exceeding levels achieved by our previous vaccine regimens in primates. Together, these data suggest a potential advantage of B cell priming followed by a rest interval and protein boosting to present JRFL Env spikes to the immune system to better generate HIV-1 cross-clade neutralizing antibodies.
INTRODUCTION
The human immunodeficiency virus type 1 (HIV-1) exterior envelope glycoprotein, gp120, and the transmembrane glycoprotein, gp41, are derived from the cleavage of the gp160 precursor protein and are the only virally encoded proteins on the surface of the virus. These noncovalently associated glycoproteins form the trimeric functional spikes that mediate viral entry. The gp120 subunit binds the primary receptor, CD4, and following gp120 association with the coreceptor, usually CCR5, the gp41 subunit participates to accomplish virus-to-cell membrane fusion and entry of viral genomic information into the target cell (1–12).
Neutralizing antibodies administered passively at high systemic concentrations before, or immediately after, exposure to chimeric simian-human immunodeficiency virus (SHIV) can protect against viral challenge, confirming their importance in the context of a prophylactic vaccine (13–17). Approximately 10 to 20% of HIV-1 chronically infected individuals display “neutralization breadth” mediated by antibodies present in their serum. The broad neutralizing activity elicited during this natural infection process can sometimes be mapped to distinct subregions of Env (18–22). Recently, a plethora of new broadly neutralizing antibodies (bNabs) were isolated from HIV-1-infected individuals directed against the CD4 binding site and glycan-shielded regions of Env (22–25). Very recently, the highly potent, non-self-reactive, gp41-directed and membrane proximal external region (MPER)-specific bNab, 10E8, was isolated (26). Additionally, recent data suggest that a cocktail of bNabs might suppress viremia in vivo in the absence of highly active antiretroviral therapy (HAART) (27).
Numerous attempts have been made using Env immunogens in either monomeric (gp120) or in trimeric (gp140, gp41) contexts to elicit HIV-1 broadly neutralizing antibodies. To date, however, most of these Env immunogens do not fully mimic the properties of the functional spike and have not efficiently elicited broad neutralization (reviewed in reference 28). Due to this lack of success, one approach to develop vaccine candidates against HIV-1 is to design versions of trimeric Env immunogens that maintain structural and physiochemical properties similar to Env present on infectious viruses. The rationale is that presentation of a more faithful mimetic of the spike might be a better vaccine candidate to elicit antibodies capable of neutralizing diverse HIV-1 isolates. To date, however, soluble trimers that faithfully mimic the antigenic properties of the functional spike have not been forthcoming. Therefore, alternative means to present functional spikes to the immune system are worthy of investigation.
Previously, we demonstrated that, following transient transfection of plasmid DNA, primary isolate Env glycoproteins expressed at the cell surface are oligomeric and predominantly trimeric (29). Subsequently, using fluorescence-activated cell sorting (FACS)-based cell surface staining, coupled with direct biochemical analysis, we documented the efficient precursor cleavage of Env derived from the primary isolate, JRFL, and assembly into functional spikes on the cell surface (30). In this system, the efficient cleavage of JRFL Env requires use of the virally encoded, long terminal repeat (LTR)-driven env gene expression, along with Tat in trans. To date, this is the only such wild-type HIV-1 Env that demonstrates apparently complete cleavage when transiently expressed from plasmid DNA in 293F cells and in the absence of exogenous furin, for reasons that are not fully clear. Importantly, if JRFL Env is expressed in this manner, the functional, cleaved spikes are efficiently recognized only by bNabs but not by non-bNabs or nonneutralizing monoclonal antibodies (MAbs). In several studies using both gp120-directed and gp41-directed neutralizing and nonneutralizing antibodies, we and others show that there is a substantial difference in binding by antibody/CD4 to the cleaved functional spike compared to an uncleaved version of the same spike (30, 31). That is, the recognition of nonneutralizing antibodies increased markedly when the cleavage site between gp120 and gp41 was eliminated through site-directed mutagenesis, suggesting the exposure of nonneutralizing epitopes in the context of uncleaved Env.
A reasonable strategy to attempt to elicit broadly neutralizing antibodies therefore is to present cleaved, full-length functional spikes through genetic means, in vivo. Therefore, we primed nonhuman primates (NHPs) by electroporation (EP) using plasmid DNAs encoding cleavage-competent primary isolate JRFL Env expressed from non-codon-optimized or codon-optimized env genes. Plasmid DNA encoding soluble JRFL trimers was also included to assess if DNA priming with homologous Env trimers followed by trimer protein boosting provided an advantage over trimer protein immunization alone. In this study, “priming” consisted of three DNA EP inoculations with monthly intervals between each injection. Following DNA priming with plasmids expressing either cell surface or soluble trimers, NHPs were boosted twice with trimeric JRFL gp140-F (where F stands for foldon) proteins formulated in adjuvant. These regimens presented Env to the primate immune system over 5 inoculations, paralleling our previous study that presented soluble YU2 gp140-F trimers 5 times at monthly intervals (32, 33) but introduced a long interval between the DNA prime and the protein boosts. As comparative controls, three NHPs were inoculated with JRFL gp140-F protein trimers without prior DNA priming over the 2-month interval.
Priming with either non-codon-optimized or codon-optimized cell surface or soluble Env elicited detectable enzyme-linked immunosorbent assay (ELISA) binding titers in NHPs, which greatly increased following protein boosting. The elicited antibodies displayed robust neutralizing activity against tier 1 isolates in the validated TZM-bl assay (34). Trimeric JRFL gp140-F proteins formulated in adjuvant and administered without DNA priming also elicited robust tier 1 neutralization. However, neutralization of several tier 2 isolates (more representative of circulating viruses) in the TZM-bl assay was detected only in the serum of animals primed with Env plasmid DNA followed by boosting with the soluble JRFL trimers. Collectively, these data suggest that a DNA prime-protein boost long-interval inoculation regimen can improve the generation of neutralizing Ab responses against HIV-1. However, in the more sensitive A3R5 assay (34), the sera from most animals neutralized all tier 2 isolates tested with a wide range of potencies, and the neutralization activity was independent of whether the priming was performed with full-length cleaved or uncleaved soluble Env trimers. These neutralizing responses were similar to those elicited by the soluble JRFL trimers alone but were superior to neutralizing activity elicited in our prior HIV-1 Env trimer studies performed in NHPs (32, 35, 36).
MATERIALS AND METHODS
Cell lines, antibodies, and plasmid DNAs.
TZM-bl cells (CD4+ CXCR4+ CCR5+) were obtained from the NIH AIDS Research and Reference Reagent Program, and 293T cells were purchased from ATCC. Both cell lines were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% heat-inactivated fetal calf serum (HIFCS), 20 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. The gp120 glycan-directed 2G12, PGT121, PGT128, PGT135, and PGT145 and the CD4bs MAbs, b12, VRC01, VRC03, PGV04, and VRC06, were obtained from the Vaccine Research Center (VRC) or from internal International AIDS Vaccine Initiative (IAVI) or Scripps resources. The CD4bs MAb (F105; M. Posner), the CD4i MAb (17b; J. Robinson), the anti-V3 MAb (447D; S. Zolla-Pazner, CFAR), and the anti-gp41 MAb (F240; L. Cavacini) were obtained by material transfer agreement (MTA). The Env plasmids for the non-codon-optimized (viral sequences), cell-surface, cleavage competent (+), cytoplasmic tail-deleted (ΔCT) JRFL Env is called pSVIII JRFL(+)ΔCT (30). Tat expression in vitro or in vivo was achieved by using the vector pCTat (37). The codon-optimized JRFL cell surface Env containing the heterologous CD5 leader is driven by a cytomegalovirus (CMV) promoter [pCDNA-JRFL(+)ΔCT]. The codon-optimized soluble trimeric JRFL gp140-F, also with the CD5 leader and CMV promoter driven, is called pCDNA JRFL gp140-F. The pCDNA3.1(−) vector expressing the HXBc2 coreV3S (38) with triple mutations (I423M, N425K, and G431E-TriMut), TriMut with two additional mutations at D368R and E370F (TriMut 368/370) (39), and TriMut with 368/370 and the 474A substitution were lectin and chelation affinity purified and used for the protein adsorption mapping analysis.
Flow cytometric analysis of cell surface HIV-1 Env.
