

Abstract
Paramyxovirus membrane glycoproteins F (fusion protein) and HN, H, or G (attachment protein) are critical for virus entry, which occurs through fusion of viral and cellular envelopes. The F protein folds into a homotrimeric, metastable prefusion form that can be triggered by the attachment protein to undergo a series of structural rearrangements, ultimately folding into a stable postfusion form. In paramyxovirus-infected cells, the F protein is activated in the Golgi apparatus by cleavage adjacent to a hydrophobic fusion peptide that inserts into the target membrane, eventually bringing the membranes together by F refolding. However, it is not clear how the attachment protein, known as HN in parainfluenza virus 5 (PIV5), interacts with F and triggers F to initiate fusion. To understand the roles of various F protein domains in fusion triggering and metastability, single point mutations were introduced into the PIV5 F protein. By extensive study of F protein cleavage activation, surface expression, and energetics of fusion triggering, we found a role for an immunoglobulin-like (Ig-like) domain, where multiple hydrophobic residues on the PIV5 F protein may mediate F-HN interactions. Additionally, destabilizing mutations of PIV5 F that resulted in HN trigger-independent mutant F proteins were identified in a region along the border of F trimer subunits. The positions of the potential HN-interacting region and the region important for F stability in the lower part of the PIV5 F prefusion structure provide clues to the receptor-binding initiated, HN-mediated F trigger.
INTRODUCTION
The Paramyxoviridae family is a large group of clinically and economically important viruses of humans and animals and includes parainfluenza viruses 1 to 5 (PIV1 to -5), mumps virus, Newcastle disease virus (NDV), Nipah virus, Hendra virus, measles virus, canine distemper virus (CDV), respiratory syncytial virus (RSV), and human metapneumovirus (hMPV) (1). Paramyxoviruses are enveloped viruses harboring a nonsegmented RNA genome that is negative stranded. These viruses initiate infection by fusion of the viral membrane with a cellular membrane, and the process occurs at neutral pH (except in some strains of hMPV) (2). Membrane fusion is brought about by two viral surface glycoproteins, the fusion (F) protein and the attachment protein, variously named HN, H, or G. The attachment proteins are structurally related despite the fact that HN binds to sialic acid as a receptor, whereas H and G use cell surface proteins as receptors. It is generally believed that HN (H or G) and F physically interact (3–7) and that HN (H or G) triggers the F protein for fusion in a receptor-dependent manner by lowering the activation energy of F refolding. The role of HN, H, or G protein in overcoming the energy barrier required for triggering can be supplanted by using elevated temperature as a surrogate means of activation, as has been shown for PIV5 F, CDV F, and Nipah virus F protein (8–11).
F proteins are synthesized as a precursor (F0) that assembles into a metastable homotrimer known as the prefusion form. To be biologically active, F0 has to be cleaved by a cellular protease into disulfide-linked chains F2 and F1. Cleavage releases covalent restraints on the hydrophobic fusion peptide (FP), which becomes the new N terminus of F1 and is free to relocate and insert into the target cell membrane. Membrane fusion is driven by irreversible protein refolding, involving a series of conformational changes, going from the prefusion to the postfusion form and linking fusion protein refolding to membrane merger. Ultimately, the process results in the formation of a stable, low-energy, postfusion conformation of the F protein (12–14).
Atomic structures of soluble forms of prefusion F of PIV5 and RSV show considerable similarity with a globular head situated above a membrane-proximal stalk region (15, 16). The primary sequence of F predicts two heptad repeat regions, HRA and HRB, separated by ∼250 amino acid residues, with HRA abutting the fusion peptide and HRB abutting the transmembrane domain. In the PIV5 F prefusion structure, HRB domains form a trimeric coiled coil that forms the stalk region, and the globular head is made up of domains I to III (15). Domain II contains a region that folds into an immunoglobulin-like (Ig-like) fold, comprising seven interwoven beta strands held together by disulfide bonds (Fig. 1A and B). In the prefusion form of PIV5 F, the FP lies partially solvent exposed and partially buried and is juxtaposed to the Ig-like domain II, bounded by adjacent protomers. Cleavage to expose the N-terminal hydrophobic end of the FP does not significantly change the overall structure of the PIV5 F prefusion form (17).
Fig 1.

Design of single point mutations in PIV5 F. (A) PIV5 F prefusion trimer showing the positions of mutations (purple) in Ig-like domain II and domain I, designed to disrupt the interaction of PIV5 F with PIV5 HN. One of the PIV5 F protomers is shown as a cartoon representation, while the other two are surface representations. PIV5 F trimer domains are colored as follows: domain I, yellow; domain II, red; domain III, magenta; HRB, blue; fusion peptide, light pink. (B) Enlarged view of panel A, showing the Ig-like domain in cartoon representation. (C) Ribbon representation of the PIV5 F trimer, showing positions of mutations designed to destabilize F (black box). The PIV5 F protomers are colored differently. (D) Enlarged view of the boxed area in panel C, showing the positions of the mutations designed to destabilize F (purple). The previously characterized hyperfusogenic mutation (S443P) is shown in red. (E) Sequence alignment of various paramyxovirus F proteins showing a part of domain II, including the Ig-like domain. Φ, hydrophobic stretch in the PIV5 F sequence. Strongly conserved residues are highlighted in black; moderately conserved residues are highlighted in gray. Numbers indicate PIV5 F residues. MuV, mumps virus; MeV, measles virus; NiV, Nipah virus; HeV, Hendra virus.
Analysis of the atomic structures of postfusion F of human PIV3 (hPIV3) (18), NDV (19), and RSV (20, 21) and biochemical experiments (22) indicate that refolding of the triggered F protein occurs through a series of well-coordinated large-scale conformational rearrangements resulting in translocation of the HRB domain by ∼180° relative to the prefusion head domains, refolding of much of domain III, and formation of an extremely stable 6-helix bundle (6HB). A transient intermediate with the fusion peptide inserted into the target membrane, known as the prehairpin intermediate, has been visualized by electron microscopy (23).
Biochemical and structural data on HN, H, or G from NDV, hPIV3, PIV5, measles virus, Hendra virus, and Nipah virus indicate that these attachment proteins form homotetramers, with each protomer consisting of a globular head containing the receptor binding site and a stalk participating in a 4-helix bundle (4HB) that is anchored in the membrane (24–34). Substantial evidence indicates that the F and HN (H or G) interaction takes place through the stalk domain (34–46), and recent studies indicate that the site of F and HN interaction is through a central region of the HN (or H) stalk 4HB, located in HN at the junction of a straight helix (11-mer repeat) and a left-handed supercoiled helix (7-mer repeat) (34, 36, 47, 48).
The structural data indicate that the tetrameric PIV5 and NDV HN head domain is formed by a dimer of dimers (26, 28). The PIV5 HN structure (26) is described as “four heads up” and the NDV HN structure (28) as “four heads down.” Taking together biochemical and structural studies on PIV5 and NDV HN, we proposed a simple model for fusion activation. The HN stalk harbors the region for fusion activation, but the HN heads in the 4-heads-down form protect the HN stalk, making it unable to interact with the F protein. Upon HN binding sialic acid through sites in the globular head, the heads move to attain “up” positions. This movement exposes the F activation region on the stalk and allows F to interact with the HN stalk to trigger fusion (9). A PIV5 HN stalk only (headless HN) was able to activate F, supporting this hypothesis (9). Additionally, changes in the central part of the stalk to attain an F-triggering conformation have been implicated as being critical for the fusion trigger in morbilliviruses (36, 47, 48).
The region of the PIV5 F protein that interacts with the PIV5 HN stalk is not known. In a recent study, using a homology model of the measles virus F protein based on the atomic structure of the prefusion PIV5 F protein (15), residues near the base of the measles virus F model were postulated to be involved in the H interaction (49). Certain measles virus F mutant proteins that were expressed on the surfaces of cells lost the ability to coimmunoprecipitate H protein and were unable to promote fusion, supporting the notion that the mutations were in a region of F that interacts with H. Also, in a separate study, regions of specificity of F proteins involved in HN interaction were identified based on chimeric proteins generated between PIV5 F and simian virus 41 (SV41) F proteins (50, 51).
In the study reported here, using the prefusion atomic structures of PIV5 F (15, 17) (Protein Data Bank identifiers [PDB ID] 4GIP and 2B9B) to guide mutagenesis, we investigated specific domains of the PIV5 F protein for their ability to be activated by HN. We infer that PIV5 F and HN do interact through specific regions of contact, as we show here that mutations in the Ig-like domain of PIV5 F cause a specific loss in the ability of HN to activate fusion, yet these PIV5 F mutants maintain their ability to be triggered by heat (42°C) as a surrogate for HN. We also show that the metastable nature of the F protein is, likely, in part dependent on interactions between protomers at the trimer interfaces.
