

Abstract
The human JC polyomavirus (JCPyV) causes the rapidly progressing demyelinating disease progressive multifocal leukoencephalopathy (PML). The disease occurs most often in individuals with AIDS but also occurs in individuals receiving immunomodulatory therapies for immune-related diseases such as multiple sclerosis. JCPyV infection of host cells requires the pentasaccharide lactoseries tetrasaccharide c (LSTc) and the serotonin receptor 5-hydroxytryptamine (5-HT) receptor 5-HT2AR. While LSTc is involved in the initial attachment of virus to cells via interactions with VP1, the mechanism by which 5-HT2AR contributes to infection is not clear. To further define the roles of serotonin receptors in infection, HEK293A cells, which are poorly permissive to JCPyV, were transfected with 14 different isoforms of serotonin receptor. Only 5-HT2 receptors were found to support infection by JCPyV. None of the other 11 isoforms of serotonin receptor supported JCPyV infection. Expression of 5-HT2 receptors did not increase binding of JCPyV to cells, but this was not unexpected, given that the cells uniformly expressed the major attachment receptor, LSTc. Infection of these cells remained sensitive to inhibition with soluble LSTc, confirming that LSTc recognition is required for JCPyV infection. Virus internalization into HEK293A cells was significantly and specifically enhanced when 5HT2 receptors were expressed. Taken together, these data confirm that the carbohydrate LSTc is the attachment receptor for JCPyV and that the type 2 serotonin receptors contribute to JCPyV infection by facilitating entry.
INTRODUCTION
JC polyomavirus (JCPyV) is the etiological agent of progressive multifocal leukoencephalopathy (PML), a fatal, neurodegenerative disease. JCPyV is a common human virus for which 50 to 60% healthy adults are seropositive (1–3). JCPyV is shed in urine and can be detected in untreated wastewater, which suggests that the urogenital system is involved in persistence and transmission and that JCPyV is transmitted by a fecal-oral route (4–11). JC polyomavirus has also been detected in B lymphocytes, bone marrow, and in other reticuloendothelial tissues, such as lung, lymph node, and tonsil (12–14). On the basis of these findings, it has been proposed that JCPyV establishes a life-long persistent infection in the kidney, bone marrow, lymphoid organs, and possibly on rare occasions in the central nervous system (CNS) (14–19). During immunosuppression, the virus can traffic from sites of persistence to the CNS and infect astrocytes and oligodendrocytes. JCPyV causes cytolytic destruction of the myelin-producing oligodendrocytes and ultimately leads to PML (20–22). PML is most common in individuals with HIV or AIDS where the incidence of PML is 3 to 5% (23). Recently, PML has been reported in patients undergoing immunomodulatory therapies to treat immune-related diseases such as multiple sclerosis (MS), Crohn's disease, systemic lupus erythematosus, and rheumatoid arthritis, increasing the importance of understanding this rapidly fatal disease (24–26). Natalizumab, used to treat relapsing-remitting MS and Crohn's disease, is one of the therapies that increases the risk of PML. Natalizumab is a monoclonal antibody (MAb) directed against the very late antigen 4 (VLA-4) that directs migration and infiltration of immune cells into inflamed tissues (27). The risk of developing PML when taking natalizumab varies depending on a number of factors, including prior exposure to immunosuppressive therapies, JCPyV seropositivity, and duration of treatment (28). The risk can reach 1:100, if the duration of the treatment exceeds 25 months in individuals with a record of prior immunosuppressive therapies and JCPyV-seropositive status (available for prescribing physicians at https://medinfo.biogenidec.com). PML cases have also been reported in association with two other MAbs, efalizumab, in use for psoriasis, and rituximab, in use mainly for lymphoproliferative diseases (29). There are no specific treatments for PML other than to restore immune function. However, immune restoration is associated with an immune reconstitution inflammatory syndrome (IRIS) that can be equally devastating (30). Most deaths in individuals taking natalizumab occur during IRIS (31, 32).
The receptor motif for JCPyV attachment to host cells is the α2,6-linked glycan, lactoseries tetrasaccharide c (LSTc) (33). LSTc was crystallized in complex with JCPyV capsid protein VP1, and the specific VP1 residues that interact with LSTc were identified through structural and functional studies (33). JCPyV has also been shown to require the 5-hydroxytryptamine (5-HT) receptor 5-HT2AR to infect cells (34). Blocking antibodies directed against 5-HT2AR inhibited JCPyV infection of glial cells, and transfection of 5-HT2AR in a nonpermissive cell line conferred susceptibility to JCPyV infection (34). However, the mechanism by which 5-HT2AR contributes to JCPyV infection is not fully understood. Mutational analysis of the glycosylation sites on the N terminus of 5-HT2AR revealed that LSTc is not linked to the receptor (35). Conversely, it has been reported that JCPyV infection can occur in the absence of 5-HT2AR in human brain microvascular endothelial cells (36). Moreover, pretreatment of glial cells with the specific 5-HT2 receptor inhibitors ritanserin, ketanserin, mianserin, and mirtazapine significantly reduce JCPyV infection (34, 37–41). In the clinical setting, mirtazapine has been administered to individuals diagnosed with PML, either alone or in combination with other drugs, and in several cases, it has been shown to delay the progression of this fatal disease when given at the onset of PML symptoms (42–45). In contrast, other reports have shown no effect on degeneration (44, 46). Thus, the roles of the 5-HT receptors in JCPyV infection remained unclear. We sought to clarify the roles of these receptors especially given the availability of FDA-approved serotonin receptor agonists and antagonists that could possibly be utilized in treatment of PML.
There are 15 human 5-HT receptors that can be grouped into 7 families based on sequence similarity, 5-HT1R through 5-HT7R, and each family is divided into additional subtypes indicated by a letter (e.g., 5-HT1AR or 5-HT2BR) (47, 48). Most of the 5-HT receptors are G-protein-coupled receptors (GPCRs), with the exception of 5-HT3AR, which is an ion channel receptor (47, 49). We investigated the role of each of the 5-HT receptors in JCPyV infection to determine the receptor specificity and to define the mechanism by which the 5-HT receptors increase infection. We found that overexpression of the 5-HT2 receptors either transiently or in stable cell lines increased infection of cells by JCPyV but not by simian virus 40 (SV40). The expression of 5-HT2 receptors did not increase the binding of JCPyV to cells that uniformly expressed the attachment receptor, LSTc. Consistent with earlier work, we found that the LSTc pentasaccharide competitively inhibited infection of 5-HT2 receptor-expressing cells by JCPyV. A trypan blue (TB) quenching assay showed that the presence of 5-HT2 receptors facilitated JCPyV infectious entry into a pathway that was sensitive to inhibitors known to block infection of glial cells.
MATERIALS AND METHODS
Cells, viruses, and plasmids.
