

Abstract
Polyomaviruses have repeating sequences at their origins of replication that bind the origin-binding domain of virus-encoded large T antigen. In murine polyomavirus, the central region of the origin contains four copies (P1 to P4) of the sequence G(A/G)GGC. They are arranged as a pair of inverted repeats with a 2-bp overlap between the repeats at the center. In contrast to simian virus 40 (SV40), where the repeats are nonoverlapping and all four repeats can be simultaneously occupied, the crystal structure of the four central murine polyomavirus sequence repeats in complex with the polyomavirus origin-binding domain reveals that only three of the four repeats (P1, P2, and P4) are occupied. Isothermal titration calorimetry confirms that the stoichiometry is the same in solution as in the crystal structure. Consistent with these results, mutation of the third repeat has little effect on DNA replication in vivo. Thus, the apparent 2-fold symmetry within the DNA repeats is not carried over to the protein-DNA complex. Flanking sequences, such as the AT-rich region, are known to be important for DNA replication. When the orientation of the central region was reversed with respect to these flanking regions, the origin was still able to replicate and the P3 sequence (now located at the P2 position with respect to the flanking regions) was again dispensable. This highlights the critical importance of the precise sequence of the region containing the pentamers in replication.
INTRODUCTION
Polyomaviruses are icosahedral viruses with circular genomes approximately 5 kb long. These viruses encode three capsid proteins, as well as two or three isoforms of T antigen. The longest of these, large T antigen (LT), is a multifunctional protein that is responsible for initiation of viral replication in all polyomaviruses. Historically, the replication functions of LT have been mostly studied in the simian virus 40 (SV40) system. In contrast, murine polyomavirus (Py), the subject of this study, has been studied primarily in the context of cellular transformation. In SV40, LT is primarily responsible for this activity, whereas in Py, LT is much less important than other T antigen isoforms in this respect (1).
LTs are capable of recognizing the viral origin of replication, melting the DNA, and unwinding the duplex in an ATP-dependent reaction. These proteins also recruit host factors, including replication protein A, topoisomerase I, and the polymerase alpha-primase complex, to the site of replication initiation (2). LTs are composed of functional domains that are thought to be connected via flexible linkers. The N-terminal J domain and the linker following this domain are essential for regulation of the host cell phenotype and play a role in viral DNA replication in vivo (3), but these domains are dispensable for replication in vitro (4). Recognition of the origin is largely the role of the LT origin-binding domain (OBD), which is ∼50% identical between polyomaviruses. In addition to binding DNA, the Py OBD can also activate transcription (5). The OBD is connected via a short linker to the helicase domain, which interacts with double-stranded DNA (dsDNA) before the origin is melted (6, 7) and assembles as a hexameric ring around single-stranded DNA (ssDNA) after melting. Two such rings are thought to form a head-to-head, dodecameric complex at the origin when replication begins (8–10).
The minimal origin of replication in SV40 is defined by three sequence motifs, a highly conserved AT-rich region on the late side of the origin, a central region containing GAGGC repeats, and a poorly conserved early palindrome (EP) region on the early side of the origin (Fig. 1A; see Fig. S1 in the supplemental material). The central region is normally referred to as site 1/2 in the Py literature and as site II in the SV40 literature. For clarity, we will use the older term PEN to refer to this pentamer-containing region in all polyomaviruses.
Fig 1.

Polyomavirus origin sequences. (A) Sequences from murine polyomavirus (MuPyV) human polyomavirus (HPyV), MCV, KI virus (KIPyV), WU virus (WUPyV), trichodysplasia spinulosa-associated polyomavirus (TSV), SV40, JC virus (JCV), and BK virus (BKV). The pentamer designations (P1 to P4) are based on the Py nomenclature. The underlined sequences indicate the SV40 “hidden site” that interacts with the LT OBD and the region of the EP that interacts with the LT helicase domain β-hairpin (PDB ID 4GDF). Accession numbers for all sequences are provided in Table S2 in the supplemental material. (B) Both strands of the PEN region for Py and SV40 sequences are shown. The G(A/G)GGC sequences are colored, and their directionality is indicated by arrows in both parts of the figure.
The OBDs recognize the pentameric sequence G(A/G)GGC, which appears multiple times at and near the origin of replication (Fig. 1A). Crystal structures of the OBD from SV40 and Merkel cell polyomavirus (MCV) in complex with PEN sequences have demonstrated that the OBD can interact via the major groove with all five base pairs (11–13). The GAGGC sequences in the PEN region of the SV40, BK virus (BKV), and JC virus (JCV) genomes are arranged as a pair of inverted repeats with a 1-bp spacer between the pentamers. The PEN repeats in Py are arranged as an inverted repeat, but in this case, the inner pair of repeats overlap by 2 bp. In Py, there is also a 2-bp spacer between the inner and outer pentamers instead of the 1 bp seen in SV40 (Fig. 1B).
In SV40, all four PEN repeats can be simultaneously bound by the purified SV40 OBD (12, 13), and mutations in any of the four abrogate efficient replication (2). In MCV, a human polyomavirus associated with Merkel cell carcinoma, the sequence at P3 is noncanonical; it has the sequence GGAGC rather than GGGGC. Mutations in this noncanonical repeat are tolerated in replication assays (14), and the repeat is not bound by the isolated OBD (13). The Py origin contains features of both SV40 and MCV. Like SV40, it has four perfect G(A/G)GGC repeats within the PEN region, but they are arranged with the same spacing as in MCV. Whereas only four known polyomaviruses have the “canonical” SV40-like spacing, nine viruses have the spacing seen in Py (see Fig. S1 in the supplemental material). To understand how LT interacts with such sequences, we report here the crystal structure of the Py OBD in complex with DNA from the Py PEN region.
