

Abstract
Chromatin-based regulation of herpesviral transcriptional programs is increasingly appreciated as a mechanism for modulating infection outcomes. Transcriptionally permissive euchromatin predominates during lytic infection, whereas heterochromatin domains refractory to transcription are enriched at lytic genes during latency. Reversibly silenced facultative heterochromatin domains are often enriched for histone H3 trimethylated on lysine 27 (H3K27me3), a modification catalyzed by Polycomb repressive complex 2 (PRC2). The requirement for PRC2 in suppressing the human cytomegalovirus (HCMV) lytic transcriptional program during latency has not been thoroughly evaluated. Therefore, we disrupted PRC2 activity in the highly tractable THP1 and NT2D1 quiescent-infection models by treating cells with small-molecule inhibitors of PRC2 activity. Compared to control cells, disruption of PRC2 in HCMV-infected THP1 or NT2D1 cells resulted in significant increases in viral transcript levels and the detection of viral protein. Using chromatin immunoprecipitation, we demonstrated that enrichment of H3K27me3, deposited by PRC2, correlates inversely with lytic transcriptional output, suggesting that PRC2 catalytic activity at viral chromatin directly represses lytic transcription. Together, our data suggest that PRC2-mediated repression of viral transcription is a key step in the establishment and maintenance of HCMV latency.
INTRODUCTION
Human cytomegalovirus (HCMV), a member of the Betaherpesvirinae subfamily, is a widespread human pathogen that poses a serious health risk for immunocompromised individuals, such as AIDS patients, cancer patients, and bone marrow and solid-organ transplant recipients (1). Transplacental transmission of HCMV from mother to fetus is also a major concern, since it can result in serious sequelae, ranging from sensorineural hearing loss and developmental deficits to death. As with all herpesviruses, HCMV persists for the lifetime of the host by establishing and maintaining latent infections within hematopoietic and myeloid progenitor cell populations (2–4). In latent infections, the genome is maintained, but the lytic transcriptional program is suppressed, and no infectious virus is produced. Latent HCMV infections are largely invisible to the immune system, and the virus persists for the lifetime of the host. For transplant recipients, HCMV recurrence is a major risk factor that causes pneumonia, hepatitis, and retinitis, exacerbating graft-versus-host disease and organ rejection. To address these critical issues, it is important to decipher the mechanisms involved in the establishment and maintenance of latent HCMV infections, as well as to understand the triggers of reactivation.
HCMV genomes are large, double-stranded DNA (dsDNA) molecules, and upon nuclear entry, they rapidly associate with cellular histones, forming a chromatin scaffold that regulates many viral-DNA-templated processes, such as transcription, replication, and repair (5–7). Nucleosomes represent the basic functional module of chromatin, and each nucleosome consists of 147 bp of DNA wrapped around a histone octamer, composed of two each of histones H2A, H2B, H3, and H4. Each histone tail within the nucleosome is subject to a growing list of posttranslational modifications (PTMs), such as methylation and acetylation, which create the localized structure and function of chromatin (8–10). Cellular complexes that catalyze histone PTMs, in combination with other complexes that recognize or remove histone PTMs (sometimes referred to as writers, readers, and erasers), dynamically regulate chromatin structure and function, thereby having a significant impact on the transcriptional profile of a given cell. Histone PTMs are also observed on herpesviral chromatin, and it is becoming clear that the activity of cellular complexes that write, read, and erase histone PTMs may also epigenetically regulate virus infection outcomes (11–17).
The HCMV lytic transcriptional program is a precisely controlled temporal cascade of viral gene expression that results in the production of infectious viral progeny (1). Immediate early (IE) genes are expressed from the genome first, independently of any other gene expression. The expression of viral early (E) and late (L) genes follows and relies on robust IE gene expression. The protein products of the HCMV major immediate early (MIE) promoter, namely, IE1-72 and IE2-86 (IE1 and IE2, respectively), play an essential role in potentiating the lytic replication cycle. Similarly, reactivation from latency relies on the de novo production of IE1 and IE2 (IE1/2) trans-activators to drive the lytic transcription program. The HCMV MIE enhancer/promoter (MIEEP) region is a complex cis-acting transcriptional regulator subject to epigenetic control by various chromatin-modifying complexes, such as histone deacetylases (HDACs), histone methyltransferases (HMTs), and histone chaperones (12, 18–20). DNase I hypersensitivity and chromatin immunoprecipitation (ChIP) analyses of histone PTMs during lytic and latent infections suggest that the underlying chromatin environment at the MIEEP influences transcriptional activity (21). While a strict causal role has not yet been established for HCMV, studies with other herpesviruses have provided strong evidence that local chromatin structure directly regulates viral transcriptional potential (15, 17).
Cellular chromatin-modifying activities generally act to promote one of two basic chromatin states: less-condensed, transcriptionally active euchromatin and highly condensed, transcriptionally silent heterochromatin (HC). HC is further subdivided into constitutive and facultative heterochromatin (22). While constitutive HC generally lacks the ability to revert to euchromatin, facultative HC, though transcriptionally silent, retains the potential to convert to transcriptionally active euchromatin. Reversibly silenced facultative HC regions are often enriched for histone H3 trimethylated on lysine 27 (H3K27me3), catalyzed by the multiprotein HMT complex PRC2 (Polycomb repressive complex 2) (23, 24). PRC2 is composed of EZH2, the catalytic methyltransferase subunit, along with SUZ12 and EED. PRC2 also associates with accessory proteins such as RbAp46/48 and PHF1, which regulate PRC2 targeting and catalytic activity (23, 25–29). How PRC2 acts as a transcriptional repressor is poorly defined. Some studies have provided evidence that PRC2 activity on chromatin recruits a related complex, termed Polycomb repressive complex 1 (PRC1), which inhibits transcription via further chromatin modification and compaction. Other studies have suggested that PRC2 is capable of repressing transcription in a PRC1-independent manner. PRC2 is known to regulate cellular pathways critical for cell self-renewal, differentiation, and the cell cycle (e.g., HOX genes and the INK4A locus). Interestingly, these processes are known to be tightly coupled to the molecular switch between latent and lytic HCMV infections (30–35). Since PRC2 is a transcriptional repressor, PRC2-catalyzed facultative HC domains on HCMV genomes could provide the transcriptional plasticity required for reactivation from latency, concomitant with PRC2-linked cellular differentiation cues in the latent reservoir. To date, the contribution of PRC2 to HCMV heterochromatin formation and maintenance during latency is unknown, and PRC2 involvement in directly regulating HCMV transcription has not been analyzed in any detail.
In this report, we address these issues, while raising important questions with respect to the molecular features of HCMV transcriptional repression during latency. We have previously reported a dynamic interplay between murine cytomegalovirus (MCMV) and PRC2 during permissive infection of mouse embryonic fibroblasts, where PRC2 initially targets incoming MCMV chromatin for the deposition of H3K27me3 very early in infection (36). However, this pre-immediate early modification is rapidly lost in the conversion to a euchromatic state characterized by H3K4me3 and acetylation of H3 and H4 (19, 36, 37). We also observed a viral-replication-dependent alteration in the subnuclear localization of PRC2 concomitant with global changes in H3K27me3, suggesting that PRC2 activity must be overcome in order to successfully complete the lytic replication cycle. To determine the contribution of PRC2 to repression of the HCMV lytic transcriptional program, we used chemical inhibitors to directly disrupt PRC2 activity in two widely accepted experimental models of HCMV quiescence. Undifferentiated THP1 and NT2D1 cell lines are nonpermissive for HCMV replication. However, upon differentiation, both cell lines support viral gene expression and limited genome replication (38–41). Both cell lines have been used extensively to model HCMV quiescence. Although they do not recapitulate all aspects of HCMV latency, THP1 and NT2D1 cells are highly tractable models of the transcriptional silencing that occurs during the establishment and maintenance of latent HCMV infections. These cell lines also allow us to study the differentiation-dependent regulation of viral gene expression observed in natural latent infections in vivo. We discovered that inhibition of PRC2 activity in THP1 or NT2D1 cells resulted in robust activation of the lytic transcriptional program, resulting in significant, temporally regulated increases in viral transcript levels and the detection of viral antigen. Using chromatin immunoprecipitation for H3K27me3, we were able to directly correlate increases in viral transcript levels to decreases in PRC2 activity at lytic genes, indicating that HCMV transcriptional potential is negatively regulated by PRC2. Together, our data suggest that PRC2-mediated repression of viral transcription is a key step in the establishment and maintenance of HCMV latency in vivo.
