

Abstract
Flavivirus-infected cells secrete a structurally heterogeneous population of viruses because of an inefficient virion maturation process. Flaviviruses assemble as noninfectious, immature virions composed of trimers of envelope (E) and precursor membrane (prM) protein heterodimers. Cleavage of prM is a required process during virion maturation, although this often remains incomplete for infectious virus particles. Previous work demonstrated that the efficiency of virion maturation could impact antibody neutralization through changes in the accessibility of otherwise cryptic epitopes on the virion. In this study, we show that the neutralization potency of monoclonal antibody (MAb) E33 is sensitive to the maturation state of West Nile virus (WNV), despite its recognition of an accessible epitope, the domain III lateral ridge (DIII-LR). Comprehensive epitope mapping studies with 166 E protein DIII-LR variants revealed that the functional footprint of MAb E33 on the E protein differs subtly from that of the well-characterized DIII-LR MAb E16. Remarkably, aromatic substitutions at E protein residue 306 ablated the maturation state sensitivity of E33 IgG, and the neutralization efficacy of E33 Fab fragments was not affected by changes in the virion maturation state. We propose that E33 IgG binding on mature virions orients the Fc region in a manner that impacts subsequent antibody binding to nearby sites. This Fc-mediated steric constraint is a novel mechanism by which the maturation state of a virion modulates the efficacy of the humoral immune response to flavivirus infection.
INTRODUCTION
Flaviviruses are a group of single-stranded, positive-sense, enveloped RNA viruses that annually infect over 390 million individuals worldwide (1). West Nile virus (WNV), a mosquito-transmitted virus that is a member of this genus, causes a spectrum of diseases in humans and other vertebrate animals that ranges from a self-limiting febrile illness to severe meningitis or encephalitis (2, 3). Roughly 25% of infected humans become symptomatic, and the elderly and immunocompromised are at greatest risk for severe, potentially fatal outcomes (2, 4, 5). Although WNV is typically associated with a modest number of human cases each year, intense local outbreaks with significant morbidity and mortality have occurred (6). Despite WNV's global distribution and potential to cause significant disease, there are no vaccines or therapeutics available for use in humans.
The ∼11-kb flavivirus RNA genome is contained within spherical enveloped virions that are covered by a dense arrangement of 180 envelope (E) proteins (Fig. 1A) (7, 8). The viral genomic RNA is translated as a single polyprotein and is cleaved by cellular and viral proteases into three structural proteins (E, precursor membrane [prM], and capsid [C]) and seven nonstructural proteins (9). Flaviviruses assemble at the rough endoplasmic reticulum (ER) and bud into the lumen as immature virus particles. The surfaces of the virions include 60 trimeric spikes formed from heterodimers of the prM and E structural proteins (Fig. 1A, left) (9–11). To become infectious, flaviviruses undergo a maturation process prior to egress (12, 13). When immature virus particles traffic from the ER to the low-pH environment of the trans-Golgi network, the E proteins collapse to form 90 homodimers orientated roughly parallel to the virion surface (Fig. 1A, center) (14, 15). This rearrangement exposes a site on prM that allows for cleavage by a cellular furin-like protease into pr and M (13). The cleaved pr peptide dissociates from the virion when it is released into the neutral extracellular space, resulting in the formation of a relatively smooth, infectious, mature virus particle that retains only the M peptide (14) (Fig. 1A, right). Although the cleavage of prM during maturation is necessary for acquisition of infectivity, depending on the cell type this process may be inefficient (16–21). Indeed, flavivirus-infected cells secrete a structurally heterogeneous population of infectious virions with varied degrees of prM cleavage (22–24).
Fig 1.

The WNV E protein undergoes conformational rearrangements during maturation. (A) Model of the cryo-electron single-particle reconstructions of immature flavivirus virion at near-neutral pH (left), immature flavivirus virion at acidic pH (center), and mature flavivirus virion at near-neutral pH. The WNV E protein domain I is shown in red, domain II is in yellow, and domain III is in blue. The fusion loop (FL) located within domain II is shown in green. The prM protein is shown in purple. (B) The crystal structure of the WNV E protein, with domains colored as above. In addition, the four loops comprising DIII-LR are labeled. (C) The WNV E protein domain III, displaying the 14 amino acids selected for our saturation-mutagenesis studies.
Crystal structures of several flavivirus E proteins have been solved, all of which revealed a common molecular architecture consisting of three domains (25–30) (Fig. 1B). Domain I (DI) organizes the elongated E protein structure by linking domains II and III (DII and DIII) via a β-barrel. On the distal end of DII is the hydrophobic fusion loop that facilitates the merging of viral and host lipid bilayers during viral membrane fusion (31). Finally, DIII has an immunoglobulin-like fold and contains a lateral ridge (DIII-LR), composed of four loops (DE, BC, N-terminal loop, and FG) (Fig. 1B and C), that protrudes from the surface on mature virions (32). The solvent accessibility of this region suggests a role for the DIII-LR in interacting with host attachment and/or entry receptors. Indeed, mutations within this region alter virulence and cellular tropism (33–35).
Studies from many laboratories have established that the humoral immune response is critical for the prevention and control of flavivirus infections (36–44). The majority of neutralizing antibodies are directed against the E protein; neutralizing monoclonal antibodies (MAbs) that target each of the three domains within the E protein have been characterized (reviewed in reference 45). To neutralize infection, antibodies must simultaneously engage an individual virion with a stoichiometry that exceeds a critical threshold (46). Studies with WNV E DIII-LR MAbs have suggested that approximately 30 antibodies are required for neutralization (47). Within this framework, two parameters act in concert to define conditions under which neutralization is possible. First, antibody affinity determines the fraction of accessible epitopes occupied at a given antibody concentration (48, 49). The second factor is the accessibility of the epitope on the virus particle for antibody engagement.
Differences in the arrangement of E proteins on mature, partially mature, and immature virions introduce a level of complexity with regard to the accessibility of epitopes on the virion. The prM content of infectious virions alters sensitivity to neutralization by many classes of antibodies (50). For example, the DII-FL MAb E53 only efficiently neutralizes WNV particles retaining high levels of uncleaved prM (50, 51). In contrast, the neutralization potencies of DIII-LR MAbs E16 and E24 are insensitive to the maturation state of the virus particle (50). These data are in agreement with structural models of flavivirus virions that suggest the DIII-LR epitope remains accessible to solvent irrespective of the maturation state. Unexpectedly, we recently discovered a DIII-LR MAb whose neutralization activity was modulated by the level of uncleaved prM on the surface of the virion. We investigated the functional mechanism by which the maturation state of WNV modulates the efficacy of an antibody that recognizes the accessible DIII-LR epitope of the E protein. Our study shows that the Fc region of a DIII-LR antibody can affect binding and neutralization of mature WNV virions. This provides a new mechanism by which the maturation state of a virion can impact the efficacy of the humoral immune response to flavivirus infection.
MATERIALS AND METHODS
Cell culture.
HEK-293T cells were cultured in a low-d-glucose (1 g/liter) formulation of Dulbecco's modified Eagle's medium (DMEM) containing HEPES (Invitrogen) that was supplemented with 7% fetal bovine serum (FBS; HyClone) and 100 U/ml penicillin-streptomycin (P/S; Invitrogen). Raji-DCSIGNR cells were cultured in RPMI 1640 medium containing Glutamax (Invitrogen) that was supplemented with 7% FBS and 100 U/ml P/S. Cell lines were maintained in the presence of 7% CO2 at 37°C.
Measurement of antibody functional affinities.
