

Abstract
During childhood, infections with cytomegalovirus (CMV) and Epstein-Barr virus (EBV) can occur in close temporal proximity. Active, as well as latent, CMV infection is associated with enlarged subsets of differentiated natural killer (NK) and cytotoxic T cells. How EBV infection may influence CMV-driven immune differentiation is not known. We found that EBV coinfection selectively influenced the NK cell compartment of CMV-seropositive (CMV+) children. Coinfected children had significantly higher proportions of peripheral-blood NKG2C+ NK cells than CMV+ EBV− children. Ex vivo NK cell degranulation after target cell stimulation and plasma IL-15 levels were significantly higher in CMV+ children. EBV coinfection was related to the highest levels of plasma interleukin-15 (IL-15) and IL-12p70. Remarkably, in vitro EBV infection of peripheral blood mononuclear cells (PBMC) from EBV− CMV+ children increased NKG2C+ NK cell proportions. A similar tendency was seen in cocultures of PBMC with EBV+ lymphoblastoid B-cell lines (LCL) and IL-15. After K562 challenge, NKG2C+ NK cells excelled in regard to degranulation and production of gamma interferon, regardless of whether there was previous coculture with LCL. Taken together, our data suggest that dual latency with these herpesviruses during childhood could contribute to an in vivo environment supporting differentiation and maintenance of distinct NK cell populations. This viral imprint may affect subsequent immune responses through altered distributions of effector cells.
INTRODUCTION
Epstein-Barr virus (EBV) and cytomegalovirus (CMV) are two ubiquitous and persistent herpesviruses commonly contracted during infancy. The course of primary EBV and CMV infection during childhood is typically asymptomatic, whereas infection with EBV during adolescence or adulthood is more severe and often causes infectious mononucleosis (1). After the resolution of primary infection, EBV and CMV become latent, express a highly restricted set of genes, and reside in B and myeloid cells, respectively (1, 2). EBV and CMV can reactivate from latency to produce viral progeny. However, in immunocompetent individuals, no symptoms are evident since reactivation events are tightly controlled by immune cells (1, 2).
Natural killer (NK) and CD8+ T (cytotoxic) cells play a key role in the defense against virus-infected cells. CMV, in particular, can drive the differentiation of highly mature (also known as late or terminally differentiated) cytotoxic T cells, phenotypically characterized by the lack of CD28 and expression of CD57 surface markers (3). High expression of CD57 has been linked to elevated lytic granule content in T cells (4, 5). In line with this, highly differentiated CD8+ T cells have a lower activation threshold and a strong capacity to lyse target cells and produce cytokines (3, 6).
Differentiation of NK cells is driven by multiple cytokines in addition to cell-cell interactions (7). Recent findings propose that NK cells differentiate further once they enter peripheral sites, i.e., develop to CD56dim cells from the less mature CD56bright cells, whereby they sequentially lose NKG2A, acquire killer immunoglobulin receptors, and upregulate CD57 (7–10). CMV also drives differentiation in NK cells, and NKG2C is one of the NK cell receptors specifically associated with CMV carriage (11–14). Coculture studies have shown that CMV-infected fibroblasts, together with interleukin-15 (IL-15), can induce the expansion of NKG2C+ NK cells in vitro (15). Although the precise molecular mechanism for recognition of CMV-infected cells in humans remains unclear, a specific ligand for NKG2C has been recognized as the nonclassical HLA class I molecule HLA-E (16). As for T cells, CD57 expression on NK cells has been suggested to be a marker of highly differentiated memory-like NK cells (17), which is corroborated by findings from a murine CMV infection model (18).
Acute viral infections, such as HIV-1 (19) and hantavirus (20), or chronic viruses, such as hepatitis (21), have been associated with NKG2C+ NK cell expansion in CMV-seropositive (CMV+) subjects. No significant role for EBV in driving the terminal differentiation of lymphocytes has been described (11, 22, 23). To our knowledge, however, no studies have yet focused on the possible synergistic role of EBV and CMV coinfection on antiviral effector cell maturation. Notably, earlier studies from our group have suggested a synergistic protective effect of EBV and CMV coinfection against IgE sensitization (24), and distinct modulation of NK cell gamma interferon (IFN-γ) production capacity by the two viruses (25). This raises the intriguing possibility of in vivo interplay of CMV and EBV latency and that this interplay may have a functional imprint on subsequent immune responses early in life.
We investigate here the possible effect of EBV coinfection on CMV-driven differentiation of NK and T cells and on functional responses in a cohort of 5-year-old healthy children. We demonstrate that coinfection with EBV and CMV is associated with the highest proportions of NKG2C+ NK cells, as well as memory-like CD57+ NKG2C+ NK cells, compared to single infection with CMV. Further, data herein suggest that the enrichment of NKG2C+ NK cells mediated by EBV coinfection may operate through NK cell interaction with HLA-E+ EBV+ B cells in the presence of IL-15. Based on our findings, we report a distinct role for EBV coinfection in boosting CMV-derived maturation of NK cells and their in vivo priming early in life, which may have an impact on subsequent immune responses.
MATERIALS AND METHODS
Characterization and serostatus of children.
Plasma was available from a total of 128 5-year-old children included in a study cohort described earlier (24, 26). The serostatus against EBV and CMV was known for all subjects; 50 were CMV seronegative (CMV−), and 78 were CMV seropositive (CMV+). Subdivision of the children with regard to EBV serostatus generated four groups: 31 were EBV− CMV−, 19 were EBV+ CMV−, 21 were EBV− CMV+, and 57 were EBV+ CMV+ (coinfected). Peripheral blood mononuclear cells (PBMC) were available from a smaller number of children (n = 45): 13 were CMV−, and 32 were CMV+. Further subdivision with regard to EBV serostatus generated four groups of children: 10 EBV− CMV−, 3 EBV+ CMV−, 14 EBV− CMV+, and 18 EBV+ CMV+ (coinfected). In addition, PBMC from eight CMV+ EBV− 5-year olds were used for in vitro EBV infection. All subjects were healthy and showed no clinical signs of disease at the time of blood collection. For coculture experiments, PBMC isolated from healthy adult donor buffy coats were obtained from the blood bank at Karolinska University Hospital (Stockholm, Sweden). This study was approved by the Human Ethics Committee at Karolinska University Hospital (Huddinge, Sweden [Dnr 331/02]) and the Karolinska Institute, and all of the parents provided their informed consent.