MATERIALS AND METHODS
Cells and virus.
Primary human foreskin fibroblasts (HFFs) were propagated in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS). NT2D1 embryonal carcinoma cells were cultured in DMEM containing 10% FBS, 1 mM sodium pyruvate, and 1.5 g/liter sodium bicarbonate. THP1 monocytes were cultured in RPMI medium containing 10% heat-inactivated FBS and 0.05 mM 2-mercaptoethanol. All cell lines were maintained at 37°C under a 5% CO2 atmosphere. HCMV strains AD169 and TB40E were used in this study. Virus titers in HFFs were determined by a plaque assay.
Chemical treatments.
3-Deazaneplanocin A (DZnep) was purchased from Cayman Chemical and was resuspended as a stock at 5 mg/ml in dimethyl sulfoxide (DMSO). Phorbol 12-myristate 13-acetate (PMA) was purchased from Sigma-Aldrich and was resuspended as a stock at 1 mg/ml in 100% ethanol. GSK343 was kindly provided by the Structural Genomics Consortium and was resuspended as a 50 mM stock in DMSO. Working solutions were prepared by diluting the stock into media at the final concentrations indicated in each figure legend.
Antibodies.
Antibodies against EZH2 (diluted 1:1,000 for Western blotting [WB]; 2.8 μg used for ChIP) and SUZ12 (diluted 1:1,000 for WB) were purchased from Cell Signaling. Antibodies against RING1B (diluted 1:2,500 for WB), pan-H3 (diluted 1:5,000 for WB; 5 μg used for ChIP), and H3K27me3 (5 μg for ChIP; diluted 1:200 for immunofluorescence [IF] and 1:5,000 for WB) were purchased from Millipore. Anti-HP1 (diluted 1:1,000 for WB) and anti-OCT4 (diluted 1:100 for IF and 1:1,000 for WB) were purchased from Santa Cruz. Anti-β-actin (diluted 1:15,000 for WB) and an Alexa Fluor 488-conjugated goat anti-mouse antibody (diluted 1:1,000 for IF) were purchased from Invitrogen. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit and goat anti-mouse antibodies (both diluted 1:10,000 for WB) were purchased from The Jackson Laboratory. Normal rabbit IgG (5 μg for ChIP) was purchased from Abcam. The antibody against HCMV IE1 (diluted 1:1,000 for WB and 1:200 for IF) was a kind gift from Tom Shenk at Princeton University. A phycoerythrin (PE)-conjugated antibody against CD11b/MAC1 (diluted 1:10 for flow cytometry [FC]) was purchased from BD Pharmingen.
RNA analysis.
Mock-infected and HCMV-infected cells were collected by scraping, Accutase treatment (Millipore), or centrifugation. RNA was isolated using the RNeasy Mini Kit (Qiagen). cDNA was synthesized from 250 ng of total RNA by using the QuantiTect reverse transcription kit (Qiagen). No-reverse transcriptase (no-RT) controls were included for all samples. Samples were diluted 1:20 in double-distilled water (ddH2O), and 4.5 μl of each sample was assayed by quantitative PCR (qPCR) in triplicate using a Roche LightCycler 480 system.
DNA analysis.
Mock-infected and HCMV-infected cells were collected by scraping, Accutase treatment, or centrifugation and were washed in phosphate-buffered saline (PBS)-EDTA. To remove extracellular virus, samples were treated with 0.5% trypsin-PBS-EDTA for 10 min at 37°C and were washed twice in 1 ml PBS-EDTA. Samples were then processed by using a QIAamp DNA Mini Kit (Qiagen) according to the manufacturer's recommendations. Viral DNA was quantified by qPCR in triplicate by using the UL69 primer set (see Table 1). Each sample was normalized to cellular HOXA9 DNA levels.
Table 1.
Primer and probe sequences used in this study
| Target | HCMV straina | Supplier | Sequenceb |
|---|---|---|---|
| IE1/2 | AD169 | IDT | Fwd, TGCAATCCTCGGTCACTTG |
| Rev, GACCCTGATAATCCTGACGAG | |||
| Probe, CTAAGGCCACGGAGCGAGACATC | |||
| IE1/2 | TB40E | IDT | Fwd, AGAAAGATGGACCCTGACAAC |
| Rev, AACATAGTCTGCAGGAACGTC | |||
| Probe, CCCGAGACACCCGTGACCAAG | |||
| UL54 | AD169/TB40E | IDT | Fwd, CTGGCTAAAATTCCGTTGCG |
| Rev, GTCGTACCTTTGCTGTAGTGG | |||
| Probe, TTGGGCAGGATAAAATCGCGGC | |||
| UL99 | AD169/TB40E | IDT | Fwd, GGAAGTCGGAGGGATGTTG |
| Rev, GGTGAGCCCCTGAAAGATG | |||
| Probe, TCGTAGGAGCGTAGAGACACCTGG | |||
| MIE TSS | AD169 | ABI | Fwd, CCGGTGTCTTCTATGGAGGT |
| Rev, GCAGAGCTCGTTTAGTGAACC | |||
| Probe, GCGTCTCCAAGGCGATCTGAC | |||
| MIE TSS | TB40E | IDT | Fwd, ACGCTGTTTTGACCTCCATAG |
| Rev, GCGGTACTTACGTCACTCTTG | |||
| Probe, CGGTGCATTGGAACGCGGATT | |||
| MIE enhancer | AD169 | ABI | Fwd, CAAGTGGGCAGTTTACCGT |
| Rev, TGTTCCCATAGTAACGCCAA | |||
| Probe, ACTCCACCATTGACGTCAA | |||
| MIE enhancer | TB40E | IDT | Fwd, TTGGGCATACGCGATATCTG |
| Rev, GCCTCATATCGTCTGTCACC | |||
| Probe, ACTTTGGCGACTTGGGCGATTC | |||
| MIE exon 1 | AD169 | ABI | Fwd, AGACCCATGGAAAGGAACAG |
| Rev, GGTGGAGGGCAGTGTAGTCT | |||
| Probe, CGGCAGCAACGAGTACTGCT | |||
| MIE exon 4 | TB40E | IDT | Fwd, CCCTTCACGATTCCCAGTATG |
| Rev, CACCTCACTCTTCACCTCATG | |||
| Probe, TGGCCTCATCTAACACCTGGTCC | |||
| UL69 | AD169/TB40E | IDT | Fwd, TCGGTGGGATGAATTTGGTC |
| Rev, CATGATAGCGTACTGTCCCTTC | |||
| Probe, CGTGGTTGCTGGCGGTTGTCG | |||
| β2-Microglobulin | NA | IDT | Fwd, TGGATGACGTGAGTAAACCTG |
| Rev, GGCATTCCTGAAGCTGACAG | |||
| Probe, CTAAGGCCACGGAGCGAGACATC | |||
| HoxA9 | NA | ABI | Fwd, ATTGTTTCAGAAGCCACACAGGCTG |
| Rev, GGGAGCTCGCCAACCAAACACA | |||
| Probe, TCGCCGTGGCTCCCAGCT | |||
| PCLB4 | NA | IDT | Fwd, ATAAACCCATACAGCGCCTAC |
| Rev, ACTTAATCTCTGTGGCTCTGTG | |||
| Probe, ACTGCCTCTTTTTATTTCGCGGGTCT | |||
| MYT1 | NA | IDT | Fwd, TCACCCCAAAGGCAAGTATC |
| Rev, ATCCTCAGTTTGACCAGTGC | |||
| Probe, TGAGCACAAGGACATAAATGCAAGCC |
NA, not applicable.
Fwd, forward primer; Rev, reverse primer.
Primers.
qPCR reagents and TaqMan probes were purchased from either Applied Biosystems (ABI) or Integrated DNA Technologies (IDT). TaqMan assays were designed using MacVector or Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). The primer and probe sequences used in our studies are listed in Table 1.
SDS-PAGE.