FACS staining was performed as previously described (30, 40). Forty-eight hours following transfection, the cells were harvested and washed in FACS buffer (phosphate-buffered saline [PBS], 5% heat-inactivated fetal bovine serum [HIFBS], 0.02% azide) and stained with a panel of monoclonal antibodies that were previously analyzed in the TZM-bl assay for neutralization capacity (24, 25). The monoclonal antibody-cell mixtures were washed extensively in FACS buffer, and anti-human phycoerythrin (PE) (Sigma) at a 1:200 dilution was added for 1 h, followed by extensive washing to remove unbound secondary antibody. The PE-stained cells were analyzed by flow cytometry on a BD SLRII instrument.
Expression, purification, and biophysical analysis of JRFL gp140-F trimeric Env.
The soluble JRFL trimers (gp140-F), containing a His6 purification tag, were expressed by transient transfection of the 293F cells. The glycoproteins were purified by lentil lectin affinity chromatography followed by chelating chromatography over an Ni2+ column (GE Health Care, Piscataway, NJ). The trimeric JRFL gp140-F proteins were loaded onto 10/300 GL Tricorn high-performance columns prepacked with Superdex 200 medium. The proteins were further purified by selection of fractions at the trailing edge of the trimer peak, resulting in relatively pure trimers which migrated as a single peak by size exclusion chromatography (SEC). Relevant fractions containing trimers were pooled, concentrated, dialyzed against PBS, flash-frozen in liquid nitrogen, and stored at −80°C. The purity and oligomeric status of the purified protein was assessed by SDS-PAGE and blue native gels.
Bio-layer interferometry binding assay.
Bio-layer interferometry binding assays were performed on the Octet-Red device (FortéBio, Inc., Menlo Park, CA) to determine the binding kinetics. JRFL gp140-F trimers were immobilized on amine reactive (AR2G) biosensors (FortéBio) by amine coupling according to the manufacturer's instruction. IgG or Fab of monoclonal antibodies were 2-fold serially diluted in kinetics buffer (FortéBio) and then were dispensed into 96-well microtiter plates at a volume of 200 μl per well. The operating temperature was maintained at 30°C. The AR2G biosensors were dipped into solutions of the IgG or Fab at selected concentrations with agitation for 15 min at 1,000 rpm to measure association constants. Dissociation was detected following transfer of sensors into wells containing buffer alone for 30 min. Data were collected and analyzed by Octet User software (version 7.0). For the analysis of Fab binding, a 1:1 curve-fitting model was used, while the 2:1 bivalent analyte model was used for the analysis of IgG binding.
NHP inoculation methods and regimens.
Simian immunodeficiency virus (SIV)-negative Indian rhesus macaques (Macaca mulatta) were housed at SUNY Downstate/IAVI in accordance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care and with the approval of the University of Pittsburgh's Institutional Animal Care and Use Committee (IACUC) standards and regulations. The protocols were approved and assigned the IACUC number 0803208. EP of plasmid DNA into the NHPs was performed using an Elgen 1000 (Inovio, BC, Canada) consisting of a pulse generator and a combined injection electrode device. Two parallel injection needles (22 gauge) were spaced 4 mm apart and inserted 13 to 18 mm into the quadricep muscle, and DNA was injected through the needles, which were subsequently used as electrodes. Immediately after needle/electrode insertion, electrical impulses were delivered to the quadricep muscle tissue. Two 60-ms square wave pulses at a nominal field strength of 100 to 150 V/cm and Hz were used for 5 s. The target site of administration was the muscle of the anterior hind limb. The administration site was shaved with an electric clipper and the skin cleaned with disinfectant prior to immunization. Each NHP was inoculated thrice at monthly intervals with 1 mg of plasmid DNA per inoculation time point. Control animals were immunized with the same volume of phosphate-buffered saline (PBS). Plasmid DNA mixture encoding the JRFL gp160 Env cleavage-competent, tail-deleted pSVIII JRFLΔCT(+) and pCTat with two different proportions relative to the Env plasmid (20:1 and 1:1, called Tat 1 and 2, respectively) were used. Codon-optimized JRFLΔCT(+) or JRFL gp140-F DNA does not require a Tat protein for their efficient expression. Note that the plasmid encoding the JRFL gp140-F uncleaved trimers was included and inoculated into two animals to determine if Env cell surface expression or cleavage into the bona fide spike was required to better elicit neutralizing antibodies. The animals were bled 2 weeks after every inoculation, and sera were collected and stored at −20°C. The animals received boosts with 50 μg of recombinant JRFL gp140-F protein per animal 12 and 20 weeks following the final DNA immunization in the priming phase of the regimen. The animals were bled 2 weeks after each DNA and protein inoculation.
ELISA.
Recognition of the JRFL gp140-F trimers and gp41 and the cluster 1 peptide following DNA priming or soluble protein in adjuvant was assessed using an ELISA as described previously (41). In brief, wells of MaxiSorp high-binding plates (Nunc) were coated with 100 μl of protein (1 mg/ml initial concentration) in PBS, pH 7.4, and incubated overnight at 4°C. Antisera were started at an initial 1:100 dilution and were then 3-fold serially diluted, followed by incubation with antigen and subsequent washing. After incubation with anti-human IgG-horseradish peroxidase (HRP) and additional washing, the TMB (3,3,5,5′-tetramethylbenzidine) peroxidase immunoassay substrate (Bio-Rad) was used as the colorimetric reporter reagent. The optical density (OD) was determined at 450 nm on a 96-well microtiter plate reader (Molecular Devices).
HIV-1 neutralization assays.
Pseudotyped viruses were produced by transient cotransfection of 293T cells using the HIV-1 env-deleted backbone plasmid, pSG3ΔEnv, and the Env complementation plasmid, pSVIII JRFLΔCT(+), as previously described (42) at a ratio of 3:1. Briefly, a 3:1 ratio of the transfection reagent, Fugene (Roche, Indianapolis, IN, USA), to DNA was used for transfection. Cell culture supernatants containing viruses were collected 2 days posttransfection. The TZM-bl neutralization assay using target cells expressing CD4, CXC4, and CCR5 was done as described previously (43, 44). Briefly, TZM-bl-based neutralization assays were performed with serial dilutions of heat-inactivated (56°C, 1 h) samples. Diluted samples were preincubated with virus (∼150,000 relative light unit equivalents) for 1 h at 37°C before the addition of cells. Following 48 h of incubation, cells were lysed and the Luciferase activity was determined using a microtiter plate luminometer and BriteLite Plus reagent (PerkinElmer). Neutralization titers are defined as the sample dilution at which relative luminescence units (RLU) were reduced by 50% compared to the RLU in virus control wells after subtraction of background RLU in cell control wells. To test for non-HIV-1-specific plasma effects, the samples were also tested against a pseudovirus expressing the amphotropic murine leukemia virus (MuLV) envelope. In the text and figures, we have used abbreviated names for the viruses, but the full specific names are as follows: MN, MN.3; SF162, SF162.LS; BaL, BaL.01; MW965, MW965.26; SC42, SC42261.8; and ZM109, ZM109F.PB4. The A3R5 entry assay was recently described. In brief, A3.01/CCR5 (A3R5) is a derivative of the A3.01 human lymphoblastoid cell line, CEM, that naturally expresses CD4 and CXCR4 (45) and was engineered to express CCR5 (46). Because A3R5.7 cells do not contain a reporter gene, molecularly cloned viruses that carry a reporter gene (Env.IMC.LucR viruses) are used in these assays. Again, we used abbreviated descriptions of the viruses throughout this publication; full specific isolate names are as follows: SC22, SC22.3C2.LucR.T2A.ecto; RHPA, RHPA.LucR.T2A.ecto; CH58, CH58.LucR.T2A.ecto; THRO, THRO.LucR.T2A.ecto; WITO, WITO.LucR.T2A.ecto; 1051, 1051.C22.LucR.T2A.ecto; CAP45, CAP45.2.00.G3.LucR.T2A.ecto; Ce1086, Ce1086_B2.LucR.T2A.ecto; DU151, DU151.2.LucR.T2A.ecto; and Ce2010, Ce2010_F5.LucR.T2A.ecto. As with the TZM-bl assay, diluted samples were incubated with virus (∼50,000 RLU equivalents) for 1 h at 37°C prior to the addition of cells. After incubating for 4 days, a defined portion of the cell suspension was transferred to 96-well white solid plates (Costar) for measurement of luminescence using the ViviRen live-cell substrate as described by the supplier (Promega). For each animal, pre- and postimmune serum samples were assayed side by side. Postimmune samples were scored positive for neutralizing antibody activity if the titer was >3-fold the titer of the preimmune sample. To assess the presence of CD4bs-directed neutralization in the sera, differential protein adsorptions were performed in the context of the A3R5 neutralization assay using TriMut core gp120 or TriMut 368/370 (CD4 binding-defective gp120 core) as previously described (39) or the additional CD4 binding-defective core, TriMut 368/370/474. In brief, before the addition of pseudovirus, either the VRC01 control MAb or 3-fold serial dilutions from each serum sample were preincubated with 10 μg/ml of TriMut, TriMut 368/370, TriMut 368/370/474, or cell culture medium for 1 h at 37°C. For each serum sample, three neutralization curves derived from the assays were performed in parallel to detect the presence of trimer-elicited neutralizing antibodies directed to the CD4bs by the differential inhibition of neutralization using the isogenic TriMut core probes.