Antibody functional affinities to WNV reporter virus particles (RVPs) was measured using an indirect enzyme-linked immunosorbent assay (ELISA). High-protein binding ELISA plates (Reacti-Bind; Thermo Scientific) were coated with 1 μg/ml of humanized E16 (hE16) MAb under alkaline conditions (0.2 M CO32−/HCO3−, pH 9.4) for 16 h at 4°C. ELISA plates containing immobilized hE16 were blocked with bovine serum albumin (BSA; in 1× phosphate-buffered saline [PBS], 0.05% Tween 20, 1% BSA) for at least 2 h and then washed (1× PBS, 0.05% Tween 20). Supernatants containing WNV RVPs were diluted in RPMI 1640, added to hE16-coated wells, and incubated at 25°C for 2 h. All wells were then washed (1× PBS, 0.05% Tween 20). Ten serial 2-fold dilutions of primary MAbs (MAb E16, E33, or E60) were then added and incubated at 25°C for 1 h. Following incubation, all wells were washed (1× PBS, 0.05% Tween 20) and, for secondary detection, horseradish peroxidase (HRP)-antibody conjugate (0.2 μg/ml; goat anti-mouse HRP; Thermo Scientific) was added and the mixture was incubated for 1 h. All wells were then washed (1× PBS, 0.05% Tween 20). Ultra TMB-ELISA peroxidase substrate (Thermo Scientific) was added and the mixture was incubated for approximately 10 to 15 min. Reactions were quenched with 2 M H2SO4, and absorbance at 450 nm was measured by using a SpectraMax M5 plate reader (Molecular Devices). Apparent antibody affinity was estimated using a rectangular hyperbola curve that described the equilibrium binding between WNV RVPs and an antibody as a functon of increasing antibody concentration (Prism; GraphPad Software).
Generation of WNV DIII-LR saturation mutagenesis plasmid library.
A previously described plasmid (pCBWN) carrying the New York 1999 WNV prM-E structural genes was used as a mutagenesis template (52). Primers designed to introduce a degenerate codon (NNN) at nucleotide positions corresponding to selected amino acids were synthesized (Invitrogen) and employed in site-directed mutagenesis reactions with the Pfu Ultra DNA polymerase system (Agilent Technologies). PCR mixtures were treated with DpnI (New England BioLabs) for 1 h at 37°C, transformed into Stbl2 Escherichia coli cells (Invitrogen), and spread on LB plates containing 100 μg/ml of carbenicillin. All bacterial propagation was performed at 30°C. A total of 48 colonies were selected from each reaction mixture, and their plasmids were isolated by using a Spin miniprep kit (Qiagen). Colonies were screened by sequencing the specific gene regions of interest (Macrogen Corporation, Rockville, MD). Typically, this approach yielded from 13 to 16 unique mutants for each codon, although this efficiency varied significantly as a function of the number of possible codons for each amino acid mutation. To complete the saturation mutagenesis library, a second round of PCR amplification experiments was performed with primers designed to introduce the specified mutations not isolated in the first screen. Ultimately, the full sequence of the prM-E genes from all plasmids within the library were confirmed by DNA sequencing. Furthermore, once the library was completed, 20% of plasmids within the library were selected at random and subjected to confirmatory sequencing over the region of interest.
Production of WNV RVPs.
WNV RVPs were produced as described previously (50). Furin RVPs were produced by transfection of HEK-293T cells with DNA plasmids carrying the WNV capsid pCBWN, human furin, and a WNV lineage II (strain 956) replicon expressing GFP (53) in a ratio of 30:1:10:10 by mass. NH4Cl RVPs were produced in a similar manner as described here, except the plasmid carrying human furin was replaced (on a weight-by-weight basis) with pcDNA3.1. All transfections were performed using Lipofectamine LTX and Plus reagent (Invitrogen) according to the manufacturer's instructions. For both furin and NH4Cl RVP preparations, the media were exchanged 4 h following transfection with low-glucose DMEM containing HEPES that was supplemented with 7% FBS and 100 U/ml P/S. For NH4Cl RVP preparations, culture medium was supplemented to a final concentration of 20 mM NH4Cl dissolved in 1× PBS. Furin RVPs were produced at 30°C, while NH4Cl RVPs were produced at 37°C. Culture supernatant containing WNV RVPs was harvested serially every 24 h between 24 and 96 h posttransfection, filtered through a 0.22-μm filter, and stored at −80°C. Following each serial harvest, low-glucose DMEM containing HEPES supplemented with 7% FBS and 100 U/ml P/S was added.
Measuring the titer of WNV RVPs.
Ten serial 2-fold dilutions of WNV RVPs were used to infect Raji-DCSIGNR cells at a concentration of 1 × 105 cells/well in a total volume of 200 μl. Infected Raji-DCSIGNR cells were incubated at 37°C for 36 to 48 h, after which infectivity was assessed by determining the percentage of cells expressing GFP via flow cytometry. Estimates of titers were made using only linear portions of the curve of the RVP concentration versus the percentage of infected cells. Maximal infectivity was defined as the infectivity on Raji-DCSIGNR of neat RVP-containing supernatant.
Neutralization assays.
Stocks of RVPs of a determined titer were diluted in a manner sufficient to infect between 1% and 3% of Raji-DCSIGNR cells. This condition ensured antibody excess at informative portions of the dose-response curve. Diluted RVPs were mixed with serial 4-fold dilutions of the MAb in duplicate. Antibody-RVP mixtures were incubated at 37°C for 1 h to achieve binding equilibrium, added to 1 × 105 Raji-DCSIGNR cells, and incubated at 37°C for 36 to 48 h in a total volume of 300 μl. Infectivity was assessed by determining the percentage of cells expressing GFP based on flow cytometry. The concentration of antibody required to inhibit 50% of infection (EC50) was estimated by regression analysis. Data were fit with a sigmoidal dose-response curve (variable slope), where the top of the curve was constrained to 100 and the bottom to 0 (Prism; GraphPad Software).
Production of Fab fragments.
Fab fragments were generated from purified MAbs by using a Fab fragment preparation kit (Thermo Scientific) as specified by the manufacturer. Purified MAbs were incubated for 5 to 6 h at 37°C with papain at pH 6.6 to generate Fab fragments. Fab fragments were purified by negative selection using a protein A column and concentrated by using a 30-kDa molecular mass cutoff centrifugal device (Amicon). The purities of the products were assessed via nonreducing SDS-PAGE. Fab fragments were stored at 4°C. Concentrations were measured based on the absorbance at 280 nm, with an estimated extinction coefficient (1-mg/ml solution) of 1.4.
RESULTS
Maturation state-dependent neutralization by a WNV DIII-LR antibody.
WNV RVPs are pseudo-infectious virions produced by genetic complementation of a subgenomic replicon encoding GFP with viral structural genes (C, prM, and E) expressed in trans (53). This technology has been utilized extensively to study antibody-mediated neutralization of flaviviruses (47, 50, 54–57). RVPs produced under standard conditions are inherently heterogeneous with respect to the efficiency of virion maturation, and thus they are composed of unknown ratios of mature, immature, and partially mature virus particles. To study the impact of virion maturation on antibody-mediated neutralization with more homogenous populations of virions representative of each end of the maturation spectrum, we produced RVPs under conditions that either enhanced or impaired the efficiency of prM cleavage (50, 58). To create particles that approximated mature virions, exogenous human furin protease was overexpressed in RVP-producing cells (furin RVPs). In contrast, the synthesis of RVPs approximating immature virions required the addition of NH4Cl to a final concentration of 20 mM (NH4Cl RVPs). NH4Cl raises the pH within the trans-Golgi network, which prevents the structural rearrangements of the E protein necessary for prM cleavage (59).