Mock-infected and HCMV-infected cells were collected by scraping, Accutase treatment, or centrifugation. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitor cocktail (Thermo Scientific). The cell lysate was briefly sonicated to facilitate nuclear protein release, and insoluble debris was pelleted by centrifugation. The soluble lysate was assayed for protein content by using the Bradford reagent (Bio-Rad). Twenty-five to 50 μg of protein from each sample was separated by SDS-PAGE on a 10 or 12% gel.
Western blot analysis.
Following transfer of SDS-PAGE-separated proteins to nitrocellulose membranes, blots were blocked with 5% dry milk in PBS-Tween (PBST) or Tris-buffered saline–Tween (TBST), depending on manufacturer recommendations for each antibody, for 1 to 2 h at room temperature. Blots were incubated with a primary antibody diluted in blocking buffer overnight at 4°C. After washing, blots were incubated with a secondary antibody diluted 0.1% in dry milk in PBST or TBST for 1 h at room temperature. Chemiluminescent detection was performed using the SuperSignal West Pico chemiluminescent substrate (Thermo Scientific).
Immunofluorescence.
Immunofluorescence assays (IFAs) were performed as described previously (36). Briefly, NT2D1 cells were plated on culture slides (BD Falcon). At the time of the assay, mock-infected and HCMV-infected cells were washed with PBS and were then fixed with 4% paraformaldehyde. Free aldehyde groups were then quenched using 0.1 M glycine. Cells were permeabilized using cold permeabilization buffer for 10 min. Cells were then washed 3 times, for 5 min each time, in cold PBST, followed by incubation with blocking buffer. The primary antibody was diluted in blocking buffer and was incubated with the cells in a humid chamber for 1 to 2 h at 37°C. Cells were then washed in cold PBST. Secondary antibody incubations and washes were performed as described for the primary antibody. Cells were then rinsed in PBS and were mounted using SlowFade reagent (Invitrogen) containing 4′,6-diamidino-2-phenylindole (DAPI).
Image acquisition.
Images were acquired with a Nikon TE2000-U inverted microscope. A UPLAN Fluor 10× air objective was used. The excitation source was a 100-W Hg lamp. Image collection was completed with a CoolSNAP HQ2 camera. For the DAPI filter set, the excitation wavelength was 360 to 370 nm and the emission wavelength was 420 to 460 nm; for the fluorescein isothiocyanate (FITC) filter set, the excitation wavelength was 450 to 480 nm and the emission wavelength was 535 nm. MetaMorph was used as acquisition software. Immunofluorescence data were collected from several fields of view across two independent experiments. The images collected were processed using ImageJ software (NIH).
ChIP.
ChIPs were performed essentially as described previously (36). Briefly, ∼1 × 107 mock-infected or HCMV-infected cells were fixed with 1% formaldehyde–PBS. Fixation was terminated by adding glycine-PBS and incubating for 5 min at room temperature with rocking. Cells were collected by centrifugation. The cell pellet was resuspended and was washed twice with ice-cold PBS containing protease inhibitor cocktail (Roche). Cells were lysed in immunoprecipitation (IP) lysis buffer for 10 min and the nuclei collected by centrifugation. Nuclei were resuspended in nuclear lysis buffer and were mechanically disrupted with several passages through a 22G1 syringe needle. Chromatin was sonicated to fragments of 200 to 1,000 bp by using a Bioruptor instrument (Diagenode). Insoluble aggregates were cleared by centrifugation. For each IP, ∼2 × 106 cell equivalents of cleared chromatin were diluted 1:10 in ChIP dilution buffer containing protease inhibitor cocktail supplemented with phenylmethylsulfonyl fluoride (PMSF). An antibody was added to each IP mixture, which was then incubated at 4°C overnight with rotation. The next day, 20 μl magnetic protein A beads was added to each IP mixture, which was then incubated at 4°C for 2 h with rotation. After washing, beads were resuspended in 100 μl of 10% Chelex 100 resin (Bio-Rad) and 40 μg proteinase K. After brief vortexing, samples were incubated at 57°C for 45 min on a Thermomixer (Eppendorf) with shaking. Samples were then incubated at 97°C for 30 min on a Thermomixer with shaking, after which they were cooled on ice and collected by centrifugation. The supernatant was collected and was assayed by qPCR. Aliquots (1%) of the input chromatin DNA and of the chromatin-immunoprecipitated DNA samples were assayed by qPCR in triplicate using a Roche LightCycler 480 system.
Flow cytometry.
THP1 cells were harvested 4 days after centrifugation or Accutase treatment. Cells were washed once in PBS and were incubated in 50 μl FC buffer (PBS containing 400 μM EDTA, 1% FBS, and 1% anti-Fc antibody 24G2) containing PE-conjugated anti-CD11b/MAC1 or a PE-conjugated isotype control antibody for 30 min at 4°C. Cells were washed twice in 1 ml FC buffer, and PE signals from at least 20,000 live cells were collected using a FACScalibur flow cytometer (BD Bioscience). Data were analyzed using FlowJo software, version 9.
Cell viability assay.
Four days posttreatment, THP1 and NT2D1 cells were incubated in PBS containing 15 μg/ml fluorescein diacetate (FDA) or 2 μg/ml propidium iodide (PI) for 3 min. Cells were washed once in PBS. The intracellular fluorescent cleaved product was imaged by fluorescence microscopy as described above. (Living cells actively convert the nonfluorescent FDA into the green fluorescent compound fluorescein, a positive indicator of cell health.)
RESULTS
DZnep treatment renders cells permissive for viral gene expression upon HCMV infection in experimental models of HCMV quiescence.
To assess the role of PRC2 in restricting lytic transcriptional activity in nonpermissive cells, we inhibited PRC2 activity using DZnep, a small-molecule inhibitor of EZH2 stability and catalytic activity (42). We tested the effects of DZnep in the THP1 and NT2D1 models of HCMV quiescence, since both have been successfully employed in previous studies to describe latency and reactivation determinants (38, 39, 41, 43, 44). Also, these two models represent HCMV infection of two very different cell types, allowing us to ascertain whether or not our results were cell type specific. In THP1 cells, treatment with 5 μM DZnep dramatically reduced H3K27me3 levels by 3 days posttreatment and also reduced EZH2 protein levels, as reported previously (Fig. 1A and B) (42, 45). H3K36me3 and HP1 protein levels were unaffected by DZnep, demonstrating some specificity in inhibiting HMT activity and nuclear protein stability. While we observed that DZnep treatment modestly slowed the growth of THP1 monocytes, we detected no loss of viability in response to DZnep treatment by FDA or PI staining (Fig. 1C). Additionally, we observed no evidence of DZnep-induced monocyte-to-macrophage differentiation as determined by direct observation of cells and quantification of the expression of CD11b/MAC-1, a surface marker highly expressed on macrophages (Fig. 1D and E). However, as expected, PMA treatment led to distinct cell-morphological changes consistent with a macrophage phenotype, high CD11b/MAC-1 surface expression, and no loss in cell viability.
Fig 1.

DZnep treatment inhibits PRC2 catalytic activity and decreases EZH2 protein levels in THP1 monocytes. Whole-cell lysates were prepared from THP1 monocytes treated with 5 μM DZnep, 0.2 μM PMA, or DMSO for 3 days. (A) Western blot analysis of H3K27me3 and H3K36me3. Total H3 served as a loading control. (B) Western blot analysis of PRC2 components EZH2 and SUZ12 and of the chromatin binding protein HP1. Actin served as a loading control. (C) Fluorescence microscopy of THP1 cells, treated as indicated, following FDA or PI staining for viability. (D) Bright-field microscopy of morphological changes of THP1 cells, treated as indicated. (E) CD11b/MAC-1 expression on the surfaces of THP1 cells, treated as indicated.