MATERIALS AND METHODS
Cells and antibodies.
Vero cells and 293T cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). BHK-21F cells were grown in DMEM containing 10% FBS and 10% tryptose phosphate broth. BSR-T7/5 cells were grown in DMEM containing 10% FBS, with 500 μg/ml G418 added every third passage. Antibodies specific for PIV5 F used were monoclonal antibody (MAb) F1a (52) and PIV5 F polyclonal antibody (PAb) R9176 serum, raised in rabbits against the purified PIV5 F ectodomain expressed by a recombinant baculovirus in insect cells. Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse antibodies or FITC-conjugated goat anti-rabbit antibodies (Jackson ImmunoResearch, West Grove, PA) were used as secondary antibodies for detection by flow cytometry.
Cloning and mutagenesis.
pCAGGS-HN and pCAGGS-F expression constructs harboring the PIV5 (W3A) F and HN genes were used as described previously (53). Mutations in pCAGGS-F were constructed by four-primer PCR. After introduction of the desired point mutation into the PCR fragment, the PCR fragment was digested using KpnI and SacI and ligated into a pCAGGS vector cut with the same restriction enzymes. The nucleotide sequence of the entire F open reading frame for each of the generated mutants was verified using an Applied Biosystems 3100-Avant automated DNA sequencer (Life Technologies Corp., Carlsbad, CA). pT7-luciferase was obtained from Promega Corp. (Madison, WI). The creation of the PIV5 HN “headless” stalk (HN 1-117) construct in pCAGGS was described previously (9).
Expression of PIV5 F and HN glycoproteins in mammalian cells.
PIV5 F and HN proteins were expressed from the pCAGGS-F and pCAGGS-HN constructs in Vero, BHK-21F, and 293T cells by transient transfection using the Lipofectamine Plus transfection reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Transfections were carried out in OPTIMEM media and cells incubated with DNA and transfection reagents for 5 h at 37°C, following which DMEM containing 2% FBS was added and the samples were incubated for an additional 18 h at 37°C.
Immunoprecipitation and SDS-PAGE.
293T cells were seeded onto BD PureCoat amine 6-well dishes (Becton, Dickinson, Franklin Lakes, NJ) overnight. The cells were then transfected with pCAGGS-F wt and pCAGGS-F containing mutations. Eighteen hours posttransfection, the cells were starved in DMEM deficient in cysteine and methionine for 30 min, followed by labeling with 50 μCi [35S]Promix label in the same medium for 30 min (pulse). The cells were then incubated for 90 min with complete DMEM containing 10% FBS (chase). The cells were subsequently lysed in cold radioimmunoprecipitation assay (RIPA) buffer (54) containing protease inhibitors, 50 mM iodoacetamide, and 2 mM phenylmethylsulfonyl fluoride (PMSF). The lysates were then clarified in a Beckman TLX ultracentrifuge in a Beckman TLA 120.2 rotor at 55,000 rpm for 10 min at 4°C. Clarified lysates were incubated with the PIV5 F polyclonal antibody R9176 for 2 h at 4°C, following which protein A-Sepharose beads were added and the immuno-complexes allowed to bind the beads at 4°C for 30 min. The antibody-antigen complexes were washed three times with RIPA buffer (54) containing 0.3 M NaCl, twice with RIPA containing 0.15 M NaCl, and once with 50 mM Tris-HCl, pH 7.4, 0.25 mM EDTA, and 0.15 M NaCl. The proteins were eluted from the beads by boiling them for 2 min in protein lysis buffer containing 15% dithiothreitol and separated on a 15% acrylamide gel. Radioactivity was detected using a Fuji FLA-5100 image reader with Multi Gauge v3.0 software (Fuji Medical Systems, Stamford, CT).
Flow cytometry.
To quantify the amount of PIV5 F wt and mutant F protein present on the surfaces of cells, 293T cells were transfected as described above with plasmids encoding the PIV5 wild-type (wt) F or F mutant proteins (1 μg DNA each). At 18 h posttransfection, the monolayers were washed with phosphate-buffered saline (PBS) containing 0.02% sodium azide. Nonspecific antibody binding was blocked by incubating the cell monolayers in PBS containing 1% bovine serum albumin (BSA) and 0.02% sodium azide for 1 h at 4°C. Plates were then incubated with the PIV5 F MAb F1a or PIV5 F PAb R9176 at a dilution of 1:100 in PBS containing 1% BSA. The monolayers were washed extensively with PBS to remove unbound antibody and incubated with FITC-conjugated goat anti-mouse IgG (1:200 dilution) or FITC-conjugated goat anti-rabbit IgG (1:200 dilution) (Jackson ImmunoResearch, West Grove, PA). Cells were washed extensively with PBS and resuspended in PBS containing 0.5% formaldehyde. The mean fluorescence intensity (MFI) of 10,000 cells was recorded for each sample using a FACSCanto II flow cytometer (Becton, Dickinson, Franklin Lakes, NJ). Flow cytometry data were collected using the FACSDiva software (Becton, Dickinson, Franklin Lakes, NJ) and analyzed using BD Cell Quest Pro software (Becton, Dickinson, Franklin Lakes, NJ).
Syncytium formation.
BHK-21F cells were transfected as described above using 1 μg (each) of pCAGGS-HN and pCAGGS-F or pCAGGS containing the PIV5 F point mutations. Eighteen hours posttransfection, the cells were fixed and stained using a Hema3 staining protocol (Fisher Scientific, Pittsburgh, PA) according to the manufacturer's instructions. The monolayers were photographed using an AMG EVOS xl inverted microscope (Fisher Scientific, Pittsburgh, PA).
Luciferase reporter assay.
To quantify the fusion observed in the syncytial assay, Vero cell monolayers were transfected with 1 μg (each) of pCAGGS-HN, pCAGGS-F, or pCAGGS containing the PIV5 F point mutations and pT7-luciferase, a plasmid that expresses firefly luciferase under T7 polymerase control. BSR-T7/5 cells, expressing T7 RNA polymerase, were laid over the Vero cell monolayer 15 h following transfection at a BSR-T7 cell/Vero cell ratio of 1:2 and incubated further for 7 h at 37°C or 42°C. Glo lysis buffer (Promega, Madison, WI) was used to lyse the cells. Cell debris was removed by centrifugation (13,000 rpm for 3 min), and 150 μl of the cleared lysates was then added to a 96-well dish along with 150 μl of the luciferase assay substrate (Promega, Madison, WI). The luciferase activity in relative luciferase units (RLU) was then determined using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
RESULTS
Design of single point mutations in the parainfluenza virus 5 F protein.
To understand the role of PIV5 F protein domains I and II in fusion, single point mutations were introduced into the PIV5 F protein based on the atomic structure of the prefusion PIV5 F protein (15). One set of these mutations is located within domain II, which has a characteristic immunoglobulin-like (Ig-like) fold. Mutations within this set are also located within an adjoining hydrophobic pocket formed by residues from both domain I and domain II (Fig. 1A and B). The other set of mutations were located in domain I, near the junction of protomers in the F trimer (Fig. 1C and D).
The PIV5 F Ig-like domain has a typical topology and is characterized by two β-sheets made up of 7 anti-parallel β-strands with crossover strands in between the two sheets, creating a very stable tertiary motif (15). In the majority of similar Ig-like domains, disulfide bridges provide additional stability to the domain. The PIV5 F Ig-like domain contains three disulfide bonds between residues C348 and C356, C324 and C333, and C385 and C387. It is interesting to note that the paramyxovirus fusion protein Ig-like domain is minimally altered between the PIV5 F prefusion structure and the hPIV3 F postfusion structure (15, 18). A similar observation was recently made after comparison of the matched RSV F prefusion (16) and postfusion (20) pairs. The presence of disulfide bonds and the interwoven β-strands of the Ig-like domain of paramyxovirus F proteins suggest the structural integrity of the domain.
To investigate the role of the Ig-like domain and the adjacent hydrophobic pocket in domains I and II further, PIV5 F constructs in the mammalian expression vector pCAGGS, bearing single point mutations resulting in F295L, N298D, R368L, S382A, L384T, A392T, V393T, I394N, L395N, and Q396L were created (Fig. 1A and B and Table 1). In these PIV5 F mutants, the mutations S382A, L384T, A392T, V393T, I394N, L395N, and Q396L are located within the Ig-like domain of domain II, whereas the mutations N298D, F295L, and R368L are located in domain I and form a part of the hydrophobic pocket created between domains I and II.
Table 1.