To investigate the impact of the maturation state of WNV on antibody-mediated neutralization, 20 serial 2-fold dilutions of DIII-LR-specific MAbs E16 and E33 or the DII-FL-specific MAb E60 were incubated in parallel with either furin or NH4Cl RVPs for 1 h at 37°C. The resulting immune complexes were then added to Raji-DCSIGNR cells, and the infectivities of the two populations of RVPs in the presence of each of the three MAbs were determined by flow cytometry analysis. As anticipated from prior studies (50), E16 neutralized furin and NH4Cl RVPs with a similar potency (Fig. 2A and D). In contrast, neutralization of furin RVPs required 6.2-fold more E33 (n = 36; P < 0.0001) than did prM-containing NH4Cl RVPs (Fig. 2B and E). Both E16 and E33 neutralized NH4Cl RVPs with similar efficacies. In agreement with previous findings, E60, which recognizes an epitope that is cryptic on mature virions, displayed a reduced capacity to neutralize furin RVPs compared to NH4Cl RVPs that retained significant quantities of uncleaved prM (Fig. 2C and F) (50). Analysis of the differences between the log10 EC50 values of furin and NH4Cl RVPs neutralized by E16, E33, and E60 revealed that E33 defines a novel class of maturation state-sensitive antibodies that binds a solvent-accessible epitope on the surface of the mature virion (Fig. 2G).
Fig 2.

The WNV E protein DIII-LR MAb E33 is maturation state sensitive. (A to C) Twenty 2-fold serial dilutions of MAbs E16 (A), E33 (B), and E60 (C) were incubated with either furin RVPs (blue) or NH4Cl RVPs (red) for 1 h at 37°C to achieve binding equilibrium. Immune complexes were then added to Raji-DCSIGNR cells, and infectivity was assessed 36 to 48 h postinfection by flow cytometry. Results shown were normalized to infectivity in the absence of antibody. Error bars display the standard errors of duplicate infections. Data are representative of 36 independent experiments. (D to F) The arithmetic mean log10(EC50) values for MAbs E16 (D), E33 (E), and E60 (F) for furin or NH4Cl RVPs from all 36 independent, parallel experiments are displayed. Errors bars display the standard errors. (G) Differences in log10(EC50) values between furin and NH4Cl RVPs neutralized by E16, E33, or E60. The differences were calculated from the arithmetic means of 36 independent experiments. Statistical analysis was performed using Prism software (GraphPad). The differences in log10(EC50) values between furin and NH4Cl RVPs neutralized by E16, E33, or E60 were compared by using a one-way analysis of variance followed by Tukey's multiple comparison test. The dashed line indicates the lower limit of detection. ****, P < 0.0001.
MAb E33 binds mature and NH4Cl RVPs with a similar functional affinity.
The neutralization potency of an antibody is governed in part by the affinity of epitope recognition. The affinity of a MAb to any E protein epitope is complicated by the quaternary structure of the virus particle, which may in turn be modulated by the extent of virion maturation. To determine if differences in functional affinity were responsible for maturation state-sensitive properties of E33, indirect ELISAs using furin and NH4Cl RVPs were performed. If the maturation state-sensitive property of MAb E33 were driven by differences in functional affinity for its epitope on furin and NH4Cl RVPs, we would anticipate a 6.2-fold difference in Kd values derived from our indirect ELISAs. However, E16, E33, and E60 had comparable functional affinities to both populations of RVPs (n = 5) (Table 1). These data suggest the the apparent affinity of antibody recognition alone does not govern the differential neutralization sensitivity seen with MAb E33 against furin and NH4Cl RVPs.
Table 1.
Functional affinities of MAbs E16, E33, and E60 for furin and NH4Cl RVPs
| MAb | Affinity (Kd [nM]) for RVPs ofa: |
|
|---|---|---|
| Furin | NH4Cl | |
| E16 | 0.15 ± 0.04 | 0.20 ± 0.06 |
| E33 | 0.09 ± 0.06 | 0.08 ± 0.03 |
| E60 | 0.14 ± 0.08 | 0.61 ± 0.60 |
Values are means ± standard errors.
Variants reveal DIII-LR tolerates extensive mutagenesis.
The details of the interaction between E16 and WNV DIII have been defined using biochemical, functional, and structural methodologies (40, 60). In contrast, residues on the DIII-LR that contribute to neutralization by E33 have not been extensively studied. A comparison of the E16 and E33 functional footprints, defined as those residues that when mutated impact the neutralization of virions by antibodies, might provide insight into the mechanism(s) that contributes to the maturation state sensitivity of antibodies that bind surface-accessible epitopes, such as the DIII-LR. To this end, we sought to identify residues that comprise the E33 functional footprint by using a saturation-mutagenesis approach. A total of 14 residues that are within and adjacent to the DIII-LR were selected for study and mutated to each of the remaining 19 amino acids (Fig. 1C). The selection of these residues was guided by residues previously implicated in DIII-LR recognition by antibodies (61–68), the structure of the DIII-LR antibody E16 bound to WNV DIII (40, 60, 69), and residues involved in forming the solvent-accessible loops of the DIII-LR itself (25, 29). Furthermore, because weak noncovalent interactions between the antibody and the polypeptide main chain could contribute functionally to the epitope, both surface-accessible and buried residues that aid in the formation of the overall architecture of the region were selected. Mutations were introduced into a WNV prM-E expression plasmid by site-directed mutagenesis using degenerate primers (see Materials and Methods). We created a library where all 20 amino acids for each of the 14 target residues were expressed, resulting in a collection of 266 WNV DIII-LR mutants.
Each mutant plasmid was used to create furin RVPs by complementation with plasmids expressing capsid and a subgenomic replicon encoding a GFP reporter gene (53). The infectivity of each variant was determined by titration on Raji-DCSIGNR cells and compared to the WT RVPs that were produced in parallel. In order to examine broad trends in the data, each variant was placed into one of seven categories based on the chemical identity of its mutant residue. This organization scheme revealed that the target residues of interest could be partitioned into one of three categories based on their ability to tolerate mutations. A limited number of residues (C305 and Y329) did not tolerate any mutations, as none of the mutants were infectious (Fig. 3A and D). Several residues (Y302, F309, and D333) withstood mutations to amino acids only within the same chemical group (Fig. 3B and D). However, a majority of residues (n = 9) tolerated mutation to almost any other amino acid without marked adverse effects (Fig. 3C and D).
Fig 3.

The infectivities of DIII-LR mutants partition into three patterns and reveal functional plasticity. (A to C) Ten 2-fold serial dilutions of a panel of saturation mutants at E protein residues C305 (A), Y302 (B), and Q391 (C) were added to Raji-DCSIGNR cells. The variants were divided into seven groups based on the chemical identity of the mutant side chain. The residues G and A were classified as small amino acids (red); S, T, and C are nucleophilic (light blue); V, L, I, M, and P are hydrophobic (purple); F, Y, and W are aromatic (orange); D and E are acidic (dark blue); Q and E are amides (green); H, K, and R are basic (brown). Infectivity of the mutants was assessed 36 to 48 h postinfection by flow cytometry and normalized to the maximal infectivity of the wild-type control. Results are representative of at least two independent experiments. (D) Relative infectivities of mutants, compared to the wild type. Green, yellow, and red tiles indicate a ≤2-fold, ≤10-fold, or ≥100-fold difference in maximal infectivity compared to the wild type, respectively. Blue tiles represent the wild-type infectivity data. Results were compiled from at least two independent experiments with independent RVP preparations.