HEK293A cells (ATCC) were grown in Dulbecco's minimal essential medium (DMEM) (Mediatech, Inc.) supplemented with 10% fetal calf serum (FCS) (Atlanta Biologicals) and either penicillin-streptomycin (Mediatech, Inc.) or Geneticin G418 (Teknova). Generation of the virus strains Mad-1/SVEΔ, Mad-1/SV196 was described previously (50, 51). JCPyV lysate was purified as previously described with the addition of two steps of lipid extraction using Vertrel XF cleaning agent (DuPont). JCPyV was labeled with Alexa Fluor 633 and 405 (Invitrogen), and holotransferrin (Sigma-Aldrich) was also labeled with Alexa Fluor 405 (Invitrogen) according to the manufacturers' instructions. Simian virus 40 (SV40) strain 776 was propagated in the African green monkey kidney cell line CV-1 as previously described (34). The cDNAs for human 5-HT1AR, 5-HT1BR, 5-HT1DR, 5-HT1ER, 5-HT1FR, 5-HT2AR, 5-HT2BR, 5-HT2CR, 5-HT3R, 5-HT4R, 5-HT5R, 5-HT6R, 5-HT7AR, and 5-HT7BR in pcDNA3.1 were purchased from the Missouri S&T cDNA Resource Center (www.cdna.org). -The generation of the 5-HT2AR–YFP (YFP stands for yellow fusion protein) fusion construct was previously described (35). To generate the 5-HT2BR–YFP and 5-HT2CR–YFP fusion constructs, the cDNA for both human 5-HT2BR and 5-HT2CR in pcDNA3.1 was PCR amplified with a 5′ primer containing an XhoI site followed by the Kozak sequence and the first 16 nucleotides of the open reading frame (ORF) (HT2bR F [F stands for forward] [5′-CTCGAGCACCATGGCTCTCTCTTAC-3′]) and a 3′ primer complementary to the last 19 nucleotides of the ORF with a BamHI site in place of the stop codon (HT2bR R [R stands for reverse] [5′-TGAAGAGCAAGTTAGTTATGTAaaGGATCC-3′ {lowercase letters indicate linker nucleotides}]). To generate the 5-HT2CR–YFP fusion construct, cDNA was PCR amplified with a 5′ primer containing an XhoI site followed by the Kozak sequence and the first 14 nucleotides of the ORF (HT2cR F [5′-CTCGAGCACCATGGTGAACCTGAG-3′]) and a 3′ primer complementary to the last 19 nucleotides of the ORF with a BamHI site in place of the stop codon (HT2cR R [5′-CGAAAGGATTAGCAGTGTGaaGGATCC3′]). The PCR products were cloned into the pCRII vector using the TA Cloning kit (dual promoter) (Invitrogen), digested with XhoI and BamHI and ligated into the pEYFP-N1 (EYFP stands for enhanced YFP) vector (Clontech) to fuse YFP to the C terminus of the receptor. The directionality of cloning was confirmed by sequencing using primers designed to recognize YFP (5′→3′): TGGCACCAAAATCAACGGG and CTTCAGGGTCAGCTTGCC. Sequencing reactions were performed by Genewiz and analyzed using MacVector.
Transient transfection of HEK293A cells.
HEK293A cells were plated on 1.5-mm-thick, 18-mm-diameter glass coverslips (Electron Microscopy Sciences) in 12-well plates (Fisher Scientific) in DMEM with 10% FCS overnight without antibiotics. Cells at 85% confluence were transfected using Lipofectamine 2000 (Invitrogen) with 0.4 μg of DNA per well of human 5-HT1AR, 5-HT1BR, 5-HT1DR, 5-HT1ER, 5-HT1FR, 5-HT2AR, 5-HT2BR, 5-HT2CR, 5-HT3R, 5-HT4R, 5-HT5R, 5-HT6R, 5-HT7AR, or 5-HT7BR in pcDNA3.1 or pcDNA3.1 control vector. The cells were incubated at 37°C for 4 h, and then the medium was replaced with DMEM containing 10% FCS and penicillin-streptomycin and incubated at 37°C for 20 h.
Stable transfection of HEK293A cells.
HEK293A cells plated in a six-well plate to 80% confluence were stably transfected using Fugene HD transfection reagent (Promega) with 1 μg of 5-HT2AR, 5-HT2BR, and 5-HT2CR–YFP fusion or YFP according to the manufacturers' instructions. The cells were incubated at 37°C for 48 h in DMEM containing 10% FCS. The medium was then replaced with DMEM containing 10% FCS and 600 μg of Geneticin G418 to select only for cells transfected with the YFP constructs containing the Geneticin resistance cassette. The medium was replaced every 3 days for 1 week. The cells were then expanded in a 25-cm2 flask under selective pressure for 1 week. Cells were sorted using a FACS Aria (BD Bioscience) for a 100% transfected population of cells, and cells were maintained under selective medium.
Confocal imaging of JCPyV entry.
Confocal imaging experiments were performed using an LSM-710 laser scanning confocal microscope with a 63× objective with the pinhole set to 1 Airy unit (Carl Zeiss). Cells were plated on Fluorodishes (World Precision Instrument, Inc.) to 80% confluence. HEK293A cells that were stably transfected with 5-HT2R–YFP were chilled at 4°C for 30 min. Alexa Fluor 405-labeled JCPyV virus was added to cells for 1 h at 4°C to allow virus binding and while preventing endocytosis. Cells were washed with 1× phosphate-buffered saline (PBS) and then either fixed for 30 min in 4% paraformaldehyde (PFA) at time zero, or they were shifted to 37°C for 2 h to allow the virus to enter cells. After 2 h, cells were fixed as described above and then stored in PBS. Viral entry was analyzed using a trypan blue (TB) quenching assay as previously described (52). Briefly, Z stacks were recorded in a visual field including at least 7 cells per sample. Trypan blue was added at a dilution of 1:50, and another Z stack of the same field was recorded. Images were processed and merged to show actual quenching with Image J. Fluorescence intensity was measured with Image J, and data were expressed as percentages of protected fluorescence. The same experiment was performed using transferrin labeled with Alexa Fluor 405 (transferrin-405) incubated with the cells at 37°C for 1 h. Images and data were processed as described above.
Indirect immunofluorescence assay of JCPyV infection.
Cells were infected with JCPyV at a multiplicity of infection (MOI) of 4 focus-forming units (FFU)/cell in incomplete DMEM at 37°C for 1 h, DMEM containing 10% FCS and G418 was added, and cells were incubated at 37°C. At 48 h postinfection, cells were washed with 1× PBS and fixed in ice-cold methanol (MeOH) at −20°C for at least 30 min. The cells were washed with 1× PBS, permeabilized with 1% Triton X-100 at room temperature (RT) for 5 min, and then blocked with 10% goat serum (MP Biomedicals) in PBS at RT for 40 min. JCPyV infection was assessed using an antibody specific for JCPyV large T antigen (TAg) (PAB962). The PAB962 hybridoma produces a monoclonal antibody (MAb) for JCPyV large T antigen that does not cross-react with simian virus 40 (SV40) large T antigen and was provided by S. S. Tevethia's laboratory (Penn State University). The cells were stained with undiluted PAB962 at 37°C for 1 h, washed with 1× PBS, then incubated with a goat anti-mouse Alexa Fluor 594 antibody (1:500) in PBS (Invitrogen), and washed with 1× PBS. Coverslips were mounted on slides using Vectashield with 4′,6′-diamidino-2-phenylindole (DAPI) (Vector Laboratories). Cells were analyzed by epifluorescence microscopy (Nikon E800), and infected cells were quantitated based on nuclear large T antigen expression at a magnification of ×20 or ×40, or cells were analyzed by epifluorescence microscopy (Zeiss Axio Vert) with a 20× objective, and images were captured using a CoolSNAP HQ2 camera (Photometrics) and Metamorph (Molecular Devices) imaging software. Images were visualized using Image J and counted with an Image J cell counter plug-in.