In the crystal structure, despite a canonical P3 sequence, we observe binding to only pentamers P1, P2, and P4. This stoichiometry is supported by isothermal titration calorimetry (ITC) experiments. We also report that although the Py pentamer sequences at P2 and P3 are identical, P2 is required for replication, but the P3 sequence is not necessary. These data support a model in which T antigen assembly on the Py origin proceeds via an intrinsically asymmetric mechanism, with one side of the T antigen double hexamer perhaps assembling before the other. We also find that the reversal of the PEN region causes the sequence at P3 to remain dispensable, even though it is now in what would be the P2 position of the wild-type origin (WT-ori). This highlights the crucial importance of the PEN region for replication.
MATERIALS AND METHODS
Sequence alignment.
The sequences of all polyomaviruses for which complete genomes are available were downloaded from the NCBI genome database, and in some cases, the reverse complement was generated. To find the origin, the noncoding region of each viral genome was identified via sequence alignments. The sequences were then manually aligned using Jalview (15) so that the P1 repeats of all the origins were aligned. The sequences of avian polyomaviruses, which lack GAGGC repeats and differ significantly in their OBDs, were omitted from the alignment.
Protein overexpression and purification.
The nucleotide sequence for the murine polyomavirus origin-binding domain residues 290 to 420 was cloned into pET30b with both an N-terminal and a C-terminal hexahistidine tag. The protein was overexpressed in Escherichia coli Rosetta(DE3) cells by first growing a freshly inoculated culture in 2× yeast extract-tryptone (YT) medium at 33°C to an optical density at 600 nm (OD600) of 0.5, dropping the temperature to 18°C, and finally adding IPTG (isopropyl-β-d-thiogalactopyranoside) to a final concentration of 100 μM. The cells were then grown overnight. After overexpression, cells were harvested and resuspended in 50 mM Tris, pH 8.0, 250 mM NaCl, and 5% (vol/vol) glycerol; flash-frozen; and stored at −20°C. The cells were thawed and lysed with a pneumatic microfluidizer. Additional lysis buffer and nickel column equilibration buffer was the same as the resuspension buffer. Whole-cell lysates were centrifuged at 38,000 × g for 30 min. The high-speed supernatant was applied to the nickel affinity column, and the protein was eluted with a gradient from 0 to 400 mM imidazole, pH 8.0, in the same buffer. The eluted protein was dialyzed into 50 mM Tris, pH 7.1, 250 mM NaCl, and 5% (vol/vol) glycerol; concentrated; and applied to a Sephadex S200 size exclusion column. The purified protein was concentrated to 11 mg/ml and stored at −80°C in 20 mM Tris, pH 7.1, 200 mM NaCl, 5% (vol/vol) glycerol, 1 mM EDTA, and 0.1% β-octylglucoside.
Crystallization, data collection, and structure solution.
Oligonucleotides for crystallization were purchased from IDT DNA, purified by high-pressure ion-exchange chromatography, and annealed as previously described (13). Complexes of the Py origin-containing oligonucleotide (Fig. 1B, left) and the OBD were made by mixing 60 nmol of protein and 16.7 nmol of double-stranded oligonucleotide (containing four binding sites) and concentrating to a final volume of 100 μl, resulting in a complex that was about 600 μM in concentration with respect to the protein, with a 1.12 molar excess of potential binding sites. The final buffer for the target crystallization complex was 10 mM Tris, pH 7.1, 100 mM NaCl, 3 mM dithiothreitol, and 1% glycerol. Crystal screening was done using 96-well format sitting drops.
The most productive crystal form grew in 140 mM trisodium citrate, 14% (wt/vol) polyethylene glycol (PEG) 3350 at 4°C. The crystals grew as highly elongated pyramids and were in the space group C2221. The crystals were briefly rinsed in a cryoprotective solution composed of 85% well buffer and 15% ethylene glycol and were immediately frozen in a cryostream prior to storage in liquid nitrogen. The crystals were shipped to the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory in Upton, NY, and data to 3.79-Å resolution were collected at NSLS beamline X-29. The data were reduced and scaled with HKL2000/Scalepack (16) (see Table S1 in the supplemental material).
Molecular replacement as implemented in PHASER (17) was used to solve the crystal structure. Initial search models were created by modeling the Py OBD on the SV40 OBD with Swiss-Model (18). The search model included the OBD monomer, as well as a double-stranded DNA pentamer from Protein Data Bank (PDB) entry 2NTC. PHASER readily found three OBD-DNA models for which translation and rotation functions were meaningful. The remainder of the model was built in COOT (19), and PHENIX (20) was used for refinement. The directionality of the DNA sequence within the crystal was determined based on the electron density at the DNA ends, which are different for the two DNA strands. The DNA sequence crystallized contains 3 nucleotides before the GAGGC sequence on the top strand and only 2 nucleotides before the GAGGC sequence on the bottom strand (Fig. 1B). The model was refined to 3.79-Å resolution with appropriate geometrical and crystallographic constraints. Axial bending of the DNA was analyzed with the program Curves+ (21). The protein-protein interface and the protein-DNA interactions were analyzed using PISA (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html), as well as PDBSUM (http://www.ebi.ac.uk/pdbsum/). The solvent content of crystals was estimated from the Matthew's coefficient.