THP1 monocytes were then treated with DMSO or DZnep 72 h prior to infection. Cells were then infected with HCMV at a multiplicity of infection (MOI) of 0.02 for AD169 or 0.2 for TB40E (the treatment and infection protocol is diagramed in Fig. 2A). A lower MOI was used for AD169 than for TB40E in order to keep basal lytic transcription to a minimum. Laboratory-adapted strains lack the “latency-promoting activity” of the ULb′ region and thus have a diminished ability to establish and maintain latency (46). At 1.5 h postinfection (hpi), the inoculum was replaced with fresh medium containing either DMSO or DZnep, and cells were collected at various times postinfection in order to quantify viral gene expression. Quantification of viral DNA in infected cells revealed that DZnep only modestly decreased HCMV entry from that for vehicle-treated THP1 monocytes (Fig. 2B). For RNA analysis, we measured the abundances of IE1/2, UL54, and UL99 transcripts, as representatives of the IE, E, and L phases of the lytic replication cycle. All three genes are required for the successful completion of the lytic replication cycle and are highly transcribed during lytic infection. In cells pretreated with DZnep, we detected significant activation of the lytic transcription program for both the laboratory-adapted strain AD169 (Fig. 2C) and the clinical isolate TB40E (Fig. 2D). In cells pretreated with DZnep and infected with AD169, we detected a 100-fold increase in IE1/2 transcript abundance over that in DMSO-treated monocytes at 3 hpi, and the level continued to rise to a peak of ∼800-fold at 12 hpi. UL54 transcript levels peaked at ∼400-fold at 12 hpi. Finally, statistically significant increases in UL99 expression were not observed until 24 hpi, when transcript abundance was ∼50-fold higher that that in DMSO-treated monocytes. Furthermore, at 24 hpi, we detected a DZnep-dependent increase in AD169 IE1 protein levels over those in DMSO-treated monocytes (Fig. 2E). Similarly, the TB40E lytic transcription program was significantly activated in DZnep-treated THP1 monocytes, as evidenced by the 22-, 10-, and 9-fold increases in the abundances of IE1/2, UL54, and UL99 transcripts, respectively, over those in DMSO-treated monocytes. These increases in viral transcript levels were not as robust as those observed for AD169, which may be due to differences in transcriptional kinetics between the two strains or in the MOIs used, leading to increased basal transcription within the DMSO-treated monocyte population. As an additional comparison for lytic gene transcription, we also infected PMA-stimulated THP1 cells with AD169 at an MOI of 0.02. PMA treatment results in the differentiation of THP1 cells into adherent macrophages, and while a quiescent, latent-like infection predominates in the undifferentiated THP1 monocytes, PMA-induced THP1 macrophages are fully permissive for infection and execute the full lytic transcriptional cascade, DNA replication, and, for some virus strains, virion production (47). PMA treatment had little effect on HCMV infectivity in THP1 cells (Fig. 2B). As expected, PMA-treated THP1 cells supported robust activation of the lytic transcriptional program (Fig. 2C) and IE1 protein production (Fig. 2E).
Fig 2.

DZnep treatment primes nonpermissive THP1 cells for HCMV AD169 lytic mRNA expression. (A) Schematic diagram of the treatment and infection protocol. Cells were infected with AD169 at an MOI of 0.02. d, day. (B) Quantification of viral copy numbers in infected treated cells. Total DNA was prepared at 2 hpi, and relative viral copy numbers were quantified by qPCR. The relative genome copy number is reported as the mean fold change from the copy number in DMSO-treated cells for three biological replicates. Error bars, standard errors of the means. (C) Gene expression analysis in HCMV AD169-infected THP1 cells pretreated with DMSO, DZnep, or PMA. The expression levels of the major immediate early genes IE1/2, the early gene UL54, and the late gene UL99 were measured by qRT-PCR at the indicated times postinfection and are represented as the mean fold changes from the expression levels in DMSO-treated cells. Data are averages for three biological replicates, and error bars reflect the standard errors of the means of the normalized expression levels. Asterisks indicate P values of <0.05 (*), <0.01 (**), <0.001 (***), or <0.0001 (****) by two-way ANOVA followed by Dunnett's multiple-comparison posttest. (D) THP1 cells were infected with TB40E at an MOI of 0.2 as outlined in panel A. At 24 hpi, the expression of IE, E, and L loci, as indicated, in HCMV TB40E-infected THP1 cells pretreated with DMSO or DZnep was analyzed. Data are averages for three biological replicates, and error bars reflect the standard errors of the means of the normalized expression levels. Asterisks indicate P values of <0.05 (*), <0.01 (**), or <0.001 (***) by paired, two-tailed t tests. (E) Western blot analysis of AD169 IE1 protein accumulation in DZnep-treated THP1 cells. THP1 cells were treated as described in the legend to Fig. 1 and were infected with AD169 at an MOI of 1.0. Whole-cell lysates were prepared at 24 hpi, and IE1 protein expression was detected by Western blotting. HP1 served as a loading control.
To confirm our results, we carried out a similar experimental strategy in a second model of HCMV quiescence, NT2D1 cells. As with THP1 cells, we measured reduced levels of H3K27me3 and EZH2 protein in DZnep-treated NT2D1 cells (data not shown). DZnep treatment had no effect on NT2D1 viability or differentiation as determined by FDA or PI staining and by OCT4 expression, respectively (Fig. 3A and B). We did detect a reduction in the level of HCMV entry into DZnep-treated cells from that for vehicle-treated cells (Fig. 3C). This may be a result of alteration of the cell surface protein repertoire by DZnep. NT2D1 cells were infected with AD169 or TB40E at an MOI of 1.0, and viral transcript abundance was quantified at 24 hpi (the treatment and infection protocol is diagramed in Fig. 3D). In contrast to DMSO pretreatment, DZnep pretreatment resulted in robust activation of the lytic transcription program upon infection for both strains of HCMV (Fig. 3E). Additionally, IE1 protein production in DZnep-treated NT2D1 cells increased as measured by quantification of IE1-positive cells by immunofluorescence (Fig. 3F and G). Together, these initial experiments demonstrate that DZnep, a potent inhibitor of PRC2 activity, primes normally nonpermissive cells for robust activation of the HCMV lytic transcription program. Furthermore, our data suggest that the ability of PRC2 to immediately repress lytic transcription from incoming HCMV templates represents a key step in the establishment of a latency-like state in these experimental models.
Fig 3.

DZnep treatment primes pluripotent NT2D1 cells for HCMV AD169 lytic gene expression. (A) Fluorescence microscopy of NT2D1 cells, treated as indicated, following FDA or PI staining for viability. (B) Differentiation analysis. (C) Quantification of viral copy numbers in infected treated cells. Total DNA was prepared at 2 hpi, and the relative viral copy number was quantified by qPCR. The relative genome copy number is reported as the mean fold change in the copy number from that in DMSO-treated cells for three biological replicates. Error bars, standard errors of the means. (D) Schematic diagram of the treatment and infection protocol. Cells were infected at an MOI of 1.0. (E) At 24 hpi, gene expression in cells pretreated with DMSO or DZnep and infected with AD169 or TB40 was analyzed as described in the legend to Fig. 2. Asterisks indicate P values of <0.05 (*) or <0.01 (**) by paired, two-tailed t tests. (F) IE1 protein accumulation in DZnep-treated NT2D1 cells. The expression of IE1 protein in NT2D1 cells infected with AD169 or TB40E at an MOI of 1.0 was analyzed by immunofluorescence at 48 hpi. (G) Quantification of IE1-positive cells from the immunofluorescence assay for which results are shown in panel F. Bars represent the mean percentages of cells staining positive for IE1 protein within a 10× field of magnification. Results are from four random fields of view. Error bars, standard errors of the means.
DZnep treatment is sufficient to activate lytic gene expression following the establishment of a quiescent HCMV infection in THP1 and NT2D1 cells.
Since DZnep prevented HCMV from effectively establishing a quiescent infection in both THP1 and NT2D1 cells, we asked whether DZnep could disrupt preexisting repression of HCMV lytic transcripts in nonpermissive infected cells. To address this question, we infected THP1 monocytes with AD169 at an MOI of 1.0 and allowed the virus to establish a quiescent infection. At 5 days postinfection (dpi), cells were resuspended in a medium containing either DMSO, DZnep, or reduced levels of serum. At 72 h posttreatment, cells were collected for gene expression analysis (the treatment and infection protocol is diagramed in Fig. 4A). DZnep treatment of HCMV-infected THP1 cells resulted in significant increases in the levels of all lytic transcripts assayed over those obtained with DMSO treatment (Fig. 4B). Importantly, serum starvation of THP1 cells, which we used as a control for DZnep-induced changes in cell proliferation, did not result in increases in viral transcript levels, suggesting that potential viral genome dilution in normally proliferating control cells does not contribute to DZnep-induced viral transcript abundances.
Fig 4.