RESULTS
FACS-based binding to cell surface JRFL Env shows efficient recognition by bNabs.
Before initiating immunogenicity studies with the immunogens and regimen described for Fig. 1, we sought to characterize further the cell surface JRFL trimeric Env immunogens, which were to be used for priming of B cell responses in NHPs. Therefore, we analyzed the cell surface staining of cells transfected with the plasma membrane-anchored JRFL-cleaved trimers. To efficiently express the cleaved trimers, we transiently transfected 293T cells with an LTR-driven plasmid that coexpresses Rev in cis, along with a second plasmid expressing Tat in trans (see Materials and Methods). We then assessed recognition of the cell surface JRFL trimers with a panel of neutralizing and nonneutralizing antibodies. As previously reported, when analyzing recognition of the cleaved JRFL trimers by gp120-directed MAbs, only the MAbs such as 2G12 and b12 that efficiently neutralize the JRFL virus efficiently recognized the JRFL Env cell surface-expressed trimers. Again consistent with our previous reports, there were clear differences in the recognition of gp120-directed neutralizing antibodies compared to recognition of the nonneutralizing antibodies of the cleaved JRFL cell surface Env trimers. Therefore, in this study, we extended our analysis to the newly discovered bNabs directed to glycan on the V1V2V3 regions and to the CD4bs to determine if they would also efficiently recognize the cleaved JRFL trimers in comparison to representative nonneutralizing Env-directed antibodies.
Fig 1.
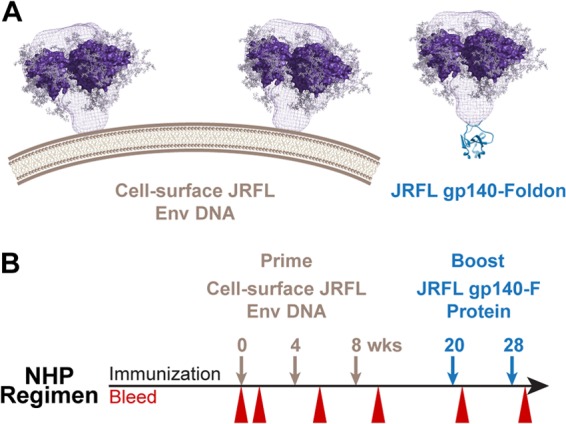
Schematics of JRFL trimers and inoculation regimen. (A) Left, JRFL cleaved, cell surface trimers are modeled on the surface of a transiently transfected 233T cell. The trimer density (blue mesh) is based upon the work of Liu et al. (60), and the crystal structure of the core gp120 is modeled as a trimer into this density. Right, schematic depiction of the soluble JRFL gp140-F trimer with the genetically fused foldon (F) motif in place of the HIV-1 Env gp41 transmembrane region. (B) Timeline and parameters of the EP plasmid DNA prime (brown) and trimer boost (blue) regimen in NHPs are shown with inoculations and bleeds to isolate sera for analysis as indicated.
The mean fluorescence intensities (MFI) of different concentrations of antibodies specifically interacting with cell surface Env were derived from the flow cytometry histograms, and these data were used to generate the binding curves shown in Fig. 2. As expected, the neutralizing and glycan- and gp120-directed antibody, 2G12, bound to cleavage-competent Env expressed on the 293T cell surface with the highest relative affinity (Fig. 2A). From the FACS-based binding curves shown in Fig. 2, it is clear that the gp120-directed antibodies bound to the cleaved JRFL Env spikes with various efficiencies. Generally, most glycan-related bNabs capable of neutralizing JRFL bound with reasonable efficiency to the cell surface JRFL Env trimers. The glycan-related bNab, PGT128, bound second most efficiently, followed by PGT121 (Fig. 2A). The bNab PGT135 bound at low levels to the JRFL Env compared to those for the other glycan-related MAbs, consistent with its reduced capacity to neutralize JRFL (24). PGT145, which cannot neutralize JRFL, displayed minimal binding. The CD4bs-directed bNabs recognized the JRFL cell surface spikes in a similar manner and roughly equivalent to PGT121 (Fig. 2B), whereas the non-bNab CD4bs MAb F015 did not recognize these spikes (Fig. 2C). The nonneutralizing and gp41-directed antibody F240 also did not bind efficiently to the cleaved functional Env spikes. The V3-directed MAb 447-52D can neutralize JRFL at relatively high concentrations, exceeding 50 μg/ml, and did recognize the spikes better than the nonneutralizing MAbs, such as F105 or the cluster I, gp41-directed MAb, F240. There appeared to be a semiquantitative level of binding, roughly exceeding 900 MFI at the highest antibody concentration used in the assay, which correlated with the ability to neutralize JRFL (Fig. 2D and E).
Fig 2.

FACS-based binding curves of MAbs to the cell surface JRFL-cleaved trimers. (A) The glycan-related bNabs; (B) the CD4bs-directed bNabs; (C) nonneutralizing or non-bNabs; (D and E) an arbitrary MFI value at approximately 900 maximal MFI at the highest MAb concentrations tested (dashed lines) correlated with the ability of PGT135 and 447-52D to weakly neutralize JRFL pseudovirus. The other MAbs that bind at levels below the observed cutoff MFI shown cannot neutralize JRFL, a clear correlate of binding with MAb neutralization capacity.
Binding kinetics to the JRFL gp140-F trimers.
As shown in Fig. 3A, stable and homogenous JRFL gp140-F glycoproteins were purified from the supernatants of the 293F cells as described previously. In brief, following lectin affinity chromatography, chelation affinity chromatography, and SEC, the purified gp140-F oligomers were homogenous and trimeric as verified by blue native PAGE. We sought to determine the recognition of the purified JRFL gp140-F trimers by selected Env-specific MAbs displaying neutralizing and nonneutralizing capacity. Therefore, we performed kinetic binding measurements on the JRFL gp140-F trimers captured on the surface of the Octet red probe with selected monomeric Fabs in solution as analytes. Using this assay format, it is more straightforward to accurately assess on rate, off rate, and affinity to trimer Env in the absence of the often confounding avidity effects when using bivalent IgG as the analyte.
Fig 3.
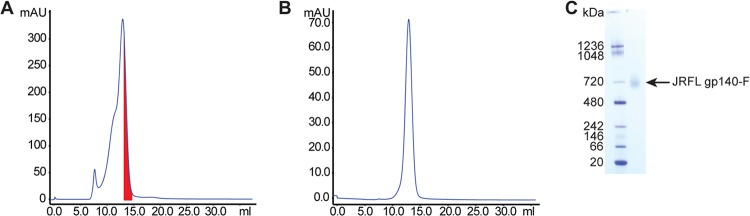
Purification of JRFL gp140-F trimers. (A) The SEC profile of affinity-purified JRFL gp140-F(His6) trimers; the fractions of the major peak containing the trimeric proteins are indicated in red. (B) These fractions were pooled and rerun by SEC to confirm that the purity of the JRFL gp140-F proteins were near homogeneity. (C) The blue native gel analysis of JRFL gp140-F trimers (right lane) with the molecular mass markers (left lane).
High-affinity binding was observed for the glycan-related bNab, PGT128, and most of the CD4bs-directed antibodies examined (Fig. 4). These data suggest that the structure of the Env critical for binding to these antibodies is intact and in some ways approximates the native trimeric spike conformation. The on rate for the Fab derived from CD4i antibody, 17b, was markedly slower than that for the other Fabs examined, such as the CD4bs-directed bNabs, VRC01, PGV04, and b12. Therefore, we assessed 17b binding as a full IgG along with the bNabs PGV04 and VRC03, from which we had difficulty generating functional Fabs. The 17b binding results suggest that the JRFL trimers (in a pre-CD4 bound state) do not well expose the nonneutralizing 17b epitope, which overlaps with the coreceptor binding site. This binding profile is consistent with the lack of coreceptor exposure or formation on the native viral trimer. Furthermore, 17b recognition of the trimers could be induced by sCD4, also consistent with a well-folded trimer (see Fig. S1 in the supplemental material). However, the JRFL virus nonneutralizing CD4bs antibodies, F105 and b6 (Fig. 4; see also Table S1 in the supplemental material), efficiently recognized these trimers, suggesting that the JRFL gp140-F trimers are not optimal mimetics of the functional spike. This is in contrast to the cleaved JRFL cell surface trimers, which display a more restricted antigenic profile, as shown above.