Sample handling, cell culture conditions, and in vitro EBV infection.
After the collection of venous blood, plasma was separated after centrifugation and stored at −86°C. PBMC were isolated by density gradient centrifugation (Ficoll-Paque; Pharmacia Diagnostics AB, Sweden) and cryopreserved in liquid nitrogen. For flow cytometric analyses, the PBMC were thawed, washed three times, and resuspended in culture medium (RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum [HyClone Laboratories, Inc., USA]), l-glutamine (2 mmol/liter), penicillin G sodium (100 U/ml), and streptomycin sulfate (100 μg/ml; Merck, Darmstadt, Germany) to a concentration of 106 cells/ml and cultured at 37°C and 5% CO2. In target cell assays, PBMC were cultured for 20 h, followed by 4 h of incubation with medium only or the cell lines Daudi (Burkitt's lymphoma, BL), K562, uncoated P815, or P815 coated with anti-CD3 (S4.1; Invitrogen, USA) or anti-CD16 (3G8; BD Biosciences, USA) monoclonal antibodies. Fluorochrome-conjugated anti-CD107a was added at the start of 4-h cultures, and monensin and brefeldin A (GolgiStop and GolgiPlug; BD Biosciences) were added for the last 3 h of the assay.
For in vitro EBV infections, PBMC were thawed, washed three times, and then incubated with B95-8 virus-containing supernatant for 1.5 h with shaking every 30 min. The virus titer of B95-8 EBV was 2.5 × 105 Ramos infectious units (RaIU). The RaIU count was determined by infection of the EBV-negative Burkitt lymphoma B-cell line Ramos, followed by an anti-complement immunofluorescent assay to detect the number of infected cells. Successful EBV infection was monitored by changes in morphology, upregulation of the memory marker CD27, and proliferation of the B cell population, as previously described (27). Thereafter, the cells were washed and resuspended in culture medium and cultured for 7 or 14 days, at which time they were analyzed by flow cytometry. Culture supernatants were collected and stored at −86°C prior to measuring the cytokines. For coculture experiments, adult PBMC were thawed, washed, and depleted from CD3+ cells (depletion was 96 to 99% successful; StemCell Technologies, Inc., United Kingdom) and then plated for a 3-day culture alone or together with either a mitomycin C-treated EBV+ lymphoblastoid cell line (LCL05.08) or an EBV− Burkitt lymphoma cell line (Ramos) at a 1:2 target/effector cell ratio. In these three culture conditions, the cells were either in culture medium alone or in culture medium containing recombinant human IL-12p70 (1 ng/ml), IL-15 (10 ng/ml), or both or with blocking IL-12p70 (1 μg/ml; MT3279H; Mabtech, Sweden) and IL-15 (1 μg/ml; 34505.11; R&D Systems, United Kingdom) monoclonal antibodies in combination. Prior to coculture, supernatants from LCL or Ramos cells cultured alone at a concentration of 106cells/ml for 3 days were collected and stored at −86°C. A portion of the PBMC was cultured with cell culture medium spiked with 50% LCL or Ramos supernatant. Thereafter, the cells were resuspended in medium and stimulated with K562 targets cells for 4 h in the presence of fluorochrome-conjugated anti-CD107a antibodies and monensin for flow cytometric analysis.
Flow cytometry.
For flow cytometry, the following fluorochrome-conjugated specific antibodies from BD Biosciences (if not otherwise stated) were used: CD3 (SK7 or UCHT1 [Dako, Denmark] or S4.1 [Invitrogen]), CD4 (SK3 or S3.5; BD and Invitrogen), CD8 (3B5; Invitrogen), CD14 (MϕP9), CD16 (3G8), CD19 (SJ25C1 or HIB19), CD45RA (MEM-56; Invitrogen), CD56 (NCAM 16.2), CD57 (HNK-1; BD or BioLegend, USA), CD69 (TP1.55.3; Beckman Coulter, USA), CD107a (H4A3), CD226-FITC (DX11), HLA-E (3D12; BioLegend) ki-67 (B56), NKG2A (Z199; Beckman Coulter), NKG2C (catalog no. 134591; R&D Systems), NKG2D FITC (1D11; AbD Serotec, United Kingdom), NKp30 APC (p30-15), 2B4 PE (C1.7; Beckman Coulter), MIP-1β (D21-1351), IFN-γ (25723.11, 4S.B3, or B27), and tumor necrosis factor (TNF; Mab11).
A fixable dead cell marker (Invitrogen) was used to exclude nonviable cells from the analysis. Data from in vitro infections and coculture experiments were acquired through BD FACSCalibur and BD FACSVerse, while all of the remaining data were acquired with BD LSR Fortessa and analyzed by FlowJo software (TreeStar, USA). Gating was performed on live CD14− and CD19− single cells within the lymphocyte gate, which were CD3+ or CD3− CD56+.
Cytokine detection.
Plasma or culture supernatant IFN-α, IFN-γ (Mabtech), and IL-15 (R&D Systems) were measured by sandwich enzyme-linked immunosorbent assay (ELISA) according to standard procedures. ELISA kits were used for the detection of IL-18 (MBL, Japan) and IL-12p70 (high sensitivity; eBioscience, USA) according to the manufacturer's guidelines. The detection limits were as follows: IFN-α and IFN-γ, 10 pg/ml; IL-15, 3 pg/ml; IL-18, 12.5 pg/ml; and IL-12p70, 0.1 pg/ml.
Statistical analyses.
Statistical differences between the CMV− and CMV+ groups were evaluated by using the Mann-Whitney U nonparametric test. A further comparison between multiple groups was made only after the establishment of significance and was performed by using the Kruskal-Wallis analysis of variance (ANOVA) test. Then, given significance in the Kruskal-Wallis ANOVA, further pairwise comparisons were tested. Correlations were evaluated either with the Spearman's rank or the Kendall's tau-b test. Differences between control and EBV-infected cultures were evaluated by using the Wilcoxon matched-pair test. P values below 0.05 were considered statistically significant. Analyses were carried out using Statistica (Statsoft Software, Inc., USA) or Stata (StataCorp LP, USA).