(A and B) DZnep treatment activates the AD169 lytic transcription program in quiescently infected THP1 monocytes. (A) Schematic diagram of the treatment and infection protocol. Cells were infected with AD169 at an MOI of 1.0. (B) HCMV lytic mRNA expression. qPCR was performed, and data were analyzed and graphed, as described in the legend to Fig. 2. Asterisks indicate P values of <0.05 (*) or <0.01 (**) by paired, two-tailed t tests. (C and D) DZnep treatment reactivates the HCMV lytic transcription program in pluripotent NT2D1 cells. (C) Schematic diagram of the treatment and infection protocol. Cells were infected with AD169 or TB40E at an MOI of 1.0. (D) AD169 or TB40E lytic gene mRNA expression. qPCR was performed, and data were analyzed and graphed, as described for Fig. 2. Asterisks indicate P values of <0.05 (*), <0.01 (**), or <0.001 (***) by paired, two-tailed t tests.
To confirm our results, we carried out a similar experimental strategy with NT2D1 cells (the treatment and infection protocol is diagramed in Fig. 4C). At 48 h posttreatment, cells were collected for gene expression analysis. The results were similar to those we obtained with THP1 monocytes; DZnep treatment of HCMV-infected NT2D1 cells resulted in significant increases in the levels of all lytic transcripts assayed (Fig. 4D). Collectively, these experiments demonstrate that DZnep treatment is sufficient to activate the lytic transcriptional program following the establishment of a quiescent infection in nonpermissive cells. These experiments also suggest that PRC2 activity is necessary to maintain the repression of lytic transcription, indicating that HCMV transcription may be poised for activation with changes in PRC2 activity on chromatin.
PRC2-catalyzed H3K27me3 enrichment on the HCMV genome correlates inversely with lytic transcription.
To relate lytic transcript abundance with PRC2 activity at HCMV lytic genes, we measured H3K27me3 enrichment on HCMV chromatin by chromatin immunoprecipitation (ChIP) in THP1 monocytes. We assayed H3K27me3 enrichment at several loci within the AD169 MIEEP, the UL69 locus, and the cellular MYT1 and PLCB4 promoters (Fig. 5). THP1 monocytes were treated with DMSO or 5 μM DZnep 72 h prior to infection. Cells were then infected with AD169 at an MOI of 1.0. At 48 hpi, cells were harvested for ChIP analysis using H3K27me3- and pan-H3-specific antibodies. In DMSO-treated THP1 monocytes, all HCMV loci assayed demonstrated 4- to 7-fold enrichment of H3K27me3 over the level for the negative-control region, PLCB4. DZnep treatment of THP1 cells reduced the level of H3K27me3 enrichment across all viral loci assayed. We also observed that H3K27me3 enrichment was dramatically reduced at all viral loci assayed in permissive THP1 macrophages (differentiated with PMA), nearing the level of enrichment observed at the PLCB4 locus. These results demonstrate that DZnep treatment of THP1 cells inhibits the PRC2-related formation of facultative HC on newly assembled viral chromatin, permitting lytic transcription. These data also highlight a sharp contrast in the PRC2-related viral chromatin landscape between permissive and nonpermissive infections.
Fig 5.

PRC2 activity correlates inversely with AD169 lytic transcriptional levels in THP1 monocytes. (A) Schematic diagram of the MIE and UL69 loci of AD169. Open boxes represent regions probed for H3K27me3 enrichment: Enh., the enhancer region; TSS, the transcriptional start site of the IE1/2 transcriptional unit; Exon, exon 2 within the open reading frame of the IE1/2 locus. Bent arrows indicate +1 transcriptional start sites. The distal enhancer region is indicated. Shaded arrows represent exons. (B) THP1 monocytes were treated as described in the legend to Fig. 1 and were infected with AD169 at an MOI of 1. At 48 hpi, ChIP was performed using anti-H3K27me3, anti-pan-H3, and normal rabbit IgG. The material recovered was analyzed by qPCR using primer-probe sets specific for the indicated loci. Each bar represents the IgG-normalized H3K27me3/H3 ratio for each viral locus relative to that for the negative-control region, PLCB4.
We also used the NT2D1 model to test whether PRC2 targets HCMV chromatin for modification. We quantified H3K27me3 enrichment at both viral and cellular loci by ChIP with cells infected with AD169 or TB40E at an MOI of 1.0 (Fig. 6). We observed significant enrichment of H3K27me3 for all viral loci over the level for the negative-control region, PLCB4, suggesting that in NT2D1 cells, incoming viral genomes are rapidly targeted by PRC2 for facultative HC formation, suppressing lytic transcription. Together, the data from the ChIP experiments further strengthen the argument that PRC2 directly represses viral transcription by modifying genome-associated histones and altering viral chromatin structure.
Fig 6.
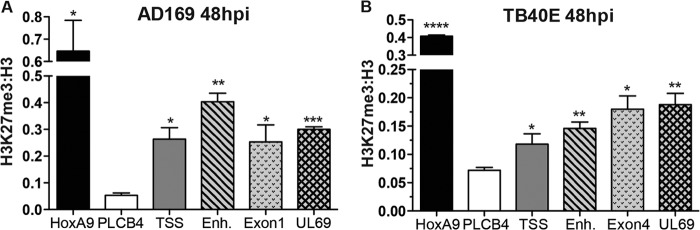
PRC2 rapidly targets HCMV chromatin for H3K27me3 during infection of pluripotent NT2D1 cells. NT2D1 cells were infected with AD169 (A) or TB40E (B) at an MOI of 1.0. At 48 hpi, ChIP was performed using anti-H3K27me3, anti-pan-H3, and normal rabbit IgG. The material recovered was analyzed by qPCR using primer-probe sets specific for the indicated loci (abbreviations explained in the legend to Fig. 5A). Each bar represents the IgG-normalized H3K27me3/H3 ratio for the viral locus queried, the negative-control region, PLCB4, or the positive-control region, HOXA9. Ratios are means of data collected from at least three biological replicates; error bars represent standard errors of the means. Asterisks indicate P values of <0.05 (*), <0.01 (**), <0.001 (***), or <0.0001 (****) by paired, two-tailed t tests.
GSK343 inhibition of EZH2 activates lytic gene expression in quiescently infected THP1 monocytes.
There is widespread interest in developing cancer therapies targeted to epigenetic regulators. Recently, several highly specific and effective inhibitors of PRC2 have been synthesized and validated in a variety of cell types (48–52). These chemical probes outcompete S-adenosyl methionine (SAM) for cofactor binding of EZH2 and globally reduce H3K27me3 levels by 3 days posttreatment. We tested whether one of these new and highly specific probes, GSK343, could potentiate the HCMV lytic transcriptional program in THP1 monocytes. We infected monocytes with AD169 at an MOI of 1.0 and allowed viral transcription to become silenced over a 5-day period, after which we administered GSK343 or DMSO for 3 days. At 3 days posttreatment, we performed gene expression analysis. As shown in Fig. 7, inhibition of PRC2 by GSK343 resulted in significant increases in the levels of all lytic transcripts assayed, with no effect on THP1 cell viability or differentiation (data not shown). Remarkably, GSK343 treatment stimulated HCMV lytic RNA expression to higher levels than DZnep treatment, demonstrating that highly specific and direct inhibition of PRC2 activity is sufficient to activate the HCMV lytic transcriptional program in the context of a quiescent infection. To confirm that GSK343 activated the lytic transcription program through a reduction in PRC2 activity, we performed ChIP on THP1 monocytes 5 dpi, prior to GSK343 treatment, and 8 dpi, 3 days into GSK343 treatment. Treatment with GSK343 for 3 days dramatically reduced H3K27me3 levels at the MYT1 promoter from those prior to treatment, and these reduced levels approximated those at the negative-control region, PLCB4. This result strongly suggests that GSK343 activates lytic gene transcription through specific inhibition of PRC2 activity, and it further suggests a direct role for PRC2 in repressing HCMV lytic gene expression throughout quiescent infection.
Fig 7.
