

Abstract
Human immunodeficiency virus type 1 (HIV-1) Vpu enhances the release of viral particles from infected cells by interfering with the function of BST-2/tetherin, a cellular protein inhibiting virus release. The Vpu protein encoded by NL4-3, a widely used HIV-1 laboratory strain, antagonizes human BST-2 but not monkey or murine BST-2, leading to the conclusion that BST-2 antagonism by Vpu is species specific. In contrast, we recently identified several primary Vpu isolates, such as Vpu of HIV-1DH12, capable of antagonizing both human and rhesus BST-2. Here we report that while Vpu interacts with human BST-2 primarily through their respective transmembrane domains, antagonism of rhesus BST-2 by Vpu involved an interaction of their cytoplasmic domains. Importantly, a Vpu mutant carrying two mutations in its transmembrane domain (A14L and W22A), rendering it incompetent for interaction with human BST-2, was able to interact with human BST-2 carrying the rhesus BST-2 cytoplasmic domain and partially neutralized the ability of this BST-2 variant to inhibit viral release. Bimolecular fluorescence complementation analysis to detect Vpu–BST-2 interactions suggested that the physical interaction of Vpu with rhesus or chimpanzee BST-2 involves a 5-residue motif in the cytoplasmic domain of BST-2 previously identified as important for the antagonism of monkey and great ape BST-2 by simian immunodeficiency virus (SIV) Nef. Thus, our study identifies a novel mechanism of antagonism of monkey and great ape BST-2 by Vpu that targets the same motif in BST-2 used by SIV Nef and might explain the expanded host range observed for Vpu isolates in our previous study.
INTRODUCTION
BST-2 (also referred to as tetherin, CD317, or HM1.24) is an interferon-inducible transmembrane (TM) protein that was originally identified in terminally differentiated human B cells of patients with multiple myeloma (1, 2). The protein consists of approximately 180 amino acids that include a short N-terminal cytoplasmic domain, a TM domain, and a rod-like α-helical ectodomain with the propensity, at least in vitro, to form a rod-like coiled-coil structure (3–8). The BST-2 C terminus is made up of a stretch of hydrophobic residues that, in rat BST-2, has been determined to function as a glycosylphosphatidylinositol (GPI) anchor signal (4). Recent experimental evidence suggests that in human BST-2 (huBST-2), this C-terminal hydrophobic domain may constitute a second TM domain rather than a GPI anchor signal (9). As far as the BST-2 ectodomain is concerned, it contains three cysteine residues important for the formation of covalent cysteine-linked dimers, as well as two N-linked carbohydrate side chains whose functional importance is currently unclear (1, 2, 4, 10, 11). BST-2 associates with lipid rafts at the cell surface and on internal membranes (4, 12–15).
While the physiological function of BST-2 in uninfected cells remains unclear, BST-2 has recently been shown to inhibit the release of human immunodeficiency virus type 1 (HIV-1) (16, 17). In fact, inhibition of virus release by BST-2 is not HIV-1 specific but affects other lentiviruses, such as HIV-2 and simian immunodeficiency virus (SIV), as well as a wide variety of enveloped viruses, including Ebola virus, Lassa virus, Marburg virus, Kaposi sarcoma-associated herpesvirus (KSHV), porcine endogenous retrovirus (PERV), and endogenous betaretrovirus of sheep (18–23). Three different lentiviral proteins, HIV-1 Vpu, HIV-2 Env, and SIV Nef, have been found to antagonize the activity of their natural host, BST-2, thus allowing for efficient particle release from BST-2-expressing cells. They are either TM proteins, like BST-2 (Vpu, Env), or are membrane associated by means of a myristic acid modification (Nef). All of them are thought to interfere with BST-2 function through direct physical interaction (24–33). Aside from these lentiviral proteins, the KSHV-encoded ubiquitin ligase K5 has been found to antagonize BST-2 by inducing ubiquitin-mediated endocytosis and lysosomal degradation of BST-2 (34, 35).
A current model suggests that BST-2 tethers mature virions to the cell surface by means of its N-terminal TM region and C-terminal membrane anchor. Indeed, immune electron microscopy identified BST-2 on virions tethered to the cell surface and on particles tethered to each other (11, 36–39). Interestingly, artificial tetherin consisting of the N-terminal TM region of transferrin receptor, a coiled-coil ectodomain of the cytoplasmic dimeric protein dystrophia myotonica protein kinase, and a GPI anchor signal derived from urokinase plasminogen activator receptor was capable of inhibiting the release of HIV-1 virions tethered to the cell surface (11). This suggests that the secondary structure rather than the primary amino acid sequence of BST-2 is important for its tethering function. Indeed, our own studies investigating the importance of structural elements in the BST-2 ectodomain revealed surprising flexibility in the size of the BST-2 ectodomain. Both increases and decreases in the size of the ectodomain, as well as replacement of the C-terminal hydrophobic domain by heterologous TM domains, were functionally tolerated (9, 40).
The widely used HIV-1 laboratory strain NL4-3 encodes a Vpu protein that is able to target human BST-2 but not murine or monkey BST-2, leading to the notion that Vpu antagonism of BST-2 is species specific (41–46). More-recent evidence, however, indicates that other Vpu proteins, such as the Vpu encoded by the clinical isolate HIV-1DH12, have a less restricted phenotype and are able to antagonize not only human BST-2 but rhesus BST-2 (rhBST-2) as well (47). Transfer of the TM domain from HIV-1DH12 Vpu was sufficient to render HIV-1NL4-3 Vpu capable of antagonizing rhesus BST-2 (47). The importance of the TM domain in Vpu for antagonism of BST-2 was affirmed by the finding that a single amino acid change in the TM domain of rhesus BST-2 (I48T) rendered the protein sensitive to HIV-1NL4-3 Vpu in a gain-of-function analysis (48). Similarly, the change of a single amino acid in the TM domain of human BST-2 to the corresponding residue found in the BST-2 protein of the Tantalus monkey (T45I) rendered the protein insensitive to HIV-1 Vpu in a loss-of-function study (42). Thus, all available evidence points to a functional interaction of the TM domains of Vpu and rhesus BST-2 as necessary and sufficient for functional neutralization of rhesus BST-2.
In an extension of these studies, we designed experiments to confirm that the expanded host range associated with the transfer of the DH12 Vpu TM domain to NL4-3 Vpu was due to a gain of interaction between Vpu and rhesus BST-2. Surprisingly, binding studies revealed that the nonfunctional NL4-3 Vpu interacted with rhesus BST-2 as efficiently as the functional Vpu variant, VputmDH12, suggesting that the interaction of Vpu and BST-2 was necessary but not sufficient for the functional neutralization of BST-2 and may involve sequences in the cytoplasmic domain of Vpu. Indeed, we found that while HIV-1 Vpu, as expected, interacted with human BST-2 primarily through their respective TM domains, the physical association with rhesus BST-2 was, at least in part, mediated by their cytoplasmic domains. Importantly, a Vpu mutant carrying two mutations in its TM domain (A14L and W22A) rendering it incompetent for interaction with human BST-2 was capable not only of interacting with human BST-2 carrying the rhesus BST-2 cytoplasmic domain (huBST-2CYTrh) but also of partially antagonizing huBST-2CYTrh in a functional assay. Analyses of additional BST-2 variants revealed that the physical interaction of Vpu and rhesus BST-2 involves a 5-residue motif in the cytoplasmic domain of rhesus BST-2. The same motif is conserved in chimpanzee BST-2 (chimpBST-2) and is critical for the interaction with Vpu. Interestingly, this motif was previously identified as important for antagonism of rhesus and chimpanzee BST-2 by SIV Nef (43, 44, 49–51). Thus, our data identify a novel mechanism of antagonism of rhesus BST-2 by Vpu that targets the same motif in the cytoplasmic domain of BST-2 used by SIV Nef.
MATERIALS AND METHODS
Plasmids.
The construction of plasmid pNL4-3, encoding the laboratory-adapted full-length molecular HIV-1 isolate NL4-3 (52), its Vpu-deficient variant pNL4-3/Udel (53), and pNL4-3 Vpu tmDH12, encoding a chimeric Vpu (47), have been described previously. Vectors for the expression of untagged human and rhesus BST-2 (huBST-2 and rhBST-2, respectively) have been described previously (48, 54). In addition, rhBST-2I48T, carrying a mutation in its TM domain, rhBST-2CYThu, a chimera encoding the human cytoplasmic domain, and rhBST-2TMhu, carrying the human TM domain, have been described previously (48). Plasmids pKGC-huBST-2, pKGN-Vpu, pKGN-VpuRD, and pKGNstop, for analyzing the interaction of Vpu and BST-2 in live cells, have been described previously (25). All mutants were created by overlapping extension PCR, and the resulting DNA fragments were subcloned into the pNL4-3, pcDNA3.1(−) (Invitrogen Corp., Carlsbad, CA), phmKGC-MC, or phmKGN-MN (MBL International, Woburn, MA) vector backbone. The nucleotide sequence of each construct was verified by sequence analysis. The open reading frame of codon-optimized SIVcpzMB897 Vpu was generated by single-step assembly of 6 oligodeoxyribonucleotides (55) according to the algorithm reported for codon-optimized NL4-3 Vpu (56) and was subcloned into phmKGN-MN. The chimpanzee BST-2 open reading frame (ORF) was amplified from pCGCG-chimpanzee BST-2 (provided by Frank Kirchhoff [43]).
Cell culture and transfection.
HEK293T (293T) is a human kidney cell line lacking expression of endogenous BST-2. 293T cells were cultured in Dulbecco's modified Eagle's minimal essential medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS). For transfection, cells were grown to about 80% confluence. Cells were transfected by using TransIT LT-1 (Mirus, Madison WI) transfection reagent according to the manufacturer's recommendations.
Flow cytometric analysis and immunoblotting for BiFC assay.
