

Abstract
Hepatitis C virus (HCV) remains a challenging public health problem worldwide. The identification of viral variants establishing de novo infections and definition of the phenotypic requirements for transmission would facilitate the design of preventive strategies. We explored the transmission of HCV variants in three cases of acute hepatitis following needlestick accidents. We used single-genome amplification of glycoprotein E1E2 gene sequences to map the genetic bottleneck upon transmission accurately. We found that infection was likely established by a single variant in two cases and six variants in the third case. Studies of donor samples showed that the transmitted variant E1E2 amino acid sequences were identical or closely related to those of variants from the donor virus populations. The transmitted variants harbored a common signature site at position 394, within hypervariable region 1 of E2, together with additional signature amino acids specific to each transmission pair. Surprisingly, these E1E2 variants conferred no greater capacity for entry than the E1E2 derived from nontransmitted variants in lentiviral pseudoparticle assays. Mutants escaping the antibodies of donor sera did not predominate among the transmitted variants either. The fitness parameters affecting the selective outgrowth of HCV variants after transmission in an immunocompetent host may thus be more complex than those suggested by mouse models. Human antibodies directed against HCV envelope effectively cross-neutralized the lentiviral particles bearing E1E2 derived from transmitted variants. These findings provide insight into the molecular mechanisms underlying HCV transmission and suggest that viral entry is a potential target for the prevention of HCV infection.
INTRODUCTION
The World Health Organization has estimated that 150 million people are chronically infected with hepatitis C virus (HCV) worldwide, with 3 to 4 million new infections occurring annually (1). About 30% of acute HCV infections resolve spontaneously, but most infections become chronic, with a strong tendency to progress toward life-threatening liver diseases, such as cirrhosis and hepatocellular carcinoma (2, 3).
HCV is transmitted mostly via direct blood-to-blood contact (4). Occupational hepatitis C is occasionally reported in health care workers (5, 6), and cases resulting from well-monitored needlestick accidents provide rare opportunities for tracking the entire transmission process from donor to recipient (7, 8). In vivo, HCV replicates rapidly, using an error-prone viral RNA polymerase. This results in the generation of a group of related, but genetically different, viral variants within each infected individual (9). The genetic bottleneck generally observed after transmission indicates that productive infections may be initiated by one or a small number of viral variants (10–13). These variants are then subjected to constant immune pressure, and the role of the immune response in the clearance of HCV infection or the establishment of chronic hepatitis C has been thoroughly investigated. However, little is known about the transmitted variants responsible for the spread of the disease, principally because it is difficult to recruit patients early enough in acute infection for such studies. Moreover, studies of the viruses transmitted in humans have essentially focused on genetic identification of the transmitted/founder (T/F) variants and their early diversification, through phylogenetic and mathematical approaches, without direct comparison with donor virus populations (10–12). A key question in the rational design of strategies for preventing HCV infection concerns the phenotypic determinants conferring fitness for transmission in T/F viruses. The HCV envelope glycoproteins, which are involved principally in virus attachment and entry into target cells, are likely candidates for such transmission-related signatures.
We addressed this issue by exploring HCV transmission in health care workers (the recipients) who developed acute hepatitis C after needlestick accidents, comparing findings from these individuals with those from the corresponding chronically infected patients from whom the virus was transmitted (the donors). We mapped the genetic bottleneck leading to productive clinical infection by single-genome amplification of viral envelope glycoprotein E1E2 sequences, direct amplicon sequencing, and phylogenetic analyses (12, 14). The recovery of full-length E1E2 sequences from donors and recipients made it possible to investigate the phenotypic properties of these proteins potentially relevant to transmission. Our data provide insight into the selective transmission of HCV variants and early stages of infection, during which the virus may be most vulnerable to elimination by preventive vaccines or immunotherapies.
MATERIALS AND METHODS
Study participants.
Three health care workers (recipients RA1, RA2, and RB), infected with HCV genotype 1b through documented needlestick exposure to blood from patients with chronic hepatitis (donors DA and DB), were enrolled in the study. All the subjects other than DB were female. Recipients RA1 and RA2 shared the same donor, DA, but were infected 10 months apart. They received similar bitherapy with pegylated interferon and ribavirin to achieve viral clearance. Recipient RB was contaminated by donor DB and displayed spontaneous viral clearance. This study was approved by the Institutional Review Board of Tours University Hospital (Comité de Protection des Personnes [CPP]), and written informed consent was obtained, in accordance with French regulations.
HCV RNA and antibody assays.
HCV antibody testing was performed with the qualitative Abbott Architect anti-HCV chemiluminescent microparticle immunoassay. Quantitative HCV RNA detection was performed with an Abbott HCV RealTime assay (lower limit of detection, 12 IU/ml for a sample volume of 0.5 ml).
Single-genome amplification and sequencing of HCV E1E2 envelope glycoprotein genes.
Full-length E1E2 sequences (encoding a region including the last 22 amino acids of the core through the end of E2) were amplified by single-genome amplification (SGA) from the plasma of donors and recipients. For each sample, viral RNA was extracted with the QIAamp viral RNA minikit (Qiagen). The extracted RNA was reverse transcribed to generate cDNA, with the antisense primer ExtAS, 5′-GAGCAGGAGCAGCGGCCAT-3′ (nucleotides [nt] 2720 to 2738; all primer locations are indicated relative to the H77 reference genome [GenBank accession no. NC_004102]), and SuperScript III (Invitrogen). The cDNA was serially diluted and amplified in 96-multiwell plates by nested PCR of the full-length E1E2 sequence to identify the dilution, giving a maximum frequency of 3/10 PCR-positive reactions. At this dilution, most of the wells contain amplicons derived from a single cDNA molecule (12, 14). Nested PCR was carried out with Platinum Taq high fidelity polymerase (Invitrogen), as previously reported (12), with the following primers: first-round sense primer ExtS, 5′-CGGCGTGAACTATGCAACAGG-3′ (nt 821 to 841), and antisense primer ExtAS (see above), second-round sense primer IntS, 5′-TCTGATGGGTTGCTCTTTCTCTATCTTCC-3′ (nt 845 to 873), and antisense primer IntAS, 5′-AATCAGGCCTCAGCCTGGGCTATCAG-3′ (nt 2559 to 2584).
All products were directly sequenced with BigDye Terminator chemistry, using an ABI 3130 capillary sequencer (Applied Biosystems). Electropherograms were manually inspected, and amplicons displaying mixed bases (double peaks), suggesting amplification from multiple templates or a Taq polymerase error, were excluded from further analysis.