Maturation-sensitive MAb E33 recognizes a distinct functional epitope on the DIII-LR.
We utilized this panel of WNV mutants to determine the functional epitope of MAb E33. To do so, we generated furin RVPs of panels of viable mutants at positions 306, 307, 310, 311, 330, 332, 366, 390, and 391. These positions were selected from the original 14 residues of interest since amino acids from all seven different chemical groups could be expressed without markedly altering infectivity compared to WT. Parallel neutralization studies with E16, E33, and E60 were performed on each mutant. To ensure that our saturation-mutagenesis approach could unambiguously determine a functional footprint, E16 was included in each experiment as a positive control. Furthermore, the maturation state-sensitive MAb E60 also was included as a control to ensure that mutations introduced within or adjacent to the DIII-LR did not perturb either the quaternary structure of the virion or the efficiency of prM cleavage. Since E60 recognizes an epitope that includes the DII-FL, we anticipated that the EC50 for this antibody toward any of the DIII-LR variants would be comparable to that for the WT. All neutralization studies were performed in parallel, at least two times, using independently produced furin RVPs. A representative neutralization study using the library of S306 variants is shown in Fig. 4. Mutations at this position increased, decreased, or had no effect on neutralization potencies of E16 or E33. Analysis of the E60 data revealed that none of the mutations markedly altered the EC50 of this MAb, suggesting that the quaternary structure of the E proteins in the presence of these mutations was not altered compared to the WT. Furthermore, our MAb E60 control ensured that all of the variants tested possessed comparable maturation states.
Fig 4.

Parallel neutralization studies of MAbs E16, E33, and E60 against a panel of 19 S306 mutants. Ten 4-fold serial dilutions of MAbs E16, E33, and E60 were incubated with the S306 panel of mutant WNV RVPs that were produced under conditions of furin overexpression (i.e., furin RVPs) for 1 h at 37°C to achieve binding equilibrium. Immune complexes were added to Raji-DCSIGNR cells, and infectivity was assessed 36 to 48 h postinfection by flow cytometry. Results were normalized to infectivity in the absence of antibody. Results are representative of at least three independent experiments performed in parallel. Shown are neutralization curves for each of the 19 mutants at S306 and MAb E16 (top row), E33 (middle row), and E60 (bottom row). Curves for wild-type data are shown in black, and mutant curves are colored according to the scheme described in the Fig. 3 legend.
In order to utilize data collected from neutralization studies to map the functional footprint of DIII-LR antibodies, we developed an analysis metric to assign residues as major, minor, or noncontributors based on the magnitude of changes in EC50 values among variants of the different chemical groups described above. Residues were identified as “major contributors” if at least one mutant from five or more chemical groups reduced the neutralization potency of E16 or E33 by at least 10-fold. “Minor contributors” were residues where at least one mutant from five or more chemical groups impaired the neutralization sensitivity of MAb E16 or E33 by at least 2-fold. Residues that did not meet the aforementioned criteria were classified as noncontributors to the epitope. By utilizing the benchmark of five chemical groups, we aimed to ensure a rigorous analysis metric. We reasoned that if a residue contributed to the footprint of an antibody, alterations to several different side chain chemistries should perturb the neutralization sensitivity. We excluded from these analysis criteria the entire chemical group to which the WT residue belonged, because those amino acid substitutions do not significantly alter the chemistry of the side chain. Such mutants should not affect the neutralization potency of either E16 or E33.
Based on these analysis criteria, the residues S306, K307, and T332 were implicated in the functional footprints of both E16 and E33 (Fig. 5A to C). While structural studies suggested that E16 engages residues within all four loops of the DIII-LR, our results highlight that only a subset of these crystallographically defined contacts are functionally important for antibody-mediated neutralization. Additionally, our functional data closely corroborated epitope mapping data for E16 derived from yeast surface display studies (40, 60). Although S306, K307, and T332 comprised the functional epitopes of E16 and E33, these two antibodies did not recognize all three residues in a comparable manner. For instance, aromatic (F, Y, and W) and basic (H, K, and R) mutations at residue S306 increased the neutralization potency of MAb E33 but decreased neutralization by MAb E16 (Fig. 5A). Furthermore, any mutation at residue K307 abrogated the neutralization potency of MAb E33 by at least 1,000-fold. In contrast, E16 had only diminished neutralization efficacy when assayed using the panel of K307 variants; most impaired neutralization by approximately 10-fold or less (Fig. 5B). On the contrary, both E16 and E33 were comparably sensitive to mutations at position T332, as evidenced by similar EC50 changes for the mutants studied (Fig. 5C).
Fig 5.

Mapping reveals that MAbs E16 and E33 recognize distinct functional epitopes on the surface of DIII-LR. Ten 4-fold serial dilutions of MAbs E16, E33, and E60 were incubated with the indicated panel of mutant WNV RVPs that were produced under conditions of furin overexpression. RVPs and antibody were incubated for 1 h at 37°C to achieve binding equilibrium. Immune complexes were added to Raji-DCSIGNR cells, and infectivity was assessed 36 to 48 h postinfection by flow cytometry. Shown are mean fold differences in the EC50 compared to that for the wild type, using E16 (top row) and E33 (bottom row) to neutralize mutants at positions 306 (A), 307 (B), 332 (C), 310 (D), 330 (E), 366 (F), 391 (G), 311 (H), and 390 (I). Error bars display the standard errors of at least two parallel, independent experiments. Bold dashed lines indicate a 2-fold difference, which was the limit of detection. Fine dashed lines indicate a 10-fold difference. Mutants that were noninfectious or had low levels of infectivity (i.e., >100-fold difference in maximal infectivity compared to wild type) were omitted from the functional mapping studies.
Whereas only three residues within the DIII-LR were implicated as important to virion neutralization by MAb E16, four additional residues were found to contribute to the MAb E33 functional footprint: K310, T330, T366, and Q391 (Fig. 5D to G). Residues T330 and T366 were major contributors to the E33 functional footprint, as several mutations reduced the neutralization sensitivity by greater than 10-fold. Not all DIII-LR target residues tested impacted E16 and E33 neutralization; mutation of F311 and E390 had no functional consequences on E16 and E33 potencies (Fig. 5H and I). From this analysis, E33 possesses a functional footprint that includes residues from all four loops of the DIII-LR. This functional footprint is subtly different from that of MAb E16.
Mutations at position 306 reverse the maturation state-sensitive properties of MAb E33.
Our neutralization studies revealed that aromatic substitutions at residue S306 increased the neutralization efficacy of MAb E33 (Fig. 5A). These effects were comparable in magnitude to the difference in the EC50 of MAb E33 for furin versus NH4Cl RVPs. To investigate whether changes in residue S306 might affect the maturation state-sensitive properties of E33, neutralization studies were performed using MAbs E16, E33, and E60 and both furin and NH4Cl RVPs with S306F, S306Y, or S306W E protein variants incorporated. Aromatic substitutions at position S306 unexpectedly conferred maturation state sensitivity to MAb E16 (1.7- to 3.2-fold difference; n = 6; P < 0.05) (Fig. 6A). In contrast, all three aromatic mutations at S306 abrogated the maturation state-sensitive properties of MAb E33 (6.2- to 1.8-fold difference; n = 6; P < 0.0001) (Fig. 6B). Although MAb E60 is sensitive to the maturation state of the virion, the aromatic mutations at residue S306 did not affect the neutralization efficacy of E60 (Fig. 6C). This MAb was able to neutralize S306F, S306Y, and S306W NH4Cl RVPs approximately 200-fold more effectively than furin RVPs, and these results were comparable to those obtained with WT furin and NH4Cl RVPs.