293T cells grown in 6-well plates were transfected with appropriate pairs of constructs (1 μg each) tagged with the N- or C-terminal fragment of Kusabira green (KGN or KGC, respectively) and 2 μg of an mCherry-expressing plasmid as a transfection marker (provided by Juan Martin-Serrano). Forty-eight hours posttransfection, cells were washed with phosphate-buffered saline (PBS), scraped, and resuspended in 2 ml of PBS. One fraction (0.5 ml) of the cell suspension was analyzed on a FACSCalibur flow cytometer (BD Biosciences Immunocytometry Systems, Mountain View, CA) as described previously (25, 57). The mean fluorescent intensities (MFI) of bimolecular fluorescence complementation (BiFC) signals in mCherry-positive cells are shown in the figures. The remaining cells (1.5 ml) were pelleted, resuspended in 100 μl of PBS, and mixed with an equal volume of 2× sample buffer (4% sodium dodecyl sulfate, 125 mM Tris-HCl [pH 6.8], 10% 2-mercaptoethanol [ME], 10% glycerol, 0.002% bromophenol blue). Samples were heated at 95°C for 10 min with occasional vortexing. Cell lysates were subjected to SDS-PAGE; proteins were transferred to polyvinylidene difluoride membranes and were reacted with an anti-KGN or anti-KGC monoclonal antibody (MAb) (MBL International, Woburn, MA). Membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences, Piscataway, NJ), and proteins were visualized by enhanced chemiluminescence (Amersham Biosciences).
Virus release assay.
The release of newly produced virions was measured by determining the efficiency of release of metabolically labeled viral Gag proteins. Cells were grown in 25-cm2 flasks and were transfected as described above with constant amounts of proviral vectors and increasing amounts of nontagged BST-2-encoding vectors. Total amounts of transfected DNA were kept constant in all samples of a given experiment (typically 5.2 μg) by adding empty-vector DNA as appropriate. Twenty-four hours later, cells were washed with PBS, scraped, and suspended in 3 ml of a labeling medium lacking methionine and cysteine (Millipore Corp., Bedford, MA). Cells were incubated for 10 min at 37°C to deplete the endogenous methionine pool. Cells were then suspended in 400 μl of the labeling medium together with 150 μCi of Expre35S35S protein labeling mix (Perkin-Elmer, Shelton, CT). Cells were labeled for 90 min at 37°C. Cells and virus-containing supernatants were then separated by centrifugation and were processed separately for immunoprecipitation. To identify HIV-1-specific proteins, cells were lysed in 600 μl of NP-40–deoxycholate (DOC) lysis buffer (20 mM Tris [pH 7.5], 2 mM EDTA, 20 mM NaCl, 1% Igepal, 0.5% sodium deoxycholate) and were incubated on ice for 5 min. Insoluble material was pelleted at 13,000 × g for 2 min, and supernatants were used for immunoprecipitation. Virus-containing supernatants were treated with 200 μl of 3× NP-40–DOC lysis buffer to disrupt viral membranes. Cell and virus lysates were adjusted to a 1.1-ml total volume with PBS containing bovine serum albumin (BSA; final concentration, 0.1%) and were incubated on a rotating wheel for 1 h at 4°C with protein A-Sepharose coupled with serum from an HIV-positive patient. Beads were washed three times with wash buffer (50 mM Tris [pH 7.4], 300 mM NaCl, 0.1% Triton X-100). Bound proteins were eluted by heating in 1× sample buffer (2% sodium dodecyl sulfate, 62.5 mM Tris-HCl [pH 6.8], 5% 2-mercaptoethanol, 5% glycerol, and 0.001% bromophenol blue) for 10 min at 95°C, separated by SDS-PAGE, and visualized by fluorography. Virus release was quantified by phosphorimage analysis using a Fujifilm FLA7000 system. The fraction of p24 released in the absence of BST-2 relative to the total amount of intra- and extracellular Gag protein was defined as 100%.
Pulse-chase analysis.
The pulse-chase assay is similar to the virus release assay except that viral release kinetics are determined by collecting samples at multiple time points over an observation period as described previously (58). Briefly, cells grown in 25-cm2 flasks were transfected with 5 μg of pNL4-3/Udel, 0.01 μg of pKGC-chimpBST-2, and 0.5 μg of constructs expressing KGN-tagged Vpu or the KGN tag only. Twenty-four hours later, cells were labeled for 30 min at 37°C as described for the virus release assay. The isotope was then removed, and cells were chased in complete DMEM–FBS for the times indicated in Fig. 6C. At each time point, cells and supernatant were collected separately, lysed in NP-40–DOC lysis buffer, and immunoprecipitated as described for the virus release assay.
Fig 6.

The cytoplasmic domain of HIV-1 Vpu renders a SIVcpz Vpu capable of interacting with and antagonizing chimpanzee BST-2. (A) (Top) Amino acid alignment of the cytoplasmic domains of the BST-2 variants used in this experiment. Identical sequences are shown in black; residues in blue are conserved in chimpanzee and rhesus BST-2; residues in red are conserved in human and chimpanzee BST-2; a residue in brown is specific to chimpanzee BST-2. (Bottom) Schematic representation of Vpu variants employed in the experiment. (B) The physical interaction of the chimpBST-2 variants with SIVcpz Vpu variants was assessed by flow cytometry as described for Fig. 1B. The BiFC signal obtained for the interaction of Vpu with wild-type chimpBST-2 (bar 2) was defined as 100%. ***, P < 0.01; n.s., P > 0.05. Bars outlined in red represent combinations where the respective Vpu variant was able to antagonize the indicated BST-2 variant (as assessed in panel C). The expression of the KGC-tagged and KGN-tagged variants was assessed by immunoblotting as described for Fig. 1B. The KGN fragment encoded by the “tag-only” vector has a faster mobility and is not visible here. Faint bands in lanes 1 and 5 are nonspecific background. (C) The ability of SIVcpz VpucytNL to antagonize chimpanzee BST-2 via a cytoplasmic domain interaction was assessed by pulse-chase analysis as described in Materials and Methods. Open circles, empty vector; filled circles, NL4-3 Vpu; open squares, SIVcpz Vpu; filled squares, SIVcpz VpucytNL. Data are means ± standard errors of the means from two independent experiments. The statistical significance of the difference in virus release for SIVcpz Vpu and SIVcpz VpucytNL was determined by two-way ANOVA. ***, P < 0.01.
TZM-bl assay.
The TZM-bl assay was carried out essentially as described previously (40). Virus stocks were prepared by transfection of 293T cells. Virus-containing supernatants were harvested 24 h after transfection. Cellular debris was removed by centrifugation (3 min, 1,500 × g). One hundred microliters of viral supernatant was used to infect 5 × 104 TZM-bl cells in a 24-well plate in a total volume of 1.1 ml containing 1 μM ritonavir (NIH AIDS Research and Reference Reagent Program, catalog no. 4622). Infection was allowed to proceed for 48 h at 37°C. The medium was removed; cells were lysed in 300 μl of 1× reporter lysis buffer (Promega Corp., Madison, WI); and lysates were frozen at −80°C for a minimum of 30 min. To determine the luciferase activity in the lysates, 5 μl of each lysate was combined with 20 μl of a luciferase substrate (Steady-Glo; Promega Corp., Madison, WI), and light emission was measured using a Modulus II microplate reader (Turner BioSystems Inc., Sunnyvale, CA).
Statistical analysis.
To determine statistical significance, Student's t tests and two-way analyses of variance (ANOVA) were carried out using GraphPad Prism (version 6) for the BiFC interaction assay and the functional assays (the virus release, pulse-chase, and TZM-bl assays), respectively.
RESULTS
The interaction of rhesus BST-2 with HIV-1 Vpu involves the cytoplasmic domain.
To gain a better understanding of the physical and functional interaction between Vpu and BST-2, we chose to carry out a live-cell protein-protein interaction assay based on bimolecular fluorescence complementation (BiFC) (59, 60). The BiFC approach is based on complementation between two fragments of a fluorescent protein such as Kusabira green (KG) (61). Individual fragments such as the N- and C-terminal fragments of KG (referred to as KGN and KGC, respectively) are not fluorescent by themselves; however, reconstitution of a complex by these fragments brought together by the association of two interaction partners fused to the fragments restores fluorescence and allows for quantitative analysis of protein complexes using flow cytometry (25, 57).
We started out by assessing the molecular interaction between HIV-1 Vpu and BST-2 by using the BiFC assay. For that purpose, we used N-terminally KGC-tagged BST-2 constructs expressing huBST-2 (25), rhBST-2, or rhBST-2I48T, as shown at the top of Fig. 1A. We also used C-terminally KGN-tagged NL4-3 Vpu constructs expressing wild-type (WT) Vpu (25), a mutant with a scrambled TM domain, VpuRD (25), or VputmDH12 (Fig. 1A, bottom). After confirmation of protein expression by immunoblotting (Fig. 1B, top), the intensities of BiFC signals resulting from different levels of protein-protein interaction were measured by flow cytometry (Fig. 1B, bottom). Bars framed in red indicate combinations previously found to result in functional antagonism of BST-2 (47, 48). A strong BiFC signal was observed in cells transfected with huBST-2 and NL4-3 Vpu but not in cells expressing huBST-2 and the KGN tag only (Fig. 1B, compare bars 1 and 2). In addition, a scrambled-TM-domain mutant of Vpu, VpuRD, which is unable to antagonize BST-2 (62), produced only background signals in our BiFC assay (Fig. 1B, compare bars 1 and 3). These results demonstrate that the physical interaction of Vpu and BST-2 in the context of KGC- and KGN-tagged variants faithfully replicates previous results from studies using coimmunoprecipitation (co-IP) (26–28, 63), fluorescence resonance energy transfer (FRET) (64), nuclear magnetic resonance (NMR) spectroscopy (31), or BiFC (25). Finally, VputmDH12, carrying the TM domain of DH12 Vpu, which we previously found to antagonize both human and rhesus BST-2 (47), interacted with huBST-2, although the BiFC signal was not as strong as that observed with NL4-3 Vpu (Fig. 1B, compare bars 2 and 4). Nevertheless, the BiFC signal was significantly higher than that observed for VpuRD or the “tag-only” control (Fig. 1B, compare bar 4 with bar 3 or 1). The failure of VpuRD to produce significant BiFC signals when expressed in combination with huBST-2 indicates that the signal observed in conjunction with wild-type Vpu (Fig. 1B, bar 2) is specific and is not the result of random encounters of two membrane proteins in a 2-dimensional plane. Taken together, these results validate our BiFC assay as a suitable method for the assessment of molecular interactions between HIV-1 Vpu and host BST-2 in live cells.