Sequence analysis.
Nucleotide sequences were aligned using ClustalW in MEGA 5 (15) and then manually checked to improve the alignments, according to the codon translation. The overall phylogenetic relationships between sequences were analyzed by constructing a neighbor-joining tree by the Tamura three-parameter method. The number of viral variants amplified from each subject and their genetic diversity (Hamming score) were determined at the nucleotide and amino acid levels. The changes in virus population composition following transmission were investigated with the Highlighter tool (http://hcv.lanl.gov/content/sequence/HIGHLIGHT/highlighter_top.html). We checked for specific signature sequence variations with the viral epidemiology signature pattern analysis (VESPA) program (http://hcv.lanl.gov/content/sequence/VESPA/vespa.html) and default settings (16). Sequence logos were computed with WebLogo (http://weblogo.berkeley.edu/logo.cgi) (17).
Pseudoparticle production, infection, and neutralization assays.
E1E2 SGA products selected for further phenotypic analyses were inserted into the pcDNA3.1 mammalian expression vector (Invitrogen). Lentiviral pseudoparticles were generated by the cotransfection of 293-T cells with pNL4-.Luc.R−E− and expression vectors encoding E1E2 glycoproteins derived from donors and recipients, vesicular stomatitis virus glycoprotein (VSV-G), the UKN1B5.23 HCV envelope, or the no-envelope (ΔE1E2) control, as previously reported (13, 18, 19). Viral supernatants were collected 48 h later and purified by passage through a filter with 0.45-μm pores. E1E2 incorporation was assessed on sucrose cushion-purified HCV pseudoparticles (HCVpp). We centrifuged 600 μl of each supernatant through a 30% (wt/vol) sucrose cushion in TBS (Tris-buffered saline) at 125,000 × g for 2 h. Virus pellets were then analyzed by Western blotting with the 3/11 anti-E2 monoclonal antibody (MAb) (20) and with a specific rabbit antiserum against HIV-1 p24 (ARP432; Programme EVA Centre for AIDS Reagents).
For HCVpp infectivity assays, Huh7.5 cells were used to seed 96-well plates at a density of 5 × 103 cells per well on the day before assays were performed. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% heat-inactivated fetal calf serum (FCS) and antibiotics (100 IU of penicillin and 100 μg/ml streptomycin; Invitrogen). Viral infectivities were determined by infecting the Huh7.5 cells with serial 5-fold dilutions of p24-normalized viral supernatants (Innotest HIV antigen MAb kit; Innogenetics). Each experiment was performed in triplicate. The cells were harvested at 72 h postinfection, and relative light units (RLU) were measured within the cell lysates with the luciferase assay system (Promega). RLU were quantified with a Centro LB 960 luminometer (Berthold Technologies). The detection limit for positive luciferase reporter protein expression was 10 × 103 RLU/assay, corresponding to the mean ± 3 SD of background levels obtained with cells infected with ΔE1E2 pseudoparticles.
The antibody-mediated neutralization of HCVpp was assessed by determining the decrease in luciferase activity in Huh7.5 cells infected with HCVpp in the presence of human sera or neutralizing monoclonal antibodies. These experiments were restricted to E1E2 sequences conferring a high level of infectivity on HCVpp (signal-to-background ratio, >10), to minimize variability between assays and errors in the calculation of antibody titers attributable to background infectivity. HCVpp were mixed with dilutions of donor sera, genotype-specific serum pools (consisting of pools of three sera containing antibodies specific for HCV genotype 1a, 1b, 2a, or 3a), a control serum (consisting of a pool of three anti-HCV antibody-negative sera), anti-E2 MAb AR3A, anti-E2 MAb HC-11, or the irrelevant isotype control IgG R04 (21, 22). The mixtures were incubated for 1 h at 37°C and added to Huh7.5 cells used to seed 96-well plates at a density of 5 × 103 cells per well on the day before the assay. Each experiment was performed in triplicate. The cells were harvested at 72 h postinfection, and RLU were quantified for the cell lysates, as described above. For each dilution, the percentage of neutralization was calculated as follows: 100 − [100 × (infectivity of HCVpp in the presence of serum or MAb/infectivity of HCVpp in the presence of anti-HCV-negative control sera or irrelevant isotype control IgG)] (23). The neutralization titer of the sera was defined as the reciprocal of the dilution resulting in a 50% decrease in HCVpp infectivity. This 50% inhibitory dilution value was calculated by linear interpolation, taking into account two observations (i.e., the last dilution resulting in a decrease in HCVpp infectivity of at least 50% and the first dilution resulting in a decrease in HCVpp infectivity of less than 50%). The MAb concentrations that decreased HCVpp infectivity by 50% (IC50) were assessed by testing antibodies at concentrations of 20, 2, 0.2, or 0.02 μg/ml.
RESULTS
Study subjects and single-genome sequencing.
We studied the genotypic and phenotypic E1E2 envelope glycoprotein determinants underlying HCV transmission by investigating the differences in virus population composition between three health care workers (recipients RA1, RA2, and RB) and the corresponding chronically infected patients (donors DA and DB) (Fig. 1). Single-genome amplification was performed on donor plasma samples obtained at the time of the needlestick accident for the transmission pairs DA/RA1 and DB/RB. No plasma sample was available from donor DA at the time of the needlestick accident for the transmission pair DA/RA2, with this transmission event occurring 10 months after that between DA and RA1. For recipients RA1 and RB, SGA was conducted before antibody seroconversion on the first viremic plasma sample, collected 25 and 14 days postinfection, respectively. For RA2, E1E2 sequences were successfully amplified only from the second sequential sample collected 44 days postinfection, at the time of peak viremia and antibody seroconversion. In total, 155 full-length E1E2 nucleotide sequences were derived by single-genome sequencing (i.e., SGA followed by direct amplicon sequencing) from the two donors and three recipients (range, 24 to 37 per subject) (Table 1).
Fig 1.

Virological course of recipients RA1, RA2, and RB after needlestick exposure to infected blood. Viremia was monitored after the needlestick injury by sequential plasma samples, repeated until the virus could no longer be detected. The solid line with circled values corresponds to plasma viral RNA quantified with the Abbott HCV RealTime assay (see Materials and Methods). The dotted line indicates the lower limit of detection. The filled circles correspond to the recipient viremic time points analyzed by SGA, and plus signs denote positivity for anti-HCV antibody. An arrow indicates the start of treatment for subjects RA1 and RA2.