RESULTS
Enriched populations of NKG2C+ NK cells in EBV-coinfected CMV+ children.
To establish whether CMV-driven differentiation of T and NK cells was present also in CMV+ children in our cohort, we phenotyped PBMC ex vivo. Corroborating previous studies (3, 11–14), we found markedly higher proportions of CD8+ CD57+ T cells and NKG2C+, CD57+ NKG2C+, and CD57bright NKG2C+ among CD56dim NK cells (from here on referred to as NK cells) in the CMV+ group compared to the CMV− controls (Fig. 1A to E). To assess whether EBV coinfection influenced this differentiation, we subdivided the CMV− and CMV+ groups on the basis of EBV serostatus. The highest percentages of NKG2C+, CD57+ NKG2C+, and CD57bright NKG2C+ NK cells, but not CD8+ CD57+ T cells, were associated with EBV and CMV coinfection (Fig. 1F to I). The frequencies of NKG2C+ cells expressing NKG2A were similar in all groups (Fig. 1J and K). A significant positive correlation between the percentages of NKG2C+ NK cells and CD8+ CD57+ T cells, as well as between CD57+ NKG2C+ NK cells and CD8+ CD57+ T cells, was found (Fig. 1L and M). As an additional measure of NK cell differentiation, the relation between CD57 and NKG2A expression on NKG2C+ NK cells was assessed. There was an inverse correlation between CD57bright and NKG2A+ cell frequencies among NKG2C+ NK cells considering all subjects (n = 45, P < 0.05, r = −0.36), which upon subgrouping showed that the CMV+ group (n = 32, P < 0.05 r = −0.43) but not the CMV− group (n = 13, P = 0.60, r = 0.16) contributed to this association.
Fig 1.

Phenotype of T and NK cells in relation to CMV and EBV serostatus. PBMC from CMV− (n = 13) and CMV+ (n = 32) 5-year-old children were phenotyped ex vivo. (A) Representative example of flow cytometric gating of viable CD14− CD19− CD8+ CD3+ and CD14− CD19− CD3− CD56dim lymphocytes for the proportion of CD57 and NKG2C expressing cells. (B to K) Proportions of CD57+ CD8+ T cells (B and F), NKG2C+ NK cells (C and G), CD57+ NKG2C+ NK cells (D and H), CD57bright NKG2C+ NK cells (E and I), and) NKG2A+ NKG2C+ NK cells (J and K) in CMV− and CMV+ groups and after subdivision with regard to EBV serostatus. (L and M) Spearman analysis of correlation between the proportions of CD57+ CD8+ T cells and NKG2C+ NK cells (L) or between CD57+ CD8+ T cells and CD57+ NKG2C+ NK cells (M). Filled circles depict outlier values.
Further, CMV+ children had a lower expression intensity of the signaling lymphocyte activation receptor 2B4 on NK cells than CMV− children, which was not further influenced by EBV serostatus (P < 0.05 and P = 0.19, respectively [data not shown]). Expression of CD8, CD16, CD45RA, CD57, CD69, DNAM-1, and NKG2D did not significantly differ between the CMV− and CMV+ groups, neither when total CD56dim NK cells nor when only NKG2C+ NK cells were considered (data not shown).
There were no differences in bulk frequencies of T and NK cells between the CMV+ and CMV− groups (Table 1 and 2). However, CMV+ children had a somewhat larger fraction of CD56bright cells and thus fewer CD56dim NK cells (Table 2).
Table 1.
Frequencies of bulk T-cell subsets in CMV− and CMV+ 5-year-old children
| Serostatus | No. of children | Median % frequency (range)a |
||
|---|---|---|---|---|
| CD3+ | CD3+ CD4+ | CD3+ CD8+ | ||
| CMV− | 13 | 89 (77–93) | 65 (52–77) | 27 (14–40) |
| CMV+ | 22 | 88 (77–94) | 64 (50–79) | 26 (14–38) |
For CD3+, the values for the frequencies among CD14− CD19− living, single-cell, lymphocytes are shown. For CD3+ CD4+ and CD3+ CD8+, the values for the frequencies among CD3+ cells are shown.
Table 2.
Frequencies of bulk NK cell subsets in 5-year-old children seronegative and seropositive for CMV and/or EBV
| Serostatus | No. of children | Median % frequency (range)a |
|
|---|---|---|---|
| CD3− CD56+ | CD3− CD56dim | ||
| CMV− | 13 | 8 (3–15) | 91 (85–95) |
| CMV+ | 32 | 6 (2–18) | 89 (66–97)* |
| CMV− EBV− | 10 | 7 (3–15) | 91 (85–95) |
| CMV− EBV+ | 3 | 8 (6–9) | 91 (91–95) |
| CMV+ EBV− | 14 | 5 (2–13) | 89 (66–92)†‡ |
| CMV+ EBV+ | 18 | 6 (3–18) | 89 (80–97) |
For CD3− CD56+, the frequencies among CD14− CD19− living single lymphocytes are shown. For CD3− CD56dim, the frequencies among CD3− CD56+ cells are shown. *, P < 0.05 (compared to the CMV− group); †, P < 0.05 (compared to the CMV− EBV− group); ‡, P < 0.05 (compared to the CMV− EBV+ group).
NK cells from CMV+ children have enhanced cytotoxic potential.
Since EBV and CMV coinfection appeared to have significant impact on the NK cell compartment, we chose to further investigate NK cell functionality. In mice, herpesvirus latency has been suggested to prime NK cells for efficient cytotoxicity (28). To understand whether herpesvirus carriage also associated with priming the NK cell function in humans, we induced NK cell activation through different pathways in PBMC from a group of children. We used K562 and Daudi target cells to induce activation via NK cell receptors, anti-CD16 antibodies to induce antibody-dependent cell-mediated cytotoxicity, and the addition of peptidoglycan (a Toll-like receptor 2 agonist), together with IL-15, to mimic accessory cell-mediated NK cell activation (25).