Fig 1.
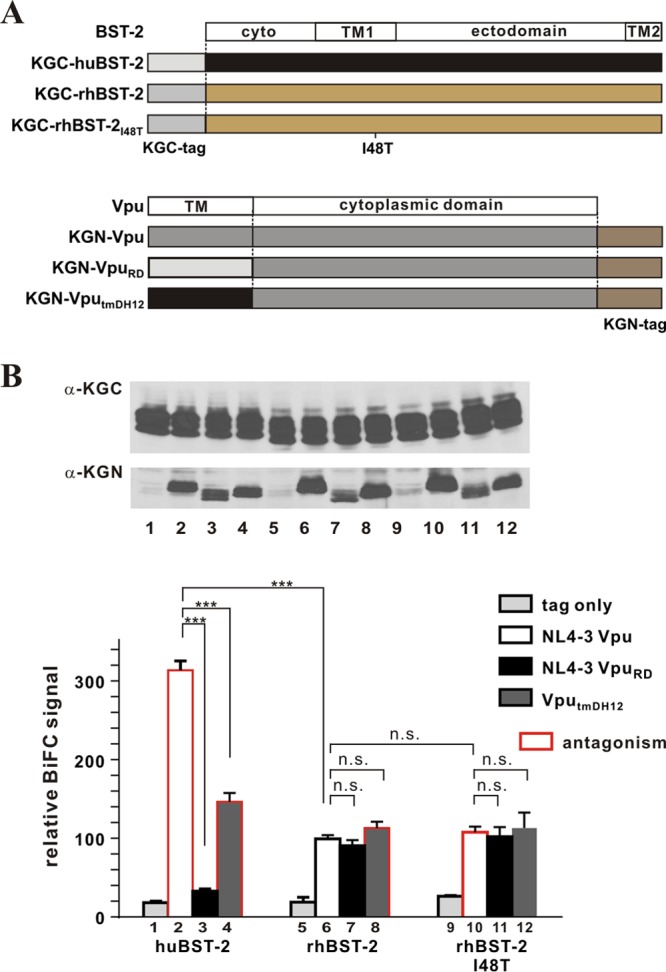
Physical interaction is not sufficient for BST-2 antagonism by Vpu. (A) Schematic representation of the BST-2 and Vpu constructs used for assessing physical interaction through bimolecular fluorescence complementation (BiFC). The KGC tag was fused in frame to the N terminus of BST-2 (huBST-2, rhBST-2, and rhBST-2I48T). The KGN tag was fused in frame to the C terminus of Vpu and its mutants. The cartoon is not drawn to scale (the KGC tag, the KGN tag, human BST-2, and NL4-3 Vpu consist of 51 residues, 168 residues, 180 residues, and 81 residues, respectively). (B) (Top) The expression of KGC-tagged BST-2 variants and KGN-tagged Vpu variants was assessed by immunoblotting using antibodies to KGC and KGN, respectively. The KGN fragment encoded by the “tag-only” vector has a faster mobility and is not visible here. Faint bands in lanes 1, 5, and 9 are nonspecific background. Lane numbers in the immunoblot correspond to bar numbers in the graph below. (Bottom) The physical interaction of BST-2 and Vpu was quantified by a BiFC assay. 293T cells transfected with pKGC-huBST-2, pKGC-rhBST-2, or pKGC-rhBST-2I48T, and with the indicated pKGN-Vpu variants, were analyzed by flow cytometry as described in Materials and Methods. The MFI of NL4-3 Vpu with rhBST-2 (bar 6) was empirically defined as 100%. Results are expressed as the relative MFI (BiFC) plus the standard error of the mean calculated from three independent experiments. ***, P < 0.01; n.s., not significant (P > 0.05). Bars outlined in red represent combinations where the Vpu variant was able to antagonize the BST-2 variant (47, 48).
We next assessed the interaction of Vpu with rhBST-2 in our BiFC assay (Fig. 1B, bars 5 to 8). NL4-3 Vpu does not antagonize rhBST-2 in a functional assay but was nevertheless found to interact with rhBST-2, even though the BiFC signal was lower than that for huBST-2 (Fig. 1B, compare bars 2 and 6). Of note, NL4-3 Vpu interacted with rhBST-2 almost as efficiently as VputmDH12, which is capable of functionally antagonizing rhBST-2 (Fig. 1B, compare bars 6 and 8). This suggests that VputmDH12 antagonizes rhBST-2 without increasing the physical interaction between the proteins. Surprisingly, VpuRD interacted with rhBST-2 as efficiently as NL4-3 Vpu (Fig. 1B, compare bars 6 and 7), even though it was unable to interact with huBST-2. To confirm this result, we also assessed the interaction between NL4-3 Vpu and rhBST-2I48T, because we reported previously that mutation of I48T in the TM domain of rhesus BST-2 renders the protein sensitive to NL4-3 Vpu (48). Interestingly, NL4-3 Vpu produced similar BiFC signals when coexpressed with wild-type rhBST-2 or with rhBST-2I48T (Fig. 1B, compare bars 6 and 10), suggesting that the I48T mutation rendered rhesus BST-2 sensitive to NL4-3 Vpu without a noticeable increase in the physical interaction between the proteins. The same was true for VpuRD and VputmDH12, which produced very similar BiFC signals when coexpressed with wild-type rhBST-2 (Fig. 1B, bars 7 and 8) or rhBST-2I48T (Fig. 1B, bars 11 and 12). Given that the Vpu variants analyzed so far differed only in their TM domains, these results support the notion that Vpu interaction with rhBST-2 involves domains other than—or in addition to—the TM domain. Taking our findings together, we conclude that a physical interaction between Vpu and BST-2 may be necessary but is not sufficient for functional antagonism of BST-2 by Vpu, suggesting that physical interaction is only one step in a series of events that ultimately lead to antagonism of BST-2.
To test whether the interaction of Vpu with rhBST-2 involves additional sequences in the cytoplasmic domain, we generated KGC-tagged rhBST-2 variants carrying the human cytoplasmic domain (CYThu) or the human TM domain (TMhu). The constructs are shown schematically in Fig. 2A. After confirming the expression of the proteins by immunoblotting (Fig. 2B, top), we assessed the relative levels of protein-protein interaction by BiFC (Fig. 2B, bottom). As expected, the interaction of NL4-3 Vpu with rhBST-2TMhu was significantly stronger than that with wild-type rhBST-2 (Fig. 2B, compare bars 4 and 6), because the TM domain of human BST-2 contributes prominently to the interaction with NL4-3 Vpu. Nevertheless, the BiFC signal for rhBST-2 and NL4-3 Vpu was significantly above background (Fig. 2B, compare bars 1 and 4). Of note, the BiFC signal for NL4-3 Vpu and rhBST-2CYThu was lower than the signal for NL4-3 Vpu and rhBST-2 (Fig. 2B, compare bars 4 and 5), suggesting involvement of the rhBST-2 cytoplasmic domain in the interaction with Vpu. Consistent with these results, we found that the cytoplasmic domain of rhBST-2 also contributes to the interaction with VpuRD (Fig. 2B, compare bars 7 and 8).
Fig 2.
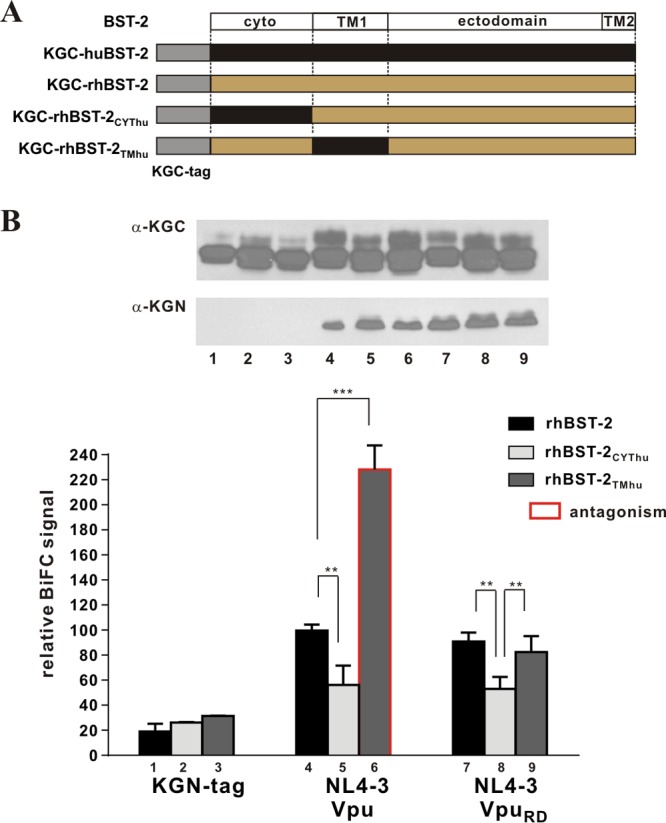
The interaction of rhesus BST-2 with Vpu involves their cytoplasmic domains. (A) Schematic representation of the BST-2 variants used in this analysis. The cartoon is not drawn to scale (the KGC tag and human BST-2 comprise 51 and 180 residues, respectively). (B) The physical interaction between NL4-3 Vpu or VpuRD and rhBST-2 variants was quantified by flow cytometry as described for Fig. 1B. Error bars reflect the standard errors of the means from three independent experiments. ***, P < 0.01; **, 0.01 < P < 0.05. The expression of KGC-tagged BST-2 variants and KGN-tagged Vpu mutants was assessed by immunoblotting as described for Fig. 1B. The KGN fragment encoded by the “tag-only” vector in lanes 1, 2, and 3 has a faster mobility and is not visible here.
The cytoplasmic domain of rhesus BST-2 is critical for functional antagonism by Vpu.