Table 1.
Composition and diversity of the HCV E1E2 sequences derived by SGA from donor and recipient viral quasispecies
| Patienta | Total no. of E1E2 SGA sequences analyzedb | No. of viral variants | Mean genetic diversity (%) (Hamming score) |
|---|---|---|---|
| Donor DA | |||
| nt | 33 | 33 | 1.57 |
| aa | 31 | 20 | 0.819 |
| Recipient RA1 | |||
| nt | 37 | 16 | 0.05 |
| aa | 36 | 8 | 0.081 |
| Recipient RA2 | |||
| nt | 28 | 14 | 1.49 |
| aa | 27 | 11 | 1.012 |
| Donor DB | |||
| nt | 33 | 33 | 1.94 |
| aa | 30 | 19 | 1.859 |
| Recipient RB | |||
| nt | 24 | 7 | 0.05 |
| aa | 23 | 3 | 0.032 |
Abbreviations: nt, nucleotides; aa, amino acids.
The differences between the number of nucleotide and amino acid sequences result from the occurrence of stop codons or deletions altering the open reading frame.
Change in HCV variant distribution between donors and recipients.
The E1E2 nucleotide sequences derived from donor and recipient subjects were first subjected to neighbor-joining phylogenetic analysis (see Fig. S1 in the supplemental material). Infection with a 1b genotype was confirmed for all the subjects enrolled in the study. Nucleotide sequences corresponding to each transmission pair clustered together with high bootstrap support, confirming the epidemiological link. In particular, the E1E2 nucleotide sequences of recipient RA2 were interspersed with those of donor DA and recipient RA1, whereas the sequences of recipients RA1 and RB formed a lineage characterized by extremely low diversity (bootstrap support, ≥98%). These phylogenetic data were consistent with the mean within-subject genetic diversities, which were 1.49% for RA2 sequences and 0.05% for RA1 or RB sequences (Table 1). As expected, sequences from the chronically infected subjects DA and DB displayed broader genetic diversities, with mean within-subject values of 1.57% and 1.94%, respectively. Thus, there was a decrease in viral genetic diversity after transmission, which was very pronounced in recipients RA1 and RB.
For further characterization of the transmission process, we first focused on the pattern of nonsynonymous substitutions (amino acid changes) within each transmission pair. These amino acid changes were analyzed by a combination of neighbor-joining phylogenetic tree reconstruction and Highlighter plots (http://hcv.lanl.gov/content/sequence/HIGHLIGHT/highlighter_top.html). Polymorphisms within the E1E2 amino acid sequences derived from donor DA and recipient RA1 are represented in Fig. 2A. The sequence used as the reference, at the top of the tree and the Highlighter plot, corresponds to the dominant variant of donor DA, which was not transmitted to recipient RA1. Twenty-nine of the 36 amino acid sequences obtained from RA1 were identical, with the remaining sequences differing by only one or two randomly distributed substitutions. This strong genetic homogeneity suggests that a single T/F virus was likely responsible for productive infection in RA1. The T/F virus E1E2 amino acid sequence was found to be identical to that of a minor variant present among donor DA variants. The corresponding nucleotide sequences could be distinguished on the basis of patterns of polymorphisms, with synonymous mutations specific to DA or RA1 (see Fig. S2 in the supplemental material). We then extended this analysis to recipient RA2, using the same dominant variant sequence from donor DA as a reference (Fig. 2B; see also Fig. S3 in the supplemental material). The 27 amino acid sequences derived from recipient RA2 clearly formed five lineages, with consensus sequences differing by three or more nonsynonymous substitutions. This suggests that at least five genetically different T/F viruses were responsible for initiating productive infection in RA2. No E1E2 amino acid sequence common to donor DA and recipient RA2 was identified, but the consensus sequences of the transmitted lineages differed by only one to four amino acids from those of the closest variants present in the donor virus population. The genetic relationships between the E1E2 sequences present in donor DB and recipient RB are depicted in Fig. 2C. The sequence used as the reference, at the top of the tree and the Highlighter plot, is that of one of the two major variants from donor DB, which was not transmitted to recipient RB. Twenty-one of the 23 amino acid sequences derived from recipient RB were identical, with the remaining sequences differing by only one random substitution. This strong genetic homogeneity suggests that a single T/F virus was likely responsible for productive infection in recipient RB. However, in this case, the T/F virus E1E2 amino acid sequence was identical to that of a major variant from donor DB. Thus, the donor virus populations went through a strong genetic bottleneck, with only a single variant from the inoculum generating productive infection in two of the three recipients studied. In these two cases, the E1E2 amino acid sequences of the transmitted variant were fully conserved during transmission, although synonymous substitutions nevertheless occurred in the corresponding nucleotide sequences. The larger number of T/F viruses detected in the second recipient (RA2) infected by donor DA suggests that a less stringent selective process occurred in this case. Alternatively, the infecting inoculum may have been numerically more complex than that for RA1, resulting in the transmission of a larger number of variants.
Fig 2.

Phylogenetic relationships and patterns of substitution in HCV E1E2 amino acid sequences between the transmission pair variants. E1E2 amino acid sequences derived from recipient RA1 (A), RA2 (B), and RB (C) (red) were analyzed by neighbor-joining trees (left) and Highlighter plots (right), with pretransmission donor sequences included (blue). Polymorphisms are indicated by a colored tick mark specific for each amino acid, according to the color scheme of BioEdit (http://www.mbio.ncsu.edu/bioedit/bioedit.html). A schematic diagram of the E1E2 proteins, showing the location of HVR1 of E2 in light blue, is provided above the Highlighter plots. The scale bar represents 1 amino acid (aa). The E1E2 sequences identified on the left side of the Highlighter plots were selected for further phenotypic analyses. The sequence indicated by an asterisk in the Highlighter plots corresponds to the transmitted variant V1 of recipient RA1.