Summary of properties of F point mutantsa
| Serial no. | F protein mutation | Surface expression | Cleavage (% of wt F cleavage) | Fusion |
|||
|---|---|---|---|---|---|---|---|
| With HN | At 37°C | At 42°C | With the HN stalk | ||||
| 1 | A392T | ***** | 105.09 | ** | ** | ***** | ****** |
| 2 | V393T | ***** | 104.44 | * | * | ***** | — |
| 3 | I394N | — | ND | — | — | — | — |
| 4 | L395N | ***** | 105.94 | * | * | ***** | ****** |
| 5 | Q396L | — | ND | — | — | — | — |
| 6 | A392T/L395N | ***** | 103.48 | * | * | ***** | ** |
| 7 | S382A | **** | 34.63 | **** | * | ******* | ****** |
| 8 | L384T | ***** | 71.16 | * | * | ***** | ** |
| 9 | N298D | ***** | 105.52 | * | * | ***** | ***** |
| 10 | F295L | — | ND | — | — | — | — |
| 11 | R368L | — | ND | — | — | — | — |
| 12 | T312A | ***** | 61.69 | * | — | * | ND |
| 13 | S314A | ***** | 41.12 | **** | * | **** | ND |
| 14 | Q339A | ***** | 54.47 | *** | — | * | ND |
| 15 | V340A | ***** | 54.34 | ******* | ******* | ND | ND |
| 16 | V362A | ***** | 59.53 | ******* | ******* | ND | ND |
| 17 | S364A | ***** | 36.72 | ******* | ******* | ND | ND |
List of point mutations generated in the PIV5 F protein, along with the surface expression of each protein measured by flow cytometry as fusion in the presence of HN and fusion with F alone in cells transfected at 37°C and 42°C. The fusion of F point mutants with the HN headless stalk is also shown. Cleavage as a percentage of wt F cleavage was determined as follows: total quantified cleaved F protein (F1 + F2)/total F protein (F0 + F1 + F2). Asterisks denote approximate fusion relative to that of wt F and wt HN (percentages), as follows: *******, 120 to 160%; *****, 90 to 100 %; ****, 70 to 90%; ***, 50 to 70%; **, 30 to 50%; and *, 10 to 30%. + signs denote approximate percentages of expression of mutant F proteins on cell surfaces, in comparison to wt F surface expression, as follows: +++++, 90 to 100%; ++++, 70 to 90%; +++, 50 to 70%; ++, 30 to 50%; and +, 10 to 30%. —, not expressed on the cell surface; ND, not determined.
Interestingly, residues 391 to 395 of PIV5 F form a stretch of significantly hydrophobic residues (Φ) in the Ig-like domain (Fig. 1B), which, rather unusually, are solvent exposed. When a sequence alignment with F proteins of other paramyxoviruses was done (Fig. 1E), an isoleucine (I394) within this stretch and a glutamine at position 396, adjacent to the hydrophobic stretch, was found to be highly conserved. However, there is considerable variation in sequence between the other residues within this hydrophobic stretch among paramyxovirus F proteins. The unusual hydrophobic nature of this exposed stretch of residues within the Ig-like domain suggests a potential protein-protein interaction surface, and the sequence variability across paramyxoviruses may indicate a specificity of this interaction.
To examine the unusual nature of the surface-exposed hydrophobic stretch, Φ (residues 391 to 395), these residues were changed to polar or charged amino acids, and each mutated amino acid was chosen to be the closest in size to the parent amino acid to minimally interfere with structure and folding. The highly conserved polar residue at the end of this stretch (Q396) was mutated to a leucine to study the effect of lengthening the hydrophobic stretch. The roles of residues in the adjoining hydrophobic pocket of domain I were investigated by changing a wt, charged polar residue within the hydrophobic pocket to a hydrophobic residue (R368L), by reversing the charge of a polar residue (N298D) or by changing the physical dimensions of a hydrophobic residue (F295L) to probe the nature of a possible protein-protein interaction in this region.
In recent studies with the measles virus F protein, residues near the base of the F protein head were implicated to interact with the measles virus receptor binding protein H (49). In this study, the PIV5 F prefusion atomic structure was used to model a measles virus prefusion F protein and guide mutagenesis. It was found that some of the measles virus F mutants that expressed well on cell surfaces and disrupted F-H interactions were located in the Ig-like domain or near the hydrophobic pocket created by the Ig-like domain in the measles virus F prefusion model. Other residues postulated to disrupt measles F-H interactions were located along the base of the measles virus F head model in domain I. Also, in a separate study, switching domain I fragments of PIV5 F with those of SV41 F converted the HN specificity of PIV5 F, resulting in partial triggering of this PIV5-SV41 chimeric F protein by the SV41 HN protein (51). Thus, to probe further the role of domain I of the PIV5 F prefusion structure in HN interaction and fusion promotion, mutations were generated within domain I, near the region of interaction between two protomers. Accordingly, the following PIV5 F mutants in pCAGGS were created: the T312A, S314A, Q339A, V340A, V362A, and S364A mutants (Fig. 1C and D and Table 1).
Expression of PIV5 F mutants on the cell surface as measured by flow cytometry.
To investigate surface expression of the PIV5 F point mutants in transiently transfected 293T cells, flow cytometry was performed using antibodies specific to the PIV5 F protein. A monoclonal antibody specific to the PIV5 F protein in its prefusion form (F1a) (52) or a polyclonal PIV5 F antibody, R9176, was used to detect PIV5 F wt and mutant proteins on the cell surface 18 h posttransfection (Fig. 2A and Table 1). The N298D, S328A, L384T, A392T, V393T, and L395N mutants were expressed at levels slightly higher than or comparable to that of wt F (Fig. 2A), as detected by either antibody, whereas the I394N, Q396L, R368L, and F295L mutants failed to express at the cell surface (Table 1). Interestingly, out of the four mutant proteins that failed to reach the cell surface, three of them harbor mutations at positions I394, Q396, and R368 that are highly conserved among paramyxovirus F proteins (Fig. 1E). This suggests that these residues may be involved in stabilizing the local structure within these proteins. Changes at these residue positions may severely compromise the ability of PIV5 F to fold correctly, hence, its failure to be trafficked to the cell surface. Mutants that failed to reach the cell surface were not examined further. The remainder of the mutants appeared to be correctly folded and exhibited levels of the prefusion form of F and total protein comparable to that of wt F on the cell surface.
Fig 2.
Expression of PIV5 F mutants on the surfaces of 293T cells. (A) Detection of proteins by flow cytometry at the surfaces of 293T cells transfected with wt PIV5 F or PIV5 F point mutants. Surface proteins were detected using the prefusion PIV5 F-specific F1a MAb (gray bars) or a PIV5 F R9176 polyclonal antibody (PAb) (white bars). PIV5 F-positive cells were labeled using a fluorescein-conjugated goat anti-mouse secondary antibody or a fluorescein-conjugated goat anti-rabbit secondary antibody. The mean fluorescence intensity (MFI) of 10,000 cells is shown as a percentage of wt protein levels. Results are from three independent experiments. (B) Detection of PIV5 F mutant proteins F-V340A, F-V362A, and F-S364A with the PIV5 F polyclonal antibody R9176 by flow cytometry. Fluorescein-conjugated goat anti-rabbit antibody was used to fluorescently label F protein-positive cells. Flow cytometry was performed 18 h posttransfection upon incubation of transfected cells at 37°C (gray bars) or 33°C (white bars). ns, not significantly different (P > 0.05). Results are from three independent experiments.
Of the F mutants with mutations in domain I, near the protomer junction, the T312A, S314A, and Q339A mutants had cell surface expression levels equivalent to those of wt F (Table 1), whereas F-V340A, F-V362A, and F-S364A were expressed at the cell surface at slightly lower levels (approximately 65 to 80% of wt-F levels) (Fig. 2A and Table 1), when they were detected using the F1a monoclonal antibody, specific to the prefusion form of F. This reduction in surface expression, compared to wt-F expression, was statistically significant for mutants F-V340A and F-S364A. However, as the F1a antibody specifically detects the prefusion form of the PIV5 F protein, it was thought possible that even in the absence of HN expression, there may be a reduction in the prefusion F population on the cell surface at 37°C due to spontaneous conversion of F to the postfusion form. This seems likely to be the case, as the mutant proteins F-V340A, F-V362A, and F-S364A were present at the cell surface at wt-F levels when they were detected with the polyclonal antibody to PIV5 F (R9176) that recognizes both the pre- and postfusion forms (Fig. 2A). Additionally, to minimize damage to the cell monolayer due to a possible spontaneous triggering and fusion of F-V340A and F-V362A mutants, the flow cytometry was also carried out after incubating the cells at 33°C (Fig. 2B, white bars). For mutant F-V340A, there was an increase in surface expression compared to that in the same sample incubated at 37°C (Fig. 2B, gray bars); however, this increase was not statistically significant (P = 0.0523). Thus, overall, these data suggest that even though comparable amounts of F-V340A and F-V362A mutant proteins are transported to the surfaces of 293T cells, there may be less of these proteins in the prefusion form than wt F.