Treatment of cells with chlorpromazine.
Treatment with chlorpromazine (CPZ) was performed as described previously (53). Briefly, cells at 70% confluence were pretreated with chlorpromazine (Enzo Life Sciences) at 30 μM concentration for 1 h in complete medium at 37°C. The cells were then infected in the presence of the drug with JCPyV at an MOI of 4 for 2.3 h in incomplete DMEM at 37°C. The cells were washed with 1× PBS and warmed DMEM with anti-JCPyV serum at 1:1,000 dilution, and 10% FCS and G418 were added. The cells were stained for TAg after 48 h as described above.
Flow cytometry.
Cells were washed with 1× PBS and removed from plates using a cell stripper (Cellgro). The cells were pelleted, washed, and incubated in 100 μl of 1× PBS with 2.5 μg of JCPyV labeled with Alexa Fluor 633 (JCPyV-633) for 2 h on ice with occasional agitation. The cells were pelleted and washed with 1× PBS twice, then fixed in 1% paraformaldehyde (PFA), and analyzed for virus binding using a BD FACSCalibur (BD Bioscience). Compensation controls were set up for both Alexa Fluor 633 and 514 fluorophores. Data were analyzed using FlowJo software (Tree Star, Inc.). Biotinylated Polyporus squamosus lectin (PSL) (EY Laboratories) was incubated with cells for 1 h on ice with occasional agitation. The cells were pelleted and washed twice with 1× PBS and incubated with streptavidin labeled with Alexa Fluor 633 (streptavidin-633) on ice for 1 h. Samples were washed, fixed, and analyzed for binding as described above.
LSTc inhibition assay.
JCPyV at an MOI of 4 was pretreated with 5 mM LSTb or LSTc (V Labs, Inc.) (diluted in sterile distilled H2O [diH2O]) in 25 μl of incomplete medium on ice for 1 h. HEK293A cells stably transfected with 5-HT2R–YFP were plated to 80% confluence in 96-well plates and prechilled at 4°C for 30 min. JCPyV-LST complexes were added to cells and incubated at 4°C for 1 h. The cells were washed with 1× PBS and medium containing 10% FCS and 500 μg G418 was added, and cells were incubated at 37°C for 48 h. The cells were fixed and stained by indirect immunofluorescence as described above.
RESULTS
5-HT2AR, 5-HT2BR, and 5-HT2CR expression in HEK293A cells significantly increases susceptibility to JCPyV infection.
Previous studies from our laboratory have shown that 5-HT2AR is required for JCPyV infection (34). Therefore, we sought to determine whether other isoforms of serotonin receptor could also support JCPyV infection. Poorly permissive HEK293A cells were transiently transfected with 14 different isoforms of 5-HT receptors in a pcDNA3.1 vector. After transfection, expression levels of cells transfected with each isoform were evaluated by real-time PCR and found to be similar (data not shown). The cells were then infected with JCPyV, and infection was scored by indirect immunofluorescence for nuclear JCPyV large T antigen (TAg) (Fig. 1A). All members of the 5-HT2 family of serotonin receptors significantly increased the susceptibility of HEK293A cells to JCPyV infection, while the other 5-HT receptor (5-HTR) isoforms did not. We then established HEK293A cells stably expressing 5-HT2AR–YFP, 5-HT2BR–YFP, 5-HT2CR–YFP, and YFP alone to further analyze the roles of the 5-HT2 receptors in JCPyV infection (Fig. 1B). The expression of these receptors increased susceptibility to JCPyV but not the related polyomavirus SV40 infection, suggesting that the 5-HT2 receptors are specific facilitators of JCPyV infection (Fig. 1C). As previously shown, the fusion of the YFP tag to the C terminus did not affect viral infection (35).
Fig 1.
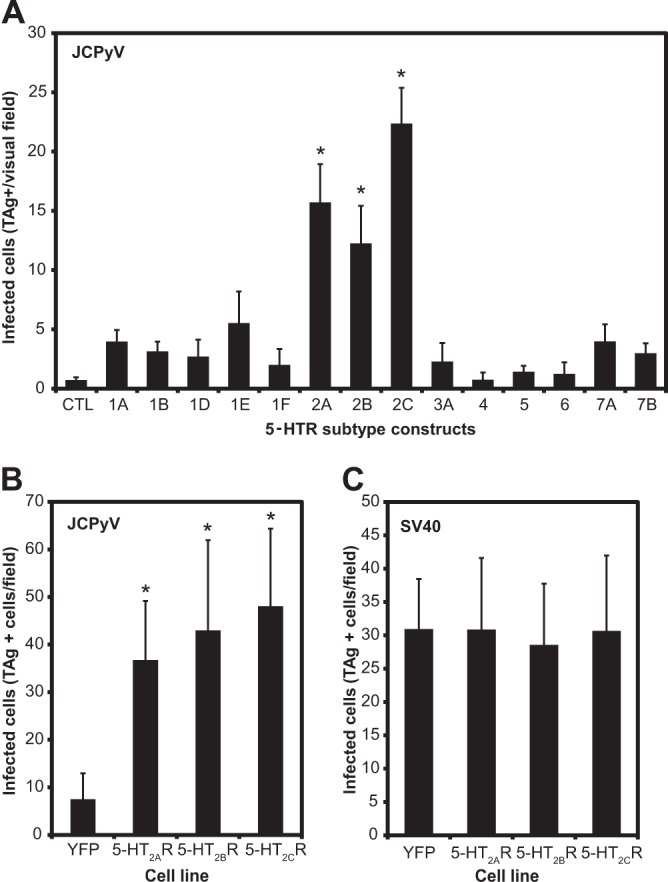
5-HT2AR, 5-HT2BR, and 5-HT2CR overexpression in HEK293A cells significantly increases susceptibility to JCPyV but not to SV40 infection. (A) HEK293A cells were transfected for 24 h with 14 different isoforms of 5-HTR cloned in the pcDNA 3.1 vector. CTL, control. (B) HEK293A cells were stably transfected with YFP, 5-HT2AR–YFP, 5-HT2BR–YFP, and 5-HT2CR–YFP and sorted for a population of cells uniformly expressing either receptor. The cells were infected with JCPyV or with SV40 at 37°C for 1 h. Samples were fixed and stained for either JCPyV TAg or SV40 TAg 48 h postinfection. Staining was detected using anti-mouse Alexa Fluor 488 antibody (A) and with Alexa Fluor 594 (B and C). The cells were analyzed by epifluorescence microscopy, and cells expressing nuclear TAg were scored as infected cells. Data represent the average number of infected cells in a visual field of view at a magnification of ×40 (A) or ×20 (B and C) for five fields of view for triplicate samples. The bars represent the averages of three independent experiments. Error bars indicate standard deviations (SD). Values that are significantly different (P < 0.05) from the value for the control (CTL or YFP) are indicated by an asterisk.
Expression of 5-HT2 receptors in HEK293A cells does not affect cell surface α-2,6-linked sialic acid expression.