We checked that no T/F virus was missed due to our focus on E1E2 amino acid sequences by also examining the pattern of nucleotide substitutions by the same methodological approach (i.e., using a combination of neighbor-joining phylogenetic tree reconstruction and Highlighter plots). Polymorphisms within the E1E2 nucleotide sequences are represented in Fig. S2 in the supplemental material. As previously observed with the amino acid analysis, most of the nucleotide sequences from RA1 were identical, with the remaining sequences differing by only one or two nucleotides. The RB nucleotide sequences were also highly homogeneous, with two main groups of identical sequences distinguished by a single shared synonymous substitution, with the remaining sequences differing by only one nucleotide from these two main groups of sequences. The observed shared polymorphism may result from a polymerase error early in infection being retained in the population (12). There is, therefore, no difference in the estimates of T/F virus numbers for recipients RA1 and RB obtained in analyses of amino acid and nucleotide sequences. A similar result was obtained for four of the five amino acid sequence lineages previously associated with T/F viruses in recipient RA2, with the nucleotide sequences differing from the consensus sequence of each lineage by no more than two nucleotide substitutions. However, in the fifth lineage, one of the three variants differed by six nucleotides from the consensus sequence. This difference suggests that two different T/F viruses may have generated this specific lineage of sequences (12), indicating that six T/F viruses in total are likely to have initiated the infection in recipient RA2.
Molecular determinants underlying HCV transmission.
For identification of the signature of molecular determinants related to the observed genetic bottleneck, we investigated amino acid differences between the sequences derived from donors and those obtained from the corresponding recipients with VESPA (16). VESPA calculates the frequency of each amino acid at each position in an alignment for the query (recipient) and reference set (donor) and selects the positions for which the most common character in the query set differs from that in the background set. Six putative signature sites were identified in the E2 glycoprotein, three of which were located in hypervariable region 1 (HVR1) (Fig. 3). The signature site located at position 394 in HVR1 (R394 or Y394) was common to the transmitted variants in all three transmission pairs (Fig. 3A). Variants harboring a histidine residue at position 394 were detected only in donors, with the exception of a single E1E2 sequence derived from recipient RA2. The frequencies of each combination of signature amino acids are summarized in Fig. 3B. A specific combination of signature amino acids characterized the E1E2 sequences of the transmitted variants identified in recipients RA1 and RB (i.e., R394-R445-D641 for recipient RA1 and Y394-F399-K401-E476 for recipient RB). In recipient RA2, combinations of signature amino acids identified four of the five genetic lineages previously identified with the Highlighter tool (amino acid sequence analysis); two of these lineages shared the same combination of signature amino acids (i.e., Y394-R445). These data suggest that, during transmission or early in infection, a putative key amino acid located at position 394 in HVR1, together with additional signature amino acids specific to each transmission pair, may confer a fitness advantage on the transmitted variants studied here.
Fig 3.

Identification of the key residues of the E1E2 envelope glycoprotein characterizing the recipient virus populations. (A) Sequence logo depiction of signature amino acids specific to the transmitted E1E2 variants. A schematic representation of the E1E2 proteins, showing the location of hypervariable region 1 (light blue), is provided at the top. The positions of the signature amino acids, identified with VESPA (16) in the 3 transmission pairs, are indicated below the schematic representation of E1E2 proteins. The numbers indicate positions relative to the H77 polyprotein (GenBank accession no. NC_004102). The 1b reference sequence logo (top row) was obtained with an alignment of 340 full-length HCV subtype 1b sequences from different sources (Los Alamos National Laboratories HCV database). Sequence logos indicating the variability of each amino acid in sequences derived from donors and recipients are shown below (17). The height of each single-letter amino acid code is proportional to the representation of that amino acid at the position concerned. (B) Frequencies of putative key residue combinations circulating within donors and recipients in each transmission pair.
Influence of viral entry and donor neutralizing antibodies on variant selection.
We investigated the mechanism of selection operating during HCV transmission by determining the relative entry efficiency conferred by E1E2 amino acid sequences representative of the main genetic lineages previously identified in donor and recipient virus populations. A few additional E1E2 sequences derived from minor variants were also studied (Fig. 2; see also Fig. S4 in the supplemental material). Lentiviral HCVpp bearing E1E2 glycoproteins were generated, and their ability to infect Huh7.5 cells was assessed, as described in Materials and Methods. Expression of 15 of 19 selected E1E2 sequences resulted in the production of infectious HCVpp (Fig. 4). Levels of E1E2 glycoprotein incorporation into HCVpp were similar in each case (see Fig. S5 in the supplemental material). The E1E2 sequences corresponding to the recipient RA1 T/F virus or the two main genetic lineages identified among the recipient RA2 variants (i.e., RA2-V1 and RA2-V3) conferred high levels of infectivity but of the same order of magnitude as that observed for the variants not transmitted to recipients (including the dominant sequence from DA, V3) (Fig. 4A). These data suggest that the putative effect of residue 394 is not directly linked to greater infectivity in the HCVpp system. This finding was supported by results for the transmission pair DB/RB, for which the E1E2 sequence derived from the recipient RB T/F virus had a level of infectivity at 1 order of magnitude lower than that of the infectious DB variants bearing the H394 residue (Fig. 4B).
Fig 4.
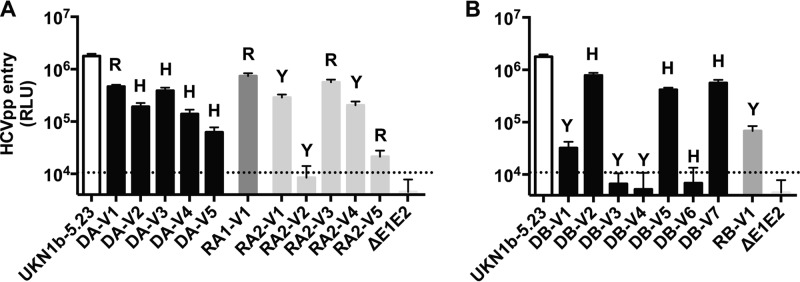
Infectivities of the HCVpp bearing donor and recipient E1E2 envelope glycoproteins. The infectivity of HCVpp conferred by major or minor nontransmitted E1E2 variants was compared with that conferred by the variants transmitted in the transmission pairs DA/RA1 or DA/RA2 (A) and DB/RB (B). Infection assays with the luciferase reporter gene were performed with target Huh7.5 cells. Similar amounts of viral particles were used in each experiment. Results are expressed in relative light units (RLU) plotted on a logarithmic scale. The UKN1B5.23 envelope was used as an external reference. The dotted line indicates the threshold for detectable infection in this system. The detection limit for positive luciferase reporter protein expression was 10 × 103 RLU/assay, corresponding to the mean ± 3 SD of the background levels obtained with cells infected with ΔE1E2 pseudoparticles. Means ± SD from four independent experiments (performed in triplicate) are shown. The letter above each bar indicates the amino acid at position 394 of each E1E2 variant.