Protein expression and cleavage activation of PIV5 F point mutants.
To examine F protein expression and F protein cleavage, the PIV5 F single point mutations were expressed in 293T cells, labeled metabolically with [35S]Promix label, and immunoprecipitated from cell lysates using F-specific polyclonal antibody R9176. Immunoprecipitated polypeptides were analyzed on 15% SDS-PAGE gels. Most PIV5 F mutant proteins bearing mutations in the Ig-like domain were cleaved to F1 and F2 to an extent similar to that in wt PIV5 F (Fig. 3A and Table 1). Mutants F-S382A and F-L384T, however, had lower cleavage efficiencies (Table 1). However, despite the defect in cleavage, especially for F-S382A, there was no major variation in the levels of surface expression of these mutants (Fig. 2A).
Fig 3.
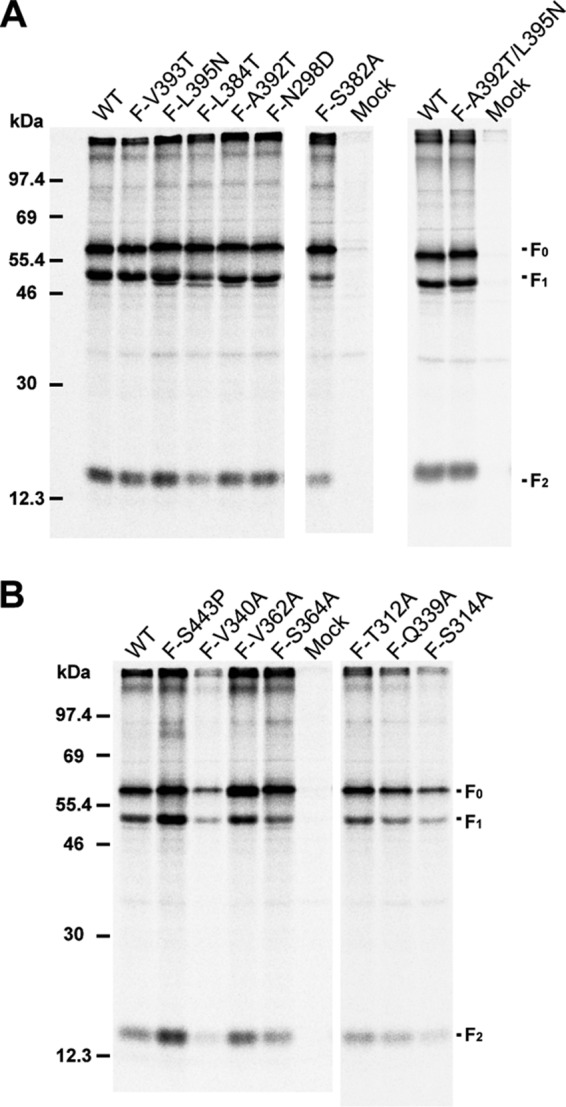
Expression and cleavage of PIV5 F mutant proteins. PIV5 wt F or F point mutants expressed in 293T cells were immunoprecipitated. The cells were labeled with [35S]TranS label for 30 min (pulse) in medium lacking methionine and cysteine, followed by incubation in complete medium (chase) for 90 min. Cells were then lysed in RIPA buffer, and proteins were immunoprecipitated using the PIV5 F polyclonal antibody R9176. Polypeptides were analyzed by SDS-PAGE. Numbers indicate molecular masses (kDa). The positions of the uncleaved F protein (F0) and F protein cleavage products F1 and F2 are indicated. (A) PIV5 F wt and F point mutants with mutations located in the Ig-like domain and the adjacent hydrophobic pocket. (B) PIV5 F wt and point mutants with mutations located in domain I near the F-protomer junction.
Analysis of PIV5 F mutants located in domain I indicated that they were expressed in 293T cells at levels comparable to those of wt F (Fig. 3B and Table 1), except for the V340A mutant. Cleavage of all domain I mutants were reduced to some extent (to ∼37 to 62% of wt F cleavage) (Table 1). Mutant V340A showed an overall reduction in the intensity of bands. However, due to the cytopathic nature (syncytium formation) of some of these mutant F proteins at 37°C in the absence of HN, parts of the 293T cell monolayers were lost. Hence, the total protein yields from cell lysate generated from single wells of a 6-well dish varied significantly. Thus, the band intensity of F species in Fig. 3B may not reflect the expression level of the mutant protein F-V340A in comparison to that of wt F protein. Measurements using a flow cytometer depend on a fixed number of cells of relatively uniform size per sample, and thus, this effect was not observed in Fig. 2A. Expression at a lower temperature (33°C) to prevent selective exclusion of multinucleated fused cells in flow cytometry also did not seem to affect the protein expression of F-V340A in a statistically significant way (Fig. 2B).
PIV5 F mutants in the Ig-like domain do not exhibit fusion promotion by HN coexpression but are triggered for fusion using heat as a surrogate for HN.
To understand the role of the PIV5 F point mutations located within the Ig-like domain and those located in the hydrophobic pocket adjacent to the Ig-like domain in fusion activation, the mutant proteins were coexpressed with or without wt PIV5 HN in BHK-21 cells and incubated at 37°C. These mutant F proteins were also expressed alone (without HN) and were incubated at 42°C (Fig. 4A and Table 1). The cells were fixed and stained, and cell-cell fusion was observed in the form of syncytia. wt F protein caused significant syncytium formation when coexpressed with HN at 37°C, and mutant F-S382A caused moderate syncytium formation when coexpressed with wt HN. For the A392T F mutant, some syncytia were detectable over background levels. The other mutants, F-N298D, F-L384T, F-V393T, and F-L395N, which are located either within the Ig-like domain or in the adjacent hydrophobic cavity, had no detectable fusion activity when coexpressed with HN (Fig. 4A). In the absence of HN expression, neither wt F nor the F mutants could be triggered at 37°C to cause fusion beyond the low level observed previously for wt F (4, 55). However, when cells expressing the N298D, S382A, L384T, A392T, V393T, and L395N F mutants were incubated at 42°C in the absence of HN, they produced significant amounts of fusion on BHK-21 cells (Fig. 4A). The F-S382A mutant, though producing fusion upon coexpression with HN at 37°C, produced much more extensive syncytia at 42°C in the absence of HN. Thus, these data suggest that even though the single point mutants with mutations either within or near the Ig-like domain could not be activated specifically by PIV5 HN, their mutations did not render these F proteins incapable of causing fusion, and they could be triggered at 42°C.
Fig 4.

Fusion mediated by the PIV5 F point mutants in baby hamster kidney cells. Baby hamster kidney (BHK-21F) cells transfected with PIV5 F or its point mutants alone or cotransfected with PIV5 HN or the PIV5 HN 1-117 headless stalk were incubated at 37°C or 42°C. At 18 h posttransfection, cells were fixed, stained, and imaged. (A) Point mutations in PIV5 F located in the Ig-like domain and in an adjacent hydrophobic pocket. (B) Locations of residues A392, V393, L395, and L384 within the Ig-like domain in the PIV5 F prefusion structure. Green, single mutations at these residues disrupt the HN interaction; blue, combined mutations at these residues disrupt the HN interaction. (C) Syncytial assay of point mutations in PIV5 F that are located in domain I near the protomer-protomer junction of the F trimer; cells were transfected or not transfected with PIV5 HN.
To investigate further the role of individual residues identified in the above-described HN interaction, these F point mutants were cotransfected with the PIV5 HN 1-117 headless stalk (9) and cell-cell fusion was observed as syncytia (Fig. 4A, bottom panel). Interestingly, the N298D, A392T, and L395N mutants caused an increased amount of fusion with the HN 1-117 stalk but were incapable of fusion when triggered by the full-length HN protein. A possible explanation for this observation is that, as the PIV5 HN 1-117 headless stalk lacks its regulatory head domain, it is a more potent activator of F because of the longer dwell time of F on the HN stalk (9). Thus, the greatly increased number of F-HN stalk interaction events may be able to trigger the N298D, A392T, and L395N F mutant proteins. On the other hand, mutants F-V393T and F-L384T could be activated efficiently only by heat, and neither the full-length PIV5 HN nor the headless stalk of PIV5 HN could activate these mutant proteins, suggesting that these residues may have a more central role in the HN interaction. The locations of residues V393 and L384 are close to each other on two outer β-strands of the Ig-like domain (Fig. 4B), suggesting that together they may constitute a specific HN interaction interface.