JCPyV attachment to host cells requires engagement of the α2,6-linked glycan LSTc (33). To determine whether these cells expressed LSTc, we used a lectin specific for the Neu5Acα2-6Galβ found on LSTc (Polyporus squamosus lectin [PSL]). Cells expressing the 5-HT2 receptor were incubated with biotinylated PSL, and binding was detected using streptavidin labeled with Alexa Fluor 633 (streptavidin-633) by flow cytometry (Fig. 2A and B). PSL binding was equivalent in all cell types compared to the control.
Fig 2.

Biotinylated PSL (Polyporus squamosus lectin) binds equally to HEK293A cells stably expressing YFP or 5-HT2 receptors. (A) Cells were incubated with PSL for 2 h. Binding was detected by incubation with streptavidin labeled with Alexa Fluor 633 (streptavidin-633) for 1 h. Cells were fixed in 1% PFA, and binding was analyzed by flow cytometry. Cell counts are shown as a percentage of the maximum number of cells. (B) The histogram shows the average values of the mean fluorescence intensities (MFI) of three independent experiments; each replicate represents 10,000 events. Error bars indicate SD.
Expression of 5-HT2AR–YFP, 5-HT2BR–YFP, and 5-HT2CR–YFP does not increase JCPyV binding to cells.
We next tested whether the overexpression of the 5-HT2 receptors had an effect on JCPyV attachment. Purified viral particles were labeled with Alexa Fluor 633, and cells expressing 5-HT2AR–YFP, 5-HT2BR–YFP, and 5-HT2CR–YFP were incubated with the virus for 2 h, and binding was assessed by flow cytometry (Fig. 3A and B). JCPyV bound equally to 5-HT2 receptor-expressing cells compared to YFP-expressing cells, suggesting that expression of 5-HT2 receptors at the cell surface does not enhance viral attachment.
Fig 3.
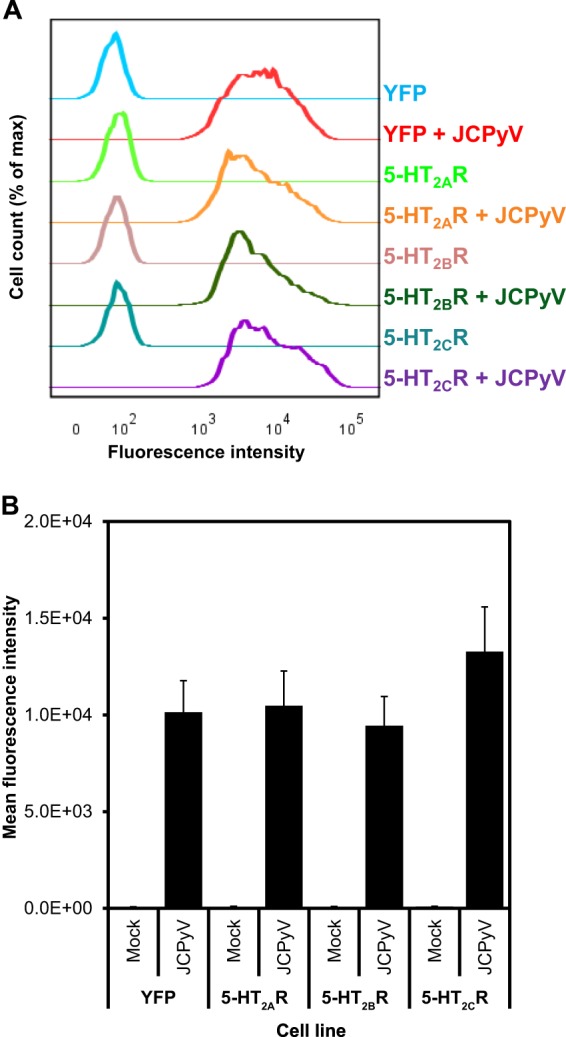
JCPyV-633 binds equally to HEK293A cells stably expressing 5-HT2 receptors compared to the control. (A) Purified JCPyV labeled with Alexa Fluor 633 was incubated with cells on ice for 2 h. Cells were fixed in 1% PFA, and binding was tested by flow cytometry. (B) The histogram shows the average values of the mean fluorescence intensity (MFIs) of three independent experiments; each replicate represents 10,000 events. Error bars indicate SD.
Pretreatment of JCPyV with LSTc pentasaccharide significantly inhibits infection.
To further define which receptors are necessary for JCPyV attachment and infection, we tested whether the inhibition of LSTc binding motifs on the surface of the JCPyV capsid would still allow JCPyV to infect cells expressing the 5-HT2 receptor. To test this, we preincubated JCPyV with 5 mM LSTc pentasaccharide and then infected 5-HT2R-expressing cells (33). For a control, we incubated JCPyV with LSTb, as it was previously shown that it is not a functional receptor motif for the virus (33) (Fig. 4). Preincubation of the virus with LSTc, but not with LSTb, significantly inhibited JCPyV infection of 5-HT2 receptor-expressing cells. Our results suggest that the virus must bind to LSTc to infect host cells, and the presence of 5-HT2 receptors is likely not sufficient to initiate the JCPyV life cycle.
Fig 4.

Pretreatment of JCPyV with LSTc significantly inhibits infection. JCPyV was preincubated on ice for 1 h with either LSTb or LSTc. The complexes were then added to cells at 4°C for 1 h. Cells were fixed and stained for TAg 48 h postinfection (PI). Data are the average numbers of infected cells in a field of view at a magnification of ×20 for five fields of view for triplicate samples. The bars are the averages of two independent experiments. Error bars indicate SD. *, P < 0.05.
5-HT2 receptors direct JCPyV entry.
Our data suggest that the expression of 5-HT2 receptors does not increase expression of LSTc at the cell surface or enhance JCPyV binding. Furthermore, inhibition of LSTc binding sites on the VP1 capsid protein blocked the ability of JCPyV to infect 5-HT2R-expressing cells. These findings suggest that JCPyV does not interact with 5-HT2 receptors at the cell surface. To determine whether 5-HT2 receptors enhance the ability of JCPyV to enter cells, we performed confocal microscopy using a trypan blue (TB) quenching assay to analyze virus entry (52). Trypan blue is a nonpermeable dye and quenches fluorescence quickly following addition to cells. Given the impermeable nature of TB, internalized fluorescent proteins are not quenched by TB. The cells were infected with JCPyV labeled with Alexa Fluor 405 (JCPyV-405) for 1 h, and two time points were analyzed: 0 and 2 h postinfection. Z-stacks of infected cells were recorded before and after the addition of trypan blue. At time zero postinfection, JCPyV-405 is bound to the outer membrane, and it is sensitive to quenching by the addition of trypan blue. When the virus is internalized by 2 h postinfection, JCPyV-405 is protected and cannot be quenched (Fig. 5A). Confocal imaging revealed that at 0 h postinfection, viral particles bound equivalently to the plasma membranes of the cells expressing 5-HT2R–YFP and control cells expressing YFP, and the particles were quenched by TB. However, at 2 h postinfection, there is an increased amount of viral particles internalized into 5-HT2R-expressing cells compared to the control cells expressing YFP, and JCPyV-405 is protected from quenching (Fig. 5A). Fluorescence intensity pre- and postquenching was measured using Image J software, and results are expressed as percentage of protected fluorescence (Fig. 5B). These data suggest that the presence of the 5-HT2 receptors facilitates more-efficient internalization of the virus, indicating that 5-HT2 receptors direct JCPyV entry. To further confirm that the increase in JCPyV entry in cells stably transfected with 5-HT2R is not due to nonspecific internalization of ligands, we performed a trypan blue quenching experiment using Alexa Fluor 405-labeled transferrin. Cells were incubated with transferrin-405 at 37°C for 1 h, fixed, and analyzed for transferrin internalization by TB quenching (Fig. 5C). Transferrin entry in cells expressing the 5-HT2 receptor was equivalent to that of the control cells. Taken together, these results suggest that serotonin 2 receptors facilitate entry of JCPyV in a specific manner.