We reported previously that VputmDH12, but not NL4-3 Vpu, was able to antagonize rhBST-2 (47). Given that the mutation of the Vpu TM domain in VpuRD inhibited binding to huBST-2 (Fig. 1B) and resulted in a loss of antagonizing activity toward huBST-2, we next investigated the relative importance of the cytoplasmic domain in rhBST-2 for functional antagonism by Vpu. The BST-2 constructs used in this experiment are shown schematically in Fig. 3A. We first tested the ability of VputmDH12 to interact with rhBST-2 or rhBST-2CYThu (Fig. 3B). Protein expression was verified by immunoblotting (Fig. 3B, inset). BiFC analysis indeed demonstrated that coexpression of VputmDH12 with wild-type rhBST-2 produced a stronger BiFC signal than coexpression with rhBST-2CYThu, carrying the human cytoplasmic domain (Fig. 3B, compare bars 3 and 4). The change in the BiFC signal caused by the replacement of the rhBST-2 cytoplasmic domain with the huBST-2 cytoplasmic domain provides further evidence that the cytoplasmic domain of rhBST-2 contributes to the interaction with VputmDH12 (Fig. 3B).
Fig 3.

Cytoplasmic interactions are necessary for functional antagonism of rhesus BST-2 by Vpu. (A) Schematic representation of rhBST-2 variants employed in the experiment. The cartoon is not drawn to scale (the KGC tag and human BST-2 comprise 51 and 180 residues, respectively). (B) The physical interaction of VputmDH12 and rhBST-2 variants was quantified by flow cytometry as described for Fig. 1B. The BiFC signal for the interaction between VputmDH12 and rhBST-2 (bar 3) was defined as 100%. Error bars reflect the standard errors of the means from three independent experiments. ***, P < 0.01. (Inset) The expression of KGC-tagged BST-2 variants and KGN-tagged Vpu proteins was assessed by immunoblotting as described for Fig. 1B. The protein band corresponding to the tag-only fragment runs below the edge of the gel slice shown here. (C) 293T cells (5 × 106) were transfected with 5 μg of either pNL4-3 (WT), pNL4-3/Udel, or pNL4-3 VputmDH12 in the absence of BST-2 or in the presence of 0.01 μg, 0.02 μg, or 0.05 μg of pcDNA rhBST-2 (left) or pcDNA rhBST-2CYThu (right). Metabolic labeling was carried out as described in Materials and Methods, and cell lysates and cell-free supernatants were subjected to immunoprecipitation using HIV-positive pooled human serum. Gag-specific cellular and viral proteins (Pr55 and p24) were quantified by phosphorimage analysis. Filled circles, NL4-3 (WT); filled squares, NL4-3 VputmDH12; open circles, NL4-3/Udel. Virus release in the absence of BST-2 (0 μg) was defined as 100%. Results are means plus standard errors of the means from three independent experiments. Statistical significance was determined by two-way ANOVA. ***, P < 0.01; n.s., P > 0.05. (D) The physical interaction between NL4-3 Vpu and rhBST-2I48T or rhBST-2CYThu I48T was quantified as described for panel B. The BiFC signal for the interaction of NL4-3 Vpu with rhBST-2I48T (bar 3) was defined as 100%. ***, P < 0.01. (Inset) The expression of KGC-tagged BST-2 variants and KGN-tagged proteins was assessed by immunoblotting as described for Fig. 1B. (E) The ability of Vpu to antagonize rhBST-2I48T or rhBST-2CYThu I48T was assessed as described for panel C.
The ability of VputmDH12 to functionally antagonize the antiviral activity of rhBST-2CYThu was tested in a virus release assay (Fig. 3C). The contribution of the rhBST-2 cytoplasmic domain to Vpu responsiveness was tested by comparing the relative inhibition of virus release for wild-type (WT) NL4-3, NL4-3/Udel, and NL4-3 VputmDH12. In these experiments, Vpu was expressed in the context of full-length viral genomes. In agreement with our previous observations (47), VputmDH12, but not NL4-3 Vpu, was able to antagonize rhBST-2 (Fig. 3C, left, compare filled squares with open circles) (P, 0.0003). In contrast, neither VputmDH12 nor NL4-3 Vpu was able to antagonize rhBST-2CYThu (Fig. 3C, right, compare filled squares and filled circles with open circles) (P, 0.36). These results indicate that even though rhBST-2 carrying the cytoplasmic domain of huBST-2 (rhBST-2CYThu) is able to inhibit HIV-1 release effectively, the presence of a human cytoplasmic domain abolished sensitivity to VputmDH12 due to the reduced physical interaction.
To further confirm the importance of the rhBST-2 cytoplasmic domain for the interaction with and antagonism by Vpu, we carried out similar experiments using rhBST-2I48T, carrying a mutation in its TM domain that renders it sensitive to NL4-3 Vpu (48). We also analyzed a derivative of rhBST-2I48T, rhBST-2CYThu I48T, additionally encoding the human cytoplasmic domain. We generated KGC-tagged as well as untagged forms of rhBST-2I48T and rhBST-2CYThu I48T (Fig. 3A). Protein expression was verified by immunoblotting (Fig. 3D, inset). BiFC analysis indicated that NL4-3 Vpu indeed interacted with rhBST-2I48T (Fig. 3D, bar 3). Again, the BiFC signal resulting from the coexpression of NL4-3 Vpu and rhBST-2CYThu I48T was weaker than that with rhBST-2I48T (Fig. 3D, compare bars 3 and 4), suggesting a significant contribution of the BST-2 cytoplasmic domain to the physical interaction of rhBST-2 and NL4-3 Vpu. Analysis of these constructs in our virus release assay confirmed that NL4-3 Vpu antagonizes rhBST-2I48T (Fig. 3E, left) (P, 0.0003), as reported previously (48). However, replacing the rhBST-2 cytoplasmic domain with the huBST-2 sequence caused a complete loss of Vpu sensitivity (Fig. 3E, right) (P, 0.679). From these loss-of-function studies, we conclude that the rhBST-2 cytoplasmic domain is important for the physical interaction with Vpu as well as for functional sensitivity to Vpu.
We next employed a gain-of-function approach to assess whether interaction between BST-2 and Vpu through their cytoplasmic domains is necessary for functional antagonism. For that purpose, we made use of an inactive NL4-3 Vpu mutant, Vpu14/22, carrying two amino acid changes (A14L, W22A) in its TM domain. This mutant was previously shown to be unable to enhance virus release, to coimmunoprecipitate huBST-2, or to downregulate cell surface expression of huBST-2 (65). BiFC analyses indicate that huBST-2 indeed interacts only inefficiently with Vpu14/22 compared to NL4-3 Vpu (Fig. 4A, top, compare bars 2 and 3). Comparable expression of NL4-3 Vpu and Vpu14/22 as well as huBST-2 was verified by immunoblotting (Fig. 4A, bottom). The ability of Vpu14/22 to antagonize huBST-2 was assessed by measuring its effect on virus release using full-length NL4-3 carrying the mutations at amino acids 14 and 22 in Vpu (NL4-3 Vpu14/22). In agreement with the results reported by Vigan and Neil (65), we found that unlike wild-type Vpu, Vpu14/22 was unable to antagonize huBST-2 in our virus release assay (Fig. 4B, compare solid circles to solid squares). These results were confirmed in an independent assay employing infection of TZM-bl cells as a means to quantify virus release (Fig. 4C). The results are consistent with those of the virus release assay shown in Fig. 4B, demonstrating that wild-type NL4-3 Vpu (WT), but not Vpu14/22, enhances virus release in the presence of huBST-2 (Fig. 4C, compare solid circles and solid squares).
Fig 4.
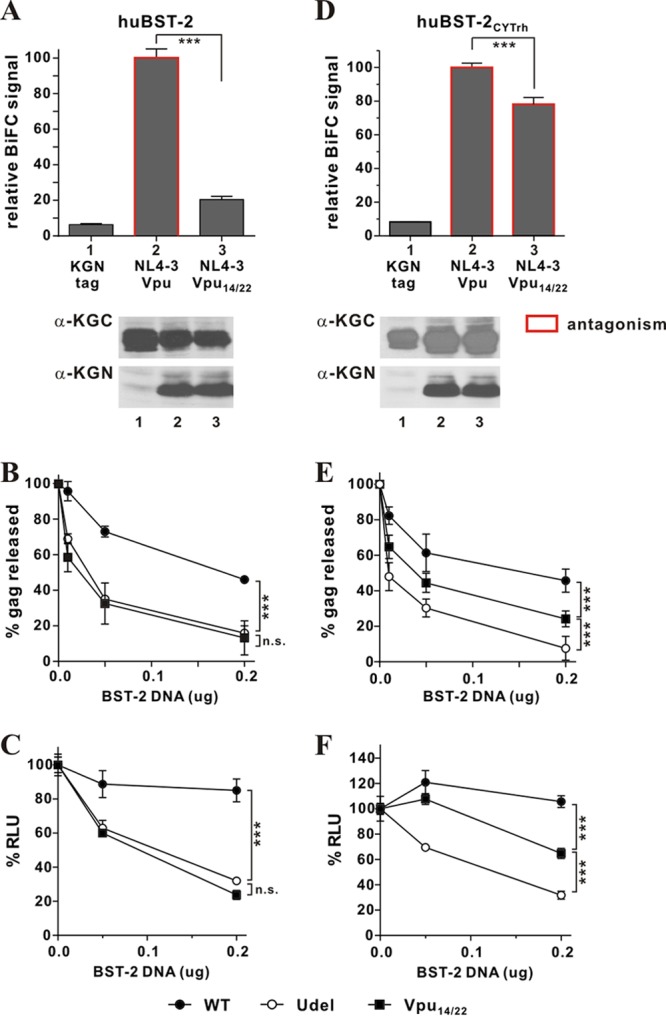
Gain-of-function analysis of cytoplasmic interactions between Vpu and BST-2. (A to C) Mutation of residues 14 and 22 in the TM domain of NL4-3 Vpu results in functional inactivation. (A) The physical interaction of huBST-2 and Vpu14/22 was quantified by flow cytometry as described for Fig. 1B. The BiFC signal measured for the interaction of wild-type Vpu and huBST-2 (bar 2) was defined as 100%. ***, P < 0.01. The expression of KGC-tagged huBST-2 and KGN-tagged proteins was assessed by immunoblotting as described for Fig. 1B. The KGN fragment encoded by the “tag-only” vector has a faster mobility and is not visible here. Faint bands in lane 1 are nonspecific background. (B) The functional antagonism of human BST-2 by Vpu variants was assessed by metabolic labeling as described for Fig. 3C. Filled circles, NL4-3 (WT); filled squares, NL4-3 Vpu14/22; open circles, NL4-3/Udel. Data are means plus standard errors of the means from three independent experiments. Statistical significance was determined by two-way ANOVA. ***, P < 0.01; n.s., P > 0.05. (C) The functional antagonism of human BST-2 by Vpu variants was assessed by a TZM-bl assay. 293T cells (5 × 106) were transfected with 5 μg of either pNL4-3 (WT), pNL4-3/Udel, or pNL4-3 Vpu14/22 in the absence of BST-2 or in the presence of 0.05 μg or 0.2 μg of pcDNA-huBST-2. The TZM-bl assay was carried out as described in Materials and Methods. Virus release in the absence of BST-2 (0 μg) was defined as 100%. Data are means plus standard errors of the means from three independent experiments. Statistical significance was determined by two-way ANOVA. ***, P < 0.01; n.s., P > 0.05. (D to F) Cytoplasmic interactions are sufficient to restore the BST-2-antagonizing activity of Vpu14/22. (D) The physical interaction between huBST-2CYTrh and Vpu or Vpu14/22 was quantified as described for Fig. 1B. The BiFC signal obtained for the interaction of NL4-3 Vpu and huBST-2CYTrh (bar 2) was defined as 100%. ***, P < 0.01. Bars outlined in red represent combinations where the respective Vpu variant was able to antagonize huBST-2CYTrh (as assessed in panels E and F). The expression of KGC-tagged huBST-2CYTrh and KGN-tagged proteins was assessed by immunoblotting as described for Fig. 1B. (E) The ability of Vpu14/22 to antagonize huBST-2CYTrh via cytoplasmic domain interaction was assessed by metabolic labeling as described for Fig. 3C. Data are means plus standard errors of the means from three independent experiments. Statistical significance was determined by two-way ANOVA. ***, P < 0.01. (F) The ability of Vpu14/22 to antagonize huBST-2CYTrh was also assessed by a TZM-bl assay as described for panel C. Data are means plus standard errors of the means from three independent experiments. Statistical significance was determined by two-way ANOVA. ***, P < 0.01.