Interestingly, after K562 target cell stimulation, there was a small but significant difference in the percentage of CD107a+ NK cells with higher ratios in CMV+ children compared to their CMV− peers (Fig. 2A), regardless of EBV serostatus (Fig. 2B). Similar results were seen when NK cells were regrouped into CD57− and CD57+ cells (data not shown). CMV− and CMV+ groups did not differ in their NK cell TNF, IFN-γ, or MIP-1β production ability upon stimulation with K562 cells (Fig. 2C to E). No consistent relation was found between the expression of the activation marker CD69 and CD107a, TNF, IFN-γ, or MIP-1β, and no differences were seen between CMV− and CMV+ children in NK cell functionality following activation by the other investigated stimulation pathways (data not shown).
Fig 2.
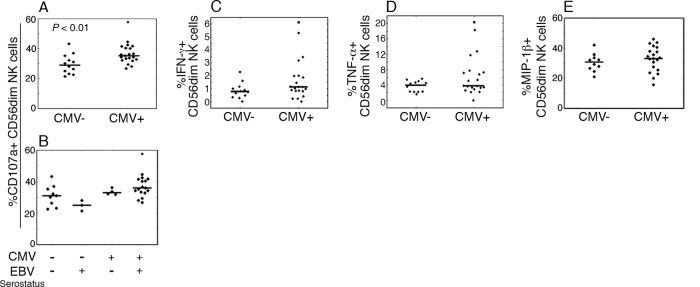
Relation between herpesvirus carriage and functional capacity of NK cells. PBMC from CMV− (n = 12) and CMV+ (n = 21) children were rested for 20 h, followed by culture during 4 h in medium alone or with K562 target cells. Subsequently, viable, CD14− CD19− CD3− CD56dim lymphocytes were analyzed for the proportions of CD107a (A and B), IFN-γ (C), TNF (D), and MIP-1β (E) cell positivity by flow cytometry. (B) Proportions of CD107a+ CD56dim NK cells upon stratification of CMV− and CMV+ groups based on EBV serostatus. Filled circles and crosses depict outlier and extreme values, respectively. Functional assay values, where from statistical data were acquired, correspond to values from nonstimulated cultures subtracted from stimulated cultures.
Cytokines required for NK cell development and differentiation are increased in CMV+ children.
Latent herpesvirus carriage in mice has been linked to elevated systemic IFN-γ and boosted innate immune defense (28, 29). We sought to determine whether the higher cytotoxic potential of NK cells in CMV+ children could be a result of in vivo priming by cytokines. We measured the antiviral and NK cell priming cytokines IFN-α, IFN-γ, IL-12, IL-15, and IL-18 in plasma from the children in our study cohort. The groups did not differ in circulating IFN-α or IL-18 levels (Fig. 3A and F and Fig. 3E and J), but the IFN-γ levels were significantly higher in the CMV+ group regardless of the EBV serostatus (Fig. 3B and G).
Fig 3.
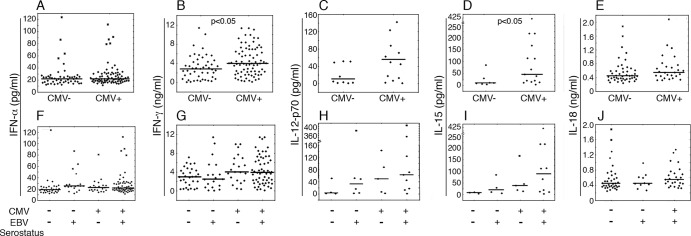
Relation between systemic cytokine profiles and herpesvirus carriage at 5 years of age. Plasma from 5-year-old children (n = 128 for all cytokines except IL-12p70 and IL-18, where 80 and 71 samples were tested, respectively) with various EBV and CMV serostatuses was assessed by ELISA for a range of cytokines related to antiviral and NK cell activity. The levels of IFN-α (A), IFN-γ (B), IL-12p70 (C), IL-15 (D), and IL-18 (E) were compared between CMV− and CMV+ children or after subdivision of CMV− and CMV+ groups with regard to EBV serostatus (F to J, respectively). Values below detection limits are not shown in the figure.
No significant differences in IL-12p70 levels between CMV− and CMV+ children were observed (Fig. 3C). Interestingly, however, CMV+ children had markedly higher levels of IL-15 (Fig. 3D). Stratification on the basis of EBV serostatus did not yield any differences, although the IL-12p70 and IL-15 levels from coinfected children tended to be the highest (Fig. 3H and I). Moreover, there was a significant positive correlation between plasma IL-12p70 and IL-15 levels (n = 63 total, P < 0.001, r = 0.69).
In CMV+ children NKG2C+ NK cells are enriched and activated after EBV infection in vitro.
Since we observed that EBV coinfection was associated with the highest percentages of NKG2C+ and CD57+ NKG2C+ NK cells, we wanted to directly address the effect of EBV infection on their proportions. We used PBMC samples from a group of EBV-naive CMV+ 5-year-old children and infected the samples with EBV in vitro. The percentage of NKG2C+ and CD57+ NKG2C+ CD56dim NK cells was assessed by flow cytometry on days 7 and 14 postinfection (Fig. 4A to D). Interestingly, EBV-infected cultures on day 14 contained higher percentages of NKG2C+ NK cells than control cultures, although the difference did not reach statistical significance (Fig. 4C, P = 0.08). At the same time point, EBV-infected cultures contained NK cells that produced IFN-γ: median = 4.0% and range = 0 to 8.3 for NKG2C+ and median = 1.9% and range = 0 to 9.6 for NKG2C− NK cells. No clear changes in the CD57+ NKG2C+ NK cell proportions were found (Fig. 4D).
Fig 4.

EBV infection in vitro induces NKG2C+ NK cell enrichment. PBMC from 5-year-old CMV+ children (n = 3 to 5) were subjected to in vitro EBV infection and cultured for 14 days, when the NK cell population was phenotyped by flow cytometry. (A) Gating strategy for NKG2C expression on CD3− CD19− CD56dim cells. (B and C) Percentages of NKG2C+ CD56dim NK cells on day 7 (B) and day 14 (C) postinfection. (D) Percentage of CD57+ NKG2C+ CD56dim NK cells on day 14 postinfection. Numbers in the diagrams indicate the fold change that was calculated by dividing the values from infected cultures by those for noninfected cultures.