Fig 4.

Binding kinetics of selected Fabs and IgGs to the JRFL gp140-F trimers. Recognition of purified JRFL gp140-F trimeric protein by the Fabs of the glycan-directed PGT128, the CD4bs-neutralizing VRC01 and b12 and nonneutralizing F105, and the CD4i-directed 17b was assessed by Octet using bio-layer interferometry; representative curves are shown. We had difficulty generating functional Fabs for the bNabs PGV04 and VRC03, so we assessed binding of the full IgGs for PGV04 and VRC03 compared to 17b as assessed by Octet bio-layer interferometry as described above.
EP of Env plasmid DNA elicits IgG binding titers that are boosted by protein trimers.
To evaluate the capacity of cleaved trimers to prime for the elicitation of broadly neutralizing antibody responses, we inoculated NHPs at weeks 0, 4, and 8 by electroporation (EP) with plasmid DNA encoding JRFL cell surface, cleavage-competent Env [pSVIII JRFL(+)ΔCT]. Plasmid DNA encoding HIV Tat was coadministered in two ratios relative to the concentration of the Env plasmid (1:1 and 1:20), since we had not yet evaluated this parameter in vivo. Tat expression is required for efficient transcription of the Env structural mRNA from the HIV-1 LTR promoter used in this vector. Note that both plasmids would need to enter the same cell following EP to mediate efficient cell surface JRFL Env expression, another aspect of this approach not previously evaluated in vivo. In other groups of animals, we assessed plasmid DNA expressing codon-optimized cell surface JRFL Env under the control of the CMV promoter and with a heterologous CD5 leader, a means of Env expression that doesn't result in efficient cleavage in vitro. The CMV promoter-driven plasmid vector expressing soluble JRFL gp140-F trimers was used to assess priming by the uncleaved soluble gp140 trimers compared to the cell surface gp160 Env. For the latter two plasmids, Tat expression is not required for efficient transcription and elongation of Env mRNA. The animals in each group received three DNA inoculations, which for the purpose of this study are considered a “prime,” before receiving JRFL gp140-F protein trimers as two boosts in adjuvant. The final group of animals, comprised of three NHPs, did not receive any Env plasmid DNA priming but did receive the two inoculations with JRFL gp140-F trimer protein formulated in adjuvant. Preimmune bleeds were obtained and analyzed similarly as negative controls for the study, and we also included 2 NHPs that were inoculated with PBS instead of Env (see Fig. S5 in the supplemental material).
Following completion of the inoculation regimens, serum samples were collected from animals 2 weeks after each inoculation of DNA EP and were tested for the presence of antibodies recognizing JRFL gp140-F protein by ELISA. Virtually no ELISA binding titers of JRFL gp140-specific IgG antibodies could be detected following the first Env plasmid DNA inoculation (Fig. 5); however, IgG antibody titers increased after subsequent immunizations in all of the DNA EP-inoculated animals (Fig. 5). Furthermore, the antibody titer increased by approximately 20-fold after a single JRFL gp140-F trimeric protein boost. Antibody titers in the sera from the animals inoculated a single time with JRFL protein trimers were relatively low but detectable (as indicated in Fig. 5, bottom left). These data suggest that DNA priming by EP resulted in JRFL Env expression in vivo and induction of Env-specific antibody responses. Similar levels of antibody titers were observed in animals that received two inoculations of JRFL gp140-F trimers (Fig. 5, underlying red curves) and in animals that received three DNA inoculations followed by two protein boosts, where the responses were roughly 100-fold-increased over the three DNA priming inoculations alone.
Fig 5.
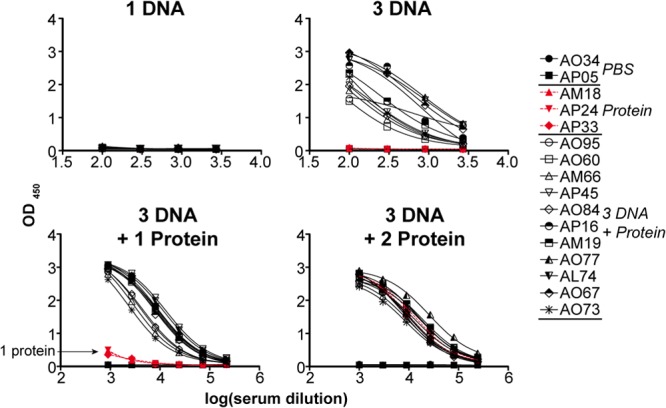
ELISA binding titers to JRFL gp140-F trimers elicited by DNA EP of cell surface trimers and protein boosting. The presence of anti-Env-specific antibody in serum samples derived from the trimer-inoculated NHPs was detected by binding to trimeric JRFL gp140-F immobilized to the solid phase by ELISA. The sera from NHPs inoculated with PBS were included as negative controls (A034 and AP05), and there was no detectable binding of prebleed sera. Top left and right panels depict the ELISAs performed with antisera collected after 1 and 3 EP DNA immunizations, respectively. The bottom left and right panels show the results of ELISA performed with antisera collected after 3 DNA immunizations followed by 1 and 2 trimeric protein boosts, respectively. The symbols and the animal numbers shown on the right designate the individual NHPs from which serum was isolated.
One concern when presenting the trimeric, cleaved but labile HIV-1 spike by genetic means is that the spike might dissociate over time and generate soluble gp120 or gp41 six-helix bundle remnants on the cell surface. These dissociation products might then be presented to the immune system and drive irrelevant antibody specificities. We therefore examined if the DNA priming with the cleaved spikes would generate gp41-directed antibody responses, indicative of such dissociation events. To test for this possibility, we examined serum binding to recombinant gp41 by ELISA and binding to a peptide derived from the immunodominant cluster 1 region of gp41, an epitope region that is exposed when gp41 acquires the six-helix bundle conformation. After the three EP DNA primes, the responses to the recombinant gp41 were detectable but low, perhaps not surprising since the overall gp140 binding titers were relatively low following DNA inoculation alone (see Fig. S2 to S4 in the supplemental material). After one protein boost, the cross-reactive gp41 titers increased considerably, and this increased to even higher levels after two protein boosts. Surprisingly, even though there was detectable overall gp41 binding elicited following the protein boost, responses to the cluster 1 peptide, which is quite well recognized by the human cluster 1 MAb, 7B2, remained low following all JRFL Env trimer inoculations, although this is reputed to be the most immunogenic gp41 epitope during infection.
Neutralization of pseudoviruses by Env-elicited antisera.
Next, we performed analysis of the sera derived from the JRFL trimer-immunized NHPs by both TZM-bl and A3R5 neutralization assays. The TZM-bl assay is a fully validated and standardized HIV-1 neutralization assay (44); the A3R5 assay (34) is more sensitive and is currently undergoing formal validation. Results from both assays are summarized in Fig. 5 and 6.
Fig 6.

TZM-bl ID50 values from the sera of trimer-inoculated NHPs. (A) ID50 neutralization values compared to the corresponding prebled sera of different groups of immunized NHP sera were tested against HIV-1 isolates in a standard TZM-bl assay. Lower-dose values were obtained with sera collected after 3 DNA inoculations, and increasingly higher neutralization titers were obtained with sera collected after 1 protein boost or 2 protein boosts. ID50 values from 50 to 99 are coded in yellow, those from 100 to 999 are highlighted in orange, and the values above 1,000 are marked in red. Neutralization values above the black bar are derived from DNA-primed, trimer protein-boosted animals, whereas values below the black bar are derived from animals immunized with trimer protein alone. Sera derived from PBS-inoculated animals were used as negative controls for HIV-1 neutralization specificity, and the sera derived from Env-inoculated animals was confirmed not to neutralize virus pseudotyped with murine leukemia virus (MuLV) Env (see Fig. S5 in the supplemental material). (B) Left, scatter plot of ID50 values comparing 2 JRFL trimer protein-elicited neutralization values to 3 DNA and 2 trimer protein-elicited neutralization values. Right, scatter plot comparing JRFL trimer-elicited titers (2 and 5 inoculations) to YU2 gp140-F trimer-elicited titers after 5 inoculations (short intervals of 1 month) on viruses MN, HXBc2, SF162, BaL, SS1196, JRFL, YU2, 6535, and MW965. The plotted JRFL values are derived from the data shown in panel A, and the YU2 trimer-elicited TZM-bl titers were previously described (32). The horizontal bars indicate the geometric mean of the ID50 values.