Fig 5.

JCPyV entry is more efficient in cells expressing 5-HT2AR–YFP, 5-HT2BR–YFP, and 5-HT2CR–YFP, and transferrin entry is not enhanced in 5-HT2R-expressing cells. (A and B) Cells were prechilled at 4°C for 30 min, and then JCPyV-405 was incubated with cells on ice for 1 h. Virus entry was analyzed at 0 and 2 h PI by confocal microscopy before and after the addition of trypan blue. (A) Representative confocal micrograph images recorded at 2 h PI are shown. (B) Quantitation of images recorded at 0 and 2 h PI is shown. (C) Cells were incubated with transferrin-405 at 37°C for 1 h, transferrin entry was analyzed, and quantitation is shown. Z stacks were recorded at a magnification of ×63, and fluorescence intensity was measured for at least five cells per visual field using Image J software. Data are expressed as a percentage of protected fluorescence, and the values are averages of three independent experiments. Error bars represent the standard errors of the means (SEM). *, P < 0.05.
Treatment of cells expressing the 5-HT2 receptor with chlorpromazine significantly inhibits JCPyV infection.
To confirm that JCPyV enters HEK293A cells expressing the 5-HT2 receptor in a manner similar to human glial cells, we used chlorpromazine (CPZ), a drug known to inhibit JCPyV entry into human glial cells (53, 54). Cells were pretreated with 30 μM CPZ for 1 h prior to infection and then infected in the presence of CPZ (Fig. 6). Treatment of 5-HT2R-expressing cells with CPZ significantly inhibited JCPyV infection.
Fig 6.
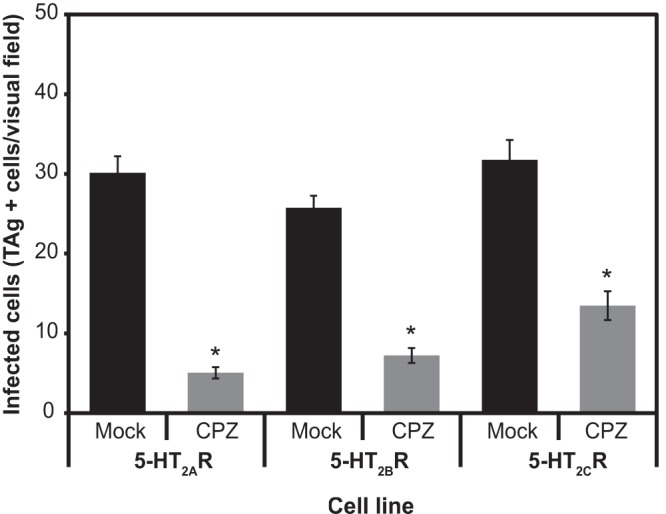
Treatment of cells expressing 5-HT2 receptors with chlorpromazine (CPZ) significantly inhibits JCPyV infection. Cells were pretreated with 30 μM CPZ for 1 h and then infected in the presence of CPZ at 37°C for 3 h. Complete medium was added with JCPyV antiserum to neutralized, not internalized, virus. Cells were fixed and stained for TAg at 48 h PI. Data are the average numbers of infected cells in a field of view at a magnification of ×20 for five fields of view for triplicate samples. The bars represent the averages of three independent experiments. Error bars indicate SEM. *, P < 0.05.
DISCUSSION
In the present study, we found that the 5-HT2 receptors 5-HT2AR, 5-HT2BR, and 5-HT2CR increase susceptibility to JCPyV but not to SV40 infection (Fig. 1). We generated cells stably expressing 5-HT2R as an in vitro model system to further explore their role in JCPyV infection. The overexpression of the 5-HT2 receptors does not affect the expression of α-2,6-linked sialic acid at the cell surface and does not affect virus attachment (Fig. 2 and 3). JCPyV infection is dependent on engagement of the functional receptor motif LSTc, and the presence of 5-HT2 receptors is not sufficient to initiate an infectious cycle (Fig. 4). These data indicate that 5-HT2 receptors function as facilitators of JCPyV entry into a pathway that is chlorpromazine sensitive (Fig. 5 and 6).
The functional receptor motif LSTc can be found on glycoproteins and on glycolipids (55). Mutation of the five glycosylation sites on the N terminus of the 5-HT2AR does not affect JCPyV infection, suggesting that the α-2,6-linked sialic acid is likely expressed on an alternate receptor (35). 5-HT2BR has only one glycosylation site in the N terminus, and mutation of the asparagine residue does not affect JCPyV infection (unpublished data). These findings lend further support to the idea that JCPyV may not directly interact with the 5-HT2 receptors at the plasma membrane. We hypothesize that JCPyV binding to LSTc causes clustering of viral particles at the plasma membrane and also of 5-HT2 receptors in adjacent regions, and this activates a specific signaling cascade leading to receptor and viral internalization. The molecule on which LSTc is located might be crucial to signal 5-HT2 receptor clustering and activation, yet this remains to be determined. Previously published data showed that JCPyV and 5-HT2A receptor colocalize both at 5 and 30 min postinfection, suggesting an early involvement of these receptors in the entry process (34). However, we cannot completely exclude the possibility that a transient interaction might also take place on the cell surface. SV40 polyomavirus is known to bind to GM1 at the plasma membrane and to use integrins to direct entry (56). Interestingly, it has recently been shown that SV40 interacts transiently with the TAM (Tyro3, Axl, and Mer) receptor Axl immediately after adsorption (57). It has been speculated that this process is crucial to initiate signaling events that promote SV40 entry and infection (57). Similarly, JCPyV may recognize LSTc, and it may transiently interact with 5-HT2R early in the entry process. This event could potentially lead to clustering, signal activation, and subsequent entry.
Interestingly, there are many other examples of viruses that use different mechanisms for attachment and entry, including mammalian reovirus and human papillomavirus type 16 (HPV16) (54, 58, 59). Mammalian reovirus directly interacts with cell surface carbohydrate receptors and junctional adhesion molecule A (JAM-A) to direct attachment, and β1-integrins direct viral entry but do not function in viral attachment (54, 60). HPV16 uses different cellular factors to mediate attachment and entry (61). Among others, HPV16 L1 capsid protein binds to the heparan sulfate proteoglycans (HSPGs) at the plasma membrane, subsequently, cyclophilin B (CyPB) mediates conformational changes to the capsid which then employs α6/β4 integrin to direct entry (62, 63). JCPyV may also require additional cellular factors to complete the mechanism of viral entry into cells to start an infectious cycle.