Next, we assessed the ability of Vpu14/22 to physically interact with and functionally counteract huBST-2 carrying the rhBST-2 cytoplasmic domain (huBST-2CYTrh). BiFC analysis revealed significant interaction of Vpu14/22 with huBST-2CYTrh compared to the vector control (Fig. 4D, compare bars 1 and 3), although the BiFC signal was slightly weaker than that observed for wild-type Vpu (Fig. 4D, compare bars 2 and 3). These results show that the cytoplasmic domain of rhBST-2 renders the protein capable of interacting with NL4-3 Vpu in the absence of TM domain interaction. To assess whether the interaction of Vpu14/22 with BST-2 via its cytoplasmic domain is sufficient for Vpu14/22 to functionally antagonize BST-2, we carried out our virus release assay (Fig. 4E), as well as the TZM-bl infection assay (Fig. 4F). Both assays demonstrated that the level of virus release from cells expressing huBST-2CYTrh in the presence of Vpu14/22 is significantly higher than that in the absence of Vpu (Fig. 4E and F, compare filled squares with open circles) (P, 0.007 and <0.0001, respectively). However, the antagonism of huBST-2CYTrh by Vpu14/22 was less efficient than that seen for wild-type Vpu (Fig. 4E and F, compare filled circles with filled squares) (P, 0.002 and <0.0001, respectively). The results from this gain-of-function analysis therefore demonstrate that (i) a physical interaction between Vpu and BST-2 is necessary for functional antagonism and (ii) the interaction of Vpu with the cytoplasmic domain of rhBST-2 contributes at least in part to the expanded host range of Vpu.
A 5-amino-acid sequence in the rhesus BST-2 cytoplasmic domain accounts for the interaction with Vpu and for rhesus BST-2 antagonism by HIV-1 Vpu.
One of the most prominent differences between the cytoplasmic domains of rhesus and human BST-2 is a 5-amino-acid segment [(G/D)DIWK] that is present in rhBST-2 but is missing in huBST-2 (Fig. 5A). This segment was previously identified as critical for the antagonism of monkey and great ape BST-2 by SIV Nef (43, 44, 49). To investigate the importance of the (G/D)DIWK element for the interaction with NL4-3 Vpu, we deleted these five residues in rhBST-2 (Fig. 5A, rhBST-2del5). To control for the potential effect of the deletion, we also mutated these five residues to alanine (Fig. 5A, rhBST-25AA). Finally, since the cytoplasmic domains of human and rhesus BST-2 also differ in six positions at their extreme N termini, we replaced the six differing residues in rhBST-2 with the corresponding human sequences (Fig. 5A, rhBST-2Nhu). All mutants were transferred into the BiFC (KGC) vector, and expression of the KGC-tagged BST-2 variants and KGN-tagged Vpu was verified by immunoblotting (Fig. 5B, right). All proteins were expressed at comparable levels, except rhBST-2del5, whose expression was about 2-fold higher in this experiment (Fig. 5B, top right, lane 4). As expected from our results shown in Fig. 2B (bars 4 and 5), we detected a strong BiFC signal for the interaction of NL4-3 Vpu with wild-type rhBST-2 (Fig. 5B, bar 2) and a reduced signal for the interaction of Vpu with rhBST-2CYThu (Fig. 5B, bar 3). Deletion of the DDIWK motif or its replacement with alanine resulted in similar reductions of the BiFC signals (Fig. 5B, bars 4 and 6, respectively). In contrast, replacement of six N-terminal residues by the human sequence did not significantly alter the BiFC signal (Fig. 5B, bar 5). These results indicate that the (G/D)DIWK motif in the cytoplasmic domain of rhBST-2 represents the cytoplasmic component responsible for the interaction with HIV-1 Vpu.
Fig 5.
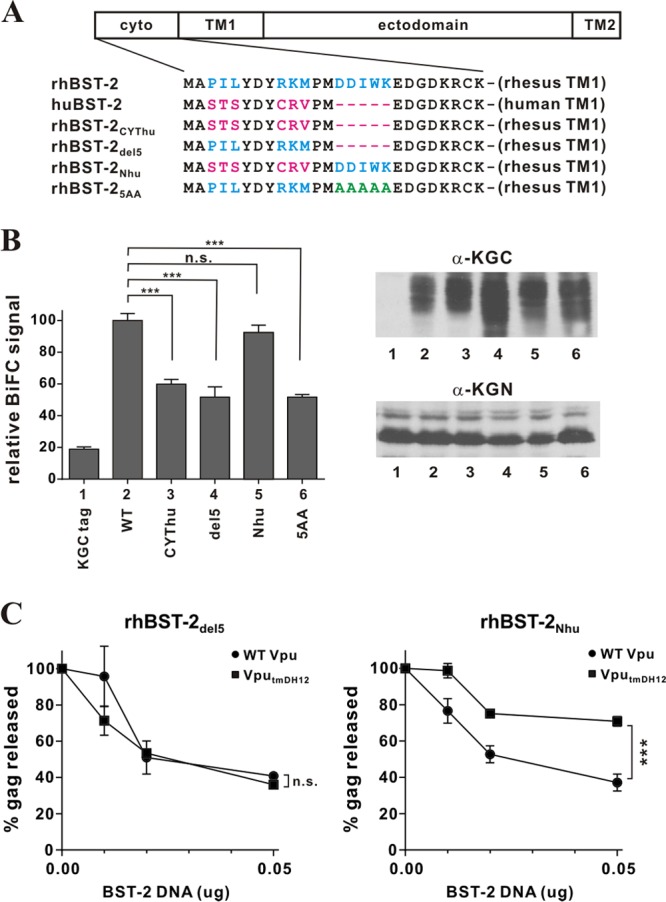
The 5-residue (G/D)DIWK motif in the cytoplasmic domain of rhesus BST-2 is important for interaction with Vpu. (A) Amino acid alignment of the cytoplasmic domains of the BST-2 variants used in this experiment. Identical sequences are shown in black. Residues in blue are rhesus BST-2-specific sequences; residues in red are human BST-2-specific sequences. The DDIWK-to-AAAAA substitution in rhBST-25AA is indicated in green. (B) (Left) The physical interaction of the rhBST-2 variants with NL4-3 Vpu was assessed by flow cytometry as described for Fig. 1B. The BiFC signal obtained for the interaction of Vpu and rhBST-2 (WT) (bar 2) was defined as 100%. ***, P < 0.01; n.s., P > 0.05. (Right) The expression of the KGC-tagged BST-2 variants and KGN-tagged Vpu was assessed by immunoblotting as described for Fig. 1B. The protein band corresponding to the KGC tag alone has a faster mobility and is not visible here. (C) The functional antagonism of rhesus BST-2 variants by Vpu was assessed in a virus release assay by metabolic labeling as described for Fig. 3C. Circles, NL4-3 (WT); squares, NL4-3 VputmDH12. Data are means ± standard errors of the means from three independent experiments. Statistical significance was determined by two-way ANOVA. ***, P < 0.01; n.s., P > 0.05.
Although NL4-3 Vpu can interact with rhBST-2 via its cytoplasmic domain, it is unable to functionally antagonize rhBST-2 unless it also encodes the TM domain of the DH12 isolate of Vpu (47). To assess whether the (G/D)DIWK motif similarly contributes to the functional antagonism of rhBST-2 by VputmDH12, we carried out our virus release assay using NL4-3 VputmDH12 in the presence of either rhBST-2del5 (Fig. 5C, left) or rhBST-2Nhu (Fig. 5C, right). We found that VputmDH12, but not NL4-3 wild-type Vpu, was capable of functionally antagonizing rhBST-2Nhu (Fig. 5C, right) (P, 0.0015). In contrast, neither Vpu variant was able to antagonize rhBST-2del5 (Fig. 5C, left) (P, 0.44). These results indicate that the 5-amino-acid DDIWK motif in the rhBST-2 cytoplasmic domain is indeed important not only for the physical interaction but also for functional antagonism by Vpu. Overall, we conclude that a physical interaction of Vpu with rhBST-2 via their cytoplasmic domains is necessary but not sufficient for functional antagonism, which depends on determinants within the Vpu TM domain.