As seen in Fig. 6, in the TZM-bl assay, there was considerable neutralization detectable against tier 1A viruses. In fact, for some sera, one can detect neutralization after the three plasmid EP DNA inoculations alone (see Fig. S5 in the supplemental material). Comparison of the 50% infective dose (ID50) neutralization values between the different immunization regimens is shown in Fig. 6 (see Materials and Methods). For ease of visual comparison, the values are color coded as described. Essentially, the “hotter” the color (i.e., red), the more the serum could be diluted and still inhibit 50% of viral entry. For example, the neutralization activity increased substantially when the DNA priming was followed by one protein boost in most animals (see Fig. S5). After the single protein boost, reasonable titers of neutralization were detected against the clade B and clade C tier 1 viruses MN, HXBc2, SF162, BaL, and MW965 in most animals from each of the experimental groups receiving this regimen (see Fig. S5). As expected, no neutralization was detectable in animals AM18, AP24, and AP33 after a single protein inoculation, as these animals had not received the Env plasmid DNA prime. In contrast, most animals inoculated with DNA three times displayed neutralizing activity following one protein inoculation that further increased in potency and breadth following the second protein inoculation (Fig. 7; see Fig. S5 in the supplemental material). Note that against the more sensitive viruses in the TZM-bl assay, following DNA and two protein boosts, some ID50 titers approached or exceeded a dilution factor of 50,000. Additionally, there were substantial titers detected against the generally more resistant tier 1B isolates, such as S1196 and BaL, against which robust neutralizing responses are not usually achieved (32). These data suggest that the DNA primed the animals for elicitation of potent neutralizing antibodies after a single protein boost, which was boosted further by the second trimer protein inoculation.
Fig 7.

Tier 2 A3R5 ID50 values from the sera of trimer-inoculated NHPs. (A) ID50 neutralization values of JRFL Env-immunized NHP sera were tested against tier 2 HIV-1 isolates and one tier 1 isolate (CAP45) in the A3R5 assay. IC50s from 20 to 99 are coded in yellow, and those from 100 to 999 are highlighted in orange, and the values above 1,000 are marked in red. Neutralization values above the black bar are derived from DNA-primed, trimer protein-boosted animals, whereas values below the black bar are derived from animals immunized with trimer protein alone. Sera from the PBS-inoculated animals and prebleed sera were used as negative controls for neutralization specificity. (B) Scatter plot of A3R5 ID50 values comparing JRFL-elicited sera following 5 inoculations (3 DNA and 2 protein) to YU2 trimer protein-elicited antisera following 5 inoculations of protein for the five viruses tested: SC22, RHPA, CH58, Ce1086, and DU151 (see the numeric values in Fig. S7 in the supplemental material). The horizontal bars indicate the geometric mean of the ID50 values.
Interestingly, sporadic but modest neutralization activity was detected against the tier 2 isolates JRFL, SC422, and ZM109 but only and exclusively in the animals that received the DNA priming and only following the two additional JRFL trimer protein boosts. Although the tier 2 neutralization is infrequent, for the six tier 2 viruses analyzed, there is detectable neutralization in 16 of the 66 instances (Fig. 6A, above the black bar). This contrasts to the protein-alone group in which there was no detectable tier 2 virus neutralization detected in the 18 cases tested (below the black bar), and this difference is statistically significant (P = 0.027: Fig. 6B, left). Compared to our previous immunization studies in NHPs with recombinant YU2 gp140-F trimers inoculated at shorter monthly intervals, this is the first time that we have observed this level of neutralization in the TZM-bl assay against the more resistant tier 2 isolates (32). If one evaluates the data by each group individually, there is a statistically significant difference between groups (P = 0.022), but this difference cannot be attributed to any specific regimen (see Fig. S6 in the supplemental material). Note that no apparent difference was observed between the non-codon-optimized env DNA and the codon-optimized env cell surface or genes encoding the soluble JRFL gp140-F trimers each used for priming the Env-specific B cell responses. Additionally, if we compared JRFL trimer-elicited neutralization to YU2 trimer-elicited neutralization in the TZM-bl assay, the JRFL trimers also generated higher titers for several clade B isolates (P < 0.0001) and one clade C isolate (see Fig. 6B).
Encouraged by detectable tier 2 neutralization in the TZM-bl assay, we next analyzed the serum neutralization capacity against several tier 2 isolates in the more sensitive A3R5 assay. In the A3R5 assay, we observed much more potent and consistent neutralization of both tier 2 clade B and tier 2 clade C HIV-1 isolates and one tier 1 clade isolate, CAP45 (Fig. 7; see also Fig. S7 in the supplemental material). This tier 2 neutralization activity was not dependent upon DNA priming, as such neutralization was observed even in the three NHPs that received two protein inoculations in adjuvant without prior Env DNA inoculation. Note that, importantly, and to place the A3R5 neutralization results in context, the responses that are reported here, measured against tier 2 viruses in the same A3R5 assay and performance laboratory, are superior to the responses detected in immune sera generated from human clinical trials of Env candidates, including those assessed in Vax003 and RV144 (34). For the latter human clinical trial, which resulted in modest efficacy of protection (47), there was essentially no detectable neutralization of tier 2 isolates generated from vaccinee sera (34). Similarly, the primate neutralizing responses elicited by the JRFL trimers as reported here exceeded those generated in both the Vax004 and HVTN 049 human clinical trials (D. Montefiori, personal communication).
To assess further the context of the JRFL trimer-elicited A3R5 tier 2 neutralizing activity, we compared neutralizing activity that we had previously elicited by 5 inoculations of the YU2 gp140-F trimers formulated in adjuvant into NHPs (32). As seen in the Fig. 7B scatter plot comparisons, the sera elicited by 5 inoculations of the JRFL trimers displayed significantly higher geometric mean titers (GMT) than did the YU2 trimers when analyzed on the same virus isolates (P = 0.037 for clade B and P = 0.003 for clade C), suggesting that the JRFL trimers elicit more potent neutralizing titers of the tier 2 isolates examined. The precise neutralization titers elicited by the YU2 gp140-F trimers in the A3R5 assay used to generate the scatter plot in Fig. 7 are shown in Fig. S8 in the supplemental material.
JRFL trimer-elicited A3R5 tier 2 neutralization maps to the HIV-1 CD4bs.
In our previous NHP studies, we were able to map neutralization elicited by the YU2 gp140-F trimers to the CD4bs using the HXBc2 virus (32, 48), which we confirmed with selected sera in our current study. To expand this analysis, we sought to determine if any of the regimens used here elicited CD4bs-directed Nab responses against a tier 2 isolate in the A3R5 assay. To detect CD4bs-directed neutralization, we performed differential protein competition assays using a system previously described that employs modified gp120 core glycoproteins (39). In brief, we used a pair of probes, TriMut and TriMut 368/370 gp120 cores, which contain three mutations in the gp120 bridging sheet region [423(I/M), 425(N/K), and 431(G/E)] that eliminate CD4 binding but do not affect recognition by any of the known CD4bs antibodies (39, 49). Because the TriMut core proteins are rendered CD4 binding defective by these modifications, they can be directly added into the HIV entry assay and do not interfere with the CD4-dependent virus entry process. The first probe, TriMut 368/370, contains two additional mutations in the CD4 binding loop [368(D/R) and 370(E/F)] which specifically eliminate recognition by most, but not all, CD4bs-directed antibodies. Recognition by all known CD4bs MAbs can be eliminated by a third change in the CD4 binding region at residue 474 (50), which was used to generate TriMut 368/370/474. Specificity to detect CD4bs-directed neutralization by direct addition to the neutralization assay was verified using VRC01, a broadly neutralizing human antibody directed to CD4bs. For each serum sample, three neutralization assays were performed with TriMut, TriMut 368/370, or cell-culture medium (mock) added to the neutralization assay. As a control, and as seen, VRC01-meditated neutralization of SC22 could be absorbed by the TriMut core but not by the TriMut 368/370 or TriMut 368/370/474 cores, validating the differential neutralization absorption process (Fig. 8A). Using this assay with the tier 2 isolate, SC22, CD4bs-directed neutralization of selected sera was mapped to the CD4bs by this differential absorption process (Fig. 8). Note that in serum sample AL74, the TriMut core could adsorb out all neutralization; however, following 368/370 adsorption, some residual neutralization remained, indicating some of the activity was directed against this site. The 368/370/474 failed to adsorb out any neutralization, indicating that for most sera tested, neutralization at the CD4bs is 368/370 focused; however, for this serum sample, there was some activity also directed against the 474 residue-containing portion of the general CD4 binding region.