The A392T and L395N F mutants were not activated by wt HN but, counterintuitively, were activated by the HN 1-117 stalk (Fig. 4A). We thought that the most likely explanation for this observation is that F-A392T and F-L395N contain mutations sufficient to weaken the interaction of wt HN so that it does not activate F. In contrast, because the HN 1-117 stalk interacts constitutively with F mutants, the prolonged but weakened interaction still permits F activation. To test this hypothesis, a double mutant, F-A393T/L395N, was constructed and expressed with wt HN or the HN 1-117 stalk. The double mutant F-A392T/L395N was expressed, cleaved, and transported to the surfaces of cells at levels similar to those of wt F (Fig. 2A and 3A and Table 1). As predicted, the F double mutant failed to be activated (Fig. 4A) by wt HN or the HN 1-117 stalk, suggesting that the combined effect of mutations A392T and L395N is sufficient to overcome the increased dwell time with the headless stalk and prevent fusion activation. The double mutant F-A392T/L395N, however, can still be activated by heat at 42°C.
Mutants in domain I of PIV5 F are hyperfusogenic, and fusion is HN independent.
The proteins with single point mutations in PIV5 F domain I were tested for their ability to cause cell-cell fusion in BHK-21 cells in the presence and absence of HN expression. As a comparison, mutant F-S443P, described previously as a hyperfusogenic mutant of PIV5 F, was also tested (53). Mutants F-V340A, F-V362A, and F-S364A showed a very strong hyperfusogenic phenotype, yielding more-extensive syncytia than wt F when coexpressed with HN (Fig. 4C). The extent of fusion produced by these mutants in the presence of HN is comparable to that of the F-S443P mutant and probably reflects the location of these mutations, which is close to S443P in domain I, bordering the junction between two protomers in the F trimer (Fig. 1D). In addition to producing significantly larger syncytia than wt F on cotransfection with HN, mutants F-V340A and F-V362A produced large syncytia even in the absence of HN, similar to the properties of the mutant F-S443P. This suggests that these mutations destabilize F to such an extent that at 37°C, the activation energy of these mutants is low enough to allow conversion to the postfusion form even in the absence of HN. Mutant F-S364A was triggered at 37°C without HN, though unlike mutants F-V340A and F-V362A, it did not appear to be completely independent of HN.
The other mutants with mutations in domain I, i.e., the T312A, S314A, and Q339A mutations, had variable phenotypes (Table 1). Mutation S314A did not affect the extent of fusion activation of the F protein, and this mutant had fusion properties similar to those of wt F. On the other hand, the Q339A and T312A mutants failed to be triggered to wt levels of fusion by HN and were not triggered by heat at 42°C when expressed in the absence of HN, suggesting that the F-refolding process was probably disrupted in these mutants (Table 1).
Results of the quantitative luciferase fusion assay of PIV5 F mutants reflect the results of the syncytial assay only upon receptor engagement.
To quantify the extent of fusion mediated by the PIV5 F mutant proteins in the presence of PIV5 HN, a quantitative luciferase reporter assay was used. Vero cells were transfected with PIV5 wt F or mutant F plasmids together with PIV5 HN and with a luciferase reporter gene under T7 polymerase control. The T7 polymerase is provided by BSR-T7 cells, which are laid over transfected Vero cells. Upon receptor binding by HN and subsequent fusion of the two cell types, the luciferase reporter is activated. F mutants with mutations near the Ig-like domain showed little luciferase reporter activity when cotransfected with HN, with the exception of mutants F-S382A and F-A392T (Fig. 5A). Mutant F-S382A produced fusion at about 75% of the level of the wt, whereas mutant F-A392T showed only a small amount of fusion. Thus, most of these mutants could not be triggered specifically upon interaction with HN, reflecting the results observed in the syncytial assay in Fig. 4A.
Fig 5.
Quantitative luciferase reporter assay of cell-cell fusion activities of PIV5 F point mutants. The fusion activities of PIV5 F point mutants were characterized by using a quantitative luciferase reporter assay. wt PIV5 F or its point mutants were coexpressed with PIV5 HN and luciferase under the control of the T7 promoter in Vero cells, which were overlaid with BSR-T7 cells, 15 h posttransfection. Seven hours postoverlay, the cells were lysed and luciferase activity (expressed in RLU) was determined. Results are from three independent experiments. (A and B) Fusion activities of PIV5 F point mutants in the presence of PIV5 HN. The fusion-inducing capabilities of the F point mutants are shown, with those of wt F and HN considered 100%. (A) Mutants located in the Ig-like domain and the adjoining hydrophobic pocket. (B) Mutants located in domain I near the protomer-protomer junction of the F trimer. (C) Fusion activities of PIV5 F point mutants with mutations located in the Ig-like domain hydrophobic pocket. Cells were transfected without HN at 37°C (white bars) and 42°C (gray bars). The fusion of F point mutants is shown; wt F fusion at 37°C was considered 100% fusion. (D) Fusion activation of PIV5 F hyperfusogenic mutants with (gray bars) or without (white bars) PIV5 HN or with the PIV5 HN 1-117 stalk construct (black bars). (E) Fusion activation of PIV5 F hyperfusogenic mutants in the presence (white bars) or absence (gray bars) of an independent nonactivating receptor-binding protein, influenza virus Udorn HA. Fusion is expressed in RLU as a percentage of wt F fusion in the absence of a receptor-binding protein. *, P = 0.01 to 0.05; **, P = 0.001 to 0.01. Results in all panels constitute data from at least three independent experiments.
The luciferase reporter assay for hyperfusogenic mutants in domain I reflected those of the syncytial assay (Fig. 4C) when these mutants were cotransfected with HN (Fig. 5B). Mutants F-V340A, F-V362A, and F-S364A exhibited levels of fusion 1.5 to 3 times that of wt F, levels similar to those produced by the previously characterized mutant, F-S443P.
When the F point mutations within the Ig-like domain (S382A, L384T A392T, V393T, and L395N) and that within the adjoining hydrophobic cavity (N298D) were tested for fusion promotion without HN in the luciferase reporter assay, only small differences of fusion activation between samples incubated at 37°C and 42°C were observed (Fig. 5C). These differences did not correspond to the dramatic variation of fusion activation at the two temperatures observed in Fig. 4A. The explanation thought most likely is that there is a major difference between syncytium formation in cells in confluent monolayers and that in the luciferase assay, where one cell type is laid over another cell type. In the latter assay, luciferase reporter activity is generated only when BSR-T7 cells in the overlay fuse, upon attachment with Vero cells in the adherent monolayer below. Thus, in the absence of receptor-mediated attachment, F triggering on the surfaces of Vero cells may not coincide with fusion peptide insertion into the overlaying BSR-T7 cells, and thus a large proportion of F proteins may be triggered but fail to mediate a successful membrane merger. This is in comparison to cells within a confluent monolayer in a syncytial assay, which are involved in significant cell-cell contacts throughout the duration of the assay. Hence, even though qualitative syncytial assays agree with quantitative luciferase reporter fusion assays when HN is present to bind the receptor (Fig. 5A and B), they do not do so in the absence of receptor binding (Fig. 5C and D). This was illustrated when fusion activation of the PIV5 F domain I hyperfusogenic mutants were tested in the presence of HN, in the absence of HN, or in the presence of the HN 1-117 stalk domain (Fig. 5D). The previously characterized PIV5 HN 1-117 stalk domain cannot bind the receptor (9). Thus, in samples where there was no receptor binding, namely, transfections without HN or with the PIV5 HN 1-117 stalk, the hyperfusogenic mutants exhibited much-reduced fusion activity, in contrast to the fusion activity observed in Fig. 4C (and data not shown). On the other hand, cotransfection with full-length, receptor-binding-capable PIV5 HN yielded fusion 1.5 to 3 times that of wt F and wt HN, as observed in Fig. 5B.
It was observed that fusion promotion in samples expressing F-V340A and F-V362A with the HN 1-117 stalk was slightly lower than in samples expressing the F-V340A and F-V362A mutant proteins alone (Fig. 5D). It is possible that the PIV5 HN 1-117 stalk protein prematurely triggers the F-V340A and F-V362A proteins upon reaching the surface, as a result of which fusion observed in cotransfected samples is slightly lower than that of the F-V340A and the F-V362A proteins expressed without HN. However, due to potential surface expression and protein transport differences between cells transfected with single proteins and those transfected with multiple proteins, this conclusion cannot be confirmed from the current data.
To illustrate further the importance of receptor binding in the luciferase reporter assay overlay, wt F and the HN-independent mutants F-V340A, F-V362A, and F-S364A were cotransfected with or without a non-cognate receptor-binding protein. In this case, the influenza A virus (A/Udorn/72) hemagglutinin (HA) protein, which binds sialic acid but does not activate PIV5 F, was used as the non-cognate receptor-binding protein (Fig. 5E). The assay is carried out at neutral pH to prevent membrane fusion by the influenza A HA protein. It was observed that luciferase reporter activity, indicating cell-cell fusion, was significantly increased in the presence of the influenza A virus HA protein, suggesting that fusion peptide insertion and membrane merger increases upon receptor-mediated attachment of overlaying cells.