Isothermal titration calorimetry.
ITC data were collected with an ITC-200 Calorimeter (Microcal, Northhampton, MA). For the four-pentamer oligonucleotide experiment, i.e., the wild type, the top strand had the sequence 5′-AAGCAGAGGCCGGGGGCCCCTGGCCTCCGCTT-3′ and the bottom strand was 5′-AAGCGGAGGCCAGGGGCCCCCGGCCTCTGCTT-3′. For the P1-only experiment, the top strand was 5′-AAGCAGAGGCCGATA-3′, where the boldface indicates the mutation to abolish the P2 PEN. The bottom strand was 5′-TATCGGCCTCTGCT-3′.
The data were analyzed with Origin software provided by the manufacturer. Prior to the experiment, the dsDNA oligonucleotide and protein were buffer exchanged into 10 mM sodium phosphate buffer, pH 7.0, and 50 mM NaCl, using PD-10 columns (GE Healthcare). Protein and DNA concentrations were determined spectrophotometrically, using calculated extinction coefficients from the ProtParam Web server (http://ca.expasy.org/tools/protparam.html) and the IDT DNA website, respectively.
Transient-replication assay.
Plasmid pFLORI40, which contains a firefly luciferase (Fluc) reporter under the control of the cytomegalovirus (CMV) promoter in cis with the polyomavirus origin of replication (modified from reference 22), was used to perform the replication assays. Point mutations were introduced in the “core” polyomavirus origin of the plasmid using site-directed mutagenesis with the QuikChange mutagenesis kit (Agilent) and standard PCR techniques based on Pfu1 polymerase (Stratagene). The primers were as follows: for mutant P1 ori, 5′-ATTAAGCAGATCCCGGGGGCTC-3′; for P2 ori, 5′-AAGCAGAGGCCGATAGCCCCTGGCCTC-3′; for P3 ori, 5′-GAGGCCGGGGGCTATTGGCCTCCGCTT-3′; for P4 ori, 5′-GGGCTCCTGGAGTCCGCTTACTC-3′. To make the origin with SV40 spacing, the primer 5′-GAGGCCGGGGGCCGCCCCTGGCCTCCG-3′ and its reverse complement were used. To make an origin inverted with respect to the AT-rich region, the primer 5′-TGTTTTTTTTAGTATTAAGCGGAGGCCAGGGGCCCCCGGCCTCTGGCTTACTCTGGAGA-3′ was used. All DNA constructs were verified by sequencing. Transient transfections using N,N-bis[2-hydroxyethyl]-2-aminoethanesulfonic acid (BES)-buffered saline were done using NIH 3T3 cells (23). Forty percent confluent cells were transfected with 3 μg of WT-ori Fluc, mutant P1-ori Fluc, mutant P2-ori Fluc, mutant P3-ori Fluc, or mutant P4-ori Fluc, together with 1 μg of polyomavirus LT (5), in 100-mm-diameter plates. Cells were harvested, and low-molecular-weight DNA was extracted 48 h posttransfection using the Hirt extraction protocol (24). The DNA was digested with 5 units of DpnI (New England BioLabs) overnight in a 37°C water bath to remove methylated input DNA. The primers that were used to amplify a 435-bp portion of pFLORI40, including polyomavirus origin sequence, were as follows: pCI, 5′-TGGCGTAATAGCGAAGAG-3′, and PY, 5′-GAAGAGAGGCTTCCAGAG-3′. PCRs were performed using the following PCR amplification conditions: 95°C for 30 s, 37°C for 1 min, and 68°C for 18 min for 10 or 15 thermal cycles. The amplicon was detected using ethidium bromide staining of 1.5% agarose gels and quantified using ImageJ software (http://imagej.nih.gov/ij/).
Replication was also assessed by the expression of luciferase from pFLORI40 using an established procedure (22). CMV beta-galactosidase was also used as a transfection control. After approximately 40 h, cell lysates were prepared and assayed for both luciferase and beta-galactosidase activity.
Protein structure accession number.
Atomic coordinates and structure factors for the Py OBD-DNA cocrystal reported here have been deposited with the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank. The accession code is 4FB3.
RESULTS
The OBD from murine polyomavirus (residues 291 to 420) was crystallized in complex with duplex DNA encoding the PEN sequence, with all four of the G(A/G)GGC repeats at the Py origin. A notable feature of these crystals is their exceptionally large solvent channels. These channels are approximately 90 Å across. The structure, which contains one DNA duplex and three copies of the Py OBD in the asymmetric unit, was solved by molecular replacement and refined to 3.79-Å resolution (Fig. 2A; see Table S1 in the supplemental material). Extensive efforts to obtain better-diffracting crystals of the complex were unsuccessful. It is important to note that the 69.5% solvent content of the crystals (Matthews coefficient = 4.0) resulted in significantly more data per atom than is typical at this resolution. Therefore, these data yielded surprisingly clear electron density maps (see Fig. S2 in the supplemental material).
Fig 2.

Structure of the Py OBD-DNA complex. (A) Ribbon representation of the protein and DNA within the asymmetric-unit cell of the crystal structure. (B) Molecule in panel A rotated to highlight the 30° bend in the DNA.