Fig 8.
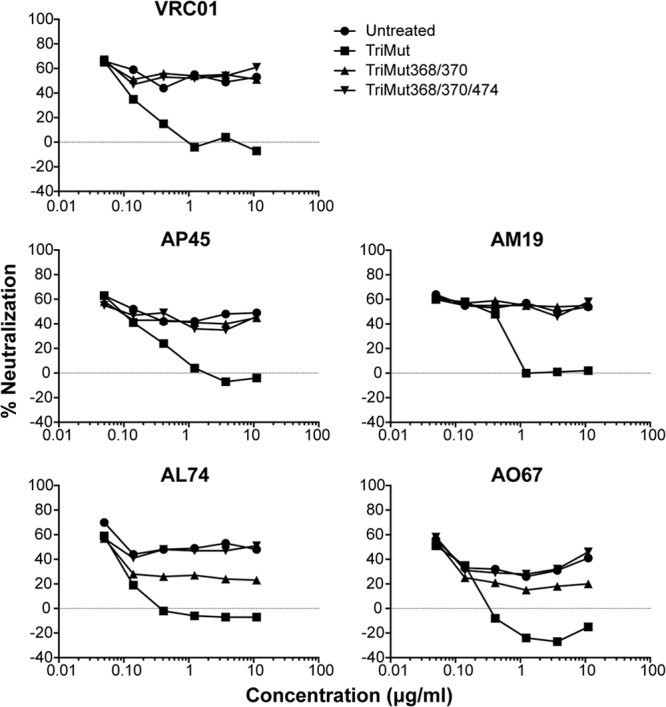
CD4bs mapping of selected NHP sera. Shown are differential protein inhibition curves of neutralization using the TriMut core containing the triple mutations (I423M, N425K, and G431E), TriMut 368/370 core containing two additional mutations (D368R and E370F), and a third TriMut 368/370/474 core containing the 368/370 and 474 mutations as designated. The different protein probes were serially diluted and added to the MAb or serum to either a fixed concentration of VRC01 (top) or a fixed dilution of sera. Following incubation, the mixtures were assessed in the neutralization assay to determine the capacity to block the neutralization either by no protein (blank) or TriMut core or the CD4bs knockout cores TriMut 368/370 or TriMut 368/370/474. The differential protein inhibition effects were evaluated by the extent of neutralization detected in the A3R5 assay.
DISCUSSION
In this study, we assessed the capacity of HIV-1 Env trimers based upon the primary isolate, JRFL, to elicit broadly neutralizing antibodies in primates. Thereby, we inoculated NHPs with DNA encoding fully cleaved cell surface JRFL trimers to preferentially elicit B cells bearing B cell receptors capable of accessing conserved and shielded neutralizing epitopes on these cleaved spikes. In contrast to two forms of cell surface JRFL Env, we also included a DNA prime consisting of soluble, uncleaved gp140-F trimers. We then boosted the DNA-primed Env-specific antibody responses by two inoculations of soluble JRFL spikes administered in adjuvant. Interestingly, infrequent but detectable tier 2 neutralizing activity was observed in the TZM-bl assay, and this activity was dependent upon plasmid DNA priming and the longer interval used in this prime-boost regimen than the shorter regimen consisting of two protein inoculations alone. The tier 2 neutralizing capacity of the elicited sera did not appear to be dependent upon priming with cell surface-cleaved JRFL Env, as priming with soluble trimer DNA also generated such activity. Consistent with these data, previous studies have demonstrated improved neutralization following HIV-1 Env DNA prime-protein boost regimens compared to inoculation of Env gp120 protein alone (51–55). In the more sensitive A3R5 assay, we observed high to modest tier 2 neutralization of all clade B and clade C isolates tested in the serum of most trimer-inoculated animals. However, A3R5-detectable neutralizing activity was not dependent upon Env DNA priming, as two inoculations with the soluble JRFL trimers alone elicited such activity, which was superior to our previous study using YU2-foldon trimers. This improvement might be due to the longer prime-boost interval, the Adjuplex adjuvant used in the current study, or the strain differences in the derivation of the Env trimers.
The binding data of the cell surface JRFL trimers presented in Fig. 2 reinforce our previous observations that these cleaved trimers are selectively recognized by bNabs but not by nonneutralizing or non-broadly neutralizing, gp120-specific MAbs. This association implies that the neutralizing antibodies have relatively rapid on rates and relatively slow off rates to the functional spike compared to those of the non-bNabs and the nonneutralizing MAbs. That MAb recognition above the threshold level identified in our binding analysis (Fig. 2B) correlated with general neutralization capacity of these antibodies supports the notion that MAb recognition of the cleaved JRFL cell surface trimers is essentially a surrogate binding assay that can predict neutralization capacity of gp120-directed MAbs.
The binding kinetics of selected Fabs and IgGs to the soluble JRFL gp140-F trimers presented in Fig. 4 indicated that the JRFL trimers possess some properties consistent with the native Env spike. However, that these trimers are recognized by nonneutralizing antibodies F105 and b6 indicates that these trimers are not optimal mimetics of functional, cleaved Env spikes. However, our hypothesis was that since we primed B cell responses with the cleaved spikes by DNA EP, which presents only epitopes recognized by the bNabs, such cell surface-presented trimers might activate B cells possessing antibody specificities capable of penetrating the spike shields, which can be expanded upon boosting with the soluble trimers even if they are less faithful mimics of the functional spike. That Env proteins expressed from the non-codon-optimized cleavage-competent Env vector, which is cleaved in 293T cells, and the codon-optimized cleavage competent Env, which is not cleaved in 293 cells, were roughly equivalent suggests that the role of Env cleavage in this study requires further investigation. A recent study demonstrates more complete gp160 precursor cleavage in human osteosarcoma (HOS) cells compared to expression of the same Envs in 293 cells, indicating that in the muscle cells transfected in vivo by EP, there could be more precursor cleavage ongoing than anticipated from our in vitro data (56, 57).
The ELISA binding data shown in Fig. 5 clearly demonstrate that the EP DNA trimer priming elicits Env-specific binding titers, beyond the usual capacity of plasmid DNAs delivered by other means, such as needle injection of DNA or gene gun delivery (57). These other DNA delivery means prime B cell responses that can be boosted by subsequent protein inoculation, but with lower titers than detected here. That two proteins in adjuvant elicited similar binding titers to a single boost of protein primed by DNA suggested that a DNA prime was essentially equal to a protein prime at the quantitative level of binding antibodies, as has been observed in several other studies using a similar DNA prime-protein boost format (51–55).
The neutralization data presented in the validated TZM-bl assay, in contrast with the neutralization data presented in the A3R5 assay, are the most critical results that are comparatively assessed in this study. A recent previous report analyzing antisera generated by repeated clade C trimer inoculation into guinea pigs also reported detectable clade C-matched tier 2 neutralizing activity in the A3R5 assay (58), consistent with the cross-clade neutralization observed in our current study. The current JRFL trimer-elicited titers exceeded previous peak neutralization detected in the A3R5 assay in the modestly successful human clinical trial RV144 and the higher responses in the nonefficacious trial, Vax003 (34). Although there are many different variables between the human clinical trials and the primate preclinical study described here, the comparative data performed in the same laboratory with sera from each of the studies provide some context to interpret the JRFL Env-elicited A3R5 neutralization data. In the analysis of these previous studies, the short-lived kinetics of the response to Env was also examined. Here, we did not examine the durability of the B cell response; however, in similar Env immunogenicity studies, gp120-directed antibody responses did wane over time (59) and would be expected to do so as well here.
One interesting observation is that although the binding titers elicited by the three DNA priming inoculations were of relatively low magnitude, in a few sera, HIV-1 neutralization could be detected. This indicates that although EP of plasmid DNA may not elicit antibodies of high magnitude, the level of neutralizing activity might be higher relative to the elicited quantitative binding titers. These results suggest that priming with plasmid DNA expressing cleaved, cell surface-presented (JRFL) trimers, which selectively present broadly neutralizing determinants, might more efficiently promote neutralizing antibodies, especially if this means of presentation can be rendered more immunogenic. However, as noted previously, priming with DNA encoding uncleaved gp140-F trimers, followed by protein boosting, also elicited detectable tier 2 neutralizing activity. Finally, mapping studies indicate that for the tier 2 clade B isolate, SC22, most of the neutralizing activity could be mapped to the CD4bs, consistent with our previous study where approximately 10% of the B cell response to the YU2 gp140-F trimers is directed toward this region (33). Such results bode well for eliciting neutralizing antibodies against this conserved neutralizing surface of HIV-1 Env.