DISCUSSION
The nature of the F protein trigger, which is initiated by the attachment protein upon receptor binding and which leads to destabilization of the F protein to cause membrane fusion, is as yet unclear. To understand presumed interactions between PIV5 F and HN and the associated triggering of the metastable prefusion F protein, we designed proteins with single point mutations in the Ig-like domain II and in a hydrophobic pocket at the interface between domain I and domain II of PIV5 F, based on the prefusion PIV5 F atomic structure (15). The rationale for investigating the role of the Ig-like domain in HN interaction is 3-fold.
First, Ig-like domains are known to be structurally robust, forming protein-protein interaction interfaces. Multiple examples are found in the interleukin family of cell-signaling molecules and in their interactions with interleukin receptors. Such interactions represent contacts between Ig-like domains of interleukin receptor molecules and the 4HB of interleukin ligands. The atomic structures of multiple such receptor-ligand pairs have been determined (56–58). This draws an interesting parallel to a possible interaction between the 4HB of the paramyxovirus HN stalks and the Ig-like domains of F proteins. Second, the Ig-like domain of PIV5 F contains a hydrophobic loop (Φ) (residues 391 to 395) that is entirely solvent exposed, suggesting that this loop may be involved in a protein-protein interaction. Additionally, there is a hydrophobic pocket adjacent to the Ig-like domain, which may also be involved in such contacts. Third, as is evident from the prefusion F structure, the Ig-like domain protrudes from the molecule such that the fusion peptide is sequestered immediately behind the Ig-like domain. We suggest that interaction of the Ig-like domain loop with the HN stalk may lead to a possible release of the fusion peptide.
The mutant PIV5 F proteins with single residue changes in the Ig-like domain and the adjacent hydrophobic pocket created by the Ig-like domain resulted in an almost complete loss of the ability of PIV5 F to be triggered by coexpressing HN. However, when PIV5 F was expressed in the absence of PIV5 HN and the cells were shifted to 42°C, extensive cell-cell fusion occurred (Fig. 4A). These data suggest that even though the ability of the mutant F proteins to be triggered by HN was lost, the mutant F proteins could be triggered by heat. For mutant F-S382A, triggering by heat was more effective than reduced triggering by HN, perhaps suggesting a debilitated F-HN interaction. Coexpression of the mutant F proteins with a headless PIV5 HN 1-117 stalk domain further narrowed the potential region of interaction, implicating residues V393 and L384 in the outer loops of the Ig-like domain as major contact areas. A double mutation of two residues of the hydrophobic loop (F-A392T/L395N) showed that a combined effect of these two mutations was sufficient to overcome triggering by the HN 1-117 headless stalk, further indicating the importance of the hydrophobic stretch (Φ) within the Ig-like domain in HN interaction. As the F-HN interaction block in mutant F-N298D (adjacent to the hydrophobic pocket) could be overcome by coexpression of F and the HN 1-117 stalk, presumably by increasing the dwell time of the HN stalk-F interaction, residues in the adjacent hydrophobic pocket probably play a more indirect role in the F-HN interaction. It is also possible that these residues are involved in an overall movement of the Ig-like domain with respect to domain I upon interaction of HN with the Ig-like domain. Notably, residues L384, A392, V393, and L395, which are believed to be involved directly in HN interaction, are all located within the outer two strands of the Ig-like domain (Fig. 4B), suggesting that these residues can form a putative interaction interface.
A recent study using a model of the measles virus F protein based on the PIV5 F atomic structure suggested that single point mutations in the Ig-like domain of the measles virus F model completely ablated fusion activation by the measles virus H protein and prevented the coimmunoprecipitation of measles F and H proteins (49). A comparison of the measles virus F protein model (49) with the PIV5 F protein prefusion structure (15) and sequence alignment highlight important similarities and differences in the locations and functions of mutated residues between the two viruses (Fig. 6 and Table 2). Interestingly, L394 of measles virus F, one of the major residues implicated in H interaction (49), corresponds to residue L384 of PIV5 by sequence alignment, a residue implicated in PIV5 F-HN interaction (Fig. 4A and Table 2), suggesting a commonality between the interaction interfaces of the two proteins. For residue Q383 of measles virus F, which has been implicated in H interaction, the corresponding PIV5 F residue (D373) abuts the Ig-like domain. On the other hand, though residues Q322 and Y349 of measles virus F have been implicated in H interaction, corresponding residues of PIV5 F domain I (T312 and Q339) do not affect fusion promotion significantly. It is possible that the contacts between F protein domains I and II of the two viruses are different, resulting in different positioning of the HN-H interaction sites, or that the overall region of paramyxovirus F and HN, H, or G interactions may be reasonably broad, as suggested by the extent of mutations that presumably affect HN interaction, ranging from the hydrophobic pocket of the Ig-like domain and extending across domain I (49, 51). The fact that the fusion activity of the PIV5-SV41 chimeric F protein induced by SV41 F amounts to only ∼20% of the activities of wt SV41 F and HN indicates that there may be a larger surface of interaction on paramyxovirus F proteins. However, an analysis of exact residue-to-residue or domain-to-domain comparison between measles virus F and PIV5 F requires an atomic structure of the measles virus F protein in its prefusion form.
Fig 6.

Model suggesting the role of Ig-like domains of PIV5 F in interactions with the HN stalk. Images are reconstructed from atomic structures by aligning the PIV5 HN stalk structure (PDB ID 3TSI) (right panel) with the PIV5 HN 4-heads-up structure (PDB ID 1Z4X). The distance between the last residue visible in the HN stalk structure and the viral or cellular membrane (gray bar) is estimated to be ∼28.5Å, if the assumption is made that these 20 residues are helical (dotted lines). Cleaved PIV5 F-GCNt (PDB identfier 4GIP) was modeled adjacent to the constructed PIV5 HN in the 4-heads-up conformation described above. The distance between the last residue before GCNt in the PIV5 F-cleaved structure and the viral or cellular membrane (gray bar) is estimated to be ∼10Å, if the assumption is made that these residues are helical (dotted lines). The model predicts that the PIV5 F Ig-like domain corresponds in height to the middle region of the PIV5 HN stalk, which has been implicated to be involved in F triggering (34). The adjoining key shows the color labeling of PIV5 F or measles virus F residues, which are according to function and sequence alignment.
Table 2.
Comparison of HN-H interaction residues of PIV5 F and measles virus Fa
| Serial no. | Mutated PIV5 F residue | Corresponding measles virus F residue | Location in PIV5 F prefusion structure | Possible PIV5 HN-interacting residue | Corresponding measles virus H-interacting residue |
|---|---|---|---|---|---|
| 1 | A392 | T402 | Domain II (Ig-like) | Y | ND |
| 2 | V393 | I403 | Domain II (Ig-like) | Y | ND |
| 3 | I394 | I404 | Domain II (Ig-like) | ND | |
| 4 | L395 | N405 | Domain II (Ig-like) | Y | ND |
| 5 | Q396 | Q406 | Domain II (Ig-like) | ND | |
| 6 | S382 | S392 | Domain II (Ig-like) | Y | ND |
| 7 | L384 | L394 | Domain II (Ig-like) | Y | Y |
| 8 | N298 | S308 | Domain I (h.p.) | Y | ND |
| 9 | F295 | N305 | Domain I (h.p.) | ND | |
| 10 | R368 | R378 | Domain I (h.p.) | ND | |
| 11 | T312 | Q322 | Domain I (h.p.) | N | Y |
| 12 | S314 | Y324 | Domain I (h.p.) | N | ND |
| 13 | Q339 | Y349 | Domain I (h.p.) | N | Y |
| 14 | V340 | P350 | Domain I | N | ND |
| 15 | V362 | S372 | Domain I | N | ND |
| 16 | S364 | S374 | Domain I | N | ND |
List of point mutants generated in PIV5 F and the corresponding residues of measles virus F as shown by sequence alignment. The residues in PIV5 F or measles virus F (49) that are implicated in interactions with HN or H are indicated. PIV5 F residues shown in gray do not reach the surfaces of cells. Y, yes (possible interaction); N, no (possibly not involved in direct interaction); h.p., hydrophobic pocket adjacent to the Ig-like domain (Ig-like); ND, not determined.