The program Curves+ (25) indicated that the DNA in the crystal is bent by 30° between the P1 and P2 binding sites (Fig. 2B). The bending does not significantly alter the conformation of the DNA at the potential P3 binding site, where the DNA is essentially B form. Interestingly, though the OBDs from Py and MCV are very similar (root mean square deviation [RMSD], ∼1.2 Å), the DNAs in the respective costructures are bent differently. The bending in the MCV OBD-DNA costructure causes the head-to-head-oriented OBDs at P2 and P4 to contact one another, forming a small protein-protein interface that has little effect on DNA binding when mutated (13). In the Py structure, the DNA in this region is straighter and the OBDs do not touch, a situation similar to that seen in SV40 (11, 12).
The OBDs in the crystal are positioned over the P1, P2, and P4 repeats. Though the P3 repeat has the same sequence (GGGGC) as P2, there is no OBD associated with this pentamer. The OBD seems to be missing from this site because the OBD bound to P2 occludes access to the P3 site. Modeling reveals that the side chains of N363 and N307 of a hypothetical P3 OBD clash in the major groove of the DNA with side chains Y305 and R357, respectively, of the OBD at P2. Steric occlusion of P3 in the major groove was also noted in the crystal structure of the MCV OBD in complex with the analogous region of the MCV origin (13). In the earlier structure, the interpretation was somewhat more complicated, because in MCV, the P3 site does not contain a canonical OBD-binding sequence. In addition, since the hypothetical side chain clash involving OBDs at P2 and P3 was relatively minor, it was difficult to determine if small changes in the DNA and/or protein structure might allow both OBDs to bind if the DNA contained a canonical binding site at P3.
In the crystal structure reported here, the structural environments at P2 and P3 are essentially identical. Thus, this structure does not explain why the site at P2 is preferred over P3, although the functional data discussed below show this to be the case. No base-specific contacts outside the G(A/G)GGC sequences are observed. Flanking sequences may exert a subtle effect on the DNA conformation, resulting in a difference in binding affinity. Alternatively, crystal-packing forces could help dictate which of the mutually exclusive sites is occupied. Notably, though the contents of the unit cells in the MCV and Py structures were similar (three OBDs and two 26-mer DNA oligonucleotides), there is no similarity in crystal packing between the Py and MCV OBD structures with their respective PEN sequences. As discussed below, binding studies show that in both MCV and Py, only three OBDs bind the DNA. The failure to see OBD binding to P3 is superficially at odds with older methylation interference results showing that the third and fourth Gs of P3 are important in Py LT binding (26). However, examination of the crystal structure reveals that occupancy of the OBD at P2 (which partially overlaps P3) would be affected by these methylations.
The limited resolution of the crystal structure curtails our ability to make definitive statements regarding specific atomic contacts between the DNA and the protein. There are small differences among the three crystallographically unique OBD-DNA complexes, but overall, the models are quite similar, with an RMSD of 0.55 Å on all Cα atoms. The consensus of the three OBD-DNA interactions observed in this structure is summarized in Fig. 3, left. The contacts to the DNA are similar to those seen in the analogous crystal structures of the SV40 and MCV OBDs in complex with DNA from the equivalent regions of the respective viral origins (11–13). The OBD sequences are approximately 50% identical among the three viruses, and the DNA consensus sequences are the same for all three.
Fig 3.
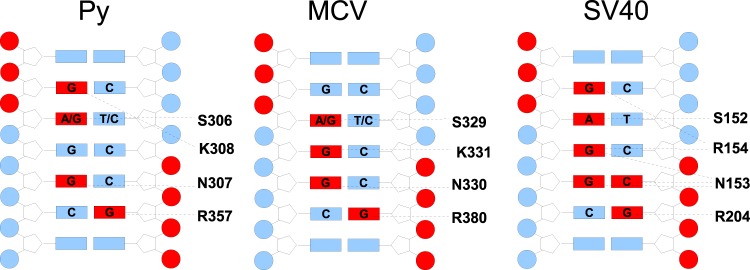
Contacts between the OBD and DNA. The interaction data derived from crystal structures of the SV40 and MCV OBDs in complex with DNA sequences from the origins of these viruses is compared to that of Py. In each case, bases and phosphates colored red indicate protein contacts. Amino acids that make base-specific interactions are noted.
We used ITC to test whether the stoichiometry of binding is the same in solution as in the crystals (Fig. 4). The titration of the Py OBD and the entire PEN region was performed three times, with similar results each time. Consistent with the crystallographic result, the ITC data fit best to a model with three noninteracting binding sites overall. One of these sites exhibits ∼21 nM affinity for the OBD. The remaining two sites exhibit ∼625 nM affinity. We were surprised to see two different types of sites. In MCV and SV40, the three and four sites, respectively, that are present in this region of the origin can be easily modeled using the assumption that they have similar affinities (13). MCV has affinity similar to those of the lower-affinity Py sites, while SV40 affinity is more similar to that of the high-affinity Py site. The affinities and stoichiometries observed in all three viruses are presented in Table 1. Additional ITC experiments were performed to better understand the roles of individual pentamers in OBD binding. P1 on its own clearly binds quite tightly (KD [dissociation constant], ∼37 nM) (Fig. 4B). However, an analogous isolated P4 oligonucleotide has similar affinity (data not shown). Moreover, experiments involving the mutated PEN sequences P2 and P3 do not paint a consistent picture, since mutation of some sites seems to alter the affinity of others (data not shown).
Fig 4.