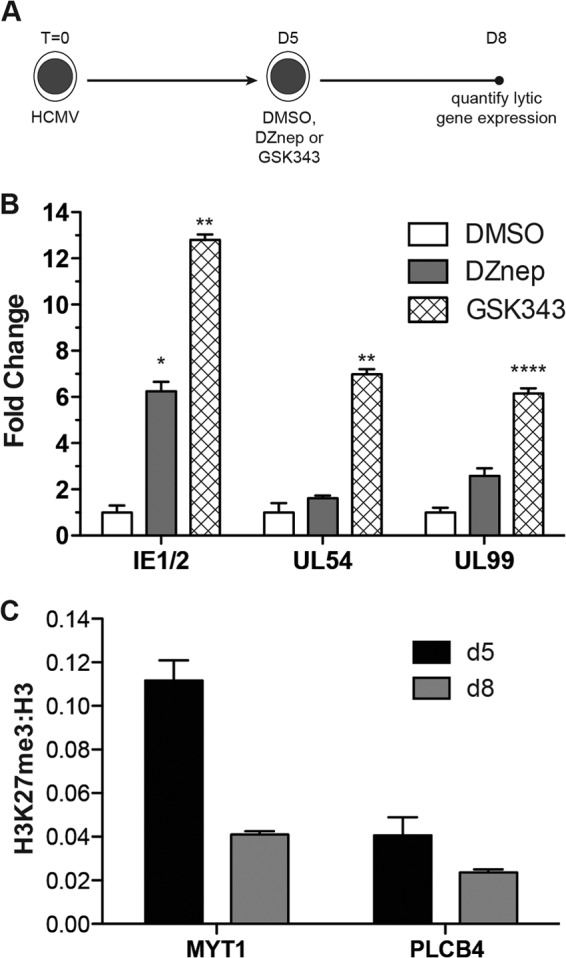
Treatment with the EZH2-specific inhibitor GSK343 reactivates the AD169 lytic transcription program in THP1 monocytes. (A) Schematic diagram of the treatment and infection protocol. Cells were infected with AD169 or TB40E at an MOI of 1.0; infected cells were treated daily with either DMSO, 5 μM DZnep, or 10 μM GSK343. (B) HCMV lytic mRNA expression. qPCR was performed, and data were analyzed and presented, as described above. Significance was calculated using paired, two-tailed t tests. Asterisks indicate P values of <0.05 (*), <0.01 (**), or <0.0001 (****). (C) ChIPs were performed using anti-H3K27me3, anti-pan-H3, and normal rabbit IgG. The material recovered was analyzed by qPCR using primer-probe sets specific for the indicated cellular loci, before and after GSK343 treatment. Each bar represents the IgG-normalized H3K27me3/H3 ratio for the locus indicated.
DISCUSSION
Upon entry into the host nucleus, CMV DNA is recognized and converted into a chromatin template that acts as a canvas for a variety of chromatin writers, readers, and erasers to act upon. This process is very rapid, and ultimately, the initial formation of heterochromatin at critical lytic promoters renders incoming viral genomes poor templates for productive replication. In permissive cells, this transcriptionally repressive chromatin state is short-lived, and through the actions of viral tegument and cellular transcription networks, viral chromatin is converted into a robust, temporally regulated transcriptional unit. However, during the infection of nonpermissive cells, this conversion fails to occur; instead, transcriptionally refractory heterochromatin is maintained at lytic genes throughout the latency period. HCMV survival in the host requires continual suppression of lytic gene expression throughout the latency period. Inappropriate viral antigen production could lead to the identification of infected cells through the adaptive immune response, resulting in the destruction of latency reservoirs. Additionally, timely reentry into lytic replication is essential for survival within the host and transmission. Therefore, identifying and understanding the chromatin-based regulatory mechanisms that control infection outcomes could lead to the development of novel antiviral therapies.
In this report, we directly tested whether PRC2 enzymatic activity represses the HCMV lytic transcriptional program in two well-defined, tractable models of HCMV quiescence. We discovered that direct inhibition of PRC2 HMT activity prior to infection primes normally nonpermissive cells for lytic transcription upon HCMV infection (Fig. 2 and 3). Interestingly, while DZnep treatment of THP1 cells significantly increased lytic transcript abundance over that with DMSO treatment, it did not drive viral transcription to the same levels observed with PMA treatment. This may indicate a requirement for specific trans-activators or signaling events induced by PMA treatment to enhance transcriptional rates. Alternatively, DZnep may inhibit additional methyltransferases responsible for transcriptional activation and/or mRNA stability, leading to decreased transcriptional rates and increased mRNA turnover. Of note, a significant increase in UL99 gene expression was observed in THP1-derived macrophages by 12 hpi (Fig. 2C). While this was unexpected, our observation may be explained, in part, by our qPCR normalization process. In DMSO-treated THP1 cells, we measured extremely low levels of UL99 RNA; therefore, the production of even a few UL99 transcripts within a subset of infected macrophages could magnify the differences in transcript abundance. Additionally, it is possible that the mRNA half-life of UL99 transcripts is longer in THP1-derived macrophages than in monocytes, so that the few transcripts produced are more readily detected. Nonetheless, our data directly implicate PRC2 as a chromatin-modifying complex essential in the initial pre-immediate early gene repression observed during CMV infection. Other studies have demonstrated that during HCMV infection of latent cell types, critical tegument-associated transactivators fail to reach the nuclear compartment (53, 54). During HCMV infection of permissive cells, these viral transactivators play central roles in disrupting the heterochromatic environment at the MIEEP, eventually leading to robust IE1/2 transcription (55). Identification of HCMV proteins that disrupt PRC2 activity will be important for a full understanding of PRC2-mediated regulation of HCMV gene expression and infection outcomes.
In this study, we also demonstrated that specific inhibition of PRC2 histone methyltransferase activity in cells in which a “latent-like” infection was established results in reanimation of the lytic transcriptional cascade (Fig. 4). Our observations held true for both laboratory-adapted strains and clinical isolates of HCMV, indicating that PRC2 targeting of incoming HCMV genomes is conserved. However, the degree to which DZnep was able to induce lytic gene expression differed between the THP1 and NT2D1 models. These differences might be explained by differences in sensitivities to DZnep, in the enrichment of PRC2-mediated heterochromatin formation at lytic promoters, or in the availability of transcriptional activators to fully drive lytic gene expression with the relief of PRC2-mediated repression. Importantly, we corroborated our DZnep findings in the THP1 model by using the recently developed, highly specific EZH2 inhibitor GSK343 (Fig. 7). In fact, GSK343 was more potent than DZnep in the reanimation of lytic gene expression. This is likely due to the high specificity of GSK343 for EZH2, which results in minimal potential for interference with off-target histone methyltransferase complexes that could be responsible for activating gene expression. The rational design and synthesis of inhibitors of chromatin-based gene regulation is progressing rapidly through computer-based protein-modeling systems and high-throughput cell-based screens. Many of these compounds show good specificity, are widely available, and, like GSK343, could prove valuable for dissecting the chromatin-based regulation of herpesviral biology. It should be noted that the development of many of these lead compounds is driven by the need for new anticancer therapies, and it will be important to weigh the impact of these drugs on host-pathogen interactions, such as those in latent herpesvirus infection. One can imagine that reactivation of HCMV through the administration of an EZH2 inhibitor designed to treat leukemia or lymphoma could have a significant health impact on the host. Therefore, aside from the use of these inhibitors as tools to elucidate chromatin-based events during infection, efforts should be made to document their potential collateral effects on HCMV reactivation.
Using ChIP, we measured significant differences in PRC2-related heterochromatin enrichment at several HCMV loci during a latent-like infection (Fig. 5 and 6). Our results are consistent with similar observations recently made with latently infected primary CD14+ monocytes (56). Importantly, our data highlight the dramatic differences in H3K27me3 enrichment between model latent, lytic, and PRC2-inhibited infections, thus providing the first specific link between PRC2-mediated heterochromatin formation and the repression of viral gene expression in nonpermissive cells. Our data also demonstrate that incoming genomes are rapidly targeted for H3K27me3, by 48 hpi, in both the THP1 and NT2D1 models. How is PRC2 targeted to HCMV chromatin? PRC2 recruitment to cellular loci is known to be mediated by several cellular proteins, including JARID2, PCL1 to -3, and RbAp46/48 (26, 57–61). Cellular long noncoding RNAs (lncRNAs) have also been implicated in the recruitment of PRC2 to heterochromatic loci. While virus-encoded lncRNAs have recently been implicated in the recruitment of PRC2 to the MIEEP, it has not been established if this involves a direct effect of viral RNAs (56, 62). lncRNAs may cooperate with PRC2 accessory factors in establishing heterochromatin on the viral genome during the establishment and maintenance of latency, and further detailed analysis of the pathways and temporal regulation of transcription are required to address this issue.