Fig 6.
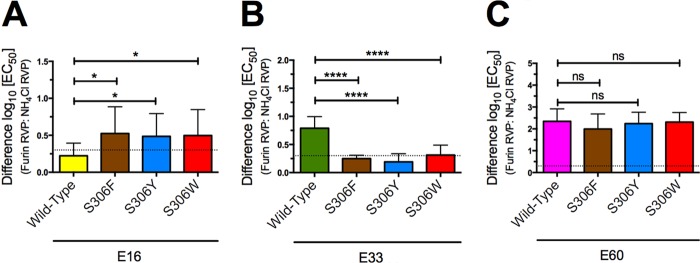
Aromatic mutations at residue 306 abrogate the maturation-sensitive properties of MAb E33. Ten 4-fold serial dilutions of MAbs E16, E33, and E60 were incubated with either furin RVPs or NH4Cl RVPs for 1 h at 37°C to achieve binding equilibrium. Immune complexes were then added to Raji-DCSIGNR cells, and infectivity was assessed 36 to 48 h postinfection by flow cytometry. Results were normalized to infectivity in the absence of antibody. The differences in log10(EC50) values between furin and NH4Cl RVPs for wild type, S306F, S306Y, and S306W neutralized by MAbs E16 (A), E33 (B), or E60 (C) are shown. Error bars display the standard errors of six independent experiments performed in parallel using at least three independent RVP preparations. Statistical analysis was performed using Prism software (GraphPad). The differences in log10(EC50) values between furin and NH4Cl RVPs neutralized by E16, E33, or E60 were compared using one-way analysis of variance followed by Tukey's multiple comparison test. The dashed line indicates the lower limit of detection. ns, P > 0.05; *, P < 0.05; ****, P < 0.0001.
To our surprise, these data demonstrated that the maturation state-sensitive property of MAb E33 is not fixed and can be modulated independently of the quantity of uncleaved prM on the virus particle. It is possible to increase the neutralization potency of MAb E33 against furin RVPs by expressing any one of three aromatic residues at S306, so that it is comparable to the efficacy observed with NH4Cl RVPs. In contrast, these mutations did not have an effect on MAb E60, whose maturation state-sensitive properties remained static. Although S306 was not classified as a major contributor to the E16 or E33 footprint, this residue can significantly abrogate the maturation state-sensitive properties of MAb E33.
Fc-mediated steric occlusion modulates maturation sensitivity of DIII-LR MAb E33.
While our mutagenesis studies revealed differences in how E16 and E33 engage the “same” DIII-LR epitope, these details do not in themselves explain differences in the maturation state dependence of these two antibodies. We next considered whether aspects beyond the epitope-paratope interface affect antibody occupancy. To investigate the role of the remainder of the antibody molecule in modulating maturation state sensitivity, we examined the extent to which furin or NH4Cl RVPs were neutralized by Fab fragments derived from E16, E33, and E60. Fab fragments generated from E16 neutralized furin and NH4Cl RVPs with comparable potencies (Fig. 7A and B). However, this was also true for fragments generated from MAb E33. The difference in neutralization potency by E33 Fab between furin and NH4Cl RVPs was 1.1-fold (n = 6). These data contrast with those for the intact E33 IgG, which had a maturation state-sensitive pattern of neutralization. Although the MAb E60 is also a maturation state-sensitive antibody, the Fab fragments did not affect this property (Fig. 7A). Similar to experiments using aromatic mutants at E protein residue 306, the maturation state-sensitive properties of E33 could be modulated without changes to the prM content of the particles; E60 IgG remained maturation state sensitive. Our data suggest that the presence of the Fc domain on MAb E33 confers the property of differential neutralization between furin and NH4Cl RVPs.
Fig 7.
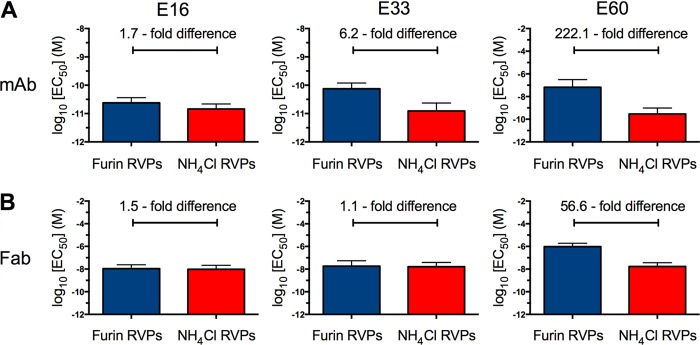
Fab fragments generated from MAb E33 do not show maturation-sensitive neutralization. Ten 4-fold serial dilutions of MAb or Fab fragments were incubated with either furin (blue) or NH4Cl RVPs (red) for 1 h at 37°C to achieve binding equilibrium. Immune complexes were then added to Raji-DCSIGNR cells, and infectivity was assessed 36 to 48 h postinfection by flow cytometry. Shown are the mean log10(EC50) values for MAbs E16 (left graphs), E33 (middle graphs), and E60 (right graphs) neutralization of furin and NH4Cl RVPs using MAb (A) or Fab (B) fragments.
DISCUSSION
Flaviviruses are structurally heterogeneous targets for antibody recognition (54, 70). While antibody affinity for viral antigens is an important parameter that helps define the neutralization potency of an antibody, the accessibility of epitopes for antibody binding is also a critical and variable factor. In particular, many epitopes recognized by neutralizing antibodies are not predicted to be accessible using existing models of the mature virus particle. This limits the neutralizing potential of many classes of antibodies, even at saturating concentrations (47, 50, 71).
We have shown previously that the inefficiency of virion maturation alters the landscape for antibody recognition by increasing the accessibility of otherwise cryptic determinants (50). Increasing the efficiency of the maturation process decreases the potency of neutralization of several classes of antibodies, including those binding the DII fusion loop. We proposed that the process of maturation reduces the number of accessible epitopes on the average virion, often below the minimum required to exceed a stoichiometric threshold. This would result in a large fraction of viruses that are no longer sensitive to neutralization, even with saturating concentrations of antibody (47, 50). However, in this study, we identify a MAb (E33) that was sensitive to the extent of virion maturation yet bound an epitope predicted to be accessible on E proteins of both mature and immature virus particles. The functional affinities of MAb E33 toward its epitope on furin and NH4Cl RVPs were comparable; therefore, the maturation state sensitivity cannot be explained by affinity alone.
To investigate the underlying mechanism for the maturation state-sensitive pattern of E33, we created a functional map of its epitope. This was accomplished by first generating a large library of virions with mutations incorporated at 14 residues of the DIII-LR. Analysis of the infectivity of these mutants revealed that the DIII-LR could tolerate extensive variation. At nine of the positions selected for study, it was possible to produce infectious virus particles that had incorporated almost any amino acid substitution without deleterious effects on infectivity. It is important to note that the functional studies described within were performed using a cell line for which virus attachment is mediated by interactions with the c-type lectin DC-SIGNR. As DIII has been directly implicated in interactions of flaviviruses with cellular factors during binding and entry (72, 73), it will be of interest to repeat the analysis of the infectivity of this panel of viruses on other cellular substrates for which the mode of attachment may differ. Furthermore, our recent studies suggest that not all cell types are equally sensitive substrates for detection of maturation state-dependent patterns of neutralization, due to the impact of uncleaved prM on the efficiency of virus-cell interactions (S. Mukherjee and T. Pierson, unpublished data).