Isothermal titration calorimetry of the Py OBD and DNA. (A) Titration using the entire PEN region. (B) Titration using a DNA oligonucleotide containing only P1. In both cases, the calorimetric trace is shown in the inset, with time (min) on the x axis and power (μcal/s) on the y axis. The stoichiometry and association constants are determined from curve fitting of the integrated calorimetric trace and are reported in Table 1.
Table 1.
Isothermal titration calorimetry
| Protein | DNA | KDa(nM) | n |
|---|---|---|---|
| Py | P1-P2-P3-P4 | 21 and 625 | 0.87 and 2.30 |
| Py | P1 only | 38 | 1.06 |
| MCV | P1-P2-P3-P4 | 741 | 2.79 |
| SV40 | P1-P2-P3-P4 | 93 | 3.57 |
KD, equilibrium dissociation constant n, stoichiometry.
The crystallography and ITC results raise questions about the arrangement of the repeats in the core origin and, in particular, the functional role of P3. To assess this, we analyzed the effects of mutations in the origin sequence using transient-replication assays. Two different wild-type Py strains, A2 and NG59RA, were compared. NG59RA has thymine substituted for one of the cytosine bases in P3. Site-directed mutagenesis was used to create mutant origins P1 to P4 in the context of each strain (Fig. 5A). A2 and NG59RA were found to replicate with similar efficiencies in these assays, and for both, the P3 mutant origin yielded replicated DNA at levels comparable to those of the wild-type ori-containing plasmid (Fig. 5B). In contrast, mutated replication origins P1, P2, and P4 showed little DNA replication, despite similar levels of LT expression. This confirms the importance of these sites for replication. A second type of replication assay (22) was used to confirm these results. In this assay, replication is tested by luciferase expression from a viral origin-containing plasmid. Amplification of the plasmid leads to increased luciferase-catalyzed light production. LT that is defective in replication has little effect on luciferase-generated light, while inhibition of DNA replication by mimosine blocks the increase in luciferase caused by LT (not shown). Whereas mutations in P1, P2, and P4 caused a significant decrease in replication, as measured by luciferase activity, mutation of P3 had little effect (Fig. 5C). Our results clearly validate this simpler assay. Both of the experimental approaches are consistent with our structural and biophysical findings, showing that the repeat at P3 appears to play only a very minor role in Py replication.
Fig 5.

Replication assays. (A) Origin sequences of wild-type and mutant A2 and NG59RA origin regions. (B) (Top) NIH 3T3 cells were transiently transfected with wild-type A2 or the NG59RA polyomavirus origin-containing plasmid pFLORI40 or their respective mutants P1, P2, P3, or P4, together with LT, for 48 h. Low-molecular-weight DNA prepared according to the Hirt protocol was digested with DpnI. Replicated DNA was amplified using PCR analysis with specific primers and visualized by ethidium bromide (EtBr) staining of the 1.5% agarose gel. (Middle) The signals from replicated DNAs from 10-thermal-cycle PCRs (Top) were quantified using Image J software. (Bottom) Lysates from the replication assay (Top) were analyzed by Western blotting for expression of LT. Lysates from cells transfected with control vectors that have no LT were used as the negative control. Actin was used as a loading control. A 500-bp marker is shown in lane m. (C) Extracts from NIH 3T3 cells transiently transfected with wild-type A2, NG59RA, or their mutant (P1, P2, P3, and P4) origin luciferase reporter vectors and large T or control vectors, along with CMV beta-galactosidase, were tested for luciferase and beta-galactosidase activities approximately 40 h after transfection. The values were set to 1 for the wild-type origin and wild-type LT. The error bars represent the standard errors of the mean. (D) Extracts from NIH 3T3 cells transfected with wild-type A2 origin luciferase reporter vector or its mutants (P2 and P3) or inverse complement origin (Revori) or its mutants (RevP2 and RevP3) and large T or control vectors, along with CMV beta-galactosidase, were tested for luciferase and beta galactosidase activities approximately 40 h after transfection. The values were set to 1 for the wild-type origin and wild-type LT. The error bars represent the standard errors of the mean.
Additional mutagenesis was carried out to probe the origin structure. Since P2 and P3 overlap in Py but not in SV40, we tested the effect of separating P2 and P3. In this construct (stretched ori), P2 and P3 are separated by 1 base, as in SV40. This mutant origin was unable to replicate (Fig. 5D). Perhaps this is not surprising, since the additional DNA alters the orientation of large Ts associating with P1/P2 with respect to those associating with P3/P4. In addition, the “stretched ori” changes the relationship of the core binding sites P2 and P3 to flanking elements, such as the AT-rich region.
These data do not yet address the reason that P2 is preferred over P3. At least two possibilities exist. First, bases immediately adjacent to P2 and P3 could subtly influence the DNA structure, causing the OBD to bind one GGGGC pentamer more tightly than the other. Second, the regions outside the central core, which interact with the helicase domains of full-length LT, could be primarily responsible for the pentamer selectivity. To address this, the entire PEN region, including 1 base on either side, was inverted so that P3/P4 were on the AT-rich side, and vice versa (Fig. 5A). This construct was active in DNA replication (Fig. 5D). When mutations were introduced into the pentamers of the inverted construct, P3 remained active, while P2 was inactive. This is a very surprising result, since inverting the sequences caused P2 and P3 to swap positions relative to the remainder of the origin. These data demonstrate that it is the sequences immediately flanking the pentamers, rather than the sequences that flank the entire PEN region, that determine which pentamers are utilized during the critical step in replication.