To date, it is still unclear how PRC2-mediated heterochromatin represses transcription. Initial studies have suggested a mechanism whereby the H3K27me3 modification serves as a mark that is recognized by the CBX subunit of Polycomb repressive complex 1 (PRC1), thereby stimulating PRC1-mediated ubiquitylation of H2A on lysine 119 (H2AK119ub) (63–66). The resulting chromatin structure was refractory to SWI/SNF chromatin remodelers and interfered with transcriptional preinitiation complex formation (63–66). For HCMV, neither PRC1 nor H2AK119ub has been identified at lytic promoters; however, one of the core PRC1 components, BMI1, was demonstrated to interact with herpes simplex virus 1 (HSV-1) chromatin in latently infected mice (13). Interestingly, recent reports using high-throughput sequencing technologies have found transcriptionally repressed regions of the human genome that are PRC2/H3K27me3 enriched but not PRC1 enriched, suggesting at least that one alternative mechanism for H3K27me3-mediated transcriptional repression exists (67). Studies have demonstrated that PRC2-repressed genes often coexist with the euchromatin-associated H3K4me3 modification (68–70). Promoters targeted for these bivalent chromatin structures often have paused RNA polymerase II (Pol II) bound to the transcriptional start site, suggesting that PRC2 and H3K27me3 inhibit productive elongation (71). While we did not address bivalency and Pol II occupancy in this report, it will be important for future studies to identify these bivalent, Pol II-bound chromatin domains on HCMV genomes, since their unique transcriptional responsiveness may have dramatic implications for reactivation from latency.
Polycomb regulation of cellular gene expression is fundamental to cell lineage specification and development. Plastic by nature, PRC2-repressed cellular genes can switch to transcriptionally permissive templates in a precisely coordinated fashion as dictated by specific signaling cascades. Similarly, we have shown that during latent-like infection, HCMV genomes remain transcriptionally quiescent at lytic genes in a PRC2-dependent manner. These observations support the idea that HCMV genomes are uniquely in tune with cellular signaling that affects PRC2 activity and might utilize H3K27me3 enrichment as a rheostat that regulates the maintenance of latency or reactivation. It is likely that other chromatin-modifying complexes, with both activating and repressive functions, also control HCMV outcomes. Future large-scale screens should be useful in identifying, globally, the chromatin-based circuitry that determines HCMV infection outcomes.
ACKNOWLEDGMENTS
This work was supported by a research grant from the Cancer League of Colorado and Beginning-Grant-in-Aid 13GRNT14770053 from the American Heart Association.
We thank Courtney Fleenor for assistance with the flow cytometry analysis.
Footnotes
Published ahead of print 25 September 2013
REFERENCES
- 1.Mocarski ES, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2701–2772 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Hahn G, Jores R, Mocarski ES. 1998. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. U. S. A. 95:3937–3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodrum FD, Jordan CT, High K, Shenk T. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc. Natl. Acad. Sci. U. S. A. 99:16255–16260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sinclair J. 2008. Human cytomegalovirus: latency and reactivation in the myeloid lineage. J. Clin. Virol. 41:180–185 [DOI] [PubMed] [Google Scholar]
- 5.Nitzsche A, Paulus C, Nevels M. 2008. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J. Virol. 82:11167–11180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nevels M, Nitzsche A, Paulus C. 2011. How to control an infectious bead string: nucleosome-based regulation and targeting of herpesvirus chromatin. Rev. Med. Virol. 21:154–180 [DOI] [PubMed] [Google Scholar]
- 7.Knipe DM, Lieberman PM, Jung JU, McBride AA, Morris KV, Ott M, Margolis D, Nieto A, Nevels M, Parks RJ, Kristie TM. 2013. Snapshots: chromatin control of viral infection. Virology 435:141–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenuwein T, Allis CD. 2001. Translating the histone code. Science 293:1074–1080 [DOI] [PubMed] [Google Scholar]
- 9.Bannister AJ, Kouzarides T. 2011. Regulation of chromatin by histone modifications. Cell Res. 21:381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li G, Reinberg D. 2011. Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 21:175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nitzsche A, Steinhausser C, Mucke K, Paulus C, Nevels M. 2012. Histone H3 lysine 4 methylation marks postreplicative human cytomegalovirus chromatin. J. Virol. 86:9817–9827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nevels M, Paulus C, Shenk T. 2004. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. U. S. A. 101:17234–17239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwiatkowski DL, Thompson HW, Bloom DC. 2009. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J. Virol. 83:8173–8181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cliffe AR, Coen DM, Knipe DM. 2013. Kinetics of facultative heterochromatin and Polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. mBio 4(1):e0059012. 10.1128/mBio.00590-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toth Z, Maglinte DT, Lee SH, Lee HR, Wong LY, Brulois KF, Lee S, Buckley JD, Laird PW, Marquez VE, Jung JU. 2010. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 6:e1001013. 10.1371/journal.ppat.1001013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderton JA, Bose S, Vockerodt M, Vrzalikova K, Wei W, Kuo M, Helin K, Christensen J, Rowe M, Murray PG, Woodman CB. 2011. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin's lymphoma. Oncogene 30:2037–2043 [DOI] [PubMed] [Google Scholar]
- 17.Murata T, Kondo Y, Sugimoto A, Kawashima D, Saito S, Isomura H, Kanda T, Tsurumi T. 2012. Epigenetic histone modification of Epstein-Barr virus BZLF1 promoter during latency and reactivation in Raji cells. J. Virol. 86:4752–4761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paulus C, Nitzsche A, Nevels M. 2010. Chromatinisation of herpesvirus genomes. Rev. Med. Virol. 20:34–50 [DOI] [PubMed] [Google Scholar]
- 19.Groves IJ, Reeves MB, Sinclair JH. 2009. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic ‘pre-immediate-early' repression of viral gene expression mediated by histone post-translational modification. J. Gen. Virol. 90:2364–2374 [DOI] [PubMed] [Google Scholar]
- 20.Terhune SS, Moorman NJ, Cristea IM, Savaryn JP, Cuevas-Bennett C, Rout MP, Chait BT, Shenk T. 2010. Human cytomegalovirus UL29/28 protein interacts with components of the NuRD complex which promote accumulation of immediate-early RNA. PLoS Pathog. 6:e1000965. 10.1371/journal.ppat.1000965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sinclair J. 2010. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim. Biophys. Acta 1799:286–295 [DOI] [PubMed] [Google Scholar]
- 22.Trojer P, Reinberg D. 2007. Facultative heterochromatin: is there a distinctive molecular signature? Mol. Cell 28:1–13 [DOI] [PubMed] [Google Scholar]
- 23.Kirmizis A, Bartley SM, Kuzmichev A, Margueron R, Reinberg D, Green R, Farnham PJ. 2004. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 18:1592–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. 2002. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 16:2893–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, III, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, Reinberg D, Gamblin SJ. 2009. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461:762–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Musselman CA, Avvakumov N, Watanabe R, Abraham CG, Lalonde M-E, Hong Z, Allen C, Roy S, Nuñez JK, Nickoloff J, Kulesza CA, Yasui A, Côté J, Kutateladze TG. 2012. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat. Struct. Mol. Biol. 19:1266–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Connell S, Wang L, Robert S, Jones CA, Saint R, Jones RS. 2001. Polycomblike PHD fingers mediate conserved interaction with enhancer of zeste protein. J. Biol. Chem. 276:43065–43073 [DOI] [PubMed] [Google Scholar]
- 28.Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, Helin K. 2008. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 10:1291–1300 [DOI] [PubMed] [Google Scholar]
- 29.Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. 2004. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 23:4061–4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sparmann A, van Lohuizen M. 2006. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 6:846–856 [DOI] [PubMed] [Google Scholar]
- 31.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. 2006. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 20:1123–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morey L, Helin K. 2010. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 35:323–332 [DOI] [PubMed] [Google Scholar]