In sum, this study presents interesting results in the so far generally futile quest to elicit broadly neutralizing HIV-1 antibodies by Env vaccination into small animals or primates. The results suggest, consistent with previous studies (36, 51, 52, 54, 55), that genetic priming with Env DNA to the primate immune system may enhance the ability of soluble spike Env mimetics to elicit tier 2 broadly neutralizing antibodies. And that perhaps one pathway forward would be to enhance the immunogenicity of the Env prime by, for example, presenting more stable and antigenically well-structured Env trimers to the B cell immune repertoire.
Supplementary Material
ACKNOWLEDGMENTS
We thank Christine Corbaci for expert assistance with the figures, Joanne DeStefano and the IAVI staff for handling and coordination of the NHP experiments at SUNY, and Christopher Sundling for technical assistance with NHP sample preparation in Stockholm.
This work was supported primarily by IAVI intramural research, as well as by the NIH intramural research program, SIDA, The Bill and Melinda Gates Foundation, and the Scripps CHAVI-ID.
Footnotes
Published ahead of print 25 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01247-13.
REFERENCES
- 1.Center RJ, Leapman RD, Lebowitz J, Arthur LO, Earl PL, Moss B. 2002. Oligomeric structure of the human immunodeficiency virus type 1 envelope protein on the virion surface. J. Virol. 76:7863–7867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273 [DOI] [PubMed] [Google Scholar]
- 3.Chan DC, Kim PS. 1998. HIV entry and its inhibition. Cell 93:681–684 [DOI] [PubMed] [Google Scholar]
- 4.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. 1996. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85:1135–1148 [DOI] [PubMed] [Google Scholar]
- 5.Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312:763–767 [DOI] [PubMed] [Google Scholar]
- 6.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661–666 [DOI] [PubMed] [Google Scholar]
- 7.Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. 1999. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 99:103–115 [DOI] [PubMed] [Google Scholar]
- 8.Gallo SA, Finnegan CM, Viard M, Raviv Y, Dimitrov A, Rawat SS, Puri A, Durell S, Blumenthal R. 2003. The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta 1614:36–50 [DOI] [PubMed] [Google Scholar]
- 9.Hallenberger S, Bosch V, Angliker H, Shaw E, Klenk HD, Garten W. 1992. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 360:358–361 [DOI] [PubMed] [Google Scholar]
- 10.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430 [DOI] [PubMed] [Google Scholar]
- 11.Willey RL, Bonifacino JS, Potts BJ, Martin MA, Klausner RD. 1988. Biosynthesis, cleavage, and degradation of the human immunodeficiency virus 1 envelope glycoprotein gp160. Proc. Natl. Acad. Sci. U. S. A. 85:9580–9584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. 1996. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 384:179–183 [DOI] [PubMed] [Google Scholar]
- 13.Hessell AJ, Poignard P, Hunter M, Hangartner L, Tehrani DM, Bleeker WK, Parren PW, Marx PA, Burton DR. 2009. Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat. Med. 15:951–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hessell AJ, Rakasz EG, Poignard P, Hangartner L, Landucci G, Forthal DN, Koff WC, Watkins DI, Burton DR. 2009. Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog. 5:e1000433. 10.1371/journal.ppat.1000433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hessell AJ, Rakasz EG, Tehrani DM, Huber M, Weisgrau KL, Landucci G, Forthal DN, Koff WC, Poignard P, Watkins DI, Burton DR. 2009. Broadly neutralizing monoclonal antibodies 2F5 and 4E10, directed against the human immunodeficiency virus type 1 (HIV-1) gp41 membrane proximal external region (MPER), protect against SHIVBa-L mucosal challenge. J. Virol. 84:1302–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mascola JR, Lewis MG, Stiegler G, Harris D, VanCott TC, Hayes D, Louder MK, Brown CR, Sapan CV, Frankel SS, Lu Y, Robb ML, Katinger H, Birx DL. 1999. Protection of macaques against pathogenic simian/human immunodeficiency virus 89.6PD by passive transfer of neutralizing antibodies. J. Virol. 73:4009–4018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibata R, Igarashi T, Haigwood N, Buckler-White A, Ogert R, Ross W, Willey R, Cho MW, Martin MA. 1999. Neutralizing antibody directed against the HIV-1 envelope glycoprotein can completely block HIV-1/SIV chimeric virus infections of macaque monkeys. Nat. Med. 5:204–210 [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Migueles SA, Welcher B, Svehla K, Phogat A, Louder MK, Wu X, Shaw GM, Connors M, Wyatt RT, Mascola JR. 2007. Broad HIV-1 neutralization mediated by CD4-binding site antibodies. Nat. Med. 13:1032–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Svehla K, Louder MK, Wycuff D, Phogat S, Tang M, Migueles SA, Wu X, Phogat A, Shaw GM, Connors M, Hoxie J, Mascola JR, Wyatt R. 2009. Analysis of neutralization specificities in polyclonal sera derived from human immunodeficiency virus type 1-infected individuals. J. Virol. 83:1045–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore PL, Gray ES, Sheward D, Madiga M, Ranchobe N, Lai Z, Honnen WJ, Nonyane M, Tumba N, Hermanus T, Sibeko S, Mlisana K, Abdool Karim SS, Williamson C, Pinter A, Morris L, Caprisa 002 Study 2011. Potent and broad neutralization of HIV-1 subtype C by plasma antibodies targeting a quaternary epitope including residues in the V2 loop. J. Virol. 85:3128–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sather DN, Stamatatos L. 2010. Epitope specificities of broadly neutralizing plasmas from HIV-1 infected subjects. Vaccine 28(Suppl 2):B8–B12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TY, Pietzsch J, Fenyo D, Abadir A, Velinzon K, Hurley A, Myung S, Boulad F, Poignard P, Burton DR, Pereyra F, Ho DD, Walker BD, Seaman MS, Bjorkman PJ, Chait BT, Nussenzweig MC. 2011. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333:1633–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong CH, Phogat S, Wrin T, Simek MD, Koff WC, Wilson IA, Burton DR, Poignard P. 2011. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477:466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O'Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. 2012. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491:406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klein F, Halper-Stromberg A, Horwitz JA, Gruell H, Scheid JF, Bournazos S, Mouquet H, Spatz LA, Diskin R, Abadir A, Zang T, Dorner M, Billerbeck E, Labitt RN, Gaebler C, Marcovecchio PM, Incesu RB, Eisenreich TR, Bieniasz PD, Seaman MS, Bjorkman PJ, Ravetch JV, Ploss A, Nussenzweig MC. 2012. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature 492:118–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forsell MN, Schief WR, Wyatt RT. 2009. Immunogenicity of HIV-1 envelope glycoprotein oligomers. Curr. Opin. HIV AIDS 4:380–387 [DOI] [PubMed] [Google Scholar]
- 29.Grundner C, Mirzabekov T, Sodroski J, Wyatt R. 2002. Solid-phase proteoliposomes containing human immunodeficiency virus envelope glycoproteins. J. Virol. 76:3511–3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pancera M, Wyatt R. 2005. Selective recognition of oligomeric HIV-1 primary isolate envelope glycoproteins by potently neutralizing ligands requires efficient precursor cleavage. Virology 332:145–156 [DOI] [PubMed] [Google Scholar]
- 31.Herrera C, Klasse PJ, Michael E, Kake S, Barnes K, Kibler CW, Campbell-Gardener L, Si Z, Sodroski J, Moore JP, Beddows S. 2005. The impact of envelope glycoprotein cleavage on the antigenicity, infectivity, and neutralization sensitivity of Env-pseudotyped human immunodeficiency virus type 1 particles. Virology 338:154–172 [DOI] [PubMed] [Google Scholar]