Enrichment of NKG2C+ NK cells and their enhanced activation state after coculture with LCL is linked to HLA-E expression and IL-15, respectively.
Based on the fact that plasma levels of IL-12p70 and IL-15 paralleled the high proportions of NKG2C+ NK cells seen in coinfected children, we examined the contribution of these cytokines to enrichment of NKG2C+ NK cells upon interaction with EBV+ B cells. Since the number of PBMC samples from the children was limited, we performed an in vitro culture assay and selected three individual adult PBMC samples with large NKG2C+ and memory-like NK cell populations. In order to avoid recall responses by EBV-specific CD8+ cells, CD3+ cells were depleted prior to coculture with EBV+ LCL or EBV− Ramos (negative control) for 3 days.
In the presence of IL-15 or IL-15+IL-12p70, PBMC and LCL cocultures contained slightly increased NKG2C+ (Fig. 5A and B) and memory-like NK cell proportions (Fig. 5A and C), which was not observed with the EBV− cell line Ramos. To determine whether coculture with LCL had enhanced functionality of NKG2C+ NK cells, we assessed NK cell activity after K562 target cell challenge. Degranulation and IFN-γ and proliferative responses (ki-67+) were higher in cultures with IL-15, but coculture with LCL per se did not enhance NK cell functional capacity (Fig. 5D to F). NKG2C+ NK cells, including memory-like NK cells, were more CD107a+ and IFN-γ+ compared to NKG2C− NK cells (Fig. 5D and E). In contrast, memory-like NK cells proliferated but to a lower degree than the whole NKG2C+ or NKG2C− NK cell population (Fig. 5F).
Fig 5.

Distinct contributions of cells and cytokines to in vitro enrichment of NKG2C+ NK cells. CD3+ cell-depleted PBMC (n = 3 donors) were cultured alone or together with either an EBV+ B cell line (LCL) or an EBV− Burkitt lymphoma cell line (Ramos) at a 1:2 target/effector cell ratio. In these three culture conditions, the cells were incubated 72 h in culture medium alone or in culture medium containing recombinant human IL-12p70, IL-15, or both IL-12p70 and IL-15 or blocking (α-) IL-12p70 and IL-15 monoclonal antibodies in combination. (A) Representative flow cytometric gating strategy for live CD14− CD19− CD56dim NKG2C+ NK cells (percentage outside the lower right corner) and CD57+ NKG2C+ NK cells (percentage outside the upper right corner). (B and C) Fold change in NKG2C+ and CD57+ NKG2C+ NK cell subset proportions calculated by dividing values from treated PBMC cultures by those of the same donor PBMC in medium only (represented by a horizontal line in the graphs). After coculture as described above, PBMC were challenged for 4 h with K562 target cells, and the functional capacities of NK cells were analyzed by flow cytometry. (D and E) Proportions of degranulation (D), IFN-γ production (E), and proliferation of NKG2C−/+ and CD57+ NKG2C+ NK cell subsets (F). (G) Surface HLA-E expression by Ramos and LCL cells showing MFI values of isotype control (filled histograms) and HLA-E (overlays) and proportion of HLA-E+ cells, as determined by flow cytometry. (H) CD3+-cell-depleted PBMC were cultured with cell culture medium spiked with 50% supernatant from EBV+ LCL or EBV− Ramos cultures at a concentration of 106 cells/ml during 72 h. The proportions of NKG2C−, NKG2C+, or CD57+ NKG2C+ cells among viable CD14− CD19− CD3− CD56dim NK cells are shown.
Upregulation of HLA-E on virus-infected cells has been suggested to drive the expansion of NKG2C+ NK cells (11, 20), and we therefore assessed surface HLA-E expression on our cell lines. Both EBV+ LCL and EBV− Ramos displayed a 2-fold increase in HLA-E staining relative to isotype control (Fig. 5G). Interestingly, however, a larger proportion of LCLs were HLA-E+ (Fig. 5G). Cell-cell contact with HLA-E+ EBV-infected cells may contribute to NKG2C+ NK cell enrichment since T-cell-depleted PBMC cultured with LCL-derived supernatant alone displayed similar proportions of memory-like, NKG2C+, and NKG2C− NK cells (Fig. 5H).
DISCUSSION
Microbial agents, including viral pathogens, are important promoters of immune maturation following birth (30, 31). Most likely, a diverse array of microbial organisms contribute to proper maturation of immune system, which is believed to protect against immune-mediated diseases (30, 32, 33). There are in vivo (24, 25, 34) and in vitro (35) data suggesting synergy in latency with the two persistent herpesviruses EBV and CMV. Interestingly, our earlier observations suggested that EBV and CMV coinfection could have a synergistic influence on allergen-specific B-cell responses (24) and NK cell cytokine production (25) during infancy. We demonstrate here that EBV coinfection in children enhances CMV-driven immune maturation, as evidenced by greater frequencies of differentiated NKG2C+ and CD57+ NKG2C+ NK cell subsets.
After establishment of CMV latency, infants shed CMV for a prolonged period of time in contrast to adults (31), suggestive of a higher frequency of subclinical CMV reactivation during young age (31, 36). Successive rounds of reactivation could maintain the NKG2C+ NK cell population in CMV+ children (37), and in coinfected children additional immune responses contributing to the control over EBV infection could potentially support the maintenance of the NKG2C+ NK cells. In support of this hypothesis, we observed the accumulation of NKG2C+ NK cells from several EBV-naive CMV+ children upon in vitro EBV infection. CMV+ and coinfected children had the highest levels of plasma IL-15 and IL-12p70 and, interestingly, the combination of IL-12p70 and/or IL-15 and contact with latently EBV-infected cells sustained/enriched NKG2C+ and memory-like NK cell populations in PBMC and LCL cocultures.