5-HT2AR, 5-HT2BR, and 5-HT2CR are G-protein-coupled receptors, and when recognized by a specific ligand, they are internalized in clathrin-coated vesicles and then recycled to the plasma membrane (64, 65). It is reasonable to speculate that this mechanism of internalization can be exploited nonspecifically by JCPyV, which is known to enter cells by clathrin-dependent endocytosis (53, 66). However, in the present study, we show that JCPyV uses 5-HT2 receptors specifically, since the internalization of transferrin, a process known to be directed by clathrin, is not enhanced in cells overexpressing the receptors (Fig. 5B). In support of this, antibodies specific for the D2 dopamine receptor, a G-protein-coupled receptor that functions in clathrin-dependent internalization, have no effect on JCPyV infection of glial cells (34, 67, 68).
The distribution of 5-HT2 receptors in human tissues is consistent with JCPyV sites of infection and persistence (69) (unpublished data). Chapagain et al. reported that JCPyV infection can occur in human brain microvascular endothelial cells (HBMECs), which do not express the 5-HT2A receptor (36), suggesting that JCPyV infection of HBMECs may require additional or unique receptors. However, it is possible that primary human brain microvascular endothelial cells have different levels of 5-HT2 receptors based on individualized host factors such as 5-HT levels or use of serotonin-selective reuptake inhibitors (SSRIs). Further, these cells were not tested for the expression of the 5-HT2B and 5-HT2C receptors (36). Given that all members of the 5-HT2 receptor family can support JCPyV infection, it is possible that the 5-HT receptors serve a redundant role in JCPyV infection, or they may functionally cooperate to facilitate JCPyV entry. In fact, HEK293A cells express a very low level of these receptors (data not shown) and have been previously used as model to study 5-HT2AR by exogenous overexpression (35, 64).
PML is a severely debilitating disease, and if left untreated, it is rapidly fatal. In addition, there is no antiviral drug targeting JCPyV which makes PML and PML-IRIS devastating (30). Therefore, it is crucial to investigate pharmacological approaches to prevent PML or to treat JCPyV infection. The present study provides new insights on three different serotonin receptors which are potential drug targets. In fact, several 5-HT2 receptor inhibitors, both selective and nonselective, are approved for use by the FDA and commonly used to treat neurological disorders (70, 71). To our knowledge, only mirtazapine has been administered in patients with PML, often with positive outcomes in delaying the progression of the disease (42–45, 72), but in other cases, no effect was observed (44, 46). The use of mirtazapine is most successful if administered early at the onset of PML symptoms (44). In the present study, we found that 5-HT2 receptors act early in the viral life cycle, facilitating viral entry. Our data are consistent with more-positive outcomes in early administration of mirtazapine in individuals with PML. However, clinical studies are needed to assess the efficacy of mirtazapine in PML patients. Understanding exactly how JCPyV enters and starts an infectious cycle is crucial in order to develop pharmacological agents to prevent or treat PML. The present study provides new insights into the roles of 5-HT2 receptors in JCPyV infection and serves as a platform for the development of future treatments for PML.
ACKNOWLEDGMENTS
We thank members of the Atwood laboratory for critical discussion and review of the manuscript.
Work in our laboratory was supported by P01NS065719 (W.J.A.), R01CA071878 (W.J.A.), R01NS043097 (W.J.A.), and by Ruth L. Kirschstein National Research Service Awards F32NS070687 (C.D.S.N) and F32NS064870 (M.S.M) from the National Institute of Neurological Disorders and Stroke. Confocal microscopy analysis was completed in the Leduc Bioimaging Facility at Brown University. Transfected cells were sorted in the Flow Cytometry and Sorting Facility at Brown University. Some analyses were performed in the Center for Genomics and Proteomics at Brown University that is supported by P30RR031153 (W.J.A.).
Footnotes
Published ahead of print 2 October 2013
REFERENCES
- 1.Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C, Gosert R, Hirsch HH. 2009. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J. Infect. Dis. 199:837–846 [DOI] [PubMed] [Google Scholar]
- 2.Viscidi RP, Rollison DE, Sondak VK, Silver B, Messina JL, Giuliano AR, Fulp W, Ajidahun A, Rivanera D. 2011. Age-specific seroprevalence of Merkel cell polyomavirus, BK virus, and JC virus. Clin. Vaccine Immunol. 18:1737–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang H, Wang M, Tsai RT, Lin HS, Huan JS, Wang WC, Chang D. 2002. High incidence of JC viruria in JC-seropositive older individuals. J. Neurovirol. 8:447–451 [DOI] [PubMed] [Google Scholar]
- 4.Arthur RR, Dagostin S, Shah KV. 1989. Detection of BK virus and JC virus in urine and brain tissue by the polymerase chain reaction. J. Clin. Microbiol. 27:1174–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coleman DV, Wolfendale MR, Daniel RA, Dhanjal NK, Gardner SD, Gibson PE, Field AM. 1980. A prospective study of human polyomavirus infection in pregnancy. J. Infect. Dis. 142:1–8 [DOI] [PubMed] [Google Scholar]
- 6.Flaegstad T, Sundsfjord A, Arthur RR, Pedersen M, Traavik T, Subramani S. 1991. Amplification and sequencing of the control regions of BK and JC virus from human urine by polymerase chain reaction. Virology 180:553–560 [DOI] [PubMed] [Google Scholar]
- 7.Myers C, Frisque RJ, Arthur RR. 1989. Direct isolation and characterization of JC virus from urine samples of renal and bone marrow transplant patients. J. Virol. 63:4445–4449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Markowitz RB, Thompson HC, Mueller JF, Cohen JA, Dynan WS. 1993. Incidence of BK virus and JC virus viruria in human immunodeficiency virus-infected and -uninfected subjects. J. Infect. Dis. 167:13–20 [DOI] [PubMed] [Google Scholar]
- 9.McQuaig SM, Scott TM, Lukasik JO, Paul JH, Harwood VJ. 2009. Quantification of human polyomaviruses JC virus and BK virus by TaqMan quantitative PCR and comparison to other water quality indicators in water and fecal samples. Appl. Environ. Microbiol. 75:3379–3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamza IA, Jurzik L, Stang A, Sure K, Uberla K, Wilhelm M. 2009. Detection of human viruses in rivers of a densly-populated area in Germany using a virus adsorption elution method optimized for PCR analyses. Water Res. 43:2657–2668 [DOI] [PubMed] [Google Scholar]