The interaction of E16 with the WNV DIII-LR has been studied extensively. Crystallographic studies have demonstrated that recognition is dominated by side chain and water-mediated hydrogen bonds between E16 complementarity-determining region (CDR) loops and 18 residues within DIII (40). Remarkably, our mapping studies and analysis metrics only implicated the residues S306, K307, and T332, which are localized on the BC and N-terminal loops, as functionally important to E16 neutralization of virions. Strikingly, these data highlight that not all contacts revealed by structural studies have functional consequences and underscore the importance of cautious interpretation of such data.
Our studies with E33 suggest the functional epitope of this MAb overlaps with E16 but also extends to include amino acids in the DE and FG loops. In addition to S306, K307, and T332, our studies suggest that residues K310, T330, T366, and Q391 also contribute to the E33 functional footprint (Fig. 8). Although our mapping studies are extensive, we anticipate that further saturation-mutagenesis and parallel neutralization studies will elucidate the functional footprints with higher resolution. However, in judiciously selecting residues of interest, we covered a significant portion of the total surface area that exists within and adjacent to the DIII-LR. These studies also illustrated the complexity by which two MAbs bind to what appears at first sight to be similar epitopes on the virion. While both E16 and E33 recognized a common set of amino acids on the DIII-LR, they appeared to do so differently. Aromatic and basic mutations at S306 increased MAb E33 neutralization efficacy while having the opposite effect on MAb E16. Furthermore, any mutation at K307 impaired E33 neutralization sensitivity by at least 1,000-fold, whereas the effects of these same mutants on E16 were modest.
Fig 8.
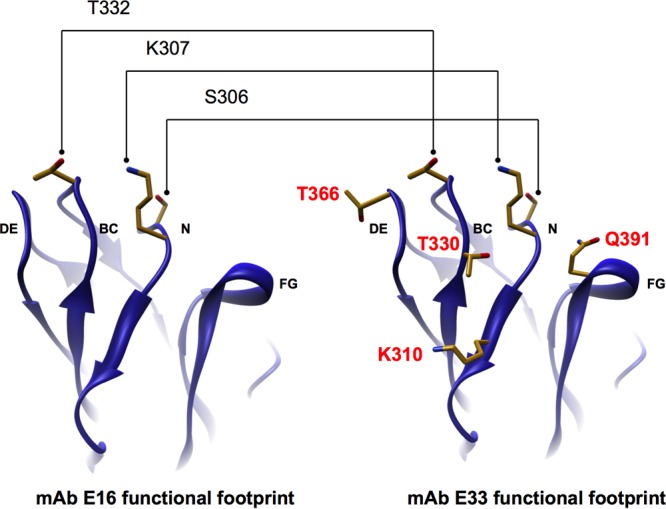
Functional footprints of MAbs E16 and E33. The WNV E protein domain III with the amino acids within MAb E16 (left) and MAb E33 (right) functional footprints displayed are shown. Residues implicated by our mapping data and analysis metrics to be part of only the MAb E33 functional footprint are highlighted in red.
Our model to explain the maturation state-dependent neutralization of E33 arose in part from differences in how E16 and E33 recognize the DIII-LR. On mature virions, the angle at which E33 IgG engages with the virion may position the antibody molecule such that it introduces steric obstacles to antibody occupancy not experienced during binding of E16 IgG on the virus particle. Aromatic substitutions at residue S306 increased the sensitivity of mature WNV RVPs to neutralization by E33 and, surprisingly, rendered the antibody insensitive to the maturation state of the virion. Since aromatic side chains are among the most bulky amino acids, it is possible that the S306F, S306Y, and S306W mutations altered the angle of engagement of E33 with the DIII-LR. Indeed, we also found that comparably bulky mutations (S306H, S306K, and S306R) rendered MAb E33 maturation state insensitive (data not shown). Furthermore, our hypothesis is supported by the observation that aromatic mutations conferred a slight maturation state sensitivity to the MAb E16. Examination of the crystal structure of the DIII-LR and E16 revealed steric constraints when aromatic side chains were modeled at position 306 (data not shown). Finally, reducing the size of the antibody molecule through removal of the Fc domain eliminated the impact of virion maturation on E33 neutralization.
Structural studies of HIV and influenza virus antibodies have demonstrated that it is possible to engage similar epitopes with markedly different modes of recognition. For example, the potent and broadly neutralizing HIV-1 antibodies 4E10 and 10E8 recognize a conserved helix at the C terminus of HIV-1 fusion protein gp41 (74, 75). While both MAbs engage a common set of six residues, 4E10 also interacts with an additional set of four residues that nest around the common core. High-resolution structural studies have revealed marked differences in the mechanism of MAb binding, including pronounced alterations in the angle of approach. We predict that a similar phenomenon occurs with the DIII-LR MAbs E16 and E33.
In summary, we have identified a MAb that recognizes the DIII-LR epitope in a maturation state-dependent manner. As this epitope is fully accessible on the virion and is the target of antibodies shown to be functionally insensitive to the prM content of the virion, this result was unexpected. Our comparison of the data between the two maturation state-sensitive antibodies, E33 and E60, underscores the incompleteness of an accessibility paradigm to describe the impact of uncleaved prM on the sensitivity of virions to neutralization. Unlike MAb E60, it is not a decrease in the quantity of solvent-accessible epitopes, per se, that governs the MAb E33 maturation state sensitivity. Our data support a model by which this sensitivity to the prM content of the virus particle is explained by steric constraints imposed by the Fc domain upon MAb binding to the DIII-LR. We propose that the E33 Fc region impairs the ability of MAbs to engage nearby epitopes that are otherwise surface accessible and thereby impacts how antibodies dock on the virion and in what numbers. This is a novel mechanism by which the maturation state of a flavivirus virion impacts the neutralization potency of an antibody that binds a solvent-exposed epitope.
ACKNOWLEDGMENTS
This work was funded by the Intramural Research Program of the National Institutes of Allergy and Infectious Diseases.
We thank members of our laboratories for critically reading the manuscript and for helpful discussions.