Despite extensive efforts, we have not been able to coimmunoprecipitate PIV5 F and HN, as has been observed for other paramyxoviruses, such as NDV, measles virus, CDV, and Nipah virus (38, 43–45, 59, 60). This failure to coprecipitate PIV5 F and HN suggests that the interaction is weak or transient in nature. Thus, even though we cannot show directly an interaction between F and HN by coimmunoprecipitation, we propose that the reason that the PIV5 F Ig-like domain mutants cannot be triggered by HN is due to the ablation of the site in the F hydrophobic loop of the Ig-like domain that interacts with the HN stalk, but notably, these mutants are able to undergo F refolding, fusion peptide insertion, and membrane merger when triggered by heat.
It has been observed for other paramyxovirus F protein mutants that when they fail to coimmunoprecipitate HN, H, or G, they are also deficient for fusion when coexpressed with the homologous attachment protein (38, 44, 49). However, there have been reports of fusion-dead F mutants that coimmunoprecipitate with the attachment protein (47, 61, 62). Thus, the correlation between the ability to cause fusion and the ability of F and the receptor-binding protein to coimmunoprecipitate still lacks a mechanistic explanation. Interestingly, the F Ig-like domain structure remains essentially unchanged between the prefusion and postfusion forms of paramyxovirus F proteins (15, 16, 18, 20). Thus, it is possible that the protein-protein interaction observed in coimmunoprecipitations persists or reforms after F refolds to the postfusion form.
A previously characterized mutation (S443P) located at the junction of the subunits within the F homotrimer in domain I conferred a hyperfusogenic phenotype to PIV5 F (53). To probe further the metastable nature of the F protein and its associated refolding dynamics, we designed single point mutations in domain I to investigate further the role of this domain in the stability of the F protein. Mutants F-V340A, F-V362A, and F-S364A showed hyperfusogenic phenotypes similar to that of F-S443P. Interestingly, as in F-S443P, these three mutations were located between adjacent protomers of the F trimer, suggesting a critical role for this region of domain I in the stability of the F protein. In fact, these mutations cause the F proteins to become unstable to such an extent that they are independent of the HN trigger and can promote significant cell-cell fusion at 37°C, when expressed in the absence of HN. Hence, unlike the Ig-like domain mutants, which are HN insensitive, these mutants with mutations in domain I are HN independent. In the study which conferred SV41 HN specificity to PIV5 F by replacing some PIV5 F segments with those of SV41 F, amino acid replacements inducing a hyperfusogenic phenotype were also found at the junction of the protomers (51).
A model was created by aligning the PIV5 F prefusion structure (15) with those of the PIV5 HN stalk 4HB (34) and the HN globular heads in the 4-heads-up conformation (26). The distances for membrane-proximal regions for which no electron density was observed were calculated under the assumption that these regions are helical in nature (Fig. 6). Interestingly, the Ig-like domain of PIV5 F lines up at approximately the same height as the central part of the HN stalk, characterized by a transition from a linear to a left-handed supercoiled region. Incidentally, this region has been recently implicated in F interactions for various paramyxoviruses, including PIV5, measles virus, and CDV (34, 36, 47, 48). The model suggests that F-triggering interactions between the proposed site of F in the Ig-like domain and the HN (H or G) stalk are plausible when HN globular heads are in the “up” position.
ACKNOWLEDGMENTS
This research was supported in part by National Institutes of Health research grants AI-23173 (to R.A.L.) and GM-61050 (to T.S.J.). M.A. is a Research Specialist and R.A.L. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Published ahead of print 2 October 2013
REFERENCES
- 1.Lamb RA. 2007. Mononegavirales, p 1357–1361 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed. Lippincott, Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 2.Schowalter RM, Chang A, Robach JG, Buchholz UJ, Dutch RE. 2009. Low-pH triggering of human metapneumovirus fusion: essential residues and importance in entry. J. Virol. 83:1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heminway BR, Yu Y, Galinski MS. 1994. Paramyxovirus mediated cell fusion requires co-expression of both the fusion and hemagglutinin-neuraminidase glycoproteins. Virus Res. 31:1–16 [DOI] [PubMed] [Google Scholar]
- 4.Horvath CM, Paterson RG, Shaughnessy MA, Wood R, Lamb RA. 1992. Biological activity of paramyxovirus fusion proteins: factors influencing formation of syncytia. J. Virol. 66:4564–4569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu X, Ray R, Compans RW. 1992. Functional interactions between the fusion protein and hemagglutinin-neuraminidase of human parainfluenza viruses. J. Virol. 66:1528–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrison T, McQuain C, McGinnes L. 1991. Complementation between avirulent Newcastle disease virus and a fusion protein gene expressed from a retrovirus vector: requirements for membrane fusion. J. Virol. 65:813–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yao Q, Hu X, Compans RW. 1997. Association of the parainfluenza virus fusion and hemagglutinin-neuraminidase glycoproteins on cell surfaces. J. Virol. 71:650–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connolly SA, Leser GP, Yin HS, Jardetzky TS, Lamb RA. 2006. Refolding of a paramyxovirus F protein from prefusion to postfusion conformations observed by liposome binding and electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 103:17903–17908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bose S, Zokarkar A, Welch BD, Leser GP, Jardetzky TS, Lamb RA. 2012. Fusion activation by a headless parainfluenza virus 5 hemagglutinin-neuraminidase stalk suggests a modular mechanism for triggering. Proc. Natl. Acad. Sci. U. S. A. 109:E2625–E2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ader N, Brindley M, Avila M, Orvell C, Horvat B, Hiltensperger G, Schneider-Schaulies J, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2013. Mechanism for active membrane fusion triggering by morbillivirus attachment protein. J. Virol. 87:314–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan YP, Lu M, Dutta S, Yan L, Barr J, Flora M, Feng YR, Xu K, Nikolov DB, Wang LF, Skiniotis G, Broder CC. 2012. Biochemical, conformational and immunogenic analysis of soluble trimeric forms of henipavirus fusion glycoproteins. J. Virol. 86:11457–11471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker KA, Dutch RE, Lamb RA, Jardetzky TS. 1999. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell 3:309–319 [DOI] [PubMed] [Google Scholar]
- 13.Lamb RA, Jardetzky TS. 2007. Structural basis of viral invasion: lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 17:427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang A, Dutch RE. 2012. Paramyxovirus fusion and entry: multiple paths to a common end. Viruses 4:613–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, Zhou T, Baxa U, Yasuda E, Beaumont T, Kumar A, Modjarrad K, Zheng Z, Zhao M, Xia N, Kwong PD, Graham BS. 2013. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340:1113–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welch BD, Liu Y, Kors CA, Leser GP, Jardetzky TS, Lamb RA. 2012. Structure of the cleavage-activated prefusion form of the parainfluenza virus 5 fusion protein. Proc. Natl. Acad. Sci. U. S. A. 109:16672–16677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin H-S, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A. 102:9288–9293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swanson K, Wen X, Leser GP, Paterson RG, Lamb RA, Jardetzky TS. 2010. Structure of the Newcastle disease virus F protein in the post-fusion conformation. Virology 402:372–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McLellan JS, Yang Y, Graham BS, Kwong PD. 2011. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 85:7788–7796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swanson KA, Settembre EC, Shaw CA, Dey AK, Rappuoli R, Mandl CW, Dormitzer PR, Carfi A. 2011. Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers. Proc. Natl. Acad. Sci. U. S. A. 108:9619–9624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russell CJ, Jardetzky TS, Lamb RA. 2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 20:4024–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim YH, Donald JE, Grigoryan G, Leser GP, Fadeev AY, Lamb RA, DeGrado WF. 2011. Capture and imaging of a prehairpin fusion intermediate of the paramyxovirus PIV5. Proc. Natl. Acad. Sci. U. S. A. 108:20992–20997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crennell S, Takimoto T, Portner A, Taylor G. 2000. Crystal structure of the multifunctional paramyxovirus hemagglutinin-neuraminidase. Nat. Struct. Biol. 7:1068–1074 [DOI] [PubMed] [Google Scholar]
- 25.Lawrence MC, Borg NA, Streltsov VA, Pilling PA, Epa VC, Varghese JN, McKimm-Breschkin JL, Colman PM. 2004. Structure of the haemagglutinin-neuraminidase from human parainfluenza virus type III. J. Mol. Biol. 335:1343–1357 [DOI] [PubMed] [Google Scholar]