DISCUSSION
The data presented here extend earlier work on SV40 and MCV to show that different polyomaviruses differ in their approaches to the very basic process of initiating DNA replication. Just as there are obvious functional differences in the properties of large and small T antigens among polyomavirus family members, there are also important differences in the origin sequences of these viruses. As part of our analysis, we manually aligned the sequences of 46 different polyomavirus origins (see Fig. S1 in the supplemental material). The polyomaviruses are ordered based on the spacing of the G(A/G)GGC repeats within the PEN region. There are only four viruses with symmetrical, SV40-like spacing. There are nine viruses with Py-like spacing, and among the remainder, the largest family is the one in which the third repeat is either imperfect or completely absent. This group includes MCV, as well as six other viruses that have also been isolated from human tissues (Fig. 1A). It is interesting that the EP region, which is the first to melt in SV40 (27), is poorly conserved, whereas the AT-rich region exhibits exceptional sequence identity. We also note that the most conserved regions of polyomavirus origins are clearly the T-rich stretch on the late side (also known as the AT-rich region) and the G(A/G)GGC repeats themselves, particularly P1. The imperfect palindrome (for which the EP region is named) is not a conserved feature. Within the EP, however, there is a weakly conserved poly(A) region that is the same distance (on the early side) from the PEN repeats as the poly(T) stretch (on the late side). Importantly, these poly(A) and poly(T) sequences are located where LT is known to distort and melt the DNA (27). Indeed, in the crystal structure of the SV40 OBD and helicase domains in complex with the early side of the origin, critical contacts are observed at precisely this spot within the EP (6) (marked in Fig. 1A), and it was proposed in earlier biochemical studies that these regions are important for LT binding (7).
The X-ray structure presented here shows that even though there are four potential binding sites in the Py PEN region, only three copies of the Py OBD are bound. This stoichiometry was confirmed by solution studies using isothermal calorimetry. Functional studies demonstrated that the third G(A/G)GGC repeat at the Py origin of replication is significantly less important in Py viral DNA replication than are the sites at P1, P2, and P4. The stoichiometry results differ from those with SV40, but they match what is seen in MCV (13, 14). As only a few viruses have SV40-like spacing of their repeats, the symmetrical OBD-DNA interactions within the PEN region observed in the SV40 system appear to be the exception rather than the rule.
SV40 has the same spacing of GAGGC repeats within the PEN region as BKV and JCV, both of which are associated with human disease. This region within SV40 has been shown to bind four copies of the OBD (13), and we predict the same binding stoichiometry for BKV and JCV. However, even for SV40, the situation with respect to what would be P3 in Py (this is referred to as P2 in the SV40 nomenclature) may be more complex. In SV40, three of the four sites are preferentially occupied by the OBD under conditions that support replication (28). Furthermore, the SV40 site analogous to P3 binds the isolated OBD 8-fold less tightly than the other three sites (29). The crystal structure of the SV40 OBD and helicase domains in complex with dsDNA from the late side of the origin (including the PEN sequences analogous to P3 and P4) was recently published (6). In this structure, the pentamer at what would be Py P3 was not engaged by the OBD. Instead of binding the GAGGC sequence, the OBD binds at a “hidden site” on the EP side of the origin (marked in Fig. 1). This hidden site is on the opposite strand and separated by approximately 1 base from the GAGGC at P4 (P1 in SV40 nomenclature). It may be that in viruses such as Py and MCV, where binding to P3 by the isolated OBD is not observed, a hidden site similar to that seen in the SV40 structure plays a role in replication initiation. Interestingly, the MCV origin sequence has a GAGGC very close to the “hidden site” position observed in SV40. This is one of two extra GAGGC sequences in the PEN region that are unique to MCV.
The PEN region is not the only DNA sequence required for replication. On the early and late sides of the origin, the AT-rich and EP regions interact with the LT helicase domains. Multiple lines of inquiry have shown that disruption of any one of the three LT binding regions interferes with replication (reviewed in reference 28). Indeed, the energetic contribution of the EP was confirmed in the SV40 system by studies in which binding to the late half of the origin (P1, P2, and the AT region) was shown to be weaker than binding to the early half (P3, P4, and the EP region) (30). We believe that different viruses may use somewhat different strategies. For example, the natures and relative importance of “hidden” sites may vary, and for Py, additional sequences representing transcriptional enhancer elements are important for replication (31).
An interesting question is how the different elements may interact with one another. In this study, we have shown that reversal of the PEN region yields a functional origin. Moreover, mutation of P3 in its new orientation again does not affect replication, while mutation of P2 in its new position stops DNA replication. It appears that subtle changes in the DNA sequence at the origin can dramatically alter the way that the DNA interacts with LT during replication.