NK cells have been shown to preferentially target EBV-infected B cells in the lytic cycle (38), which predominantly occurs in secondary lymphoid tissues. Interestingly, NK cells can be recruited to secondary lymphoid tissues, and likely also to infected lymphoid tissue (39), where they can control EBV (40). The balance of activating or inhibitory ligands on infected cells is an important factor determining NK cell activation. Higher expression of the NKG2C ligand HLA-E has previously been implicated in host responses to CMV-infected cells (11, 20, 41). Findings regarding expression of HLA class I molecules, including HLA-E, in EBV-infected B cells are contradictory since both downregulation (38) and upregulation (42) have been observed. Due to clinical sample limitation, we assessed HLA-E expression on LCL and found that roughly a third were HLA-E+, in agreement with previous studies showing heterogeneity in HLA class I expression of EBV-transformed B cells (43). The comparatively smaller effect on the NKG2C+ population enrichment/maintenance seen in LCL cocultures versus in vitro infections could also relate to differences in kinetics and sample origin, together with a more vigorous cytokine environment induced in primary in vitro EBV infection (27).
Accessory cell-derived cytokines such as IL-12p70 and IL-15 can contribute to the maintenance of differentiated NK cells (6, 44) and activate NK cells to participate in immune response against EBV-infected cells (45). Interestingly, the pattern of circulating IL-12p70 and IL-15 pointed to highest levels in coinfected children. However, despite the presence of activated myeloid cells and both innate and adaptive antiviral cytokines in supernatants of EBV-infected cultures (27), neither IL-12p70 nor IL-15 were detected in these cultures. This may partially depend on the kinetics of cytokine production and consumption in the cultures, as well as the properties by which IL-12 (46) and IL-15 (47) are delivered to NK cells. By using LCL cocultures as another approach to study contribution of accessory cell-derived cytokines to NKG2C+ NK cell maintenance, we found that the addition of IL-15 was related to higher ratios of this NK cell subset. Interestingly, the plasma IL-12p70 and IL-15 levels paralleled the ex vivo degranulation capacity by NK cells with the most potent responses in the coinfected group and, overall, the NKG2C+ NK cells were to a higher degree CD107a+ and IFN-γ+ than were the NKG2C− NK cells upon in vitro K562 challenge. After K562 target cell stimulation, the elevated NK cell degranulation capacity seen in CMV+ children may reflect an expanded NKG2C+ NK cell population in vivo (11–14) and is not likely linked to a higher NK cell 2B4 expression since K562 cells lack 2B4 ligands. Collectively, high IL-15 levels in coinfected children could maintain expanded NKG2C+ NK cell populations (6) and prime NK cell cytotoxicity (28), but the exact cellular and molecular mechanism needs further examinations.
Studies in mice have shown that latent infection with murine EBV and CMV equivalents could enhance the basal reactivity of innate cells such as NK cells (28) and macrophages via IFN-γ to protect against unrelated infections (29). Plasma IFN-γ was higher in CMV+ children, but the cellular source remains unknown, since we did not observe any differences in IFN-γ production capacity in T cells (data not shown) or NK cells between CMV− and CMV+ children. Both enriched memory CD8+ T cells (48, 49) and mature CD56dim NK cells (50) could produce IFN-γ in response to a similar cytokine environment, as seen in CMV+ children. Together, this suggests an in vivo positive-feedback loop between antiviral cell priming accessory cell-derived cytokines and IFN-γ production by antiviral cells.
This study has some limitations that should be acknowledged. We could not fully address whether EBV and CMV coinfection selectively affected the NK cell population as opposed to the T cell compartment since the numbers of CMV+ EBV− subjects were too low in the CD57+ CD8+ cell analysis for reliable conclusions. We therefore directed our attention to NK cells, but associations within the T cell population could exist and require further investigation. Furthermore, our attempts to shed light on the underlying mechanisms of how EBV coinfection could contribute to NKG2C+ NK cell enrichment in CMV+ children yielded promising, albeit preliminary results. These questions will need to be carefully considered in follow-up studies. Finally, the use of allogeneic LCL in cocultures with PBMC may have introduced an HLA mismatch. Although we cannot rule this out, higher NKC2C+ NK cell proportions were seen both in autologous in vitro infections and in allogeneic LCL cocultures and were not seen with the allogeneic EBV− cell line Ramos. Thus, it is unlikely that a potential HLA mismatch was the only reason for the redistribution of the NK cell compartment.
Our findings collectively suggest that EBV coinfection may contribute to higher NKG2C+ NK cell proportions through pathways involving HLA-E expression by infected cells, but additional signals via accessory cell cytokines seem necessary to maintain or drive the expansion of differentiated NK cells. To our knowledge, we are the first to show that a persistent EBV coinfection has a role in stimulating immune differentiation in immunocompetent CMV+ children, and the CMV-related NK cell repertoire during early life. The consequence of a functional imprint by persistent herpesviruses on subsequent heterologous immune responses may well be clinically relevant.
ACKNOWLEDGMENTS
We declare that we have no conflicting financial or commercial interests.
This study was supported by grants from the Swedish Research Council (57X-15160-07-03, 57X-15160-10-4, and K2011-80P-21806-01-4), the Swedish Association for Allergology, and the Ragnar Söderberg, Hesselman, Konsul Th C Bergh, The Golden Jubilee Memorial, Åke Wiberg, Jeanssons, Petrus and Augusta Hedlunds, and The Crownprincess Lovisa/Axel Tielman Foundations. This study was also supported by the Swedish Cancer Society, the Swedish Children's Cancer Foundation, and the Karolinska Institute Research Foundation. N.N. is a recipient of cancer research fellowships from the Cancer Research Institute (New York)/Concern Foundation (Los Angeles).