- 32.Sundling C, Forsell MN, O'Dell S, Feng Y, Chakrabarti B, Rao SS, Lore K, Mascola JR, Wyatt RT, Douagi I, Karlsson Hedestam GB. 2010. Soluble HIV-1 Env trimers in adjuvant elicit potent and diverse functional B cell responses in primates. J. Exp. Med. 207:2003–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sundling C, Li Y, Huynh N, Poulsen C, Wilson R, O'Dell S, Feng Y, Mascola JR, Wyatt RT, Karlsson Hedestam GB. 2012. High-resolution definition of vaccine-elicited B cell responses against the HIV primary receptor binding site. Sci. Transl. Med. 4:142ra96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montefiori DC, Karnasuta C, Huang Y, Ahmed H, Gilbert P, de Souza MS, McLinden R, Tovanabutra S, Laurence-Chenine A, Sanders-Buell E, Moody MA, Bonsignori M, Ochsenbauer C, Kappes J, Tang H, Greene K, Gao H, LaBranche CC, Andrews C, Polonis VR, Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Self SG, Berman PW, Francis D, Sinangil F, Lee C, Tartaglia J, Robb ML, Haynes BF, Michael NL, Kim JH. 2012. Magnitude and breadth of the neutralizing antibody response in the RV144 and Vax003 HIV-1 vaccine efficacy trials. J. Infect. Dis. 206:431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morner A, Douagi I, Forsell MN, Sundling C, Dosenovic P, O'Dell S, Dey B, Kwong PD, Voss G, Thorstensson R, Mascola JR, Wyatt RT, Karlsson Hedestam GB. 2009. Human immunodeficiency virus type 1 env trimer immunization of macaques and impact of priming with viral vector or stabilized core protein. J. Virol. 83:540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sundling C, O'Dell S, Douagi I, Forsell MN, Morner A, Lore K, Mascola JR, Wyatt RT, Karlsson Hedestam GB. 2010. Immunization with wild-type or CD4-binding-defective HIV-1 Env trimers reduces viremia equivalently following heterologous challenge with simian-human immunodeficiency virus. J. Virol. 84:9086–9095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sodroski JG, Rosen CA, Haseltine WA. 1984. Trans-acting transcriptional activation of the long terminal repeat of human T lymphotropic viruses in infected cells. Science 225:381–385 [DOI] [PubMed] [Google Scholar]
- 38.Dey B, Svehla K, Xu L, Wycuff D, Zhou T, Voss G, Phogat A, Chakrabarti BK, Li Y, Shaw G, Kwong PD, Nabel GJ, Mascola JR, Wyatt RT. 2009. Structure-based stabilization of HIV-1 gp120 enhances humoral immune responses to the induced co-receptor binding site. PLoS Pathog. 5:e1000445. 10.1371/journal.ppat.1000445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng Y, McKee K, Tran K, O'Dell S, Schmidt SD, Phogat A, Forsell MN, Karlsson Hedestam GB, Mascola JR, Wyatt RT. 2012. Biochemically defined HIV-1 envelope glycoprotein variant immunogens display differential binding and neutralizing specificities to the CD4-binding site. J. Biol. Chem. 287:5673–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koch M, Pancera M, Kwong PD, Kolchinsky P, Grundner C, Wang L, Hendrickson WA, Sodroski J, Wyatt R. 2003. Structure-based, targeted deglycosylation of HIV-1 gp120 and effects on neutralization sensitivity and antibody recognition. Virology 313:387–400 [DOI] [PubMed] [Google Scholar]
- 41.Chakrabarti BK, Kong WP, Wu BY, Yang ZY, Friborg J, Ling X, King SR, Montefiori DC, Nabel GJ. 2002. Modifications of the human immunodeficiency virus envelope glycoprotein enhance immunogenicity for genetic immunization. J. Virol. 76:5357–5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chakrabarti BK, Pancera M, Phogat S, O'Dell S, McKee K, Guenaga J, Robinson J, Mascola J, Wyatt RT. 2011. HIV type 1 Env precursor cleavage state affects recognition by both neutralizing and nonneutralizing gp41 antibodies. AIDS Res. Hum. Retroviruses 27:877–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chakrabarti BK, Walker LM, Guenaga JF, Ghobbeh A, Poignard P, Burton DR, Wyatt RT. 2011. Direct antibody access to the HIV-1 membrane-proximal external region positively correlates with neutralization sensitivity. J. Virol. 85:8217–8226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. 2005. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 79:10108–10125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Folks T, Benn S, Rabson A, Theodore T, Hoggan MD, Martin M, Lightfoote M, Sell K. 1985. Characterization of a continuous T-cell line susceptible to the cytopathic effects of the acquired immunodeficiency syndrome (AIDS)-associated retrovirus. Proc. Natl. Acad. Sci. U. S. A. 82:4539–4543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim JH, Pitisuttithum P, Kamboonruang C, Chuenchitra T, Mascola J, Frankel SS, DeSouza MS, Polonis V, McLinden R, Sambor A, Brown AE, Phonrat B, Rungruengthanakit K, Duliege AM, Robb ML, McNeil J, Birx DL. 2003. Specific antibody responses to vaccination with bivalent CM235/SF2 gp120: detection of homologous and heterologous neutralizing antibody to subtype E (CRF01.AE) HIV type 1. AIDS Res. Hum. Retroviruses 19:807–816 [DOI] [PubMed] [Google Scholar]
- 47.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH, Investigators M-T. 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361:2209–2220 [DOI] [PubMed] [Google Scholar]
- 48.Douagi I, Forsell MN, Sundling C, O'Dell S, Feng Y, Dosenovic P, Li Y, Seder R, Lore K, Mascola JR, Wyatt RT, Karlsson Hedestam GB. 2010. Influence of novel CD4 binding-defective HIV-1 envelope glycoprotein immunogens on neutralizing antibody and T-cell responses in nonhuman primates. J. Virol. 84:1683–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiang SH, Wang L, Abreu M, Huang CC, Kwong PD, Rosenberg E, Robinson JE, Sodroski J. 2003. Epitope mapping and characterization of a novel CD4-induced human monoclonal antibody capable of neutralizing primary HIV-1 strains. Virology 315:124–134 [DOI] [PubMed] [Google Scholar]
- 50.Pietzsch J, Scheid JF, Mouquet H, Klein F, Seaman MS, Jankovic M, Corti D, Lanzavecchia A, Nussenzweig MC. 2010. Human anti-HIV-neutralizing antibodies frequently target a conserved epitope essential for viral fitness. J. Exp. Med. 207:1995–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vaine M, Wang S, Crooks ET, Jiang P, Montefiori DC, Binley J, Lu S. 2008. Improved induction of antibodies against key neutralizing epitopes by human immunodeficiency virus type 1 gp120 DNA prime-protein boost vaccination compared to gp120 protein-only vaccination. J. Virol. 82:7369–7378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vaine M, Wang S, Hackett A, Arthos J, Lu S. 2010. Antibody responses elicited through homologous or heterologous prime-boost DNA and protein vaccinations differ in functional activity and avidity. Vaccine 28:2999–3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vaine M, Wang S, Liu Q, Arthos J, Montefiori D, Goepfert P, McElrath MJ, Lu S. 2010. Profiles of human serum antibody responses elicited by three leading HIV vaccines focusing on the induction of Env-specific antibodies. PLoS One 5:e13916. 10.1371/journal.pone.0013916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang S, Kennedy JS, West K, Montefiori DC, Coley S, Lawrence J, Shen S, Green S, Rothman AL, Ennis FA, Arthos J, Pal R, Markham P, Lu S. 2008. Cross-subtype antibody and cellular immune responses induced by a polyvalent DNA prime-protein boost HIV-1 vaccine in healthy human volunteers. Vaccine 26:1098–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang S, Pal R, Mascola JR, Chou TH, Mboudjeka I, Shen S, Liu Q, Whitney S, Keen T, Nair BC, Kalyanaraman VS, Markham P, Lu S. 2006. Polyvalent HIV-1 Env vaccine formulations delivered by the DNA priming plus protein boosting approach are effective in generating neutralizing antibodies against primary human immunodeficiency virus type 1 isolates from subtypes A, B, C, D and E. Virology 350:34–47 [DOI] [PubMed] [Google Scholar]
- 56.Haim H, Salas I, Sodroski J. 2013. Proteolytic processing of the human immunodeficiency virus envelope glycoprotein precursor decreases conformational flexibility. J. Virol. 87:1884–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu S, Santoro JC, Fuller DH, Haynes JR, Robinson HL. 1995. Use of DNAs expressing HIV-1 Env and noninfectious HIV-1 particles to raise antibody responses in mice. Virology 209:147–154 [DOI] [PubMed] [Google Scholar]
- 58.Kovacs JM, Nkolola JP, Peng H, Cheung A, Perry J, Miller CA, Seaman MS, Barouch DH, Chen B. 2012. HIV-1 envelope trimer elicits more potent neutralizing antibody responses than monomeric gp120. Proc. Natl. Acad. Sci. U. S. A. 109:12111–12116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sundling C, Martinez P, Soldemo M, Spangberg M, Bengtsson KL, Stertman L, Forsell MN, Karlsson Hedestam GB. 2013. Immunization of macaques with soluble HIV type 1 and influenza virus envelope glycoproteins results in a similarly rapid contraction of peripheral B-cell responses after boosting. J. Infect. Dis. 207:426–431 [DOI] [PubMed] [Google Scholar]
- 60.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. 2008. Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.