Footnotes
Published ahead of print 9 October 2013
REFERENCES
- 1.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayes EB, Gubler DJ. 2006. West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Annu. Rev. Med. 57:181–194 [DOI] [PubMed] [Google Scholar]
- 3.Sejvar JJ. 2007. The long-term outcomes of human West Nile virus infection. Clin. Infect. Dis. 44:1617–1624 [DOI] [PubMed] [Google Scholar]
- 4.Mostashari F, Bunning ML, Kitsutani PT, Singer DA, Nash D, Cooper MJ, Katz N, Liljebjelke KA, Biggerstaff BJ, Fine AD, Layton MC, Mullin SM, Johnson AJ, Martin DA, Hayes EB, Campbell GL. 2001. Epidemic West Nile encephalitis, New York, 1999: results of a household-based seroepidemiological survey. Lancet 358:261–264 [DOI] [PubMed] [Google Scholar]
- 5.Nash D, Mostashari F, Fine A, Miller J, O'Leary D, Murray K, Huang A, Rosenberg A, Greenberg A, Sherman M, Wong S, Layton M. 2001. The outbreak of West Nile virus infection in the New York City area in 1999. N. Engl. J. Med. 344:1807–1814 [DOI] [PubMed] [Google Scholar]
- 6.Beasley DW, Barrett AD, Tesh RB. 2013. Resurgence of West Nile neurologic disease in the United States in 2012: what happened? What needs to be done? Antiviral Res. 99:1–5 [DOI] [PubMed] [Google Scholar]
- 7.Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH. 2002. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukhopadhyay S, Kuhn RJ, Rossmann MG. 2005. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 3:13–22 [DOI] [PubMed] [Google Scholar]
- 9.Lindenbach BD, Rice THCM. 2007. Flaviviridae: the viruses and their replication, p 1101–1152 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Strauss SE. (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 10.Li L, Lok SM, Yu IM, Zhang Y, Kuhn RJ, Chen J, Rossmann MG. 2008. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science 319:1830–1834 [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Kaufmann B, Chipman PR, Kuhn RJ, Rossmann MG. 2007. Structure of immature West Nile virus. J. Virol. 81:6141–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elshuber S, Allison SL, Heinz FX, Mandl CW. 2003. Cleavage of protein prM is necessary for infection of BHK-21 cells by tick-borne encephalitis virus. J. Gen. Virol. 84:183–191 [DOI] [PubMed] [Google Scholar]
- 13.Stadler K, Allison SL, Schalich J, Heinz FX. 1997. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 71:8475–8481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. 2008. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 319:1834–1837 [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Zhang W, Ogata S, Clements D, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. 2004. Conformational changes of the flavivirus E glycoprotein. Structure 12:1607–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elshuber S, Mandl CW. 2005. Resuscitating mutations in a furin cleavage-deficient mutant of the flavivirus tick-borne encephalitis virus. J. Virol. 79:11813–11823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guirakhoo F, Heinz FX, Mandl CW, Holzmann H, Kunz C. 1991. Fusion activity of flaviviruses: comparison of mature and immature (prM-containing) tick-borne encephalitis virions. J. Gen. Virol. 72:1323–1329 [DOI] [PubMed] [Google Scholar]
- 18.Hanna SL, Pierson TC, Sanchez MD, Ahmed AA, Murtadha MM, Doms RW. 2005. N-linked glycosylation of West Nile virus envelope proteins influences particle assembly and infectivity. J. Virol. 79:13262–13274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khromykh AA, Varnavski AN, Westaway EG. 1998. Encapsidation of the flavivirus Kunjin replicon RNA by using a complementation system providing Kunjin virus structural proteins in trans. J. Virol. 72:5967–5977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimura T, Ohyama A. 1988. Association between the pH-dependent conformational change of West Nile flavivirus E protein and virus-mediated membrane fusion. J. Gen. Virol. 69:1247–1254 [DOI] [PubMed] [Google Scholar]
- 21.Moesker B, Rodenhuis-Zybert IA, Meijerhof T, Wilschut J, Smit JM. 2010. Characterization of the functional requirements of West Nile virus membrane fusion. J. Gen. Virol. 91:389–393 [DOI] [PubMed] [Google Scholar]
- 22.Junjhon J, Edwards TJ, Utaipat U, Bowman VD, Holdaway HA, Zhang W, Keelapang P, Puttikhunt C, Perera R, Chipman PR, Kasinrerk W, Malasit P, Kuhn RJ, Sittisombut N. 2010. Influence of pr-M cleavage on the heterogeneity of extracellular dengue virus particles. J. Virol. 84:8353–8358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis CW, Nguyen HY, Hanna SL, Sanchez MD, Doms RW, Pierson TC. 2006. West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J. Virol. 80:1290–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guirakhoo F, Bolin RA, Roehrig JT. 1992. The Murray Valley encephalitis virus prM protein confers acid resistance to virus particles and alters the expression of epitopes within the R2 domain of E glycoprotein. Virology 191:921–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanai R, Kar K, Anthony K, Gould LH, Ledizet M, Fikrig E, Marasco WA, Koski RA, Modis Y. 2006. Crystal structure of West Nile virus envelope glycoprotein reveals viral surface epitopes. J. Virol. 80:11000–11008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Modis Y, Ogata S, Clements D, Harrison SC. 2005. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J. Virol. 79:1223–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Modis Y, Ogata S, Clements D, Harrison SC. 2003. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 100:6986–6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nayak V, Dessau M, Kucera K, Anthony K, Ledizet M, Modis Y. 2009. Crystal structure of dengue virus type 1 envelope protein in the postfusion conformation and its implications for membrane fusion. J. Virol. 83:4338–4344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nybakken GE, Nelson CA, Chen BR, Diamond MS, Fremont DH. 2006. Crystal structure of the West Nile virus envelope glycoprotein. J. Virol. 80:11467–11474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. 1995. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature 375:291–298 [DOI] [PubMed] [Google Scholar]
- 31.Modis Y, Ogata S, Clements D, Harrison SC. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature 427:313–319 [DOI] [PubMed] [Google Scholar]
- 32.Mukhopadhyay S, Kim BS, Chipman PR, Rossmann MG, Kuhn RJ. 2003. Structure of West Nile virus. Science 302:248. [DOI] [PubMed] [Google Scholar]
- 33.Chu JJ, Ng ML. 2004. Interaction of West Nile virus with alpha v beta 3 integrin mediates virus entry into cells. J. Biol. Chem. 279:54533–54541 [DOI] [PubMed] [Google Scholar]
- 34.Lee E, Hall RA, Lobigs M. 2004. Common E protein determinants for attenuation of glycosaminoglycan-binding variants of Japanese encephalitis and West Nile viruses. J. Virol. 78:8271–8280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JW, Chu JJ, Ng ML. 2006. Quantifying the specific binding between West Nile virus envelope domain III protein and the cellular receptor αVβ3 integrin. J. Biol. Chem. 281:1352–1360 [DOI] [PubMed] [Google Scholar]
- 36.Ben-Nathan D, Lustig S, Tam G, Robinzon S, Segal S, Rager-Zisman B. 2003. Prophylactic and therapeutic efficacy of human intravenous immunoglobulin in treating West Nile virus infection in mice. J. Infect. Dis. 188:5–12 [DOI] [PubMed] [Google Scholar]
- 37.Camenga DL, Nathanson N, Cole GA. 1974. Cyclophosphamide-potentiated West Nile viral encephalitis: relative influence of cellular and humoral factors. J. Infect. Dis. 130:634–641 [DOI] [PubMed] [Google Scholar]
- 38.Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. 2003. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 77:2578–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diamond MS, Shrestha B, Mehlhop E, Sitati E, Engle M. 2003. Innate and adaptive immune responses determine protection against disseminated infection by West Nile encephalitis virus. Viral Immunol. 16:259–278 [DOI] [PubMed] [Google Scholar]