- 26.Yuan P, Thompson T, Wurzburg BA, Paterson RG, Lamb RA, Jardetzky TS. 2005. Structural studies of the parainfluenza virius 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure 13:803–815 [DOI] [PubMed] [Google Scholar]
- 27.Yuan P, Leser GP, Demeler B, Lamb RA, Jardetzky TS. 2008. Domain architecture and oligomerization properties of the paramyxovirus PIV 5 hemagglutinin-neuraminidase (HN) protein. Virology 378:282–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan P, Swanson KA, Leser GP, Paterson RG, Lamb RA, Jardetzky TS. 2011. Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc. Natl. Acad. Sci. U. S. A. 108:14920–14925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan P, Paterson RG, Leser GP, Lamb RA, Jardetzky TS. 2012. Structure of the Ulster strain Newcastle disease virus hemagglutinin-neuraminidase reveals auto-inhibitory interactions associated with low virulence. PLoS Pathog. 8:e1002855. 10.1371/journal.ppat.1002855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colf LA, Juo ZS, Garcia KC. 2007. Structure of the measles virus hemagglutinin. Nat. Struct. Mol. Biol. 14:1227–1228 [DOI] [PubMed] [Google Scholar]
- 31.Hashiguchi T, Kajikawa M, Maita N, Takeda M, Kuroki K, Sasaki K, Kohda D, Yanagi Y, Maenaka K. 2007. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc. Natl. Acad. Sci. U. S. A. 104:19535–19540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowden TA, Aricescu AR, Gilbert RJ, Grimes JM, Jones EY, Stuart DI. 2008. Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat. Struct. Biol. 15:567–572 [DOI] [PubMed] [Google Scholar]
- 33.Xu K, Rajashankar KR, Chan YP, Himanen JP, Broder CC, Nikolov DB. 2008. Host cell recognition by the henipaviruses: crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc. Natl. Acad. Sci. U. S. A. 105:9953–9958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bose S, Welch BD, Kors CA, Yuan P, Jardetzky TS, Lamb RA. 2011. Structure and mutagenesis of the parainfluenza virus 5 hemagglutinin-neuraminidase stalk domain reveals a four-helix bundle and the role of the stalk in fusion promotion. J. Virol. 85:12855–12866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maar D, Harmon B, Chu D, Schulz B, Aguilar HC, Lee B, Negrete OA. 2012. Cysteines in the stalk of the Nipah virus G glycoprotein are located in a distinct subdomain critical for fusion activation. J. Virol. 86:6632–6642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ader N, Brindley MA, Avila M, Origgi FC, Langedijk J, Orvell C, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2012. Structural rearrangements of the central region of the morbillivirus attachment protein stalk domain trigger F protein refolding for membrane fusion. J. Biol. Chem. 287:16324–16334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paal T, Brindley MA, St Clair C, Prussia A, Gaus D, Krumm SA, Snyder JP, Plemper RK. 2009. Probing the spatial organization of measles virus fusion complexes. J. Virol. 83:10480–10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JK, Prussia A, Paal T, White LK, Snyder JP, Plemper RK. 2008. Functional interaction between paramyxovirus fusion and attachment proteins. J. Biol. Chem. 283:16561–16572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bishop KA, Hickey AC, Khetawat D, Patch JR, Bossart KN, Zhu Z, Wang LF, Dimitrov DS, Broder CC. 2008. Residues in the stalk domain of the Hendra virus G glycoprotein modulate conformational changes associated with receptor binding. J. Virol. 82:11398–11409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bousse T, Takimoto T, Gorman WL, Takahashi T, Portner A. 1994. Regions on the hemagglutinin-neuraminidase proteins of human parainfluenza virus type-1 and Sendai virus important for membrane fusion. Virology 204:506–514 [DOI] [PubMed] [Google Scholar]
- 41.Deng R, Wang Z, Mirza AM, Iorio RM. 1995. Localization of a domain on the paramyxovirus attachment protein required for the promotion of cellular fusion by its homologous fusion protein spike. Virology 209:457–469 [DOI] [PubMed] [Google Scholar]
- 42.Deng R, Wang Z, Mahon PJ, Marinello M, Mirza A, Iorio RM. 1999. Mutations in the Newcastle disease virus hemagglutinin-neuraminidase protein that interfere with its ability to interact with the homologous F protein in the promotion of fusion. Virology 253:43–54 [DOI] [PubMed] [Google Scholar]
- 43.Melanson VR, Iorio RM. 2004. Amino acid substitutions in the F-specific domain in the stalk of the newcastle disease virus HN protein modulate fusion and interfere with its interaction with the F protein. J. Virol. 78:13053–13061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melanson VR, Iorio RM. 2006. Addition of N-glycans in the stalk of the Newcastle disease virus HN protein blocks its interaction with the F protein and prevents fusion. J. Virol. 80:623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stone-Hulslander J, Morrison TG. 1999. Mutational analysis of heptad repeats in the membrane-proximal region of Newcastle disease virus HN protein. J. Virol. 73:3630–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanabayashi K, Compans RW. 1996. Functional interaction of paramyxovirus glycoproteins: identification of a domain in Sendai virus HN which promotes cell fusion. J. Virol. 70:6112–6118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brindley MA, Takeda M, Plattet P, Plemper RK. 2012. Triggering the measles virus membrane fusion machinery. Proc. Natl. Acad. Sci. U. S. A. 109:E3018–E3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Navaratnarajah CK, Negi S, Braun W, Cattaneo R. 2012. Membrane fusion triggering: three modules with different structure and function in the upper half of the measles virus attachment protein stalk. J. Biol. Chem. 287:38543–38551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Apte-Sengupta S, Negi S, Leonard VH, Oezguen N, Navaratnarajah CK, Braun W, Cattaneo R. 2012. Base of the measles virus fusion trimer head receives the signal that triggers membrane fusion. J. Biol. Chem. 287:33026–33035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsurudome M, Ito M, Nishio M, Nakahashi M, Kawano M, Komada H, Nosaka T, Ito Y. 2011. Identification of domains on the fusion (F) protein trimer that influence the hemagglutinin-neuraminidase specificity of the F protein in mediating cell-cell fusion. J. Virol. 85:3153–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsurudome M, Nakahashi M, Matsushima Y, Ito M, Nishio M, Kawano M, Komada H, Nosaka T. 2013. Full conversion of the hemagglutinin-neuraminidase specificity of the parainfluenza virus 5 fusion protein by replacement of 21 amino acids in its head region with those of the simian virus 41 fusion protein. J. Virol. 87:8342–8350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Randall RE, Young DF, Goswami KKA, Russell WC. 1987. Isolation and characterization of monoclonal antibodies to simian virus 5 and their use in revealing antigenic differences between human, canine and simian isolates. J. Gen. Virol. 68:2769–2780 [DOI] [PubMed] [Google Scholar]
- 53.Paterson RG, Russell CJ, Lamb RA. 2000. Fusion protein of the paramyxovirus SV5: destabilizing and stabilizing mutants of fusion activation. Virology 270:17–30 [DOI] [PubMed] [Google Scholar]
- 54.Paterson RG, Lamb RA. 1993. The molecular biology of influenza viruses and paramyxoviruses, p 35–73 In Davidson A, Elliott RM. (ed), Molecular virology: a practical approach. IRL Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 55.Paterson RG, Hiebert SW, Lamb RA. 1985. Expression at the cell surface of biologically active fusion and hemagglutinin-neuraminidase proteins of the paramyxovirus simian virus 5 from cloned cDNA. Proc. Natl. Acad. Sci. U. S. A. 82:7520–7524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tamada T, Honjo E, Maeda Y, Okamoto T, Ishibashi M, Tokunaga M, Kuroki R. 2006. Homodimeric cross-over structure of the human granulocyte colony-stimulating factor (GCSF) receptor signaling complex. Proc. Natl. Acad. Sci. U. S. A. 103:3135–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chow D, He X, Snow AL, Rose-John S, Garcia KC. 2001. Structure of an extracellular gp130 cytokine receptor signaling complex. Science 291:2150–2155 [DOI] [PubMed] [Google Scholar]
- 58.Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. 2003. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science 300:2101–2104 [DOI] [PubMed] [Google Scholar]
- 59.Aguilar HC, Matreyek KA, Filone CM, Hashimi ST, Levroney EL, Negrete OA, Betrolotti-Ciarlet A, Choi DY, McHardy I, Fulcher JA, Su SV, Wolf MC, Kohatsu L, Baum LG, Lee B. 2006. N-glycans on Nipah virus fusion protein protect against neutralization but reduce membrane fusion and viral entry. J. Virol. 80:4878–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Plemper RK, Hammond AL, Gerlier D, Fielding AK, Cattaneo R. 2002. Strength of envelope protein interaction modulates cytopathicity of measles virus. J. Virol. 76:5051–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mirza AM, Iorio RM. 2013. A mutation in the stalk of the NDV HN protein prevents triggering of the F protein despite allowing efficient HN-F complex formation. J. Virol. 87:8813–8815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Corey EA, Iorio RM. 2007. Mutations in the stalk of the measles virus hemagglutinin protein decrease fusion but do not interfere with virus-specific interaction with the homologous fusion protein. J. Virol. 81:9900–9910 [DOI] [PMC free article] [PubMed] [Google Scholar]