Footnotes
Published ahead of print 2 October 2013
REFERENCES
- 1.Hislop AD, Taylor GS, Sauce D, Rickinson AB. 2007. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu. Rev. Immunol. 25:587–617 [DOI] [PubMed] [Google Scholar]
- 2.Loewendorf A, Benedict CA. 2010. Modulation of host innate and adaptive immune defenses by cytomegalovirus: timing is everything. J. Intern. Med. 267:483–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strioga M, Pasukoniene V, Characiejus D. 2011. CD8+ CD28− and CD8+ CD57+ T cells and their role in health and disease. Immunology 134:17–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chattopadhyay PK, Betts MR, Price DA, Gostick E, Horton H, Roederer M, De Rosa SC. 2009. The cytolytic enzymes granyzme A, granzyme B, and perforin: expression patterns, cell distribution, and their relationship to cell maturity and bright CD57 expression. J. Leukoc. Biol. 85:88–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiang SC, Theorell J, Entesarian M, Meeths M, Mastafa M, Al-Herz W, Frisk P, Gilmour KC, Ifversen M, Langenskiold C, Machaczka M, Naqvi A, Payne J, Perez-Martinez A, Sabel M, Unal E, Unal S, Winiarski J, Nordenskjold M, Ljunggren HG, Henter JI, Bryceson YT. 2013. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood 121:1345–1356 [DOI] [PubMed] [Google Scholar]
- 6.Sun JC, Lanier LL. 2011. NK cell development, homeostasis and function: parallels with CD8+ T cells. Nat. Rev. Immunol. 11:645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chichocki F, Miller JS, Anderson SK, Bryceson YT. 2013. Epigenetic regulation of NK cell differentiation and effector functions. Front. Immunol. 4:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, Breso V, Frassati C, Reviron D, Middleton D, Romagne F, Ugolini S, Vivier E. 2006. Human NK cell education by inhibitory receptors for MHC class I. Immunity 25:331–342 [DOI] [PubMed] [Google Scholar]
- 9.Bjorkstrom NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, Bjorklund AT, Flodstrom-Tullberg M, Michaelsson J, Rottenberg ME, Guzman CA, Ljunggren HG, Malmberg KJ. 2010. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood 116:3853–3864 [DOI] [PubMed] [Google Scholar]
- 10.Beziat V, Descours B, Parizot C, Debre P, Vieillard V. 2010. NK cell terminal differentiation: correlated stepwise decrease of NKG2A and acquisition of KIRs. PLoS One 5:e11966. 10.1371/journal.pone.0011966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guma M, Angulo A, Vilches C, Gomez-Lozano N, Malats N, Lopez-Botet M. 2004. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 104:3664–3671 [DOI] [PubMed] [Google Scholar]
- 12.Kuijpers TW, Baars PA, Dantin C, van den Burg M, van Lier RA, Roosnek E. 2008. Human NK cells can control CMV infection in the absence of T cells. Blood 112:914–915 [DOI] [PubMed] [Google Scholar]
- 13.Monsivais-Urenda A, Noyola-Cherpitel D, Hernandez-Salinas A, Garcia-Sepulveda C, Romo N, Baranda L, Lopez-Botet M, Gonzalez-Amaro R. 2010. Influence of human cytomegalovirus infection on the NK cell receptor repertoire in children. Eur. J. Immunol. 40:1418–1427 [DOI] [PubMed] [Google Scholar]
- 14.Lopez-Verges S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, York VA, Houchins JP, Miller S, Kang SM, Norris PJ, Nixon DF, Lanier LL. 2011. Expansion of a unique CD57+ NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc. Natl. Acad. Sci. U. S. A. 108:14725–14732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guma M, Budt M, Saez A, Brckalo T, Hengel H, Angulo A, Lopez-Botet M. 2006. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 107:3624–3631 [DOI] [PubMed] [Google Scholar]
- 16.Borrego F, Ulbrecht M, Weiss EH, Coligan JE, Brooks AG. 1998. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J. Exp. Med. 187:813–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez-Verges S, Milush JM, Pandey S, York VA, Arakawa-Hoyt J, Pircher H, Norris PJ, Nixon DF, Lanier LL. 2010. CD57 defines a functionally distinct population of mature NK cells in the human CD56dim CD16+ NK-cell subset. Blood 116:3865–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun JC, Lopez-Verges S, Kim CC, DeRisi JL, Lanier LL. 2011. NK cells and immune “memory.” J. Immunol. 186:1891–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guma M, Cabrera C, Erkizia I, Bofill M, Clotet B, Ruiz L, Lopez-Botet M. 2006. Human cytomegalovirus infection is associated with increased proportions of NK cells that express the CD94/NKG2C receptor in aviremic HIV-1-positive patients. J. Infect. Dis. 194:38–41 [DOI] [PubMed] [Google Scholar]
- 20.Bjorkstrom NK, Lindgren T, Stoltz M, Fauriat C, Braun M, Evander M, Michaelsson J, Malmberg KJ, Klingstrom J, Ahlm C, Ljunggren HG. 2011. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J. Exp. Med. 208:13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beziat V, Dalgard O, Asselah T, Halfon P, Bedossa P, Boudifa A, Hervier B, Theodorou I, Martinot M, Debre P, Bjorkstrom NK, Malmberg KJ, Marcellin P, Vieillard V. 2012. CMV drives clonal expansion of NKG2C+ NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur. J. Immunol. 42:447–457 [DOI] [PubMed] [Google Scholar]
- 22.Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, Ogg GS, King A, Lechner F, Spina CA, Little S, Havlir DV, Richman DD, Gruener N, Pape G, Waters A, Easterbrook P, Salio M, Cerundolo V, McMichael AJ, Rowland-Jones SL. 2002. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8:379–385 [DOI] [PubMed] [Google Scholar]
- 23.Vescovini R, Telera A, Fagnoni FF, Biasini C, Medici MC, Valcavi P, di Pede P, Lucchini G, Zanlari L, Passeri G, Zanni F, Chezzi C, Franceschi C, Sansoni P. 2004. Different contribution of EBV and CMV infections in very long-term carriers to age-related alterations of CD8+ T cells. Exp. Gerontol. 39:1233–1243 [DOI] [PubMed] [Google Scholar]