- 40.Nybakken GE, Oliphant T, Johnson S, Burke S, Diamond MS, Fremont DH. 2005. Structural basis of West Nile virus neutralization by a therapeutic antibody. Nature 437:764–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tesh RB, Arroyo J, Travassos Da Rosa AP, Guzman H, Xiao SY, Monath TP. 2002. Efficacy of killed virus vaccine, live attenuated chimeric virus vaccine, and passive immunization for prevention of West Nile virus encephalitis in hamster model. Emerg. Infect. Dis. 8:1392–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tesh RB, Travassos da Rosa AP, Guzman H, Araujo TP, Xiao SY. 2002. Immunization with heterologous flaviviruses protective against fatal West Nile encephalitis. Emerg. Infect. Dis. 8:245–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang T, Anderson JF, Magnarelli LA, Wong SJ, Koski RA, Fikrig E. 2001. Immunization of mice against West Nile virus with recombinant envelope protein. J. Immunol. 167:5273–5277 [DOI] [PubMed] [Google Scholar]
- 44.Colombage G, Hall R, Pavy M, Lobigs M. 1998. DNA-based and alphavirus-vectored immunisation with prM and E proteins elicits long-lived and protective immunity against the flavivirus, Murray Valley encephalitis virus. Virology 250:151–163 [DOI] [PubMed] [Google Scholar]
- 45.Diamond MS, Pierson TC, Roehrig JT. 2012. Antibody therapeutics against flaviviruses, p 231–255 In Shi P-Y. (ed), Molecular virology and control of flaviviruses. Caister Academic Press, Poole, United Kingdom [Google Scholar]
- 46.Burton DR, Saphire EO, Parren PW. 2001. A model for neutralization of viruses based on antibody coating of the virion surface. Curr. Top. Microbiol. Immunol. 260:109–143 [DOI] [PubMed] [Google Scholar]
- 47.Pierson TC, Xu Q, Nelson S, Oliphant T, Nybakken GE, Fremont DH, Diamond MS. 2007. The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host Microbe 1:135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klasse PJ, Moore JP. 1996. Quantitative model of antibody- and soluble CD4-mediated neutralization of primary isolates and T-cell line-adapted strains of human immunodeficiency virus type 1. J. Virol. 70:3668–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klasse PJ, Sattentau QJ. 2002. Occupancy and mechanism in antibody-mediated neutralization of animal viruses. J. Gen. Virol. 83:2091–2108 [DOI] [PubMed] [Google Scholar]
- 50.Nelson S, Jost CA, Xu Q, Ess J, Martin JE, Oliphant T, Whitehead SS, Durbin AP, Graham BS, Diamond MS, Pierson TC. 2008. Maturation of West Nile virus modulates sensitivity to antibody-mediated neutralization. PLoS Pathog. 4(5):e1000060. 10.1371/journal.ppat.1000060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cherrier MV, Kaufmann B, Nybakken GE, Lok SM, Warren JT, Chen BR, Nelson CA, Kostyuchenko VA, Holdaway HA, Chipman PR, Kuhn RJ, Diamond MS, Rossmann MG, Fremont DH. 2009. Structural basis for the preferential recognition of immature flaviviruses by a fusion-loop antibody. EMBO J. 28:3269–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davis BS, Chang GJ, Cropp B, Roehrig JT, Martin DA, Mitchell CJ, Bowen R, Bunning ML. 2001. West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzyme-linked immunosorbent assays. J. Virol. 75:4040–4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pierson TC, Sanchez MD, Puffer BA, Ahmed AA, Geiss BJ, Valentine LE, Altamura LA, Diamond MS, Doms RW. 2006. A rapid and quantitative assay for measuring antibody-mediated neutralization of West Nile virus infection. Virology 346:53–65 [DOI] [PubMed] [Google Scholar]
- 54.Dowd KA, Pierson TC. 2011. Antibody-mediated neutralization of flaviviruses: a reductionist view. Virology 411:306–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shrestha B, Austin SK, Dowd KA, Prasad AN, Youn S, Pierson TC, Fremont DH, Ebel GD, Diamond MS. 2012. Complex phenotypes in mosquitoes and mice associated with neutralization escape of a dengue virus type 1 monoclonal antibody. Virology 427:127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oliphant T, Nybakken GE, Austin SK, Xu Q, Bramson J, Loeb M, Throsby M, Fremont DH, Pierson TC, Diamond MS. 2007. Induction of epitope-specific neutralizing antibodies against West Nile virus. J. Virol. 81:11828–11839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oliphant T, Diamond MS. 2007. The molecular basis of antibody-mediated neutralization of West Nile virus. Expert Opin. Biol. Ther. 7:885–892 [DOI] [PubMed] [Google Scholar]
- 58.Mukherjee S, Lin TY, Dowd KA, Manhart CJ, Pierson TC. 2011. The infectivity of prM-containing partially mature West Nile virus does not require the activity of cellular furin-like proteases. J. Virol. 85:12067–12072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Randolph VB, Winkler G, Stollar V. 1990. Acidotropic amines inhibit proteolytic processing of flavivirus prM protein. Virology 174:450–458 [DOI] [PubMed] [Google Scholar]
- 60.Oliphant T, Engle M, Nybakken GE, Doane C, Johnson S, Huang L, Gorlatov S, Mehlhop E, Marri A, Chung KM, Ebel GD, Kramer LD, Fremont DH, Diamond MS. 2005. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat. Med. 11:522–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pitcher TJ, Gromowski GD, Beasley DW, Barrett AD. 2012. Conservation of the DENV-2 type-specific and DEN complex-reactive antigenic sites among DENV-2 genotypes. Virology 422:386–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gromowski GD, Roehrig JT, Diamond MS, Lee JC, Pitcher TJ, Barrett AD. 2010. Mutations of an antibody binding energy hot spot on domain III of the dengue 2 envelope glycoprotein exploited for neutralization escape. Virology 407:237–246 [DOI] [PubMed] [Google Scholar]
- 63.Zhang S, Bovshik EI, Maillard R, Gromowski GD, Volk DE, Schein CH, Huang CY, Gorenstein DG, Lee JC, Barrett AD, Beasley DW. 2010. Role of BC loop residues in structure, function and antigenicity of the West Nile virus envelope protein receptor-binding domain III. Virology 403:85–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matsui K, Gromowski GD, Li L, Barrett AD. 2010. Characterization of a dengue type-specific epitope on dengue 3 virus envelope protein domain III. J. Gen. Virol. 91:2249–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matsui K, Gromowski GD, Li L, Schuh AJ, Lee JC, Barrett AD. 2009. Characterization of dengue complex-reactive epitopes on dengue 3 virus envelope protein domain III. Virology 384:16–20 [DOI] [PubMed] [Google Scholar]
- 66.Gromowski GD, Barrett ND, Barrett AD. 2008. Characterization of dengue virus complex-specific neutralizing epitopes on envelope protein domain III of dengue 2 virus. J. Virol. 82:8828–8837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sukupolvi-Petty S, Austin SK, Purtha WE, Oliphant T, Nybakken GE, Schlesinger JJ, Roehrig JT, Gromowski GD, Barrett AD, Fremont DH, Diamond MS. 2007. Type- and subcomplex-specific neutralizing antibodies against domain III of dengue virus type 2 envelope protein recognize adjacent epitopes. J. Virol. 81:12816–12826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gromowski GD, Barrett AD. 2007. Characterization of an antigenic site that contains a dominant, type-specific neutralization determinant on the envelope protein domain III (ED3) of dengue 2 virus. Virology 366:349–360 [DOI] [PubMed] [Google Scholar]
- 69.Kaufmann B, Nybakken GE, Chipman PR, Zhang W, Diamond MS, Fremont DH, Kuhn RJ, Rossmann MG. 2006. West Nile virus in complex with the Fab fragment of a neutralizing monoclonal antibody. Proc. Natl. Acad. Sci. U. S. A. 103:12400–12404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pierson TC, Diamond MS. 2012. Degrees of maturity: the complex structure and biology of flaviviruses. Curr. Opin. Virol. 2:168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crill WD, Chang GJ. 2004. Localization and characterization of flavivirus envelope glycoprotein cross-reactive epitopes. J. Virol. 78:13975–13986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bhardwaj S, Holbrook M, Shope RE, Barrett AD, Watowich SJ. 2001. Biophysical characterization and vector-specific antagonist activity of domain III of the tick-borne flavivirus envelope protein. J. Virol. 75:4002–4007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chu JJ, Rajamanonmani R, Li J, Bhuvanakantham R, Lescar J, Ng ML. 2005. Inhibition of West Nile virus entry by using a recombinant domain III from the envelope glycoprotein. J. Gen. Virol. 86:405–412 [DOI] [PubMed] [Google Scholar]
- 74.Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. 2012. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491:406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cardoso RM, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, Burton DR, Wilson IA. 2005. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 22:163–173 [DOI] [PubMed] [Google Scholar]