The cytoplasmic domain of HIV-1 Vpu contributes to the physical interaction with, and functional antagonism of, chimpanzee BST-2.
The pandemic form of HIV-1 group M has been traced back to SIVcpz, a virus causing infection in chimpanzees (66). SIVcpz carries a vpu gene, but its gene product is unable to antagonize chimpanzee BST-2 (chimpBST-2), even though HIV-1 Vpu has this capacity (43, 50). Interestingly, chimpBST-2 carries the same 5-amino-acid motif in its cytoplasmic domain as rhBST-2 (Fig. 6A). To investigate why SIVcpz Vpu is functionally inactive against chimpBST-2, we studied their physical interaction. We generated N-terminally KGC-tagged chimpBST-2 and a variant carrying the human cytoplasmic domain (chimpBST-2CYThu). We chose Vpu from SIVcpzMB897 because this isolate is thought to be phylogenetically close to HIV-1 group M, which includes HIV-1NL4-3 (67). We generated C-terminally KGN-tagged expression vectors for Vpu from SIVcpzMB897 (SIVcpz Vpu) and a variant carrying the cytoplasmic domain of HIV-1NL4-3 Vpu (SIVcpz VpucytNL) (Fig. 6A). Using these constructs, we quantified the physical interaction by a BiFC assay (Fig. 6B, top) after comparable protein expression was confirmed by immunoblotting (Fig. 6B, bottom). We found that the BiFC signal resulting from the coexpression of chimpBST-2 with HIV-1NL4-3 Vpu was higher than that with SIVcpz Vpu (Fig. 6B, compare bars 2 and 3). Surprisingly, however, the coexpression of chimpBST-2 with SIVcpz VpucytNL yielded BiFC signals even higher than those observed for the interaction with SIVcpz Vpu or HIV-1NL4-3 Vpu (Fig. 6B, compare bar 4 to bars 3 and 2). These results suggest that the cytoplasmic domain of HIV-1NL4-3 Vpu contributes significantly to the physical interaction with chimpBST-2. In agreement with this conclusion, SIVcpz VpucytNL showed no detectable interaction with chimpBST-2CYThu, lacking the 5-amino-acid DDIWK motif (Fig. 6B, compare bars 7 and 8). Thus, the inability of SIVcpz Vpu to antagonize chimpBST-2 appears to be due to its inability to interact with the DDIWK motif in the cytoplasmic domain of chimpBST-2.
To further test this hypothesis, we performed a virus release experiment. Sequences encoding the cytoplasmic domain of Vpu overlap the 3′ end of the env gene. Because of that, expression of SIVcpz Vpu from a full-length molecular clone could affect Env expression. Therefore, we did not use the TZM-bl-based assay used in the preceding experiments, since potential effects on Env expression could affect the readout from this assay. Instead, we carried out pulse-chase experiments as described in Materials and Methods, using SIVcpz Vpu and, as a chimpBST-2 binding-competent control, SIVcpz VpucytNL. We used C-terminally KGN-tagged Vpu constructs as shown in Fig. 6B in conjunction with NL4-3/Udel instead of using full-length provirus constructs expressing mutant Vpu. HIV-1NL4-3 Vpu was included as a control. Virus release from transfected 293T cells was measured over a 4-h time span (Fig. 6C). In agreement with previous reports (43, 50), we found that HIV-1 Vpu, but not SIVcpz Vpu, enhanced virus release in the presence of chimpBST-2. However, transfer of the HIV-1 Vpu cytoplasmic domain to SIVcpz Vpu (SIVcpz VpucytNL) rescued the defect and rendered the resulting chimera capable of antagonizing chimpBST-2 with an efficiency similar to that of wild-type HIV-1 Vpu (Fig. 6C, compare filled squares with filled circles). Thus, the cytoplasmic domain of HIV-1 NL4-3 Vpu is critical not only for the physical interaction with chimpBST-2 but also for the ability to functionally antagonize chimpBST-2.
DISCUSSION
The ongoing HIV epidemic, which has affected millions of people worldwide, originated from the zoonotic transmission of SIVcpz, a simian immunodeficiency virus normally infecting chimpanzees (66). As a result, HIV-1 and SIVcpz are very similar in their genetic structures. In particular, they both carry a vpu gene and lack a vpx gene, while most other SIV lineages, including SIVmac, carry vpx. The two best-studied functions of HIV-1 Vpu are the degradation of CD4 and the enhancement of virus particle secretion from infected cells (for a review, see reference 68). Interestingly, SIVcpz Vpu, while capable of inducing CD4 degradation, is unable to antagonize chimpanzee or human BST-2 for efficient virus release (43). Instead, SIVcpz—like most other SIVs—relies on Nef to overcome the BST-2-imposed restriction (43, 44, 49). Nef targets a 5-amino-acid motif in the cytoplasmic domain of simian BST-2 (43, 44, 49). Of note, this motif is not found in human BST-2 and appears to have been lost more than 800,000 years ago (69). The transmission of SIV to humans is a fairly recent event, and it is interesting that during adaptation to humans, the virus somehow shifted the burden of antagonizing BST-2 from Nef to Vpu (reviewed in reference 70). This switch seems to have been successful only in group M HIV-1 isolates, since the less common HIV-1 groups N, O, and P, which arose from independent zoonotic transmissions, seemed to have failed to evolve an effective BST-2 antagonist (43, 71).
Unlike Nef, which targets BST-2 via its cytoplasmic domain, HIV-1 Vpu has evolved to engage human BST-2 primarily via its TM domain in a species-specific manner (41–46). Because of this species specificity, Vpu was not predicted to support HIV-1 replication in macaques. Indeed, HIV-1 is unable to replicate in macaques, not only because of a BST-2 restriction but also because of restrictions imposed by APOBEC3G and Trim5α (72, 73). Nevertheless, the macaque system has gained importance for HIV research and is currently the most widely used nonhuman primate model for vaccine development and the study of host immune responses. To bypass the restrictions encountered by HIV-1, investigators are using chimeric SIV variants that carry HIV-1 env sequences in conjunction with SIV gag, pol, vif, and nef. Because of the overlap of the vpu open reading frame with the env gene, simian-human immunodeficiency virus (SHIV) chimeras typically also carry the HIV-1 vpu gene. Surprisingly, experiments performed with SHIV-infected pig-tailed macaques suggested a correlation between Vpu expression and viral pathogenesis (74, 75). Of note, the increased pathogenic potential of SHIV in infected pig-tailed macaques mapped to the Vpu TM domain, pointing to BST-2 as the target of Vpu (76). Indeed, our own results gained from SHIV-infected rhesus macaques demonstrate that at least some HIV-1 Vpu proteins have an expanded host range and are capable of targeting rhesus macaque as well as human BST-2 (47).
Previous studies had used coimmunoprecipitation assays (26, 28, 29, 63, 65), FRET (64), and NMR spectroscopy (31) to demonstrate the physical interaction of Vpu with human BST-2 via their TM domains. Our approach was to assess the mechanistic basis of the expanded Vpu host range by studying Vpu-BST-2 interactions using a bimolecular fluorescence complementation (BiFC) assay as reported previously (25). Our results showing the physical interaction between HIV-1 Vpu and huBST-2 by a BiFC assay in this study faithfully reproduce previous results from co-IP studies (26–28, 63), from a FRET study (64), and from a study using NMR (31). In addition, our assay also reproduced the less-efficient interactions of VpuRD and Vpu14/22 (A14L W22A) with huBST-2 previously shown by co-IP (28, 65), by FRET (64), or by BiFC assay (25). Indeed, using this assay, we were able to document robust interactions of HIV-1 Vpu with huBST-2 via their TM domains, previously shown by other groups (26, 28, 29, 63, 65). However, our results also make clear that the binding of Vpu to BST-2 is not sufficient for BST-2 antagonism, since DH12 Vpu, but not NL4-3 Vpu, was able to antagonize rhBST-2 even though these two Vpu proteins interacted equally well with rhBST-2 in our BiFC assay (Fig. 1B). The postbinding steps required for BST-2 antagonism remain unclear. However, we found that the introduction of an I48T mutation into the rhBST-2 TM domain, which rendered the protein sensitive to antagonism by NL4-3 Vpu (48), did not affect the protein-protein interaction in our current analysis (Fig. 1B). Along the same lines, mutating a single residue in the TM domain of NL4-3 Vpu (A18L) resulted in a loss of activity against huBST-2 (65) without affecting the protein-protein interaction in our BiFC assay (data not shown). Thus, we can conclude that the binding of Vpu to BST-2 is necessary but not sufficient for functional antagonism.
As expected from our functional studies, Vpu also interacted with rhBST-2 in our BiFC assay (Fig. 1B, 2B, and 5B). Surprisingly, however, and in contrast to the interaction with huBST-2, the interaction of Vpu with rhBST-2 involved sequences in the cytoplasmic domain of BST-2. Even more surprisingly, the cytoplasmic sequences in rhBST-2 involved in the interaction with Vpu mapped to the same 5-amino-acid motif [(G/D)DIWK] that is missing from huBST-2 and that forms the target of SIV Nef. The interaction of Vpu with the cytoplasmic domain was functionally relevant and was sufficient to confer at least partial antagonistic activity on a Vpu variant (Vpu14/22) (Fig. 4E and F) previously shown to be nonfunctional due to a lack of interaction with human BST-2 (65). The results from our current study lead us to amend the previous assessment that Vpu targets BST-2 through TM domain interactions by concluding that, at least for macaque and chimpanzee BST-2, the cytoplasmic domain of BST-2 contributes to the physical interaction with Vpu. The fact that HIV-1 Vpu has the ability to interact with a sequence motif not found in human BST-2 raises interesting evolutionary questions, since the sequence is present in the BST-2 of the chimpanzee, which is the natural host for the Vpu-encoding SIVcpz. Yet Vpu from SIVcpz does not antagonize chimpanzee BST-2 (43). Instead, the five-residue motif is necessary for antagonism by SIVcpz Nef (50, 51). Our results suggest that SIVcpzMB897 Vpu fails to antagonize chimpBST-2 due to a lack of physical interaction with chimpBST-2 (Fig. 6). This is suggested by the fact that replacing the cytoplasmic domain of SIVcpz Vpu with that of HIV-1NL4-3 Vpu renders the protein capable of both physically interacting with and functionally antagonizing chimpBST-2. Taking our results together, our study revealed the importance of the Vpu cytoplasmic domain for the interaction with the (G/D)DIWK motif in monkey and great ape BST-2.