- 11.Ahmed W, Wan C, Goonetilleke A, Gardner T. 2010. Evaluating sewage-associated JCV and BKV polyomaviruses for sourcing human fecal pollution in a coastal river in Southeast Queensland, Australia. J. Environ. Qual. 39:1743–1750 [DOI] [PubMed] [Google Scholar]
- 12.Houff SA, Major EO, Katz DA, Kufta CV, Sever JL, Pittaluga S, Roberts JR, Gitt J, Saini N, Lux W. 1988. Involvement of JC virus-infected mononuclear cells from the bone marrow and spleen in the pathogenesis of progressive multifocal leukoencephalopathy. N. Engl. J. Med. 318:301–305 [DOI] [PubMed] [Google Scholar]
- 13.Major EO, Amemiya K, Elder G, Houff SA. 1990. Glial cells of the human developing brain and B cells of the immune system share a common DNA binding factor for recognition of the regulatory sequences of the human polyomavirus, JCV. J. Neurosci. Res. 27:461–471 [DOI] [PubMed] [Google Scholar]
- 14.Monaco MC, Atwood WJ, Gravell M, Tornatore CS, Major EO. 1996. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: implications for viral latency. J. Virol. 70:7004–7012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berger JR, Miller CS, Mootoor Y, Avdiushko SA, Kryscio RJ, Zhu H. 2006. JC virus detection in bodily fluids: clues to transmission. Clin. Infect. Dis. 43:e9–e12 [DOI] [PubMed] [Google Scholar]
- 16.Monaco MC, Shin J, Major EO. 1998. JC virus infection in cells from lymphoid tissue. Dev. Biol. Stand. 94:115–122 [PubMed] [Google Scholar]
- 17.Chapagain ML, Nerurkar VR. 2010. Human polyomavirus JC (JCV) infection of human B lymphocytes: a possible mechanism for JCV transmigration across the blood-brain barrier. J. Infect. Dis. 202:184–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan CS, Ellis LC, Wuthrich C, Ngo L, Broge TA, Jr, Saint-Aubyn J, Miller JS, Koralnik IJ. 2010. JC virus latency in the brain and extraneural organs of patients with and without progressive multifocal leukoencephalopathy. J. Virol. 84:9200–9209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez-Liz G, Del Valle L, Gentilella A, Croul S, Khalili K. 2008. Detection of JC virus DNA fragments but not proteins in normal brain tissue. Ann. Neurol. 64:379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Padgett BL, Walker DL, ZuRhein GM, Eckroade RJ, Dessel BH. 1971. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet i:1257–1260 [DOI] [PubMed] [Google Scholar]
- 21.Budka H, Shah KV. 1983. Papovavirus antigens in paraffin sections of PML brains. Prog. Clin. Biol. Res. 105:299–309 [PubMed] [Google Scholar]
- 22.Berger JR, Khalili K. 2011. The pathogenesis of progressive multifocal leukoencephalopathy. Discov. Med. 12:495–503 [PubMed] [Google Scholar]
- 23.Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, Major EO. 2012. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin. Microbiol. Rev. 25:471–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Major EO. 2010. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu. Rev. Med. 61:35–47 [DOI] [PubMed] [Google Scholar]
- 25.Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, Laubach J, Bawn SD, Gordon LI, Winter JN, Furman RR, Vose JM, Zelenetz AD, Mamtani R, Raisch DW, Dorshimer GW, Rosen ST, Muro K, Gottardi-Littell NR, Talley RL, Sartor O, Green D, Major EO, Bennett CL. 2009. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood 113:4834–4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleinschmidt-DeMasters BK, Tyler KL. 2005. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N. Engl. J. Med. 353:369–374 [DOI] [PubMed] [Google Scholar]
- 27.Engelhardt B, Kappos L. 2008. Natalizumab: targeting alpha4-integrins in multiple sclerosis. Neurodegener. Dis. 5:16–22 [DOI] [PubMed] [Google Scholar]
- 28.Berger JR, Houff SA, Gurwell J, Vega N, Miller CS, Danaher RJ. 2013. JC virus antibody status underestimates infection rates. Ann. Neurol. 74:84–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steiner I, Berger JR. 2012. Update on progressive multifocal leukoencephalopathy. Curr. Neurol. Neurosci. Rep. 12:680–686 [DOI] [PubMed] [Google Scholar]
- 30.Johnson T, Nath A. 2011. Immune reconstitution inflammatory syndrome and the central nervous system. Curr. Opin. Neurol. 24:284–290 [DOI] [PubMed] [Google Scholar]
- 31.Berger JR. 2011. The clinical features of PML. Cleve. Clin. J. Med. 78(Suppl 2):S8–S12 [DOI] [PubMed] [Google Scholar]
- 32.Brew BJ, Davies NW, Cinque P, Clifford DB, Nath A. 2010. Progressive multifocal leukoencephalopathy and other forms of JC virus disease. Nat. Rev. Neurol. 6:667–679 [DOI] [PubMed] [Google Scholar]
- 33.Neu U, Maginnis MS, Palma AS, Stroh LJ, Nelson CD, Feizi T, Atwood WJ, Stehle T. 2010. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 8:309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elphick GF, Querbes W, Jordan JA, Gee GV, Eash S, Manley K, Dugan A, Stanifer M, Bhatnagar A, Kroeze WK, Roth BL, Atwood WJ. 2004. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science 306:1380–1383 [DOI] [PubMed] [Google Scholar]
- 35.Maginnis MS, Haley SA, Gee GV, Atwood WJ. 2010. Role of N-linked glycosylation of the 5-HT2A receptor in JC virus infection. J. Virol. 84:9677–9684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chapagain ML, Verma S, Mercier F, Yanagihara R, Nerurkar VR. 2007. Polyomavirus JC infects human brain microvascular endothelial cells independent of serotonin receptor 2A. Virology 364:55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Hara BA, Atwood WJ. 2008. Interferon beta1-a and selective anti-5HT(2a) receptor antagonists inhibit infection of human glial cells by JC virus. Virus Res. 132:97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Croom KF, Perry CM, Plosker GL. 2009. Mirtazapine: a review of its use in major depression and other psychiatric disorders. CNS Drugs 23:427–452 [DOI] [PubMed] [Google Scholar]
- 39.Cervantes-Duran C, Pineda-Farias JB, Bravo-Hernandez M, Quinonez-Bastidas GN, Vidal-Cantu GC, Barragan-Iglesias P, Granados-Soto V. 3 December 2012. Evidence for the participation of peripheral 5-HT(2A), 5-HT(2B), and 5-HT(2C) receptors in formalin-induced secondary mechanical allodynia and hyperalgesia. Neuroscience pii:S0306-4522(12)01158-X. [Epub ahead of print.] 10.1016/j.neuroscience.2012.11.047 [DOI] [PubMed] [Google Scholar]
- 40.Peroutka SJ. 1994. Molecular biology of serotonin (5-HT) receptors. Synapse 18:241–260 [DOI] [PubMed] [Google Scholar]
- 41.Korstanje C, Sprenkels R, Doods HN, Hugtenburg JG, Boddeke E, Batink HD, Thoolen MJ, Van Zwieten PA. 1986. Characterization of flufylline, fluprofylline, ritanserin, butanserin and R 56413 with respect to in-vivo alpha 1-,alpha 2- and 5-HT2-receptor antagonism and in-vitro affinity for alpha 1-,alpha 2- and 5-HT2-receptors: comparison with ketanserin. J. Pharm. Pharmacol. 38:374–379 [DOI] [PubMed] [Google Scholar]