The finding that the PEN region is asymmetric with respect to the requirements for GAGGC sequences is not necessarily a challenge to basic views of viral DNA replication. Bidirectional replication initiated by LT is well documented for SV40, as is the dodecameric complex formed by SV40 LT at the origin of replication (8, 30, 32). The theta structures expected for such a mechanism have also been observed for Py (33). However, the asymmetry of the viral origin sequences, combined with the protein asymmetry imposed by steric constraints when the OBDs bind the major groove of the DNA, raises the question of whether all polyomavirus replication proceeds via a bidirectional mechanism. Indeed, rolling-circle replication is well documented for Py (34) and SV40 (35), as well as for papillomavirus 16, which encodes E1, an LT homolog (36). Nonetheless, in these cases, rolling-circle replication is the exception rather than the rule; it is thought to be initiated by a nicked DNA template, a stalled replication fork, or some other perturbation of the normal bidirectional mechanism (35, 36). The fact that bidirectional replication predominates in vivo in spite of the presumably asymmetrical nature of the LT dodecamer assembly process suggests significant synergy between the two hexamer rings prior to replication initiation. Were this not the case, the activities of the two forks would not be synchronized. The data presented here indicate that in some (or all) polyomaviruses, assembly of LT proceeds through an intrinsically asymmetric mechanism, most likely with one hexamer ring forming around the DNA first and this complex then facilitating final assembly of the second ring. The idea of asymmetrical, cooperative assembly of the two hexamers was proposed as early as 1991 (30). This model is supported by the fact that the structure of the DNA in the EP region differs from that in the AT-rich region upon SV40 LT binding (27, 37). Electron microscopy (EM) reconstructions of LT on the origin further highlight the asymmetry of the complex. In these reconstructions, asymmetry in the central, OBD region of the complex frequently causes the entire complex to adopt a kinked conformation in which the LT-bound DNA is bent by angles varying from 156° to 172° (38, 39).
The physical connection between the two LT rings is not completely understood. In SV40, it appears to involve residues within the B3 loop of the OBD (SV40 residues 213 to 220). Mutations within this loop interfere with double-hexamer formation, but not assembly of single hexamers (10). In SV40 crystal structures, this loop does not interact with double-stranded DNA, and it appears at the closest point between head-to-head OBDs when bound to the DNA (11, 12). It has also been proposed that these residues in SV40 interact during double-hexamer formation, though the isolated OBD in solution is monomeric (37). The head-to-head OBDs (those bound to P2 and P4) in the Py structure with DNA are 2 nucleotides closer to one another than in SV40. The helical nature of the DNA, however, causes the Py OBDs to be farther apart in space than the equivalent OBDs in SV40 (Fig. 6). Since the Py B3 loops on the head-to-head OBDs are not close to one another, interactions between the two hexamer rings are presumably mediated through other residues. The DNA-dependent variation in the OBD positions may help explain why LTs from different polyomaviruses are very sensitive to the spacing of the G(A/G)GGC repeats (40). The variation in position may also contribute to the observation that, in spite of the high degree of conservation in the DNA and protein sequences, the LTs from different viruses generally do not substitute for one another in replication assays (41).
Fig 6.

Comparison of the head-to-head OBD DNA complexes in Py and SV40. The Py DNA and protein are shown in red, with the B3 loops in yellow. The SV40 DNA and protein are shown in blue, with the B3 loops shown in green.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Howard Robinson at NSLS beamline X29 for collecting the diffraction data. We thank Lakshmi Dommeti and Jennifer Choe for help with the replication assay. In addition, we thank Jacques Archambault and Peter Bullock for helpful discussions and for their comments on the manuscript.
This work was supported by NIH R21AI082496 (A.B.) and NIH grants CA34722 and CA50661 (B.S.).
Footnotes
Published ahead of print 9 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01740-13.
REFERENCES
- 1.Cheng J, DeCaprio JA, Fluck MM, Schaffhausen BS. 2009. Cellular transformation by Simian Virus 40 and Murine Polyoma Virus T antigens. Semin. Cancer Biol. 19:218–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bullock PA. 1997. The initiation of simian virus 40 DNA replication in vitro. Crit. Rev. Biochem. Mol. Biol. 32:503–568 [DOI] [PubMed] [Google Scholar]
- 3.Campbell KS, Mullane KP, Aksoy IA, Stubdal H, Zalvide J, Pipas JM, Silver PA, Roberts TM, Schaffhausen BS, DeCaprio JA. 1997. DnaJ/hsp40 chaperone domain of SV40 large T antigen promotes efficient viral DNA replication. Genes Dev. 11:1098–1110 [DOI] [PubMed] [Google Scholar]
- 4.Weisshart K, Bradley MK, Weiner BM, Schneider C, Moarefi I, Fanning E, Arthur AK. 1996. An N-terminal deletion mutant of simian virus 40 (SV40) large T antigen oligomerizes incorrectly on SV40 DNA but retains the ability to bind to DNA polymerase alpha and replicate SV40 DNA in vitro. J. Virol. 70:3509–3516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Love TM, de Jesus R, Kean JA, Sheng Q, Leger A, Schaffhausen B. 2005. Activation of CREB/ATF sites by polyomavirus large T antigen. J. Virol. 79:4180–4190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang YP, Xu M, Machado ACD, Yu XJ, Rohs R, Chen XS. 2013. Mechanism of origin DNA recognition and assembly of an initiator-helicase complex by SV40 large tumor antigen. Cell Rep. 3:1117–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reese DK, Meinke G, Kumar A, Moine S, Chen K, Sudmeier JL, Bachovchin W, Bohm A, Bullock PA. 2006. Analyses of the interaction between the origin binding domain from simian virus 40 T antigen and single-stranded DNA provide insights into DNA unwinding and initiation of DNA replication. J. Virol. 80:12248–12259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mastrangelo IA, Hough PV, Wall JS, Dodson M, Dean FB, Hurwitz J. 1989. ATP-dependent assembly of double hexamers of SV40 T antigen at the viral origin of DNA replication. Nature 338:658–662 [DOI] [PubMed] [Google Scholar]