- 24.Nilsson C, Linde A, Montgomery SM, Gustafsson L, Nasman P, Blomberg MT, Lilja G. 2005. Does early EBV infection protect against IgE sensitization? J. Allergy Clin. Immunol. 116:438–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saghafian-Hedengren S, Sundstrom Y, Sohlberg E, Nilsson C, Linde A, Troye-Blomberg M, Berg L, Sverremark-Ekstrom E. 2009. Herpesvirus seropositivity in childhood associates with decreased monocyte-induced NK cell IFN-γ production. J. Immunol. 182:2511–2517 [DOI] [PubMed] [Google Scholar]
- 26.Saghafian-Hedengren S, Sverremark-Ekstrom E, Linde A, Lilja G, Nilsson C. 2010. Early-life EBV infection protects against persistent IgE sensitization. J. Allergy Clin. Immunol. 125:433–438 [DOI] [PubMed] [Google Scholar]
- 27.Sohlberg E, Saghafian-Hedengren S, Rasul E, Marchini G, Nilsson C, Klein E, Nagy N, Sverremark-Ekstrom E. Cytomegalovirus-seropositive children show inhibition of in vitro EBV infection that is associated with CD8+CD57+ T cell enrichment and IFN-gamma. J. Immunol., in press. 10.4049/jimmunol.1301343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White DW, Keppel CR, Schneider SE, Reese TA, Coder J, Payton JE, Ley TJ, Virgin HW, Fehniger TA. 2010. Latent herpesvirus infection arms NK cells. Blood 115:4377–4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, Miller VL, Virgin HW, 4th 2007. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 447:326–329 [DOI] [PubMed] [Google Scholar]
- 30.Wills-Karp M, Santeliz J, Karp CL. 2001. The germless theory of allergic disease: revisiting the hygiene hypothesis. Nat. Rev. Immunol. 1:69–75 [DOI] [PubMed] [Google Scholar]
- 31.Prendergast AJ, Klenerman P, Goulder PJ. 2012. The impact of differential antiviral immunity in children and adults. Nat. Rev. Immunol. 12:636–648 [DOI] [PubMed] [Google Scholar]
- 32.Vercelli D. 2006. Mechanisms of the hygiene hypothesis–molecular and otherwise. Curr. Opin. Immunol. 18:733–737 [DOI] [PubMed] [Google Scholar]
- 33.Garn H, Renz H. 2007. Epidemiological and immunological evidence for the hygiene hypothesis. Immunobiology. 212:441–452 [DOI] [PubMed] [Google Scholar]
- 34.Aalto SM, Linnavuori K, Peltola H, Vuori E, Weissbrich B, Schubert J, Hedman L, Hedman K. 1998. Immunoreactivation of Epstein-Barr virus due to cytomegalovirus primary infection. J. Med. Virol. 56:186–191 [PubMed] [Google Scholar]
- 35.Arcenas R, Widen RH. 2002. Epstein-Barr virus reactivation after superinfection of the BJAB-B1 and P3HR-1 cell lines with cytomegalovirus. BMC Microbiol. 2:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pass RF, Stagno S, Britt WJ, Alford CA. 1983. Specific cell-mediated immunity and the natural history of congenital infection with cytomegalovirus. J. Infect. Dis. 148:953–961 [DOI] [PubMed] [Google Scholar]
- 37.Malmberg KJ, Beziat V, Ljunggren HG. 2012. Spotlight on NKG2C and the human NK-cell response to CMV infection. Eur. J. Immunol. 42:3141–3145 [DOI] [PubMed] [Google Scholar]
- 38.Pappworth IY, Wang EC, Rowe M. 2007. The switch from latent to productive infection in Epstein-Barr virus-infected B cells is associated with sensitization to NK cell killing. J. Virol. 81:474–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pak-Wittel MA, Yang L, Sojka DK, Rivenbark JG, Yokoyama WM. 2013. Interferon-gamma mediates chemokine-dependent recruitment of natural killer cells during viral infection. Proc. Natl. Acad. Sci. U. S. A. 110:E50–E59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strowig T, Brilot F, Arrey F, Bougras G, Thomas D, Muller WA, Munz C. 2008. Tonsilar NK cells restrict B cell transformation by the Epstein-Barr virus via IFN-γ. PLoS Pathog. 4:e27. 10.1371/journal.ppat.0040027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomasec P, Braud VM, Rickards C, Powell MB, McSharry BP, Gadola S, Cerundolo V, Borysiewicz LK, McMichael AJ, Wilkinson GW. 2000. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 287:1031. [DOI] [PubMed] [Google Scholar]
- 42.Jochum S, Moosmann A, Lang S, Hammerschmidt W, Zeidler R. 2012. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. 8:e1002704. 10.1371/journal.ppat.1002704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hurley CK, Johnson AH. 2001. HLA type of EBV-transformed human B cell lines. Curr. Protoc. Immunol..Appendix 1G 10.1002/0471142735.ima01gs30 [DOI] [PubMed] [Google Scholar]
- 44.Sun JC, Madera S, Bezman NA, Beilke JN, Kaplan MH, Lanier LL. 2012. Proinflammatory cytokine signaling required for the generation of natural killer cell memory. J. Exp. Med. 209:947–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharif-Askari E, Fawaz LM, Tran P, Ahmad A, Menezes J. 2001. Interleukin 15-mediated induction of cytotoxic effector cells capable of eliminating Epstein-Barr virus-transformed/immortalized lymphocytes in culture. J. Natl. Cancer Inst. 93:1724–1732 [DOI] [PubMed] [Google Scholar]
- 46.Borg C, Jalil A, Laderach D, Maruyama K, Wakasugi H, Charrier S, Ryffel B, Cambi A, Figdor C, Vainchenker W, Galy A, Caignard A, Zitvogel L. 2004. NK cell activation by dendritic cells (DCs) requires the formation of a synapse leading to IL-12 polarization in DCs. Blood 104:3267–3275 [DOI] [PubMed] [Google Scholar]
- 47.Castillo EF, Schluns KS. 2012. Regulating the immune system via IL-15 transpresentation. Cytokine 59:479–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berg RE, Crossley E, Murray S, Forman J. 2003. Memory CD8+ T cells provide innate immune protection against Listeria monocytogenes in the absence of cognate antigen. J. Exp. Med. 198:1583–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sandalova E, Laccabue D, Boni C, Tan AT, Fink K, Ooi EE, Chua R, Shafaeddin-Schreve B, Ferrari C, Bertoletti A. 2010. Contribution of herpesvirus specific CD8 T cells to antiviral T cell response in humans. PLoS Pathog. 6:e1001051. 10.1371/journal.ppat.1001051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Maria A, Bozzano F, Cantoni C, Moretta L. 2011. Revisiting human natural killer cell subset function revealed cytolytic CD56dimCD16+ NK cells as rapid producers of abundant IFN-γ on activation. Proc. Natl. Acad. Sci. U. S. A. 108:728–732 [DOI] [PMC free article] [PubMed] [Google Scholar]