Investigations to determine what sequences in Vpu are involved in the interaction with the BST-2 cytoplasmic domain are ongoing. However, based on the positioning of the (G/D)DIWK motif relative to the BST-2 transmembrane domain, we would predict that the sequences in Vpu required for the interaction with the (G/D)DIWK motif map to one or both of its cytoplasmic helixes, a situation similar to that described for the interaction of Vpu with the cytoplasmic domain of CD4 (77, 78).
ACKNOWLEDGMENTS
We thank Eri Miyagi, Amy Andrew, Chia-Yen Chen, Sandra Kao, Sarah Welbourn, Robert C. Walker, Jr., Tomoko Kobayashi, Masashi Shingai, and Haruka Yoshii-Kamiyama for helpful suggestions and critical comments on the manuscript. We thank Alicia Buckler-White and Ronald Plishka for conducting nucleotide sequence analyses. We further thank F. Kirchhoff and J. Martin-Serrano for reagents.
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health (K.S.) and by grants from the Grants in-Aid for Scientific Research (24115008 and 24390112) and Research on HIV/AIDS from the Ministry of Health, Labor and Welfare of Japan (to Y.K.).
Footnotes
Published ahead of print 9 October 2013
REFERENCES
- 1.Ohtomo T, Sugamata Y, Ozaki Y, Ono K, Yoshimura Y, Kawai S, Koishihara Y, Ozaki S, Kosaka M, Hirano T, Tsuchiya M. 1999. Molecular cloning and characterization of a surface antigen preferentially overexpressed on multiple myeloma cells. Biochem. Biophys. Res. Commun. 258:583–591 [DOI] [PubMed] [Google Scholar]
- 2.Goto T, Kennel SJ, Abe M, Takishita M, Kosaka M, Solomon A, Saito S. 1994. A novel membrane antigen selectively expressed on terminally differentiated human B cells. Blood 84:1922–1930 [PubMed] [Google Scholar]
- 3.Ishikawa J, Kaisho T, Tomizawa H, Lee BO, Kobune Y, Inazawa J, Oritani K, Itoh M, Ochi T, Ishihara K, Hirano T. 1995. Molecular cloning and chromosomal mapping of a bone marrow stromal cell surface gene, BST2, that may be involved in pre-B-cell growth. Genomics 26:527–534 [DOI] [PubMed] [Google Scholar]
- 4.Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G. 2003. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 4:694–709 [DOI] [PubMed] [Google Scholar]
- 5.Hinz A, Miguet N, Natrajan G, Usami Y, Yamanaka H, Renesto P, Hartlieb B, McCarthy AA, Simorre JP, Gottlinger H, Weissenhorn W. 2010. Structural basis of HIV-1 tethering to membranes by the BST-2/tetherin ectodomain. Cell Host Microbe 7:314–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schubert HL, Zhai Q, Sandrin V, Eckert DM, Garcia-Maya M, Saul L, Sundquist WI, Steiner RA, Hill CP. 2010. Structural and functional studies on the extracellular domain of BST2/tetherin in reduced and oxidized conformations. Proc. Natl. Acad. Sci. U. S. A. 107:17951–17956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H, Wang J, Jia X, McNatt MW, Zang T, Pan B, Meng W, Wang HW, Bieniasz PD, Xiong Y. 2010. Structural insight into the mechanisms of enveloped virus tethering by tetherin. Proc. Natl. Acad. Sci. U. S. A. 107:18428–18432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swiecki M, Scheaffer SM, Allaire M, Fremont DH, Colonna M, Brett TJ. 2011. Structural and biophysical analysis of BST-2/tetherin ectodomains reveals an evolutionary conserved design to inhibit virus release. J. Biol. Chem. 286:2987–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andrew AJ, Kao S, Strebel K. 2011. C-terminal hydrophobic region in human bone marrow stromal cell antigen 2 (BST-2)/tetherin protein functions as second transmembrane motif. J. Biol. Chem. 286:39967–39981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrew AJ, Miyagi E, Kao S, Strebel K. 2009. The formation of cysteine-linked dimers of BST-2/tetherin is important for inhibition of HIV-1 virus release but not for sensitivity to Vpu. Retrovirology 6:80. 10.1186/1742-4690-6-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, Bieniasz PD. 2009. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139:499–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masuyama N, Kuronita T, Tanaka R, Muto T, Hirota Y, Takigawa A, Fujita H, Aso Y, Amano J, Tanaka Y. 2009. HM1.24 is internalized from lipid rafts by clathrin-mediated endocytosis through interaction with α-adaptin. J. Biol. Chem. 284:15927–15941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rollason R, Korolchuk V, Hamilton C, Schu P, Banting G. 2007. Clathrin-mediated endocytosis of a lipid-raft-associated protein is mediated through a dual tyrosine motif. J. Cell Sci. 120:3850–3858 [DOI] [PubMed] [Google Scholar]
- 14.Fritz JV, Tibroni N, Keppler OT, Fackler OT. 2012. HIV-1 Vpu's lipid raft association is dispensable for counteraction of the particle release restriction imposed by CD317/tetherin. Virology 424:33–44 [DOI] [PubMed] [Google Scholar]
- 15.Lopez LA, Yang SJ, Exline CM, Rengarajan S, Haworth KG, Cannon PM. 2012. Anti-tetherin activities of HIV-1 Vpu and Ebola virus glycoprotein do not involve removal of tetherin from lipid rafts. J. Virol. 86:5467–5480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neil SJ, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430 [DOI] [PubMed] [Google Scholar]
- 18.Neil SJ, Sandrin V, Sundquist WI, Bieniasz PD. 2007. An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe 2:193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jouvenet N, Neil SJ, Zhadina M, Zang T, Kratovac Z, Lee Y, McNatt M, Hatziioannou T, Bieniasz PD. 2009. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 83:1837–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. 2009. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 106:2886–2891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakuma T, Noda T, Urata S, Kawaoka Y, Yasuda J. 2009. Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 83:2382–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnaud F, Black SG, Murphy L, Griffiths DJ, Neil SJ, Spencer TE, Palmarini M. 2010. Interplay between ovine bone marrow stromal cell antigen 2/tetherin and endogenous retroviruses. J. Virol. 84:4415–4425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mattiuzzo G, Ivol S, Takeuchi Y. 2010. Regulation of porcine endogenous retrovirus release by porcine and human tetherins. J. Virol. 84:2618–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le Tortorec A, Neil SJ. 2009. Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J. Virol. 83:11966–11978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi T, Ode H, Yoshida T, Sato K, Gee P, Yamamoto SP, Ebina H, Strebel K, Sato H, Koyanagi Y. 2011. Identification of amino acids in the human tetherin transmembrane domain responsible for HIV-1 Vpu interaction and susceptibility. J. Virol. 85:932–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rong L, Zhang J, Lu J, Pan Q, Lorgeoux RP, Aloysius C, Guo F, Liu SL, Wainberg MA, Liang C. 2009. The transmembrane domain of BST-2 determines its sensitivity to down-modulation by HIV-1 Vpu. J. Virol. 83:7536–7546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mangeat B, Gers-Huber G, Lehmann M, Zufferey M, Luban J, Piguet V. 2009. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its β-TrCP2-dependent degradation. PLoS Pathog. 5:e1000574. 10.1371/journal.ppat.1000574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dubé M, Roy BB, Guiot-Guillain P, Binette J, Mercier J, Chiasson A, Cohen EA. 2010. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 6:e1000856. 10.1371/journal.ppat.1000856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Fruh K, Moses AV. 2009. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/tetherin via a βTrCP-dependent mechanism. J. Virol. 83:7931–7947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vigan R, Neil SJ. 2011. Separable determinants of subcellular localization and interaction account for the inability of group O HIV-1 Vpu to counteract tetherin. J. Virol. 85:9737–9748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skasko M, Wang Y, Tian Y, Tokarev A, Munguia J, Ruiz A, Stephens EB, Opella SJ, Guatelli J. 2012. HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J. Biol. Chem. 287:58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou J, Zhang Z, Mi Z, Wang X, Zhang Q, Li X, Liang C, Cen S. 2012. Characterization of the interface of the bone marrow stromal cell antigen 2-Vpu protein complex via computational chemistry. Biochemistry 51:1288–1296 [DOI] [PubMed] [Google Scholar]