The anti-HCV antibodies present in the inoculum might also influence the transmission process. We therefore carried out neutralization experiments with donor serum. HCVpp were incubated with serial dilutions of the donor serum; their subsequent infection levels were quantified, and neutralization titers were calculated as described in Materials and Methods. HCVpp bearing E1E2 sequences derived from transmitted variants were neutralized to an extent similar to those carrying nontransmitted variants, by the antibodies present in the corresponding donor serum (Fig. 5A). None of the donor sera neutralized HCVpp bearing the control VSV-G (data not shown). These findings indicate that the T/F viruses that predominate after transmission do not correspond to antibody escape mutants.
Fig 5.

Sensitivity of the HCVpp bearing donor and recipient E1E2 envelope glycoproteins to neutralization by donor sera (matched by transmission pair) or heterologous pools of sera. HCVpp were incubated with donor serum (A) or genotype-specific (1a, 1b, 2a, and 3a) serum pools (B) in serial dilutions for 1 h at 37°C. HCVpp-antibody complexes were then added to Huh7.5 cells, and infection assays were performed with the luciferase reporter gene. Neutralization titers were calculated as described in Materials and Methods and are indicated for each E1E2 variant derived from a donor (black) or recipient (gray). Means ± SD from two independent experiments (performed in triplicate) are shown.
Entry inhibition of transmitted variants by polyclonal or monoclonal antibodies.
In a final set of experiments, we sought to assess whether the entry of transmitted variants could be targeted with neutralizing antibodies to prevent HCV infection. HCVpp were incubated with genotype-specific (1a, 1b, 2a, 3a) serum pools, and their neutralization was studied, as described in Materials and Methods. Similar experiments were conducted with the anti-E2 broadly neutralizing MAbs AR3A and HC-11 (21, 22). The serum pools inhibited infection with HCVpp bearing E1E2 sequences derived from both transmitted and nontransmitted viral variants to various extents (Fig. 5B). The genotype 1b serum pool displayed the strongest neutralizing activities against all the variants, consistent with the known infection of both donors and recipients with HCV genotype 1b. The anti-E2 MAb also efficiently inhibited infection with HCVpp bearing E1E2 sequences derived from both transmitted and nontransmitted viral variants (IC50s of 0.1 to 1.04 μg/ml for AR3A and IC50s of 0.19 to 1.67 μg/ml for HC-11) (Fig. 6). Thus, the transmitted variants studied here were neutralized by at least two well-characterized monoclonal antibodies.
Fig 6.

Sensitivity of the HCVpp bearing donor and recipient E1E2 envelope glycoproteins to neutralization with neutralizing MAb. HCVpp neutralization sensitivities were assessed with the human anti-E2 neutralizing MAbs AR3A (A) and HC-11 (B). The percentage neutralization was calculated as described in Materials and Methods. Neutralization curves are shown for each E1E2-derived variant from donors (blue) or recipients (red). The data shown are mean values from 1 representative experiment performed in triplicate. The dotted line indicates 50% neutralization of HCVpp entry.
DISCUSSION
This study provides insight into the genotypic and phenotypic properties of the single or small number of HCV variants transmitted to a new host from the swarm of viral variants present in the donor. HCV transmission has rarely been investigated, due to the difficulty of recruiting patients at early stages of acute infection, which is usually clinically silent (2). Furthermore, only a few reports have described experimental inoculation in the chimpanzee model or accidental contamination in humans, with the availability of donor samples. Those studies were based principally on genetic analysis of HVR1 of the E2 glycoprotein gene, with no phenotypic characterization (7, 8, 24). Methods for accurately identifying T/F viruses and assessing genetic diversity at various stages of infection have also evolved with the development of SGA. This method is now the gold standard in the HIV field (14, 25–27) but has only recently been applied to HCV studies (12, 13). We used SGA for the detailed investigation of HCV transmission in three cases of acute hepatitis acquired through needlestick accidents. Phylogenetic analyses demonstrated that a change in host environment resulted in a strong genetic bottleneck, with a single T/F virus likely responsible for productive infection in recipients RA1 and RB. The genetic bottleneck observed was less stringent in the second recipient (RA2) infected by donor DA, with an estimated six T/F viruses. We do not think that sampling from RA2 at the time of seroconversion confounded this identification of T/F variants. Indeed, in a recent report, only a single instance of a potential cytotoxic T lymphocyte (CTL) escape or reversion was detected among 17 immunocompetent subjects followed during the initial 6 to 8 weeks of infection (12). In addition, the targeted epitope was located outside the E1E2 glycoproteins. Our findings for the number of T/F viruses are consistent with this report, in which 10 of the 17 acutely infected subjects examined had one to four T/F viruses. A similar number of T/F viruses was also found by 454 pyrosequencing approaches used to study seven acutely infected subjects reported by two different groups (10, 11).
These recent studies on T/F variants did not address the question of the mechanism reducing the genetic diversity of the donor virus population. By studying donor samples, we demonstrated that the T/F E1E2 amino acid sequences derived from recipients RA1 and RB were identical to those of a minor or a major variant from the DA and DB, respectively. No identical E1E2 amino acid sequence was common to the donor DA variants and the T/F viruses from recipient RA2, possibly due to the 10-month interval between sampling for DA and the needlestick accident resulting in the infection of RA2. However, the E1E2 sequences obtained from the two subjects remained very similar. It remains unclear whether the transmission of T/F viruses results from a founder effect, with one or a small number of variants being transferred between hosts, or whether it is due to early evolutionary events, with larger numbers of variants undergoing a selective sweep due to differences in fitness constraints (10). In a recent report of the experimental transmission of HCV to chimeric SCID/Alb-uPA mice with transplanted human hepatocytes, the occurrence of selective sweeps was put forward to explain the finding that undetectable inoculum variants bearing an advantageous E1E2 motif became the major variants circulating in the infected mice (13). However, the presumed small inoculum resulting from needlestick accidents and the different proportions of T/F E1E2 sequences present in donor virus populations complicate the transposition of previously described scenarios to each of our transmission pairs. Moreover, our data provide no evidence in support of a putative mechanism explaining the pattern of change observed. As things stand, these data are most consistent with a founder effect. RA1 and RA2 provided us with a rare opportunity to compare HCV transmission in two subjects with the same source of infection. The difference in the number of T/F viruses between these recipients is consistent with the source/recipient-specific clades observed in a previous study of a common-source outbreak (28).