- 9.Valle M, Gruss C, Halmer L, Carazo JM, Donate LE. 2000. Large T-antigen double hexamers imaged at the simian virus 40 origin of replication. Mol. Cell. Biol. 20:34–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weisshart K, Taneja P, Jenne A, Herbig U, Simmons DT, Fanning E. 1999. Two regions of simian virus 40 T antigen determine cooperativity of double-hexamer assembly on the viral origin of DNA replication and promote hexamer interactions during bidirectional origin DNA unwinding. J. Virol. 73:2201–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meinke G, Phelan P, Moine S, Bochkareva E, Bochkarev A, Bullock PA, Bohm A. 2007. The crystal structure of the SV40 T-antigen origin binding domain in complex with DNA. PLoS Biol. 5:e23. 10.1371/journal.pbio.0050023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bochkareva E, Martynowski D, Seitova A, Bochkarev A. 2006. Structure of the origin-binding domain of simian virus 40 large T antigen bound to DNA. EMBO J. 25:5961–5969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrison CJ, Meinke G, Kwun HJ, Rogalin H, Phelan PJ, Bullock PA, Chang Y, Moore PS, Bohm A. 2011. Asymmetric assembly of Merkel cell polyomavirus large T-antigen origin binding domains at the viral origin. J. Mol. Biol. 409:529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwun HJ, Guastafierro A, Shuda M, Meinke G, Bohm A, Moore PS, Chang Y. 2009. The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J. Virol. 83:12118–12128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otwinowski Z, Minor W. 1997. Processing of X-ray data collected in oscillation mode. Methods Enzymol. 276:307–326 [DOI] [PubMed] [Google Scholar]
- 17.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwede T, Kopp J, Guex N, Peitsch MC. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blanchet C, Pasi M, Zakrzewska K, Lavery R. 2011. CURVES+ web server for analyzing and visualizing the helical, backbone and groove parameters of nucleic acid structures. Nucleic Acids Res. 39:W68–W73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fradet-Turcotte A, Morin G, Lehoux M, Bullock PA, Archambault J. 2010. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology 399:65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen CA, Okayama H. 1988. Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques 6:632–638 [PubMed] [Google Scholar]
- 24.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365–369 [DOI] [PubMed] [Google Scholar]
- 25.Lavery R, Moakher M, Maddocks JH, Petkeviciute D, Zakrzewska K. 2009. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 37:5917–5929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cowie A, Kamen R. 1986. Guanine nucleotide contacts within viral DNA sequences bound by polyomavirus large T antigen. J. Virol. 57:505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borowiec JA, Hurwitz J. 1988. Localized melting and structural changes in the SV40 origin of replication induced by T-antigen. EMBO J. 7:3149–3158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joo WS, Kim HY, Purviance JD, Sreekumar KR, Bullock PA. 1998. Assembly of T-antigen double hexamers on the simian virus 40 core origin requires only a subset of the available binding sites. Mol. Cell. Biol. 18:2677–2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fradet-Turcotte A, Vincent C, Joubert S, Bullock P, Archambault J. 2007. Quantitative analysis of the binding of simian virus 40 large T antigen to DNA. J. Virol. 81:9162–9174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parsons RE, Stenger JE, Ray S, Welker R, Anderson ME, Tegtmeyer P. 1991. Cooperative assembly of simian virus 40 T-antigen hexamers on functional halves of the replication origin. J. Virol. 65:2798–2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veldman GM, Lupton S, Kamen R. 1985. Polyomavirus enhancer contains multiple redundant sequence elements that activate both DNA replication and gene expression. Mol. Cell. Biol. 5:649–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Enemark EJ, Joshua-Tor L. 2008. On helicases and other motor proteins. Curr. Opin. Struct. Biol. 18:243–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gourlie BB, Krauss MR, Buckler-White AJ, Benbow RM, Pigiet V. 1981. Polyoma virus minichromosomes: a soluble in vitro replication system. J. Virol. 38:805–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bjursell G. 1978. Effects of 2′-deoxy-2′-azidocytidine on polyoma virus DNA replication: evidence for rolling circle-type mechanism. J. Virol. 26:136–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li JJ, Kelly TJ. 1985. Simian virus 40 DNA replication in vitro: specificity of initiation and evidence for bidirectional replication. Mol. Cell. Biol. 5:1238–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kusumoto-Matsuo R, Kanda T, Kukimoto I. 2011. Rolling circle replication of human papillomavirus type 16 DNA in epithelial cell extracts. Genes Cells 16:23–33 [DOI] [PubMed] [Google Scholar]
- 37.Parsons R, Anderson ME, Tegtmeyer P. 1990. Three domains in the simian virus 40 core origin orchestrate the binding, melting, and DNA helicase activities of T antigen. J. Virol. 64:509–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheres SHW, Valle M, Nuñez R, Sorzano COS, Marabini R, Herman GT, Carazo J-M. 2005. Maximum-likelihood multi-reference refinement for electron microscopy images. J. Mol. Biol. 348:139–149 [DOI] [PubMed] [Google Scholar]
- 39.Scheres SHW, Gao H, Valle M, Herman GT, Eggermont PPB, Frank J, Carazo J-M. 2007. Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nat. Methods 4:27–29 [DOI] [PubMed] [Google Scholar]
- 40.Parsons R, Tegtmeyer P. 1992. Spacing is crucial for coordination of domain functions within the simian virus 40 core origin of replication. J. Virol. 66:1933–1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, Chang Y. 2008. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. U. S. A. 105:16272–16277 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.