- 33.Kueck T, Neil SJD. 2012. A cytoplasmic tail determinant in HIV-1 Vpu mediates targeting of tetherin for endosomal degradation and counteracts interferon-induced restriction. PLoS Pathog. 8:e1002609. 10.1371/journal.ppat.1002609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartee E, McCormack A, Fruh K. 2006. Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog. 2:e107. 10.1371/journal.ppat.0020107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mansouri M, Viswanathan K, Douglas JL, Hines J, Gustin J, Moses AV, Fruh K. 2009. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi's sarcoma-associated herpesvirus. J. Virol. 83:9672–9681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fitzpatrick K, Skasko M, Deerinck TJ, Crum J, Ellisman MH, Guatelli J. 2010. Direct restriction of virus release and incorporation of the interferon-induced protein BST-2 into HIV-1 particles. PLoS Pathog. 6:e1000701. 10.1371/journal.ppat.1000701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Habermann A, Krijnse-Locker J, Oberwinkler H, Eckhardt M, Homann S, Andrew A, Strebel K, Krausslich HG. 2010. CD317/tetherin is enriched in the HIV-1 envelope and downregulated from the plasma membrane upon virus infection. J. Virol. 84:4646–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hammonds J, Wang JJ, Yi H, Spearman P. 2010. Immunoelectron microscopic evidence for tetherin/BST2 as the physical bridge between HIV-1 virions and the plasma membrane. PLoS Pathog. 6:e1000749. 10.1371/journal.ppat.1000749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones PH, Mehta HV, Maric M, Roller RJ, Okeoma CM. 2012. Bone marrow stromal cell antigen 2 (BST-2) restricts mouse mammary tumor virus (MMTV) replication in vivo. Retrovirology 9:10. 10.1186/1742-4690-9-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andrew AJ, Berndsen CE, Kao S, Strebel K. 2012. The size and conservation of a coiled-coil structure in the ectodomain of human BST-2/tetherin is dispensable for inhibition of HIV-1 virion release. J. Biol. Chem. 287:44278–44288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNatt MW, Zang T, Hatziioannou T, Bartlett M, Fofana IB, Johnson WE, Neil SJ, Bieniasz PD. 2009. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 5:e1000300. 10.1371/journal.ppat.1000300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupta RK, Hue S, Schaller T, Verschoor E, Pillay D, Towers GJ. 2009. Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog. 5:e1000443. 10.1371/journal.ppat.1000443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sauter D, Schindler M, Specht A, Landford WN, Munch J, Kim KA, Votteler J, Schubert U, Bibollet-Ruche F, Keele BF, Takehisa J, Ogando Y, Ochsenbauer C, Kappes JC, Ayouba A, Peeters M, Learn GH, Shaw G, Sharp PM, Bieniasz P, Hahn BH, Hatziioannou T, Kirchhoff F. 2009. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 6:409–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, Johnson WE, Westmoreland S, Evans DT. 2009. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 5:e1000429. 10.1371/journal.ppat.1000429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goffinet C, Allespach I, Homann S, Tervo HM, Habermann A, Rupp D, Oberbremer L, Kern C, Tibroni N, Welsch S, Krijnse-Locker J, Banting G, Krausslich HG, Fackler OT, Keppler OT. 2009. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5:285–297 [DOI] [PubMed] [Google Scholar]
- 46.Tokarev A, Skasko M, Fitzpatrick K, Guatelli J. 2009. Antiviral activity of the interferon-induced cellular protein BST-2/tetherin. AIDS Res. Hum. Retroviruses 25:1197–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shingai M, Yoshida T, Martin MA, Strebel K. 2011. Some human immunodeficiency virus type 1 Vpu proteins are able to antagonize macaque BST-2 in vitro and in vivo: Vpu-negative simian-human immunodeficiency viruses are attenuated in vivo. J. Virol. 85:9708–9715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshida T, Kao S, Strebel K. 2011. Identification of residues in the BST-2 TM domain important for antagonism by HIV-1 Vpu using a gain-of-function approach. Front. Microbiol. 2:35. 10.3389/fmicb.2011.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, Munch J, Kirchhoff F, Bieniasz PD, Hatziioannou T. 2009. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6:54–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang SJ, Lopez LA, Hauser H, Exline CM, Haworth KG, Cannon PM. 2010. Anti-tetherin activities in Vpu-expressing primate lentiviruses. Retrovirology 7:13. 10.1186/1742-4690-7-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lim ES, Malik HS, Emerman M. 2010. Ancient adaptive evolution of tetherin shaped the functions of Vpu and Nef in human immunodeficiency virus and primate lentiviruses. J. Virol. 84:7124–7134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klimkait T, Strebel K, Hoggan MD, Martin MA, Orenstein JM. 1990. The human immunodeficiency virus type 1-specific protein Vpu is required for efficient virus maturation and release. J. Virol. 64:621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyagi E, Andrew AJ, Kao S, Strebel K. 2009. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. U. S. A. 106:2868–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL. 1995. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene 164:49–53 [DOI] [PubMed] [Google Scholar]
- 56.Nguyen KL, Llano M, Akari H, Miyagi E, Poeschla EM, Strebel K, Bour S. 2004. Codon optimization of the HIV-1 vpu and vif genes stabilizes their mRNA and allows for highly efficient Rev-independent expression. Virology 319:163–175 [DOI] [PubMed] [Google Scholar]
- 57.Yoshida T, Ebina H, Koyanagi Y. 2009. N-linked glycan-dependent interaction of CD63 with CXCR4 at the Golgi apparatus induces downregulation of CXCR4. Microbiol. Immunol. 53:629–635 [DOI] [PubMed] [Google Scholar]
- 58.Bour S, Akari H, Miyagi E, Strebel K. 2003. Naturally occurring amino acid substitutions in the HIV-2 ROD envelope glycoprotein regulate its ability to augment viral particle release. Virology 309:85–98 [DOI] [PubMed] [Google Scholar]
- 59.Kerppola TK. 2006. Complementary methods for studies of protein interactions in living cells. Nat. Methods 3:969–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kerppola TK. 2006. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 1:1278–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ueyama T, Kusakabe T, Karasawa S, Kawasaki T, Shimizu A, Son J, Leto TL, Miyawaki A, Saito N. 2008. Sequential binding of cytosolic Phox complex to phagosomes through regulated adaptor proteins: evaluation using the novel monomeric Kusabira-Green system and live imaging of phagocytosis. J. Immunol. 181:629–640 [DOI] [PubMed] [Google Scholar]
- 62.Schubert U, Bour S, Ferrer-Montiel AV, Montal M, Maldarell F, Strebel K. 1996. The two biological activities of human immunodeficiency virus type 1 Vpu protein involve two separable structural domains. J. Virol. 70:809–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iwabu Y, Fujita H, Kinomoto M, Kaneko K, Ishizaka Y, Tanaka Y, Sata T, Tokunaga K. 2009. HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 284:35060–35072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banning C, Votteler J, Hoffmann D, Koppensteiner H, Warmer M, Reimer R, Kirchhoff F, Schubert U, Hauber J, Schindler M. 2010. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PLoS One 5:e9344. 10.1371/journal.pone.0009344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vigan R, Neil SJ. 2010. Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. J. Virol. 84:12958–12970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao F, Bailes E, Robertson DL, Chen Y, Rodenburg CM, Michael SF, Cummins LB, Arthur LO, Peeters M, Shaw GM, Sharp PM, Hahn BH. 1999. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 397:436–441 [DOI] [PubMed] [Google Scholar]
- 67.Van Heuverswyn F, Li Y, Bailes E, Neel C, Lafay B, Keele BF, Shaw KS, Takehisa J, Kraus MH, Loul S, Butel C, Liegeois F, Yangda B, Sharp PM, Mpoudi-Ngole E, Delaporte E, Hahn BH, Peeters M. 2007. Genetic diversity and phylogeographic clustering of SIVcpzPtt in wild chimpanzees in Cameroon. Virology 368:155–171 [DOI] [PubMed] [Google Scholar]
- 68.Andrew A, Strebel K. 2010. HIV-1 Vpu targets cell surface markers CD4 and BST-2 through distinct mechanisms. Mol. Aspects Med. 31:407–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sauter D, Vogl M, Kirchhoff F. 2011. Ancient origin of a deletion in human BST2/tetherin that confers protection against viral zoonoses. Hum. Mutat. 32:1243–1245 [DOI] [PubMed] [Google Scholar]
- 70.Sauter D, Specht A, Kirchhoff F. 2010. Tetherin: holding on and letting go. Cell 141:392–398 [DOI] [PubMed] [Google Scholar]
- 71.Yang SJ, Lopez LA, Exline CM, Haworth KG, Cannon PM. 2011. Lack of adaptation to human tetherin in HIV-1 group O and P. Retrovirology 8:78. 10.1186/1742-4690-8-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gaddis NC, Sheehy AM, Ahmad KM, Swanson CM, Bishop KN, Beer BE, Marx PA, Gao F, Bibollet-Ruche F, Hahn BH, Malim MH. 2004. Further investigation of simian immunodeficiency virus Vif function in human cells. J. Virol. 78:12041–12046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kratovac Z, Virgen CA, Bibollet-Ruche F, Hahn BH, Bieniasz PD, Hatziioannou T. 2008. Primate lentivirus capsid sensitivity to TRIM5 proteins. J. Virol. 82:6772–6777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McCormick-Davis C, Zhao LJ, Mukherjee S, Leung K, Sheffer D, Joag SV, Narayan O, Stephens EB. 1998. Chronology of genetic changes in the vpu, env, and nef genes of chimeric simian-human immunodeficiency virus (strain HXB2) during acquisition of virulence for pig-tailed macaques. Virology 248:275–283 [DOI] [PubMed] [Google Scholar]
- 75.Stephens EB, McCormick C, Pacyniak E, Griffin D, Pinson DM, Sun F, Nothnick W, Wong SW, Gunderson R, Berman NE, Singh DK. 2002. Deletion of the vpu sequences prior to the env in a simian–human immunodeficiency virus results in enhanced Env precursor synthesis but is less pathogenic for pig-tailed macaques. Virology 293:252–261 [DOI] [PubMed] [Google Scholar]
- 76.Hout DR, Gomez ML, Pacyniak E, Gomez LM, Inbody SH, Mulcahy ER, Culley N, Pinson DM, Powers MF, Wong SW, Stephens EB. 2005. Scrambling of the amino acids within the transmembrane domain of Vpu results in a simian-human immunodeficiency virus (SHIVTM) that is less pathogenic for pig-tailed macaques. Virology 339:56–69 [DOI] [PubMed] [Google Scholar]
- 77.Hill MS, Ruiz A, Schmitt K, Stephens EB. 2010. Identification of amino acids within the second alpha helical domain of the human immunodeficiency virus type 1 Vpu that are critical for preventing CD4 cell surface expression. Virology 397:104–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tiganos E, Yao XJ, Froborg J, Daniel N, Cohen EA. 1997. Putative α-helical structures in the human immunodeficiency virus type 1 Vpu protein and CD4 are involved in binding and degradation of the CD4 molecule. J. Virol. 71:4452–4460 [DOI] [PMC free article] [PubMed] [Google Scholar]