Genetic and phenotypic signatures associated with the transmission of true T/F E1E2 obtained by SGA were investigated, to the best of our knowledge, only in the previously cited study on experimental transmission of HCV in a chimeric mouse model (13). This study showed that the transmitted variants harbored key substitutions in E1E2, outside HVR1. In contrast, we found that three of the six signature sites (positions 394, 399, and 401) characterizing our T/F viruses were located in HVR1 (Fig. 3). Importantly, the signature site located at position 394 was common to the T/F variants of all three transmission pairs studied here. HVR1 plays a major role in both HCV cell entry and immune evasion (29–33). This hypervariable region is globally a basic stretch of amino acids, the most basic of which are found at positions 386, 394, 397, and 410 (corresponding to positions 3, 11, 14, and 27, respectively, in HVR1) in HCV genotype 1b (34, 35). The basic residues H and R are the most frequently observed at position 394, but this does not exclude the possibility of other amino acids, including the nonbasic Y, occupying this position. The presence of basic residues in HVR1 has been reported to facilitate virus entry (34). We therefore hypothesized that the signature amino acids identified in HVR1 might be linked to phenotypic changes affecting the replicative fitness of the virus during transmission or early in acute infection (this issue is discussed in more detail below). None of the signature amino acids identified outside HVR1 (positions 445, 476, and 641) was known to participate in CD81 binding or a potential N-glycosylation site in HCV genotype 1b (36–38). Residue 476 has been identified as a potential glycosylation site in the genotype 1a reference strain H77. However, this site has a percentage of conservation below 20% in genotype 1b, and it was absent from the E1E2 sequences studied here (39). We also demonstrated that the HCVpp bearing E1E2 sequences derived from transmitted variants were neutralized to an extent similar to those carrying nontransmitted variants, suggesting that the T/F viruses that predominate after transmission are not antibody escape mutants. The residue in position 445 is part of the AR3 discontinuous epitopes (21). However, the differences at position 445 between the DA and RA1 or RA2 variants do not seem to affect the neutralization properties of these variants (Fig. 6).
In the chimeric mouse model study, the major posttransmission E1E2 variant with key substitutions outside HVR1 conferred a greater capacity for HCVpp entry (13). A similar conclusion was drawn in a previous report on HCV evolution in a liver transplant setting, in which the uPA-SCID mouse model was used to support the hypothesis that viral entry is an important determinant of relative fitness in immunodeficient hosts (23). However, we found that the E1E2 sequences corresponding to the recipient RA1 T/F virus or the two main genetic lineages identified among the recipient RA2 variants conferred levels of infectivity similar to those of most of the untransmitted variants (Fig. 4A). These data suggest that the effect of the putative signature residues identified within the RA1 or RA2 T/F envelope glycoproteins cannot be linked to an increase in entry capacity, at least in the HCVpp system. A similar observation was made for the transmission pair DB/RB (Fig. 4B). This discrepancy with previous findings may reflect differences in the settings encountered by HCV in chimeric mice and immunocompetent patients. Nevertheless, additional case studies of HCV transmission to immunocompetent patients will be required to confirm this. Another consideration regarding the absence of phenotypic traits associated with virus transmission is the extent to which the HCVpp model reflects the complexity of HCV biology. Indeed, HCV virions present in the bloodstream in vivo have to reach the polarized hepatocytes in a complex multicellular environment in which various factors, such as cytokines, inflammatory mediators, and growth factors, may play a role in regulating cell-free infection or cell-to-cell transmission (reviewed in reference 40). Moreover, there is growing evidence for the presence of lipid components within the viral particle, such as apolipoproteins B and E, which might facilitate viral entry. It is thus conceivable that the phenotypic properties conferred by a given E1E2 variant may differ in the context of the authentic viral particle and in HCVpp (41–44). Studies using chimeric JFH-1-based HCV in cell culture (HCVcc) will clearly be required to address this specific question (45, 46). However, the HCVpp system has been widely used, since 2003, for the identification of significant functional differences between E1E2 at entry and after neutralization, and even single amino acid changes in E1E2 can affect these properties (13, 23, 32, 47–50).
Finally, our findings have important implications for the development of strategies for preventing HCV transmission. Various cross-neutralizing anti-envelope glycoprotein MAbs neutralize genetically diverse HCV isolates, and some have been shown to protect against heterologous HCV challenge in an HCV animal model (21, 22, 51–54). Polyclonal anti-HCV antibodies isolated from chronically HCV-infected patients can also protect against in vivo challenge with different HCV genotypes (55, 56). We confirmed the sensitivity of HCV primary isolates, represented by our T/F variants, to cross-reactive monoclonal or polyclonal antibodies in vitro. Furthermore, our data indicate that the well-characterized anti-E2 MAbs tested here display sufficient cross-reactivity to neutralize T/F variants derived from the three recipients.
In conclusion, our findings provide insight into the molecular mechanisms underlying HCV transmission and suggest that viral entry is a potential target for the prevention of HCV infection, although the long-term protection conferred by vaccine-induced or passively transferred antibodies in groups of individuals at risk remains to be determined.
Supplementary Material
ACKNOWLEDGMENTS
We thank Charles Rice for the Huh7.5 cell line, Jonathan Ball for the UKN1B5.23 plasmid, Jane McKeating for the MAb 3/11, Mansun Law for the MAb AR3A, and Steven Foung for the MAb HC-11 and R04. The reagent ARP432 (antiserum to HIV-1 p24) was obtained from the Centre for AIDS Reagents, NIBSC, and was donated by G. Reid. The following reagent was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: pNL4-3.Luc.R−E− from Nathaniel Landau. We also thank Jean-Christophe Meunier for helpful discussions.
The study was sponsored by the CHRU of Tours and financially supported by a grant from the INSERM-DHOS Recherche Clinique Translationnelle 2010: VINTAGE VHC. Valentina D'Arienzo was supported by a fellowship from the Region Centre.
Footnotes
Published ahead of print 9 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02119-13.