- 42.Park JH, Ryoo S, Noh HJ, Seo JM, Kang HH, Shin JS, Seo SW, Na DL. 2011. Dual therapy with cidofovir and mirtazapine for progressive multifocal leukoencephalopathy in a sarcoidosis patient. Case Rep. Neurol. 3:258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lanzafame M, Ferrari S, Lattuada E, Corsini F, Deganello R, Vento S, Concia E. 2009. Mirtazapine in an HIV-1 infected patient with progressive multifocal leukoencephalopathy. Infez. Med. 17:35–37 [PubMed] [Google Scholar]
- 44.Cettomai D, McArthur JC. 2009. Mirtazapine use in human immunodeficiency virus-infected patients with progressive multifocal leukoencephalopathy. Arch. Neurol. 66:255–258 [DOI] [PubMed] [Google Scholar]
- 45.Verma S, Cikurel K, Koralnik IJ, Morgello S, Cunningham-Rundles C, Weinstein ZR, Bergmann C, Simpson DM. 2007. Mirtazapine in progressive multifocal leukoencephalopathy associated with polycythemia vera. J. Infect. Dis. 196:709–711 [DOI] [PubMed] [Google Scholar]
- 46.Iannetta M, Bellizzi A, Lo Menzo S, Anzivino E, D'Abramo A, Oliva A, D'Agostino C, d'Ettorre G, Pietropaolo V, Vullo V, Ciardi MR. 2013. HIV-associated progressive multifocal leukoencephalopathy: longitudinal study of JC virus non-coding control region rearrangements and host immunity. J. Neurovirol. 19:274–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bockaert J, Claeysen S, Becamel C, Dumuis A, Marin P. 2006. Neuronal 5-HT metabotropic receptors: fine-tuning of their structure, signaling, and roles in synaptic modulation. Cell Tissue Res. 326:553–572 [DOI] [PubMed] [Google Scholar]
- 48.Hoyer D, Martin G. 1997. 5-HT receptor classification and nomenclature: towards a harmonization with the human genome. Neuropharmacology 36:419–428 [DOI] [PubMed] [Google Scholar]
- 49.Miyake A, Mochizuki S, Takemoto Y, Akuzawa S. 1995. Molecular cloning of human 5-hydroxytryptamine3 receptor: heterogeneity in distribution and function among species. Mol. Pharmacol. 48:407–416 [PubMed] [Google Scholar]
- 50.Liu CK, Hope AP, Atwood WJ. 1998. The human polyomavirus, JCV, does not share receptor specificity with SV40 on human glial cells. J. Neurovirol. 4:49–58 [DOI] [PubMed] [Google Scholar]
- 51.Liu CK, Atwood WJ. 2001. Propagation and assay of the JC virus. Methods Mol. Biol. 165:9–17 [DOI] [PubMed] [Google Scholar]
- 52.Engel S, Heger T, Mancini R, Herzog F, Kartenbeck J, Hayer A, Helenius A. 2011. Role of endosomes in simian virus 40 entry and infection. J. Virol. 85:4198–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pho MT, Ashok A, Atwood WJ. 2000. JC virus enters human glial cells by clathrin-dependent receptor-mediated endocytosis. J. Virol. 74:2288–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maginnis MS, Forrest JC, Kopecky-Bromberg SA, Dickeson SK, Santoro SA, Zutter MM, Nemerow GR, Bergelson JM, Dermody TS. 2006. Beta1 integrin mediates internalization of mammalian reovirus. J. Virol. 80:2760–2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569 [DOI] [PubMed] [Google Scholar]
- 56.Stergiou L, Bauer M, Mair W, Bausch-Fluck D, Drayman N, Wollscheid B, Oppenheim A, Pelkmans L. 2013. Integrin-mediated signaling induced by simian virus 40 leads to transient uncoupling of cortical actin and the plasma membrane. PLoS One 8:e55799. 10.1371/journal.pone.0055799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Drayman N, Glick Y, Ben-Nun-Shaul O, Zer H, Zlotnick A, Gerber D, Schueler-Furman O, Oppenheim A. 2013. Pathogens use structural mimicry of native host ligands as a mechanism for host receptor engagement. Cell Host Microbe 14:63–73 [DOI] [PubMed] [Google Scholar]
- 58.Sourisseau M, Michta ML, Zony C, Israelow B, Hopcraft SE, Narbus CM, Parra Martin A, Evans MJ. 2013. Temporal analysis of hepatitis C virus cell entry with occludin directed blocking antibodies. PLoS Pathog. 9:e1003244. 10.1371/journal.ppat.1003244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benedicto I, Molina-Jimenez F, Bartosch B, Cosset FL, Lavillette D, Prieto J, Moreno-Otero R, Valenzuela-Fernandez A, Aldabe R, Lopez-Cabrera M, Majano PL. 2009. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J. Virol. 83:8012–8020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441–451 [DOI] [PubMed] [Google Scholar]
- 61.Raff AB, Woodham AW, Raff LM, Skeate JG, Yan L, Da Silva DM, Schelhaas M, Kast WM. 2013. The evolving field of human papillomavirus receptor research: a review of binding and entry. J. Virol. 87:6062–6072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 75:1565–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Selinka HC, Giroglou T, Nowak T, Christensen ND, Sapp M. 2003. Further evidence that papillomavirus capsids exist in two distinct conformations. J. Virol. 77:12961–12967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raote I, Bhattacharya A, Panicker MM. 2007. Serotonin 2A (5-HT2A) receptor function: ligand-dependent mechanisms and pathways, p 105–132 In A Chattopadhyay. (ed), Serotonin receptors in neurobiology. CRC Press, Boca Raton, FL: [PubMed] [Google Scholar]
- 65.Raote I, Bhattacharyya S, Panicker MM. 2013. Functional selectivity in serotonin receptor 2A (5-HT2A) endocytosis, recycling, and phosphorylation. Mol. Pharmacol. 83:42–50 [DOI] [PubMed] [Google Scholar]
- 66.Querbes W, Benmerah A, Tosoni D, Di Fiore PP, Atwood WJ. 2004. A JC virus-induced signal is required for infection of glial cells by a clathrin- and eps15-dependent pathway. J. Virol. 78:250–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paspalas CD, Rakic P, Goldman-Rakic PS. 2006. Internalization of D2 dopamine receptors is clathrin-dependent and select to dendro-axonic appositions in primate prefrontal cortex. Eur. J. Neurosci. 24:1395–1403 [DOI] [PubMed] [Google Scholar]
- 68.Kabbani N, Levenson R. 2007. A proteomic approach to receptor signaling: molecular mechanisms and therapeutic implications derived from discovery of the dopamine D2 receptor signalplex. Eur. J. Pharmacol. 572:83–93 [DOI] [PubMed] [Google Scholar]
- 69.Bonhaus DW, Bach C, DeSouza A, Salazar FH, Matsuoka BD, Zuppan P, Chan HW, Eglen RM. 1995. The pharmacology and distribution of human 5-hydroxytryptamine2B (5-HT2B) receptor gene products: comparison with 5-HT2A and 5-HT2C receptors. Br. J. Pharmacol. 115:622–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Monti JM. 2011. Serotonin control of sleep-wake behavior. Sleep Med. Rev. 15:269–281 [DOI] [PubMed] [Google Scholar]
- 71.Singh SP, Singh V, Kar N, Chan K. 2010. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br. J. Psych. 197:174–179 [DOI] [PubMed] [Google Scholar]
- 72.Lasso M, Ceron I. 2012. Mirtazapine and antiretroviral therapy in the treatment of progressive multifocal leukoencephalopathy associated with HIV-1 infection: report of a case and review of literature. Rev. Chilena Infectol. 29:217–220 [In Spanish.] [DOI] [PubMed] [Google Scholar]