- 33.Breiling A, Turner BM, Bianchi ME, Orlando V. 2001. General transcription factors bind promoters repressed by Polycomb group proteins. Nature 412:651–655 [DOI] [PubMed] [Google Scholar]
- 34.Landeira D, Sauer S, Poot R, Dvorkina M, Mazzarella L, Jorgensen HF, Pereira CF, Leleu M, Piccolo FM, Spivakov M, Brookes E, Pombo A, Fisher C, Skarnes WC, Snoek T, Bezstarosti K, Demmers J, Klose RJ, Casanova M, Tavares L, Brockdorff N, Merkenschlager M, Fisher AG. 2010. Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA polymerase II to developmental regulators. Nat. Cell Biol. 12:618–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life. Nature 469:343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abraham CG, Kulesza CA. 2012. Polycomb repressive complex 2 targets murine cytomegalovirus chromatin for modification and associates with viral replication centers. PLoS One 7:e29410. 10.1371/journal.pone.0029410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu XF, Yan S, Abecassis M, Hummel M. 2008. Establishment of murine cytomegalovirus latency in vivo is associated with changes in histone modifications and recruitment of transcriptional repressors to the major immediate-early promoter. J. Virol. 82:10922–10931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ioudinkova E, Arcangeletti MC, Rynditch A, De Conto F, Motta F, Covan S, Pinardi F, Razin SV, Chezzi C. 2006. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384:120–128 [DOI] [PubMed] [Google Scholar]
- 39.Sinclair JH, Baillie J, Bryant LA, Taylor-Wiedeman JA, Sissons JG. 1992. Repression of human cytomegalovirus major immediate early gene expression in a monocytic cell line. J. Gen. Virol. 73(Part 2):433–435 [DOI] [PubMed] [Google Scholar]
- 40.Gonczol E, Andrews PW, Plotkin SA. 1984. Cytomegalovirus replicates in differentiated but not in undifferentiated human embryonal carcinoma cells. Science 224:159–161 [DOI] [PubMed] [Google Scholar]
- 41.Meier JL. 2001. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 75:1581–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. 2007. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 21:1050–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan J, Liu X, Wu AW, McGonagill PW, Keller MJ, Galle CS, Meier JL. 2009. Breaking human cytomegalovirus major immediate-early gene silence by vasoactive intestinal peptide stimulation of the protein kinase A-CREB-TORC2 signaling cascade in human pluripotent embryonal NTera2 cells. J. Virol. 83:6391–6403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keller MJ, Wu AW, Andrews JI, McGonagill PW, Tibesar EE, Meier JL. 2007. Reversal of human cytomegalovirus major immediate-early enhancer/promoter silencing in quiescently infected cells via the cyclic AMP signaling pathway. J. Virol. 81:6669–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG, Koh TL, Yu Q, Chng WJ. 2011. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood 118:2830–2839 [DOI] [PubMed] [Google Scholar]
- 46.Goodrum F, Reeves M, Sinclair J, High K, Shenk T. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turtinen LW, Seufzer BJ. 1994. Selective permissiveness of TPA differentiated THP-1 myelomonocytic cells for human cytomegalovirus strains AD169 and Towne. Microb. Pathog. 16:373–378 [DOI] [PubMed] [Google Scholar]
- 48.Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, Gu J, Ardayfio O, Zhang JH, Yan X, Fang J, Mi Y, Zhang M, Zhou T, Feng G, Chen Z, Li G, Yang T, Zhao K, Liu X, Yu Z, Lu CX, Atadja P, Li E. 2012. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. U. S. A. 109:21360–21365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, III, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL. 2012. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492:108–112 [DOI] [PubMed] [Google Scholar]
- 50.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, Liu F, Gao C, Huang XP, Kuznetsova E, Rougie M, Jiang A, Pattenden SG, Norris JL, James LI, Roth BL, Brown PJ, Frye SV, Arrowsmith CH, Hahn KM, Wang GG, Vedadi M, Jin J. 2013. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol. 8:1324–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, Olhava EJ, Chesworth R, Moyer MP, Richon VM, Copeland RA, Keilhack H, Pollock RM, Kuntz KW. 2012. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 8:890–896 [DOI] [PubMed] [Google Scholar]
- 52.Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, Graves AP, Scherzer DA, Shu A, Thompson C, Ott HM, Van Aller GS, Machutta CA, Diaz E, Jiang Y, Johnson NW, Knight SD, Kruger RG, McCabe MT, Dhanak D, Tummino PJ, Creasy CL, Miller WH. 2012. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med. Chem. Lett. 3:1091–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Penkert RR, Kalejta RF. 2010. Nuclear localization of tegument-delivered pp71 in human cytomegalovirus-infected cells is facilitated by one or more factors present in terminally differentiated fibroblasts. J. Virol. 84:9853–9863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Penkert RR, Kalejta RF. 2013. Human embryonic stem cell lines model experimental human cytomegalovirus latency. mBio 4(3):e00298–13. 10.1128/mBio.00298-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalejta RF. 2008. Functions of human cytomegalovirus tegument proteins prior to immediate early gene expression. Curr. Top. Microbiol. Immunol. 325:101–115 [DOI] [PubMed] [Google Scholar]
- 56.Rossetto CC, Tarrant-Elorza M, Pari GS. 2013. cis- and trans-acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14+ monocytes and CD34+ cells. PLoS Pathog. 9:e1003366. 10.1371/journal.ppat.1003366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pasini D, Cloos PA, Walfridsson J, Olsson L, Bukowski JP, Johansen JV, Bak M, Tommerup N, Rappsilber J, Helin K. 2010. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 464:306–310 [DOI] [PubMed] [Google Scholar]
- 58.Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. 2010. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 24:368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brien GL, Gambero G, O'Connell DJ, Jerman E, Turner SA, Egan CM, Dunne EJ, Jurgens MC, Wynne K, Piao L, Lohan AJ, Ferguson N, Shi X, Sinha KM, Loftus BJ, Cagney G, Bracken AP. 2012. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat. Struct. Mol. Biol. 19:1273–1281 [DOI] [PubMed] [Google Scholar]
- 60.Nekrasov M, Wild B, Muller J. 2005. Nucleosome binding and histone methyltransferase activity of Drosophila PRC2. EMBO Rep. 6:348–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nowak AJ, Alfieri C, Stirnimann CU, Rybin V, Baudin F, Ly-Hartig N, Lindner D, Muller CW. 2011. Chromatin-modifying complex component Nurf55/p55 associates with histones H3 and H4 and polycomb repressive complex 2 subunit Su(z)12 through partially overlapping binding sites. J. Biol. Chem. 286:23388–23396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spitale RC, Tsai MC, Chang HY. 2011. RNA templating the epigenome: long noncoding RNAs as molecular scaffolds. Epigenetics 6:539–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bernstein E, Duncan EM, Masui O, Gil J, Heard E, Allis CD. 2006. Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol. Cell. Biol. 26:2560–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cao R, Tsukada Y, Zhang Y. 2005. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell 20:845–854 [DOI] [PubMed] [Google Scholar]
- 65.Lehmann L, Ferrari R, Vashisht AA, Wohlschlegel JA, Kurdistani SK, Carey M. 2012. Polycomb repressive complex 1 (PRC1) disassembles RNA polymerase II preinitiation complexes. J. Biol. Chem. 287:35784–35794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang L, Brown JL, Cao R, Zhang Y, Kassis JA, Jones RS. 2004. Hierarchical recruitment of polycomb group silencing complexes. Mol. Cell 14:637–646 [DOI] [PubMed] [Google Scholar]
- 67.Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, Adli M, Kasif S, Ptaszek LM, Cowan CA, Lander ES, Koseki H, Bernstein BE. 2008. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 4:e1000242. 10.1371/journal.pgen.1000242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O'Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Azuara V, Perry P, Sauer S, Spivakov M, Jorgensen HF, John RM, Gouti M, Casanova M, Warnes G, Merkenschlager M, Fisher AG. 2006. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 8:532–538 [DOI] [PubMed] [Google Scholar]
- 70.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326 [DOI] [PubMed] [Google Scholar]
- 71.Brookes E, de Santiago I, Hebenstreit D, Morris KJ, Carroll T, Xie SQ, Stock JK, Heidemann M, Eick D, Nozaki N, Kimura H, Ragoussis J, Teichmann SA, Pombo A. 2012. Polycomb associates genome-wide with a specific RNA polymerase II variant, and regulates metabolic genes in ESCs. Cell Stem Cell 10:157–170 [DOI] [PMC free article] [PubMed] [Google Scholar]