REFERENCES
- 1.WHO 2013. Hepatitis C. WHO fact sheet 164. WHO, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs164/en/index.html [Google Scholar]
- 2.Ascione A, Tartaglione MT, Di Costanzo GG. 2007. Natural history of chronic hepatitis C virus infection. Dig. Liver Dis. 39(Suppl 1):S4–S7 [DOI] [PubMed] [Google Scholar]
- 3.Maheshwari A, Ray S, Thuluvath PJ. 2008. Acute hepatitis C. Lancet 372:321–332 [DOI] [PubMed] [Google Scholar]
- 4.Averhoff FM, Glass N, Holtzman D. 2012. Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin. Infect. Dis. 55(Suppl 1):S10–S5 [DOI] [PubMed] [Google Scholar]
- 5.Yazdanpanah Y, De Carli G, Migueres B, Lot F, Campins M, Colombo C, Thomas T, Deuffic-Burban S, Prevot MH, Domart M, Tarantola A, Abiteboul D, Deny P, Pol S, Desenclos JC, Puro V, Bouvet E. 2005. Risk factors for hepatitis C virus transmission to health care workers after occupational exposure: a European case-control study. Clin. Infect. Dis. 41:1423–1430 [DOI] [PubMed] [Google Scholar]
- 6.Morin T, Pariente A, Lahmek P, Investigator Group of ANGHSPILFFNPRH 2011. Favorable outcome of acute occupational hepatitis C in healthcare workers: a multicenter French study on 23 cases. Eur. J. Gastroenterol. Hepatol. 23:515–520 [DOI] [PubMed] [Google Scholar]
- 7.Liu CH, Chen BF, Chen SC, Lai MY, Kao JH, Chen DS. 2006. Selective transmission of hepatitis C virus quasi species through a needlestick accident in acute resolving hepatitis. Clin. Infect. Dis. 42:1254–1259 [DOI] [PubMed] [Google Scholar]
- 8.Saito T, Watanabe H, Shao L, Okumoto K, Hattori E, Sanjo M, Misawa K, Suzuki A, Takeda T, Sugahara K, Ito JI, Saito K, Togashi H, Kawata S. 2004. Transmission of hepatitis C virus quasispecies between human adults. Hepatol. Res. 30:57–62 [DOI] [PubMed] [Google Scholar]
- 9.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5:453–463 [DOI] [PubMed] [Google Scholar]
- 10.Bull RA, Luciani F, McElroy K, Gaudieri S, Pham ST, Chopra A, Cameron B, Maher L, Dore GJ, White PA, Lloyd AR. 2011. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathog. 7:e1002243. 10.1371/journal.ppat.1002243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang GP, Sherrill-Mix SA, Chang KM, Quince C, Bushman FD. 2010. Hepatitis C virus transmission bottlenecks analyzed by deep sequencing. J. Virol. 84:6218–6228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Stoddard MB, Wang S, Blair LM, Giorgi EE, Parrish EH, Learn GH, Hraber P, Goepfert PA, Saag MS, Denny TN, Haynes BF, Hahn BH, Ribeiro RM, Perelson AS, Korber BT, Bhattacharya T, Shaw GM. 2012. Elucidation of hepatitis C virus transmission and early diversification by single genome sequencing. PLoS Pathog. 8:e1002880. 10.1371/journal.ppat.1002880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown RJ, Hudson N, Wilson G, Rehman SU, Jabbari S, Hu K, Tarr AW, Borrow P, Joyce M, Lewis J, Zhu LF, Law M, Kneteman N, Tyrrell DL, McKeating JA, Ball JK. 2012. Hepatitis C virus envelope glycoprotein fitness defines virus population composition following transmission to a new host. J. Virol. 86:11956–11966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 105:7552–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korber B, Myers G. 1992. Signature pattern analysis: a method for assessing viral sequence relatedness. AIDS Res. Hum. Retroviruses 8:1549–1560 [DOI] [PubMed] [Google Scholar]
- 17.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Connor RI, Chen BK, Choe S, Landau NR. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935–944 [DOI] [PubMed] [Google Scholar]
- 19.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A. 100:7271–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flint M, Maidens C, Loomis-Price LD, Shotton C, Dubuisson J, Monk P, Higginbottom A, Levy S, McKeating JA. 1999. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 73:6235–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25–27 [DOI] [PubMed] [Google Scholar]
- 22.Keck ZY, Li TK, Xia J, Gal-Tanamy M, Olson O, Li SH, Patel AH, Ball JK, Lemon SM, Foung SK. 2008. Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutralizing human monoclonal antibodies. J. Virol. 82:6061–6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fafi-Kremer S, Fofana I, Soulier E, Carolla P, Meuleman P, Leroux-Roels G, Patel AH, Cosset FL, Pessaux P, Doffoël M, Wolf P, Stoll-Keller F, Baumert TF. 2010. Viral entry and escape from antibody-mediated neutralization influence hepatitis C virus reinfection in liver transplantation. J. Exp. Med. 207:2019–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugitani M, Shikata T. 1998. Comparison of amino acid sequences in hypervariable region-1 of hepatitis C virus clones between human inocula and the infected chimpanzee sera. Virus Res. 56:177–182 [DOI] [PubMed] [Google Scholar]
- 25.Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, Parrish NF, Shaw KS, Guffey MB, Bar KJ, Davis KL, Ochsenbauer-Jambor C, Kappes JC, Saag MS, Cohen MS, Mulenga J, Derdeyn CA, Allen S, Hunter E, Markowitz M, Hraber P, Perelson AS, Bhattacharya T, Haynes BF, Korber BT, Hahn BH, Shaw GM. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 206:1273–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilen CB, Parrish NF, Pfaff JM, Decker JM, Henning EA, Haim H, Petersen JE, Wojcechowskyj JA, Sodroski J, Haynes BF, Montefiori DC, Tilton JC, Shaw GM, Hahn BH, Doms RW. 2011. Phenotypic and immunologic comparison of clade B transmitted/founder and chronic HIV-1 envelope glycoproteins. J. Virol. 85:8514–8527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, Hora B, Berg A, Cai F, Hopper J, Denny TN, Ding H, Ochsenbauer C, Kappes JC, Galimidi RP, West AP, Bjorkman PJ, Wilen CB, Doms RW, O'Brien M, Bhardwaj N, Borrow P, Haynes BF, Muldoon M, Theiler JP, Korber B, Shaw GM, Hahn BH. 2013. Phenotypic properties of transmitted founder HIV-1. Proc. Natl. Acad. Sci. U. S. A. 110:6626–6633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ray SC, Fanning L, Wang XH, Netski DM, Kenny-Walsh E, Thomas DL. 2005. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J. Exp. Med. 201:1753–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21:5017–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartosch B, Verney G, Dreux M, Donot P, Morice Y, Penin F, Pawlotsky JM, Lavillette D, Cosset FL. 2005. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol. 79:8217–8229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartosch B, Vitelli A, Granier C, Goujon C, Dubuisson J, Pascale S, Scarselli E, Cortese R, Nicosia A, Cosset FL. 2003. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 278:41624–41630 [DOI] [PubMed] [Google Scholar]
- 32.Dowd KA, Netski DM, Wang XH, Cox AL, Ray SC. 2009. Selection pressure from neutralizing antibodies drives sequence evolution during acute infection with hepatitis C virus. Gastroenterology 136:2377–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guan M, Wang W, Liu X, Tong Y, Liu Y, Ren H, Zhu S, Dubuisson J, Baumert TF, Zhu Y, Peng H, Aurelian L, Zhao P, Qi Z. 2012. Three different functional microdomains in the hepatitis C virus hypervariable region 1 (HVR1) mediate entry and immune evasion. J. Biol. Chem. 287:35631–35645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Callens N, Ciczora Y, Bartosch B, Vu-Dac N, Cosset FL, Pawlotsky JM, Penin F, Dubuisson J. 2005. Basic residues in hypervariable region 1 of hepatitis C virus envelope glycoprotein E2 contribute to virus entry. J. Virol. 79:15331–15341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Penin F, Combet C, Germanidis G, Frainais PO, Deléage G, Pawlotsky JM. 2001. Conservation of the conformation and positive charges of hepatitis C virus E2 envelope glycoprotein hypervariable region 1 points to a role in cell attachment. J. Virol. 75:5703–5710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owsianka AM, Timms JM, Tarr AW, Brown RJP, Hickling TP, Szwejk A, Bienkowska-Szewczyk K, Thomson BJ, Patel AH, Ball JK. 2006. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J. Virol. 80:8695–8704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goffard A, Callens N, Bartosch B, Wychowski C, Cosset FL, Montpellier C, Dubuisson J. 2005. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 79:8400–8409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boo I, te Wierik K, Douam F, Lavillette D, Poumbourios P, Drummer HE. 2012. Distinct roles in folding, CD81 receptor binding and viral entry for conserved histidine residues of hepatitis C virus glycoprotein E1 and E2. Biochem. J. 443:85–94 [DOI] [PubMed] [Google Scholar]
- 39.Helle F, Goffard A, Morel V, Duverlie G, McKeating J, Keck ZY, Foung SK, Penin F, Dubuisson J, Voisset C. 2007. The neutralizing activity of anti-hepatitis C virus antibodies is modulated by specific glycans on the E2 envelope protein. J. Virol. 81:8101–8111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meredith LW, Wilson GK, Fletcher NF, McKeating JA. 2012. Hepatitis C virus entry: beyond receptors. Rev. Med. Virol. 22:182–193 [DOI] [PubMed] [Google Scholar]
- 41.Liu S, McCormick KD, Zhao W, Zhao T, Fan D, Wang T. 2012. Human apolipoprotein E peptides inhibit hepatitis C virus entry by blocking virus binding. Hepatology 56:484–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Owen DM, Huang H, Ye J, Gale M., Jr 2009. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 394:99–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazumdar B, Banerjee A, Meyer K, Ray R. 2011. Hepatitis C virus E1 envelope glycoprotein interacts with apolipoproteins in facilitating entry into hepatocytes. Hepatology 54:1149–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Popescu CI, Dubuisson J. 2010. Role of lipid metabolism in hepatitis C virus assembly and entry. Biol. Cell 102:63–74 [DOI] [PubMed] [Google Scholar]
- 45.Kawaguchi K, Faulk K, Purcell RH, Emerson SU. 2011. Reproduction in vitro of a quasispecies from a hepatitis C virus-infected patient and determination of factors that influence selection of a dominant species. J. Virol. 85:3408–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gottwein JM, Scheel TK, Jensen TB, Lademann JB, Prentoe JC, Knudsen ML, Hoegh AM, Bukh J. 2009. Development and characterization of hepatitis C virus genotype 1–7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49:364–377 [DOI] [PubMed] [Google Scholar]
- 47.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haberstroh A, Schnober EK, Zeisel MB, Carolla P, Barth H, Blum HE, Cosset FL, Koutsoudakis G, Bartenschlager R, Union A, Depla E, Owsianka A, Patel AH, Schuster C, Stoll-Keller F, Doffoël M, Dreux M, Baumert TF. 2008. Neutralizing host responses in hepatitis C virus infection target viral entry at postbinding steps and membrane fusion. Gastroenterology 135:1719–1728 [DOI] [PubMed] [Google Scholar]
- 49.Fofana I, Kremer SF, Carolla P, Fauvelle C, Zahid MN, Turek M, Heydmann L, Cury K, Hayer J, Combet C, Cosset FL, Pietschmann T, Hiet MS, Bartenschlager R, Habersetzer F, Doffoël M, Keck ZY, Foung SK, Zeisel MB, Stoll-Keller F, Baumert TF. 2012. Mutations that alter use of hepatitis C virus cell entry factors mediate escape from neutralizing antibodies. Gastroenterology 143:223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.von Hahn T, Yoon JC, Alter H, Rice CM, Rehermann B, Balfe P, McKeating JA. 2007. Hepatitis C virus continuously escapes from neutralizing antibody and T-Cell responses during chronic infection in vivo. Gastroenterology 132:667–678 [DOI] [PubMed] [Google Scholar]
- 51.Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, Ploss A, Burton DR, Law M. 2012. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 109:6205–6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keck ZY, Xia J, Wang Y, Wang W, Krey T, Prentoe J, Carlsen T, Li AY, Patel AH, Lemon SM, Bukh J, Rey FA, Foung SK. 2012. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog. 8:e1002653. 10.1371/journal.ppat.1002653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edwards VC, Tarr AW, Urbanowicz RA, Ball JK. 2012. The role of neutralizing antibodies in hepatitis C virus infection. J. Gen. Virol. 93:1–19 [DOI] [PubMed] [Google Scholar]
- 54.Meunier JC, Russell RS, Goossens V, Priem S, Walter H, Depla E, Union A, Faulk KN, Bukh J, Emerson SU, Purcell RH. 2008. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the E1 glycoprotein of hepatitis C virus. J. Virol. 82:966–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meuleman P, Bukh J, Verhoye L, Farhoudi A, Vanwolleghem T, Wang RY, Desombere I, Alter H, Purcell RH, Leroux-Roels G. 2011. In vivo evaluation of the cross-genotype neutralizing activity of polyclonal antibodies against hepatitis C virus. Hepatology 53:755–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu MW, Bartosch B, Zhang P, Guo Z, Renzi PM, Shen L, Granier C, Feinstone SM, Cosset FL, Purcell RH. 2004. Neutralizing antibodies to hepatitis C virus (HCV) in immune globulins derived from anti-HCV-positive plasma. Proc. Natl. Acad. Sci. U. S. A. 101:7705–7710 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.